User login
Rigosertib falls short for high-risk myelodysplastic syndromes after failure on azacitidine or decitabine
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.

The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
THE LANCET ONCOLOGY
Key clinical point: Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
Major finding: Median overall survival was 8.2 months (95% CI, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group.
Data source: An open-label, randomized controlled trial involving 299 patients at 74 medical centers.
Disclosures: The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society..

Rigosertib falls short for high-risk myelodysplastic syndromes after failure on azacitidine or decitabine
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
A randomized phase III trial of rigosertib (NCT 02562443) is now underway in specific subgroups of high-risk patients, including patients with very high risk on the basis of the Revised International Prognostic Scoring System criteria, to determine whether the drug may benefit specific patient subgroups, according to Dr. Guillermo Garcia-Manero of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
The ONTIME study (NCT01241500) was an open-label, randomized controlled trial at 74 medical centers in the US and Europe. Patients with refractory anemia with excess blasts ([RAEB]-1, RAEB-2, RAEB-t, or chronic myelomonocytic leukemia) were enrolled based on local site assessment and treatment failure with a hypomethylating drug in the past 2 years.
Patients were randomly assigned on a 2:1 basis to receive rigosertib or best supportive care with or without low-dose cytarabine. Randomization was stratified by pretreatment bone marrow blast percentage. The 199 patients given rigosertib received 1,800 mg per 24 hours via a 72-hour continuous intravenous infusion administered every other week. Another 100 patients were assigned to best supportive care.
Median follow-up was 19.5 months. Median overall survival was 8.2 months (95% confidence interval, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group (hazard ratio, 0.87; 95% CI, 0.67-1.14; P = 0.33), the researchers reported (Lancet Oncol. 2016;17(4):496-508. doi: 10.1016/S1470-2045(16)00009-7).
The most common grade 3 or higher adverse events were anemia (18% of 184 patients in the rigosertib group and 8% of 91 patients in the best supportive care group), thrombocytopenia (19% vs 7%), neutropenia (17% vs. 8%), febrile neutropenia (12% vs 11%], and pneumonia (12% vs 11%). Adverse events led to death in 22% of 184 patients in the rigosertib group and 33% of 91 patients in the best supportive care group; three deaths were attributed to rigosertib treatment.
The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society.
On Twitter @maryjodales
THE LANCET ONCOLOGY
Key clinical point: Rigosertib failed to extend overall survival beyond that seen with best supportive care in a trial of patients who had myelodysplastic syndrome with excess blasts after failure of azacitidine or decitabine treatment.
Major finding: Median overall survival was 8.2 months (95% CI, 6.1-10.1) in the rigosertib group and 5.9 months (95% CI, 4.1-9.3) in the best supportive care group.
Data source: An open-label, randomized controlled trial involving 299 patients at 74 medical centers.
Disclosures: The study was funded by Onconova Therapeutics and the Leukemia and Lymphoma Society..

FDG-PET guides need for eBEACOPP in advanced Hodgkin’s
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).

In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).
In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
Using FDG-PET (fluorodeoxyglucose positron emission tomography) imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to a more aggressive eBEACOPP regimen, according to the results of the Southwest Oncology Group (SWOG) S0816 study.
In this large U.S. trial of PET scanning to guide treatment approach in people with high-risk stage II or stage III-IV Hodgkin’s lymphoma, progression-free survival at 2 years for those with early interim positive PET scans was 64%, which is much higher than the expected progression-free survival of 15%-30%, according to Dr. Oliver Press, a SWOG member at Fred Hutchinson Cancer Research Center and the lead author of study, which was published ahead of print in the Journal of Clinical Oncology (2016 April 11. doi: 10.1200/JCO.2015.63.1119).
In addition, just 20% of patients in the trial were exposed to eBEACOPP, which usually results in infertility, can cause sustained heart or lung damage, and increases the risk of secondary cancers.
Researchers recruited 358 Hodgkin’s patients to the trial and were able to evaluate 331 of them. All trial volunteers were given two rounds of standard ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) chemotherapy, followed by a PET scan. If the scan was negative, patients received four more cycles of ABVD. If the scan was positive, with a Deauville score of 4-5, they were advised to switch to eBEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone), a seven-drug combination used in Europe. Of 60 patients with positive interim PET scans, 11 patients declined to switch, and 49 switched as planned to six cycles of eBEACOPP.
With a median follow-up of nearly 40 months, the Kaplan-Meier estimate for 2-year overall survival was 98%, and the 2-year estimate for progression-free survival was 79%. In the subset of patients who had positive PET scans after two cycles of ABVD, the 2-year estimate for progression-free survival was 64%, more than double the expected remission rate.
At least seven phase II and III cooperative group studies are underway testing this approach in advanced-stage Hodgkin’s lymphoma, the researchers wrote. “We hope that in the future, molecular biomarker studies at initial diagnosis, or the combination of biomarkers and molecular imaging, may define patients who require more intense therapy with eBEACOPP or other novel targeted drugs with greater accuracy than is achievable with current technology.”
The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.
On Twitter @maryjodales
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Using FDG-PET imaging to gauge treatment response after the first two rounds of ABVD therapy helps to determine which patients with advanced Hodgkin’s lymphoma should be switched to the eBEACOPP regimen.
Major finding: Progression-free survival at 2 years for those with early interim positive PET scans was 64%; the historical progression-free survival for this group is 15%-30%.
Data source: Evaluations of 331 patients in the Southwest Oncology Group S0816 study.
Disclosures: The study was funded by the National Cancer Institute, the David and Patricia Giuliani Family Foundation, the Lymphoma Foundation, the Adam Spector Fund for Hodgkin Research, and the Ernest & Jeanette Dicker Charitable Foundation.

CUDC-907 enters phase II for relapsed or refractory lymphoma and multiple myeloma
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
Another oral, small-molecule therapy called CUDC-907 is emerging from phase I testing as a treatment option for patients with relapsed or refractory lymphoma and multiple myeloma.
The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule, according to Dr. Anas Younes of Memorial Sloan Kettering Cancer Center, New York, and his colleagues. A dose-expansion trial of this dose is ongoing, and the drug appears to be useful in particular for patients with refractory and relapsed diffuse large B-cell lymphoma.
The researchers tested CUDC-907, which is designed to inhibit histone deacetylase and PI3K enzyme pathways, for overall safety and response in 44 patients at four cancer centers. All participants had lymphoma or multiple myeloma and were refractory to treatment or had relapsed after two or more previous regimens.
The 44 participants were sequentially assigned to 21-day cycles of CUDC-907: 10 to once daily, 12 to twice weekly, 15 to three times weekly, and 7 to daily for 5 days followed by a 2-day break. The maximum tolerated doses were 60 mg for the once-daily schedule, 150 mg for the twice-weekly schedule, 150 mg for the three-times-weekly schedule, and 60 mg for the 5-on/2-off schedule. At data cutoff, 37 of the 44 patients had discontinued CUDC-907 because of disease progression, Dr. Younes and his associates reported in a study published online (Lancet Oncol. 2016 Mar 31. doi: 10.1016/S1470-2045(15)00584-7).
Four dose-limiting toxicities occurred in 3 of 40 evaluable patients. Grade 3 or worse adverse events occurred in 19 of 44 patients: 9 had thrombocytopenia, 3 had neutropenia, 3 had hyperglycemia. Adverse events led to dose reductions in six patients and treatment discontinuation in seven.
Of 37 response-evaluable patients, two had complete responses and three had partial responses. All five were seen in the subgroup of nine patients with diffuse large B-cell lymphoma, and three occurred in the five patients with transformed follicular disease. The 21 patients with stable disease included those with diffuse large B-cell lymphoma, Hodgkin’s lymphoma, and multiple myeloma. This ongoing trial is registered at ClinicalTrials.gov as NCT01742988.
The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
On Twitter @maryjodales
FROM THE LANCET ONCOLOGY
Key clinical point: The CUDC-907 dose to be used in phase II studies will be 60 mg on a 5-days-on/2-days-off dosing schedule.
Major finding: Two complete responses and three partial responses were seen in the subgroup of nine patients with diffuse large B-cell lymphoma; three occurred in the five patients with transformed follicular disease.
Data source: A dose-escalation study involving 44 patients at four cancer centers.
Disclosures: The study was sponsored by Curis, the maker of CUDC-907, and the Leukemia and Lymphoma Society. Five of the 15 investigators are employees of Curis.
FDA approves venetoclax for CLL with 17p deletion
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
Venetoclax has been approved for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a 17p deletion and who have been treated with a least one prior therapy, the Food and Drug Administration has announced.
The drug will be marketed as Venclexta, and is indicated for daily use after detection of a 17p deletion is confirmed through the use of the FDA-approved companion diagnostic test, the Vysis CLL FISH probe kit. A 17p deletion occurs in about 10% of patients with untreated CLL and in about 20% of patients with relapsed CLL. Venetoclax targets the B-cell lymphoma 2 (BCL-2) protein, according to the FDA press release.
“Up to half of people whose CLL progressed have 17p deletion,” Dr. Sandra Horning, chief medical officer and head of Global Product Development for Genentech, said in a press release issued by the company. Venclexta will be marketed by AbbVie and Genentech USA. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular.
“For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option,” Dr. Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research, said in a press release issued by the FDA.
The approval was based on a clinical trial of 106 patients who had CLL and 17p deletions and who had received at least one prior therapy. Trial participants took oral venetoclax daily, beginning with a 20 mg dose that was increased over a 5-week period to 400 mg. A complete or partial remission of CLL occurred in 80% of trial participants. Data on venetoclax also was presented at the annual meeting of the American Society of Hematology.
The most common side effects of venetoclax include neutropenia, diarrhea, nausea, anemia, upper respiratory tract infection, thrombocytopenia, and fatigue.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. Venclexta also received orphan drug designation.
On Twitter @maryjodales
In myelodysplastic syndrome, improved tool for predicting death after HCT
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.

“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.
“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
A new risk-stratification tool goes one better than the standard tools used to predict survival in those undergoing allogeneic hematopoietic cell transplantation (allo HCT) for myelodysplastic syndrome, based on a study published online April 4 in the Journal of Clinical Oncology.
The concordance index for the new risk-stratification tool was modestly better at 0.575, compared with 0.538 for the standard International Prognostic Scoring System (IPSS) and 0.554 for the revised IPSS (IPSS-R), according to Dr. Brian C. Shaffer of Memorial Sloan Kettering Cancer Center, New York, and his colleagues who participate in the Center for International Blood and Marrow Transplant Research (CIBMTR) network.
“The proposed system generally agrees with the IPSS-R in the very high–risk subcategory; however, a significant portion of patients in high- and very high–risk IPSS-R groups were represented in the low- and intermediate-risk proposed scoring subcategories. The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system,” the researchers wrote.
Further, the “scoring system uses readily available clinical data and can be calculated quickly, facilitating patient consultation with respect to allo HCT, and may also be used to identify high-risk populations where interventions such as post–allo HCT maintenance therapies may be of benefit,” they wrote (J Clin Oncol. 2016 April 4. doi: 10.1200/JCO.2015.65.0515).
The data were obtained from the CIBMTR, a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
The researchers applied the prognostic tool to 2,133 patients with MDS undergoing HLA-matched (n = 1,728) or -mismatched (n = 405) allo HCT. Factors prognostic of mortality were identified in a training subset (n = 1,151) of the HLA-matched cohort. A weighted score using these factors was then assigned to the validation cohort of 577 remaining patients undergoing HLA-matched allo HCT as well as to patients undergoing HLA-mismatched allo HCT. The training data set was used to develop a prognostic scoring system, and the validation data set was used to assess the prognostic ability of the scoring system, the researchers noted.
In the scoring system, 1 point was assigned for the following factors: Blood blasts greater than 3%, platelet levels of 50 × 109/L or less at transplantation, Karnofsky performance status less than 90%, comprehensive cytogenetic risk score of poor or very poor, and age 30-49 years. Two points were assigned for monosomal karyotype and age 50 years or older.
Based on the scoring system, 3-year overall survival after transplantation was 71% in patients with scores of 1 point, 49% with scores of 2-3, 41% with scores of 4-5, and 25% with scores of 6 or more. Increasing score was predictive of increased relapse and treatment-related mortality in the HLA-matched set and of relapse in the HLA-mismatched cohort.
To develop the scoring system, the researchers used a model that weighed patient age; sex; and Karnofsky performance status; disease stage at transplantation; comprehensive cytogenetic risk status; bone marrow and peripheral blood blast percentages; hemoglobin, neutrophil, and platelet counts at diagnosis and pretransplantation; lactate dehydrogenase at transplantation; pretransplantation therapy (hypomethylating agents, chemotherapy, neither, or both); time from diagnosis to transplantation; year of transplantation; conditioning regimen and regimen intensity (myeloablative v reduced intensity); donor–recipient sex match or mismatch; graft-versus-host disease prophylaxis; graft type (bone marrow vs. peripheral blood); presence of secondary myelodysplastic syndrome; and unrelated donor vs. related donor.
There were no significant differences in overall survival at 1, 3, and 5 years or in the 3-year incidences of relapse and treatment-related mortality in the training subset and the validation cohort.
Data on somatic mutations have become relevant in myelodysplastic syndrome prognostication and were missing from this analysis, the researchers wrote. “The next generation of prognostic tools will need to account for this information.”
Dr. Shaffer had no relevant financial disclosures.
On Twitter @maryjodales
FROM JCO
Key clinical point: A portion of patients with myelodysplastic syndrome in high- and very high–risk groups of the revised International Prognostic Scoring System (IPSS-R) were represented in the low- and intermediate-risk groups of the proposed scoring subcategories.
Major finding: The 3-year survival in patients classified as high risk with the IPSS-R was 75%; it was 57% in those classified as low or intermediate risk with the proposed system.
Data source: The Center for International Blood and Marrow Transplant Research (CIBMTR), a combined research program of the Medical College of Wisconsin and the National Marrow Donor Program. The CIBMTR comprises a voluntary network of more than 450 transplantation centers worldwide that contribute data on consecutive allo and autologous HCTs to a centralized statistical center.
Disclosures: Dr. Shaffer had no relevant financial disclosures.
OTX015 dose for lymphoma narrowed in phase 1 study
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
As a single agent for use in patients with lymphoma, an acceptable once-daily dose of OTX015 appears to be 80 mg on a 14 days on, 7 days off schedule, the results of a phase 1 study indicate.
The small-molecule inhibitor, which inhibits binding of bromodomain and exterminal proteins to acetylated histones, was associated with acceptable toxicity and efficacy in this regimen. The investigational drug is now being tested in expansion cohorts on a schedule of 14 days every 3 weeks, a regimen projected to allow for recovery from the drug’s toxic effects, Dr. Sandy Amorin of Hôpital Saint Louis, Paris, and associates reported.
The drug also is being evaluated in patients with acute leukemias.
Adults with nonleukemia hematologic malignancies that progressed on standard therapies participated in the open-label study, which was conducted at seven university hospital centers in Europe. Oral OTX015 was given once a day at one of five doses (10 mg, 20 mg, 40 mg, 80 mg, and 120 mg). The 3 + 3 study design permitted evaluation of alternative administration schedules. The primary endpoint was dose-limiting toxicity in the first treatment cycle (21 days). Secondary objectives were to evaluate safety, pharmacokinetics, and preliminary clinical activity of OTX015. The study is ongoing and is registered with ClinicalTrials.gov, number NCT01713582.
The study included 33 patients with lymphoma and 12 with myeloma; patients’ median age was 66 years, and they had received a median of four lines of prior therapy. No dose-limiting toxicities were seen in three patients given doses as high as 80 mg once a day. However, grade 4 thrombocytopenia occurred in five of six patients on a 21-day schedule of 40 mg twice a day. No patient tolerated various schedules of 120 mg once a day (Lancet Haematol. 2016;3[4]:e196-204).
The researchers then examined the 80 mg once a day dose on a continuous basis in four patients, two of whom developed grade 4 thrombocytopenia. In light of these and other toxicities, a regimen was proposed of 80 mg once a day on a schedule of 14 days on, 7 days off.
Thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. Other grade 3-4 events were infrequent. Anemia was seen in 41, and neutropenia in 23.
Of three patients with diffuse large B-cell lymphoma, two had complete responses at 120 mg once a day, and one had a partial response at 80 mg once a day. Six additional patients, two with diffuse large B-cell lymphoma and four with indolent lymphomas, had evidence of clinical activity, but did not meet the criteria for an objective response.
The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
On Twitter @maryjodales
FROM THE LANCET HAEMATOLOGY
Key clinical point: For lymphoma patients, a regimen has been determined for the small-molecule inhibitor OTX015 that was associated with acceptable toxicity and efficacy.
Major finding: On a regimen of 80 mg once a day on a schedule of 14 days on, 7 days off, thrombocytopenia affected 43 of 45 patients, and 26 of them had grade 3-4 events. However, other grade 3-4 events were infrequent.
Data source: The open-label study NCT01713582 was conducted at seven university hospital centers in Europe.
Disclosures: The study was funded by the developers of OTX015, Oncoethix GmbH, a wholly owned subsidiary of Merck Sharp & Dohme.
LMWH doesn’t reduce late pregnancy loss in women with thrombophilias
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
Prophylactic-dose low molecular weight heparin (LMWH), with or without aspirin, did not reduce the risk of pregnancy loss in women with inherited thrombophilia and a history of prior late or recurrent early pregnancy loss, based on a meta-analysis of randomized, controlled trials.
“To our knowledge, this is the largest study published to date that evaluates LMWH in women with inherited thrombophilia and previous pregnancy loss,” Dr. Leslie Skeith, of the University of Ottawa, and her colleagues wrote in Blood (2016 Mar 31;127[13]:1650-55). A recent Cochrane Review (2014 Jul 4;7:CD004734) similarly found no difference in live birth rates in women with or without inherited thrombophilia treated with LMWH and aspirin, compared with women given no treatment. Additionally, the Effects of Aspirin in Gestation and Reproduction [EAGeR] trial found no difference in live birth rates in women with previous pregnancy loss given aspirin or placebo (Lancet. 2014;384[9937]:29-36).
Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis. Four trials included an LMWH-plus-aspirin arm, and five trials included an LMWH-only arm. The control groups included four trials with an aspirin arm, and five trials with a placebo or no-treatment arm. The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
As there is the potential for adverse side effects and significant cost with LMWH, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia (Grade 1B, strong recommendation with moderate-quality evidence) and suggest against LMWH to prevent recurrent pregnancy loss in women with inherited thrombophilia and prior recurrent early (less than 10 weeks) pregnancy loss. (Grade 2B, weak recommendation with moderate-quality evidence.)
Given that the analysis included just 66 women with thrombophilia and prior recurrent early pregnancy loss, the researchers could not exclude a beneficial effect of LMWH in this subgroup. An ongoing randomized controlled trial, ALIFE2 (Netherlands Trial Registration Identifier: NTR3361), “is evaluating LMWH in women with inherited thrombophilia and a history of two or more miscarriages and/or intrauterine fetal death, which we hope will provide definitive answers to this question,” the researchers wrote.
They also suggest not testing for inherited thrombophilia in women with prior late or recurrent early pregnancy loss (Grade 2B, weak recommendation with moderate-quality evidence).
The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
On Twitter @maryjodales
FROM BLOOD
Key clinical point: Given the potential for adverse side effects and significant cost, the researchers advise against the use of LMWH to prevent recurrent and prior late pregnancy loss (greater than 10 weeks gestation) in women with inherited thrombophilia.
Major finding: The data indicated no difference in the treated groups and controls (relative risk of 0.81; 95% confidence interval, 0.55-1.19; P = .28).
Data source: Based on a literature search, 8 publications and 483 participants met eligibility criteria as randomized, controlled trials for the meta-analysis.
Disclosures: The study was supported by a series of university and institutional investigator research awards. Dr. Skeith received a Thrombosis Canada CanVECTOR Research Fellowship award.
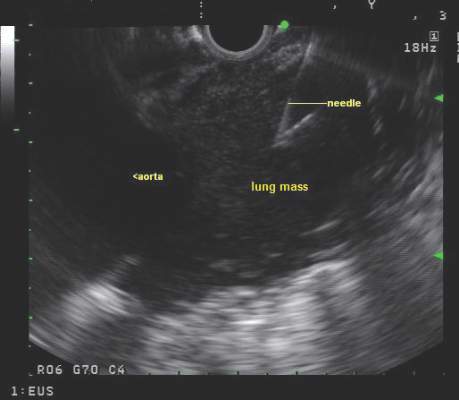
CHEST issues guidelines on EBUS-TBNA
Endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) has been recommended for diagnosis of suspected sarcoidosis or suspected tuberculosis with adenopathy and may be used as an initial diagnostic test for suspected lymphoma, according to guidelines issued by CHEST (the American College of Chest Physicians).
The guidelines, which are primarily focused on technical aspects of EBUS-TBNA, also advise obtaining additional samples for the purpose of molecular analysis in patients who undergo the procedure for the diagnosis or staging of non–small cell lung cancer.
The guidelines are based on a systematic review and critical analysis of the literature by an expert panel chaired by Dr. Momen M. Wahidi of Duke University Medical Center, Durham, N.C. Of the 12 guideline statements by the panel, 7 were graded evidence-based recommendations and 5 were ungraded consensus-based statements.
The guideline (Chest. 2016 Mar;149[3]:816-35) has been endorsed by the American Association of Bronchology and Interventional Pulmonology, American Association for Thoracic Surgery, Canadian Thoracic Society, European Association for Bronchology and Interventional Pulmonology, and Society of Thoracic Surgeons.
Use of EBUS-TBNA for diagnosis in patients with suspected sarcoidosis with mediastinal and/or hilar adenopathy is ranked Grade 1C (strong recommendation, low-quality evidence, benefits outweigh risks). The guideline writers concluded that EBUS-TBNA provides safe and minimally invasive access to the mediastinal and hilar lymph nodes with a pooled diagnostic accuracy of 79.1%. They qualified, however, that it may be difficult to use EBUS-TBNA to obtain adequate tissue from fibrotic lymph nodes and conventional bronchoscopic techniques such as transbronchial lung biopsy and endobronchial biopsy may be needed in selected patients.
One systematic review and meta-analysis study included 15 studies with a total of 553 patients with sarcoidosis. The diagnostic yield of EBUS-TBNA ranged from 54% to 93%, with the pooled diagnostic accuracy of 79% (95% confidence interval, 71-86). Ten additional studies including 573 combined patients were identified through updated searches of the systematic review, and led to a pooled diagnostic accuracy of 78.2%.
Similarly, a Grade 1C recommendation was made for using EBUS-TBNA for diagnosis in when other modalities are not diagnostic in patients with suspected tuberculosis with mediastinal and/or hilar adenopathy who require lymph node sampling. However, “it must be noted that no single study assessed the role of EBUS-TBNA for the diagnosis of TB [tuberculosis] as the primary outcome measure,” they wrote. Various techniques are available for the diagnosis of TB and should be incorporated during the diagnostic evaluation.
In patients with suspected lymphoma, EBUS-TBNA is an acceptable initial, minimally invasive diagnostic test, the guideline writers said in an Ungraded Consensus-Based Statement.
In some conditions, minimally invasive EBUS-TBNA may be preferred over surgical intervention. Repeat mediastinoscopy or surgical biopsy after treatment for relapsed lymphoma can be challenging, for example, with a lower diagnostic yield and higher complication rate.
Because treatment regimens for both non-Hodgkin and Hodgkin lymphoma depend on the specific subtype and histologic grade, a definitive diagnosis of lymphoma requires the evaluation of cell morphology, immunophenotype, and the overall architecture of the tissue. Reed-Sternberg cells, diagnostic of Hodgkin lymphoma, are usually scarce in cytologic aspirates, and it often is impossible to evaluate the overall background architecture. Currently available EBUS-TBNA needles provide only cytologic specimens, with reported high discordance between cytologic specimens and histologic specimens.
In five retrospective studies with a total of 212 patients undergoing EBUS-TBNA for suspected lymphoma, the pooled diagnostic accuracy was 68.7%, and there was heterogeneity across studies in the proportion of patients with de novo lymphoma and relapsed lymphoma. Higher diagnostic yield was noted for relapsed lymphoma, compared with de novo lymphoma. Also, the two studies with the highest yield included cases as diagnostic, even when additional tissue sampling was necessary to subclassify the lymphoma for clinical management.
The panel gave a weak recommendation (Grade 2C) based on low-quality evidence to their conclusion that moderate or deep sedation is acceptable for EBUS-TBNA, based on three studies. Moderate sedation allows patients to respond purposefully to verbal commands while maintaining a functional airway, spontaneous ventilation, and cardiovascular function. In deep sedation, patients cannot be easily aroused but respond purposefully to repeated or painful stimulation and may have compromised airway function and spontaneous ventilation; cardiovascular function usually is maintained.
In one retrospective multivariable analysis of 309 patients at two centers, deep sedation had a statistically significant benefit on diagnostic yield. In a prospective randomized, controlled study of 149 patients at a single center with a single operator, there was no difference in diagnostic yield for moderate and deep sedation. However, fewer patients in the moderate sedation group were able to complete the procedure, compared with the deep-sedation group. Patient comfort and satisfaction were similar for the two sedation groups, and no patients had major complications or needed escalation of care.
In terms of diagnostic yield, there was insufficient evidence to recommend for or against using an artificial airway when inserting the EBUS bronchoscope, the authors said. Reported practice is scattered and is largely based on expert opinion, operator comfort, sedation type, and institutional standards.
The placement of the endotracheal tube may block the ultrasonographic view of the higher paratracheal lymph nodes (lymph node stations 1, 2R, 2L, and 3P) and should be avoided if one of these lymph nodes is the sampling target of the procedure, they advised.
If using a transoral artificial airway, a bite block should be considered to protect the bronchoscope from bite damage; this approach is recommended independent of the depth of sedation. A minimum size of 8.0 should be used if placing an endotracheal tube for EBUS-TBNA to accommodate the scope diameter and leave room for gas exchange.
In an Ungraded Consensus-Based Statement, the guideline authors said that ultrasonographic features, such as size, shape, border, heterogeneity, central hilar structure, and necrosis can be used to predict malignant and benign diagnoses, but tissue samples still should be obtained to confirm a diagnosis.
Nine studies provided analysis of the characteristics of lymph nodes that predict malignancy during EBUS; however, the ultrasonographic features assessed were not the same in each study or they had varying definitions of what constituted “abnormal.” As a result, the ultrasonographic predictors of malignancy in lymph nodes are not reliable enough to forgo biopsy to obtain a definitive tissue diagnosis. However, the ultrasound features can be useful to guide sampling from lymph nodes most likely to be malignant.
A round shape, distinct margins, heterogeneous echogenicity, and a central necrosis sign were independently predictive of malignancy in one multivariate analysis that included more than 1,000 lymph nodes in nearly 500 patients. Furthermore, when all four factors were absent, 96% of the lymph nodes were benign.
In three additional studies, size criteria had conflicting results; one found size was not a reliable indicator, two others found that larger lymph nodes are more likely to harbor metastases. These studies also confirmed that round-shaped lymph nodes were more likely malignant than were triangular or draping lymph nodes. The measures used to define size may have caused the inconsistencies.
In a study that examined vascular image patterns within lymph nodes as a way to predict malignancy, nodes were considered malignant if vessel involvement increased in the node to rich flow with more than four vessels (grades 2 and 3) with a sensitivity of 87.7% and a specificity of 69.6%, suggesting that increased vascularity assessed by using power/color Doppler mode ultrasound is useful in predicting malignancy.
Two studies have assessed ultrasound features of lymph nodes in patients with sarcoidosis. In the first, lymph nodes with homogeneous echogenicity and a germinal center were more likely to indicate sarcoidosis than lung cancer. In the second, coagulation necrosis and heterogeneous echogenicity within lymph nodes were more likely to be present in tuberculosis as opposed to sarcoidosis.
In another Ungraded Consensus-Based Statement, the guideline authors said tissue sampling may be performed either with or without suction. In cases in which EBUS-TBNA is being performed with suction and the samples obtained are bloody, operators should consider switching to the use of no suction at the same sampling site. If intranodal blood vessels are visualized on EBUS imaging with or without Doppler imaging, operators should also consider obtaining samples without suction.
Needle choice should be determined by the operator, and the use of either a 21- or 22-gauge needle are acceptable options based on five trials comparing needle sizes, the authors said in a Grade 1C recommendation. No data are available on the use of 25-gauge needles.
“Future studies should investigate if ... smaller or more flexible needles would improve sampling at station 4L (known for its slightly angulated location) or if smaller needles would result in less blood contamination when sampling more vascular nodes. Studies should also examine if a particular needle size should be used depending on how the specimens are being processed (histopathology vs. cytopathology) and if needle size can affect the diagnosis of diseases that are more difficult to diagnose by EBUS-TBNA, such as lymphoma,” the authors wrote.
In the absence of rapid on-site evaluation (ROSE), the authors advised a minimum of three separate needle passes per sampling site in patients suspected of having lung cancer. The recommendation is an Ungraded Consensus-Based Statement.
Just one study of 102 patients with potentially operable non–small cell lung cancer and mediastinal adenopathy has examined the number of needle passes per sampling site. The results indicated optimal diagnostic values are reached after three passes. Each pass typically includes 5-15 agitations of the needle within the target site.
Sample adequacy was 90.1% after the first pass, 98.1% after two passes, and reached 100% after three passes. The sensitivity for differentiating malignant from benign lymph node stations was 69.8%, 83.7%, 95.3%, and 95.3% for one, two, three, and four passes, respectively.
No data exist regarding the number of needle passes required to obtain a sufficient diagnostic yield for lymphoma or nonmalignant diseases of the mediastinum.
In a Grade 1C recommendation, the authors said that tissue sampling can be performed with or without rapid on-site evaluation. ROSE does not affect the diagnostic yield in EBUS-TBNA procedures, but it may decrease the number of punctures and reduce the need for additional staging and diagnostic procedures. ROSE may be beneficial in judging the quantity of available malignant cells when testing for molecular markers is planned in patients with advanced adenocarcinoma of the lung.
In another Grade 1C recommendation, the authors said that patients undergoing EBUS-TBNA for the diagnosis or staging of suspected or known non–small cell lung cancer should have additional samples obtained for molecular analysis.
Molecular marker testing is necessary for tailoring chemotherapy to the cancer characteristics of each individual patient. The current data are insufficient to identify the number of passes needed to obtain adequate tissue for molecular marker testing, but it is strongly suggested that additional samples, over the proposed diagnostic threshold of three passes are recommended.
The guideline authors found insufficient quality of evidence to support any route of bronchoscope insertion for EBUS-TBNA over another. Translating the experience and literature from conventional flexible bronchoscopy given the size and rigidity of the EBUS bronchoscope distal tip, as well as the limited bronchoscopic view, is difficult, according to the guideline writers.
They noted that no studies were found that addressed the use of saline-filled balloons to overcome poor contact between the ultrasound probe and the bronchial wall. Although the saline-filled balloon can enhance image acquisition, it is unclear whether that translates into a better diagnostic yield, thus, no recommendations or suggestions can be made.
In a Grade 2C recommendation, they advised that low-fidelity inanimate mechanical airway models and high-fidelity computer-based electronic simulation be incorporated into training. In the three studies that compared conventional EBUS-TBNA training and simulation-based training incorporating either a low- or high-fidelity simulation tool, the same level of training could be acquired via conventional or simulation-based training; however, simulation-based training minimizes novice operators’ practice on patients.
In an Ungraded Consensus-Based Statement, the guideline authors advised that validated EBUS skills assessment tests be used to objectively assess skill level, but added that “none of the included simulation studies examined whether the skills demonstrated on a simulation assessment are transferred to an improvement in clinical skills as performed in patients.”
On Twitter @maryjodales
Endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) has been recommended for diagnosis of suspected sarcoidosis or suspected tuberculosis with adenopathy and may be used as an initial diagnostic test for suspected lymphoma, according to guidelines issued by CHEST (the American College of Chest Physicians).
The guidelines, which are primarily focused on technical aspects of EBUS-TBNA, also advise obtaining additional samples for the purpose of molecular analysis in patients who undergo the procedure for the diagnosis or staging of non–small cell lung cancer.
The guidelines are based on a systematic review and critical analysis of the literature by an expert panel chaired by Dr. Momen M. Wahidi of Duke University Medical Center, Durham, N.C. Of the 12 guideline statements by the panel, 7 were graded evidence-based recommendations and 5 were ungraded consensus-based statements.
The guideline (Chest. 2016 Mar;149[3]:816-35) has been endorsed by the American Association of Bronchology and Interventional Pulmonology, American Association for Thoracic Surgery, Canadian Thoracic Society, European Association for Bronchology and Interventional Pulmonology, and Society of Thoracic Surgeons.
Use of EBUS-TBNA for diagnosis in patients with suspected sarcoidosis with mediastinal and/or hilar adenopathy is ranked Grade 1C (strong recommendation, low-quality evidence, benefits outweigh risks). The guideline writers concluded that EBUS-TBNA provides safe and minimally invasive access to the mediastinal and hilar lymph nodes with a pooled diagnostic accuracy of 79.1%. They qualified, however, that it may be difficult to use EBUS-TBNA to obtain adequate tissue from fibrotic lymph nodes and conventional bronchoscopic techniques such as transbronchial lung biopsy and endobronchial biopsy may be needed in selected patients.
One systematic review and meta-analysis study included 15 studies with a total of 553 patients with sarcoidosis. The diagnostic yield of EBUS-TBNA ranged from 54% to 93%, with the pooled diagnostic accuracy of 79% (95% confidence interval, 71-86). Ten additional studies including 573 combined patients were identified through updated searches of the systematic review, and led to a pooled diagnostic accuracy of 78.2%.
Similarly, a Grade 1C recommendation was made for using EBUS-TBNA for diagnosis in when other modalities are not diagnostic in patients with suspected tuberculosis with mediastinal and/or hilar adenopathy who require lymph node sampling. However, “it must be noted that no single study assessed the role of EBUS-TBNA for the diagnosis of TB [tuberculosis] as the primary outcome measure,” they wrote. Various techniques are available for the diagnosis of TB and should be incorporated during the diagnostic evaluation.
In patients with suspected lymphoma, EBUS-TBNA is an acceptable initial, minimally invasive diagnostic test, the guideline writers said in an Ungraded Consensus-Based Statement.
In some conditions, minimally invasive EBUS-TBNA may be preferred over surgical intervention. Repeat mediastinoscopy or surgical biopsy after treatment for relapsed lymphoma can be challenging, for example, with a lower diagnostic yield and higher complication rate.
Because treatment regimens for both non-Hodgkin and Hodgkin lymphoma depend on the specific subtype and histologic grade, a definitive diagnosis of lymphoma requires the evaluation of cell morphology, immunophenotype, and the overall architecture of the tissue. Reed-Sternberg cells, diagnostic of Hodgkin lymphoma, are usually scarce in cytologic aspirates, and it often is impossible to evaluate the overall background architecture. Currently available EBUS-TBNA needles provide only cytologic specimens, with reported high discordance between cytologic specimens and histologic specimens.
In five retrospective studies with a total of 212 patients undergoing EBUS-TBNA for suspected lymphoma, the pooled diagnostic accuracy was 68.7%, and there was heterogeneity across studies in the proportion of patients with de novo lymphoma and relapsed lymphoma. Higher diagnostic yield was noted for relapsed lymphoma, compared with de novo lymphoma. Also, the two studies with the highest yield included cases as diagnostic, even when additional tissue sampling was necessary to subclassify the lymphoma for clinical management.
The panel gave a weak recommendation (Grade 2C) based on low-quality evidence to their conclusion that moderate or deep sedation is acceptable for EBUS-TBNA, based on three studies. Moderate sedation allows patients to respond purposefully to verbal commands while maintaining a functional airway, spontaneous ventilation, and cardiovascular function. In deep sedation, patients cannot be easily aroused but respond purposefully to repeated or painful stimulation and may have compromised airway function and spontaneous ventilation; cardiovascular function usually is maintained.
In one retrospective multivariable analysis of 309 patients at two centers, deep sedation had a statistically significant benefit on diagnostic yield. In a prospective randomized, controlled study of 149 patients at a single center with a single operator, there was no difference in diagnostic yield for moderate and deep sedation. However, fewer patients in the moderate sedation group were able to complete the procedure, compared with the deep-sedation group. Patient comfort and satisfaction were similar for the two sedation groups, and no patients had major complications or needed escalation of care.
In terms of diagnostic yield, there was insufficient evidence to recommend for or against using an artificial airway when inserting the EBUS bronchoscope, the authors said. Reported practice is scattered and is largely based on expert opinion, operator comfort, sedation type, and institutional standards.
The placement of the endotracheal tube may block the ultrasonographic view of the higher paratracheal lymph nodes (lymph node stations 1, 2R, 2L, and 3P) and should be avoided if one of these lymph nodes is the sampling target of the procedure, they advised.
If using a transoral artificial airway, a bite block should be considered to protect the bronchoscope from bite damage; this approach is recommended independent of the depth of sedation. A minimum size of 8.0 should be used if placing an endotracheal tube for EBUS-TBNA to accommodate the scope diameter and leave room for gas exchange.
In an Ungraded Consensus-Based Statement, the guideline authors said that ultrasonographic features, such as size, shape, border, heterogeneity, central hilar structure, and necrosis can be used to predict malignant and benign diagnoses, but tissue samples still should be obtained to confirm a diagnosis.
Nine studies provided analysis of the characteristics of lymph nodes that predict malignancy during EBUS; however, the ultrasonographic features assessed were not the same in each study or they had varying definitions of what constituted “abnormal.” As a result, the ultrasonographic predictors of malignancy in lymph nodes are not reliable enough to forgo biopsy to obtain a definitive tissue diagnosis. However, the ultrasound features can be useful to guide sampling from lymph nodes most likely to be malignant.
A round shape, distinct margins, heterogeneous echogenicity, and a central necrosis sign were independently predictive of malignancy in one multivariate analysis that included more than 1,000 lymph nodes in nearly 500 patients. Furthermore, when all four factors were absent, 96% of the lymph nodes were benign.
In three additional studies, size criteria had conflicting results; one found size was not a reliable indicator, two others found that larger lymph nodes are more likely to harbor metastases. These studies also confirmed that round-shaped lymph nodes were more likely malignant than were triangular or draping lymph nodes. The measures used to define size may have caused the inconsistencies.
In a study that examined vascular image patterns within lymph nodes as a way to predict malignancy, nodes were considered malignant if vessel involvement increased in the node to rich flow with more than four vessels (grades 2 and 3) with a sensitivity of 87.7% and a specificity of 69.6%, suggesting that increased vascularity assessed by using power/color Doppler mode ultrasound is useful in predicting malignancy.
Two studies have assessed ultrasound features of lymph nodes in patients with sarcoidosis. In the first, lymph nodes with homogeneous echogenicity and a germinal center were more likely to indicate sarcoidosis than lung cancer. In the second, coagulation necrosis and heterogeneous echogenicity within lymph nodes were more likely to be present in tuberculosis as opposed to sarcoidosis.
In another Ungraded Consensus-Based Statement, the guideline authors said tissue sampling may be performed either with or without suction. In cases in which EBUS-TBNA is being performed with suction and the samples obtained are bloody, operators should consider switching to the use of no suction at the same sampling site. If intranodal blood vessels are visualized on EBUS imaging with or without Doppler imaging, operators should also consider obtaining samples without suction.
Needle choice should be determined by the operator, and the use of either a 21- or 22-gauge needle are acceptable options based on five trials comparing needle sizes, the authors said in a Grade 1C recommendation. No data are available on the use of 25-gauge needles.
“Future studies should investigate if ... smaller or more flexible needles would improve sampling at station 4L (known for its slightly angulated location) or if smaller needles would result in less blood contamination when sampling more vascular nodes. Studies should also examine if a particular needle size should be used depending on how the specimens are being processed (histopathology vs. cytopathology) and if needle size can affect the diagnosis of diseases that are more difficult to diagnose by EBUS-TBNA, such as lymphoma,” the authors wrote.
In the absence of rapid on-site evaluation (ROSE), the authors advised a minimum of three separate needle passes per sampling site in patients suspected of having lung cancer. The recommendation is an Ungraded Consensus-Based Statement.
Just one study of 102 patients with potentially operable non–small cell lung cancer and mediastinal adenopathy has examined the number of needle passes per sampling site. The results indicated optimal diagnostic values are reached after three passes. Each pass typically includes 5-15 agitations of the needle within the target site.
Sample adequacy was 90.1% after the first pass, 98.1% after two passes, and reached 100% after three passes. The sensitivity for differentiating malignant from benign lymph node stations was 69.8%, 83.7%, 95.3%, and 95.3% for one, two, three, and four passes, respectively.
No data exist regarding the number of needle passes required to obtain a sufficient diagnostic yield for lymphoma or nonmalignant diseases of the mediastinum.
In a Grade 1C recommendation, the authors said that tissue sampling can be performed with or without rapid on-site evaluation. ROSE does not affect the diagnostic yield in EBUS-TBNA procedures, but it may decrease the number of punctures and reduce the need for additional staging and diagnostic procedures. ROSE may be beneficial in judging the quantity of available malignant cells when testing for molecular markers is planned in patients with advanced adenocarcinoma of the lung.
In another Grade 1C recommendation, the authors said that patients undergoing EBUS-TBNA for the diagnosis or staging of suspected or known non–small cell lung cancer should have additional samples obtained for molecular analysis.
Molecular marker testing is necessary for tailoring chemotherapy to the cancer characteristics of each individual patient. The current data are insufficient to identify the number of passes needed to obtain adequate tissue for molecular marker testing, but it is strongly suggested that additional samples, over the proposed diagnostic threshold of three passes are recommended.
The guideline authors found insufficient quality of evidence to support any route of bronchoscope insertion for EBUS-TBNA over another. Translating the experience and literature from conventional flexible bronchoscopy given the size and rigidity of the EBUS bronchoscope distal tip, as well as the limited bronchoscopic view, is difficult, according to the guideline writers.
They noted that no studies were found that addressed the use of saline-filled balloons to overcome poor contact between the ultrasound probe and the bronchial wall. Although the saline-filled balloon can enhance image acquisition, it is unclear whether that translates into a better diagnostic yield, thus, no recommendations or suggestions can be made.
In a Grade 2C recommendation, they advised that low-fidelity inanimate mechanical airway models and high-fidelity computer-based electronic simulation be incorporated into training. In the three studies that compared conventional EBUS-TBNA training and simulation-based training incorporating either a low- or high-fidelity simulation tool, the same level of training could be acquired via conventional or simulation-based training; however, simulation-based training minimizes novice operators’ practice on patients.
In an Ungraded Consensus-Based Statement, the guideline authors advised that validated EBUS skills assessment tests be used to objectively assess skill level, but added that “none of the included simulation studies examined whether the skills demonstrated on a simulation assessment are transferred to an improvement in clinical skills as performed in patients.”
On Twitter @maryjodales
Endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA) has been recommended for diagnosis of suspected sarcoidosis or suspected tuberculosis with adenopathy and may be used as an initial diagnostic test for suspected lymphoma, according to guidelines issued by CHEST (the American College of Chest Physicians).
The guidelines, which are primarily focused on technical aspects of EBUS-TBNA, also advise obtaining additional samples for the purpose of molecular analysis in patients who undergo the procedure for the diagnosis or staging of non–small cell lung cancer.
The guidelines are based on a systematic review and critical analysis of the literature by an expert panel chaired by Dr. Momen M. Wahidi of Duke University Medical Center, Durham, N.C. Of the 12 guideline statements by the panel, 7 were graded evidence-based recommendations and 5 were ungraded consensus-based statements.
The guideline (Chest. 2016 Mar;149[3]:816-35) has been endorsed by the American Association of Bronchology and Interventional Pulmonology, American Association for Thoracic Surgery, Canadian Thoracic Society, European Association for Bronchology and Interventional Pulmonology, and Society of Thoracic Surgeons.
Use of EBUS-TBNA for diagnosis in patients with suspected sarcoidosis with mediastinal and/or hilar adenopathy is ranked Grade 1C (strong recommendation, low-quality evidence, benefits outweigh risks). The guideline writers concluded that EBUS-TBNA provides safe and minimally invasive access to the mediastinal and hilar lymph nodes with a pooled diagnostic accuracy of 79.1%. They qualified, however, that it may be difficult to use EBUS-TBNA to obtain adequate tissue from fibrotic lymph nodes and conventional bronchoscopic techniques such as transbronchial lung biopsy and endobronchial biopsy may be needed in selected patients.
One systematic review and meta-analysis study included 15 studies with a total of 553 patients with sarcoidosis. The diagnostic yield of EBUS-TBNA ranged from 54% to 93%, with the pooled diagnostic accuracy of 79% (95% confidence interval, 71-86). Ten additional studies including 573 combined patients were identified through updated searches of the systematic review, and led to a pooled diagnostic accuracy of 78.2%.
Similarly, a Grade 1C recommendation was made for using EBUS-TBNA for diagnosis in when other modalities are not diagnostic in patients with suspected tuberculosis with mediastinal and/or hilar adenopathy who require lymph node sampling. However, “it must be noted that no single study assessed the role of EBUS-TBNA for the diagnosis of TB [tuberculosis] as the primary outcome measure,” they wrote. Various techniques are available for the diagnosis of TB and should be incorporated during the diagnostic evaluation.
In patients with suspected lymphoma, EBUS-TBNA is an acceptable initial, minimally invasive diagnostic test, the guideline writers said in an Ungraded Consensus-Based Statement.
In some conditions, minimally invasive EBUS-TBNA may be preferred over surgical intervention. Repeat mediastinoscopy or surgical biopsy after treatment for relapsed lymphoma can be challenging, for example, with a lower diagnostic yield and higher complication rate.
Because treatment regimens for both non-Hodgkin and Hodgkin lymphoma depend on the specific subtype and histologic grade, a definitive diagnosis of lymphoma requires the evaluation of cell morphology, immunophenotype, and the overall architecture of the tissue. Reed-Sternberg cells, diagnostic of Hodgkin lymphoma, are usually scarce in cytologic aspirates, and it often is impossible to evaluate the overall background architecture. Currently available EBUS-TBNA needles provide only cytologic specimens, with reported high discordance between cytologic specimens and histologic specimens.
In five retrospective studies with a total of 212 patients undergoing EBUS-TBNA for suspected lymphoma, the pooled diagnostic accuracy was 68.7%, and there was heterogeneity across studies in the proportion of patients with de novo lymphoma and relapsed lymphoma. Higher diagnostic yield was noted for relapsed lymphoma, compared with de novo lymphoma. Also, the two studies with the highest yield included cases as diagnostic, even when additional tissue sampling was necessary to subclassify the lymphoma for clinical management.
The panel gave a weak recommendation (Grade 2C) based on low-quality evidence to their conclusion that moderate or deep sedation is acceptable for EBUS-TBNA, based on three studies. Moderate sedation allows patients to respond purposefully to verbal commands while maintaining a functional airway, spontaneous ventilation, and cardiovascular function. In deep sedation, patients cannot be easily aroused but respond purposefully to repeated or painful stimulation and may have compromised airway function and spontaneous ventilation; cardiovascular function usually is maintained.
In one retrospective multivariable analysis of 309 patients at two centers, deep sedation had a statistically significant benefit on diagnostic yield. In a prospective randomized, controlled study of 149 patients at a single center with a single operator, there was no difference in diagnostic yield for moderate and deep sedation. However, fewer patients in the moderate sedation group were able to complete the procedure, compared with the deep-sedation group. Patient comfort and satisfaction were similar for the two sedation groups, and no patients had major complications or needed escalation of care.
In terms of diagnostic yield, there was insufficient evidence to recommend for or against using an artificial airway when inserting the EBUS bronchoscope, the authors said. Reported practice is scattered and is largely based on expert opinion, operator comfort, sedation type, and institutional standards.
The placement of the endotracheal tube may block the ultrasonographic view of the higher paratracheal lymph nodes (lymph node stations 1, 2R, 2L, and 3P) and should be avoided if one of these lymph nodes is the sampling target of the procedure, they advised.
If using a transoral artificial airway, a bite block should be considered to protect the bronchoscope from bite damage; this approach is recommended independent of the depth of sedation. A minimum size of 8.0 should be used if placing an endotracheal tube for EBUS-TBNA to accommodate the scope diameter and leave room for gas exchange.
In an Ungraded Consensus-Based Statement, the guideline authors said that ultrasonographic features, such as size, shape, border, heterogeneity, central hilar structure, and necrosis can be used to predict malignant and benign diagnoses, but tissue samples still should be obtained to confirm a diagnosis.
Nine studies provided analysis of the characteristics of lymph nodes that predict malignancy during EBUS; however, the ultrasonographic features assessed were not the same in each study or they had varying definitions of what constituted “abnormal.” As a result, the ultrasonographic predictors of malignancy in lymph nodes are not reliable enough to forgo biopsy to obtain a definitive tissue diagnosis. However, the ultrasound features can be useful to guide sampling from lymph nodes most likely to be malignant.
A round shape, distinct margins, heterogeneous echogenicity, and a central necrosis sign were independently predictive of malignancy in one multivariate analysis that included more than 1,000 lymph nodes in nearly 500 patients. Furthermore, when all four factors were absent, 96% of the lymph nodes were benign.
In three additional studies, size criteria had conflicting results; one found size was not a reliable indicator, two others found that larger lymph nodes are more likely to harbor metastases. These studies also confirmed that round-shaped lymph nodes were more likely malignant than were triangular or draping lymph nodes. The measures used to define size may have caused the inconsistencies.
In a study that examined vascular image patterns within lymph nodes as a way to predict malignancy, nodes were considered malignant if vessel involvement increased in the node to rich flow with more than four vessels (grades 2 and 3) with a sensitivity of 87.7% and a specificity of 69.6%, suggesting that increased vascularity assessed by using power/color Doppler mode ultrasound is useful in predicting malignancy.
Two studies have assessed ultrasound features of lymph nodes in patients with sarcoidosis. In the first, lymph nodes with homogeneous echogenicity and a germinal center were more likely to indicate sarcoidosis than lung cancer. In the second, coagulation necrosis and heterogeneous echogenicity within lymph nodes were more likely to be present in tuberculosis as opposed to sarcoidosis.
In another Ungraded Consensus-Based Statement, the guideline authors said tissue sampling may be performed either with or without suction. In cases in which EBUS-TBNA is being performed with suction and the samples obtained are bloody, operators should consider switching to the use of no suction at the same sampling site. If intranodal blood vessels are visualized on EBUS imaging with or without Doppler imaging, operators should also consider obtaining samples without suction.
Needle choice should be determined by the operator, and the use of either a 21- or 22-gauge needle are acceptable options based on five trials comparing needle sizes, the authors said in a Grade 1C recommendation. No data are available on the use of 25-gauge needles.
“Future studies should investigate if ... smaller or more flexible needles would improve sampling at station 4L (known for its slightly angulated location) or if smaller needles would result in less blood contamination when sampling more vascular nodes. Studies should also examine if a particular needle size should be used depending on how the specimens are being processed (histopathology vs. cytopathology) and if needle size can affect the diagnosis of diseases that are more difficult to diagnose by EBUS-TBNA, such as lymphoma,” the authors wrote.
In the absence of rapid on-site evaluation (ROSE), the authors advised a minimum of three separate needle passes per sampling site in patients suspected of having lung cancer. The recommendation is an Ungraded Consensus-Based Statement.
Just one study of 102 patients with potentially operable non–small cell lung cancer and mediastinal adenopathy has examined the number of needle passes per sampling site. The results indicated optimal diagnostic values are reached after three passes. Each pass typically includes 5-15 agitations of the needle within the target site.
Sample adequacy was 90.1% after the first pass, 98.1% after two passes, and reached 100% after three passes. The sensitivity for differentiating malignant from benign lymph node stations was 69.8%, 83.7%, 95.3%, and 95.3% for one, two, three, and four passes, respectively.
No data exist regarding the number of needle passes required to obtain a sufficient diagnostic yield for lymphoma or nonmalignant diseases of the mediastinum.
In a Grade 1C recommendation, the authors said that tissue sampling can be performed with or without rapid on-site evaluation. ROSE does not affect the diagnostic yield in EBUS-TBNA procedures, but it may decrease the number of punctures and reduce the need for additional staging and diagnostic procedures. ROSE may be beneficial in judging the quantity of available malignant cells when testing for molecular markers is planned in patients with advanced adenocarcinoma of the lung.
In another Grade 1C recommendation, the authors said that patients undergoing EBUS-TBNA for the diagnosis or staging of suspected or known non–small cell lung cancer should have additional samples obtained for molecular analysis.
Molecular marker testing is necessary for tailoring chemotherapy to the cancer characteristics of each individual patient. The current data are insufficient to identify the number of passes needed to obtain adequate tissue for molecular marker testing, but it is strongly suggested that additional samples, over the proposed diagnostic threshold of three passes are recommended.
The guideline authors found insufficient quality of evidence to support any route of bronchoscope insertion for EBUS-TBNA over another. Translating the experience and literature from conventional flexible bronchoscopy given the size and rigidity of the EBUS bronchoscope distal tip, as well as the limited bronchoscopic view, is difficult, according to the guideline writers.
They noted that no studies were found that addressed the use of saline-filled balloons to overcome poor contact between the ultrasound probe and the bronchial wall. Although the saline-filled balloon can enhance image acquisition, it is unclear whether that translates into a better diagnostic yield, thus, no recommendations or suggestions can be made.
In a Grade 2C recommendation, they advised that low-fidelity inanimate mechanical airway models and high-fidelity computer-based electronic simulation be incorporated into training. In the three studies that compared conventional EBUS-TBNA training and simulation-based training incorporating either a low- or high-fidelity simulation tool, the same level of training could be acquired via conventional or simulation-based training; however, simulation-based training minimizes novice operators’ practice on patients.
In an Ungraded Consensus-Based Statement, the guideline authors advised that validated EBUS skills assessment tests be used to objectively assess skill level, but added that “none of the included simulation studies examined whether the skills demonstrated on a simulation assessment are transferred to an improvement in clinical skills as performed in patients.”
On Twitter @maryjodales
FROM CHEST
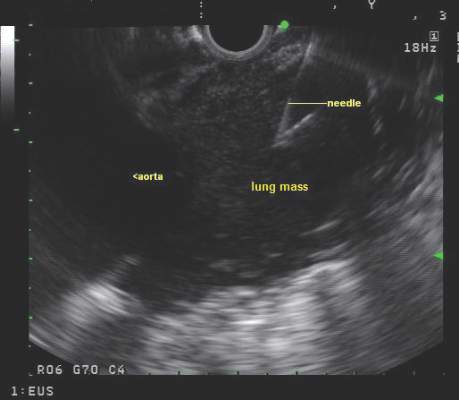
Idelalisib use halted in six combo therapy trials, FDA announces
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales
An increased rate of adverse events, including deaths, have been reported in clinical trials with idelalisib (Zydelig) in combination with other cancer medicines, the U.S. Food and Drug Administration announced.
Gilead Sciences, Inc. has confirmed that they are stopping six clinical trials in patients with chronic lymphocytic leukemia, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. The FDA is reviewing the findings of the clinical trials and will communicate new information as necessary, according to the FDA press release.
![]()
Idelalisib is not approved for previously untreated chronic lymphocytic leukemia. It is approved by the FDA for the treatment of:
• Relapsed chronic lymphocytic leukemia, in combination with rituximab, in patients for whom rituximab alone would be considered appropriate therapy due to other co-morbidities.
• Relapsed follicular B-cell non-Hodgkin lymphoma in patients who have received at least two prior systemic therapies.
• Relapsed small lymphocytic lymphoma in patients who have received at least two prior systemic therapies.
Adverse events involving idelalisib should be reported to the FDA MedWatch program, the release advised.
On Twitter @maryjodales