User login
Guidelines on asparaginase hypersensitivity and silent inactivation in ALL
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
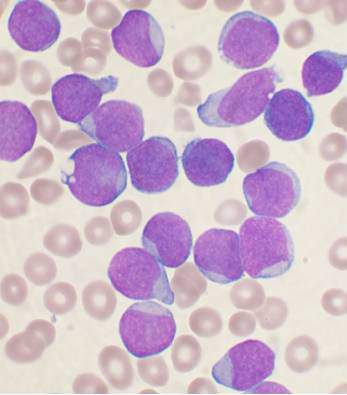
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
FROM HAEMATOLOGICA
HIV drug may overcome proteasome-inhibitor resistance in multiple myeloma
The protease inhibitor nelfinavir was used successfully to resensitize patients with proteasome inhibitor–refractory multiple myeloma for proteasome-inhibitor treatment, Dr. Christoph Driessen of Kantonsspital St. Gallen (Switzerland) and colleagues reported in Haematologica.
The researchers had hypothesized that nelfinavir would induce the unfolded protein response and would overcome proteasome-inhibitor resistance. The researchers determined a nelfinavir dose of 2,500 mg twice daily based on a dose-finding study in 12 patients with advanced hematologic malignancies.
In an exploratory extension cohort trial that followed, six patients with relapsed, bortezomib-refractory, lenalidomide-resistant myeloma were treated with 2,500 mg of nelfinavir twice daily in combination with bortezomib; three reached a partial response, two had a minor response, and one had progressive disease. All began the investigational therapy less than 2 months after progressing under therapies with bortezomib/bendamustine/dexamethasone, bortezomib/bendamustine, or bortezomib monotherapy, respectively, the researchers said (Haematologica March 2016;101:346-55).
The study protocol did not allow dexamethasone co-administration until completion of cycle three, when patients who did not achieve a minor response were allowed to co-administer 8 mg dexamethasone with bortezomib. Higher doses were excluded because of potential interactions with nelfinavir through the Cyp3A4 system.
Future research should assess whether bortezomib may be replaced by next-generation drugs like carfilzomib to avoid drug interactions and improve activity and whether nelfinavir may likewise be used in combination with novel oral proteasome inhibitors to boost their low single agent activity. “This ultimately suggests exploring the addition of HIV protease inhibitors to established combinations of proteasome inhibitors with immunomodulatory drugs, for example, in the carfilzomib/lenalidomide/dexamethasone regimen, one of the most powerful and tolerable regimens available to date for advanced multiple myeloma,” they concluded.
On Twitter @maryjodales
The protease inhibitor nelfinavir was used successfully to resensitize patients with proteasome inhibitor–refractory multiple myeloma for proteasome-inhibitor treatment, Dr. Christoph Driessen of Kantonsspital St. Gallen (Switzerland) and colleagues reported in Haematologica.
The researchers had hypothesized that nelfinavir would induce the unfolded protein response and would overcome proteasome-inhibitor resistance. The researchers determined a nelfinavir dose of 2,500 mg twice daily based on a dose-finding study in 12 patients with advanced hematologic malignancies.
In an exploratory extension cohort trial that followed, six patients with relapsed, bortezomib-refractory, lenalidomide-resistant myeloma were treated with 2,500 mg of nelfinavir twice daily in combination with bortezomib; three reached a partial response, two had a minor response, and one had progressive disease. All began the investigational therapy less than 2 months after progressing under therapies with bortezomib/bendamustine/dexamethasone, bortezomib/bendamustine, or bortezomib monotherapy, respectively, the researchers said (Haematologica March 2016;101:346-55).
The study protocol did not allow dexamethasone co-administration until completion of cycle three, when patients who did not achieve a minor response were allowed to co-administer 8 mg dexamethasone with bortezomib. Higher doses were excluded because of potential interactions with nelfinavir through the Cyp3A4 system.
Future research should assess whether bortezomib may be replaced by next-generation drugs like carfilzomib to avoid drug interactions and improve activity and whether nelfinavir may likewise be used in combination with novel oral proteasome inhibitors to boost their low single agent activity. “This ultimately suggests exploring the addition of HIV protease inhibitors to established combinations of proteasome inhibitors with immunomodulatory drugs, for example, in the carfilzomib/lenalidomide/dexamethasone regimen, one of the most powerful and tolerable regimens available to date for advanced multiple myeloma,” they concluded.
On Twitter @maryjodales
The protease inhibitor nelfinavir was used successfully to resensitize patients with proteasome inhibitor–refractory multiple myeloma for proteasome-inhibitor treatment, Dr. Christoph Driessen of Kantonsspital St. Gallen (Switzerland) and colleagues reported in Haematologica.
The researchers had hypothesized that nelfinavir would induce the unfolded protein response and would overcome proteasome-inhibitor resistance. The researchers determined a nelfinavir dose of 2,500 mg twice daily based on a dose-finding study in 12 patients with advanced hematologic malignancies.
In an exploratory extension cohort trial that followed, six patients with relapsed, bortezomib-refractory, lenalidomide-resistant myeloma were treated with 2,500 mg of nelfinavir twice daily in combination with bortezomib; three reached a partial response, two had a minor response, and one had progressive disease. All began the investigational therapy less than 2 months after progressing under therapies with bortezomib/bendamustine/dexamethasone, bortezomib/bendamustine, or bortezomib monotherapy, respectively, the researchers said (Haematologica March 2016;101:346-55).
The study protocol did not allow dexamethasone co-administration until completion of cycle three, when patients who did not achieve a minor response were allowed to co-administer 8 mg dexamethasone with bortezomib. Higher doses were excluded because of potential interactions with nelfinavir through the Cyp3A4 system.
Future research should assess whether bortezomib may be replaced by next-generation drugs like carfilzomib to avoid drug interactions and improve activity and whether nelfinavir may likewise be used in combination with novel oral proteasome inhibitors to boost their low single agent activity. “This ultimately suggests exploring the addition of HIV protease inhibitors to established combinations of proteasome inhibitors with immunomodulatory drugs, for example, in the carfilzomib/lenalidomide/dexamethasone regimen, one of the most powerful and tolerable regimens available to date for advanced multiple myeloma,” they concluded.
On Twitter @maryjodales
FROM HAEMATOLOGIA
Key clinical point: Adding HIV protease inhibitors to established combinations of proteasome inhibitors may improve outcomes in patients with multiple myeloma.
Major finding: When six patients with relapsed, bortezomib-refractory, lenalidomide-resistant myeloma were treated with 2,500 mg of nelfinavir twice daily in combination with bortezomib, three reached a partial response, two had a minor response, and disease progressed in one.
Data source: A dose-finding study in 12 patients with advanced hematologic malignancies and an exploratory extension cohort trial that followed.
Disclosures: None of the authors had financial conflicts of interest in relation to the study.
Follicular lymphoma: Quantitative PET/CT measures for detecting bone marrow involvement
Quantifying bone marrow uptake of FDG (18fluorodeoxyglucose) improved the diagnostic accuracy of PET/CT for predicting bone marrow involvement in patients with follicular lymphoma, based on the results of a retrospective study.
Visual evidence of focal increased uptake on PET/CT indicates marrow involvement in follicular lymphoma; however, diffuse uptake is a nonspecific finding. Measuring the mean bone marrow standardized uptake value (BM SUV mean) improves PET/CT diagnostic accuracy, Dr. Chava Perry and his colleagues at Tel Aviv Sourasky Medical Center reported in Medicine [(Baltimore). 2016 Mar;95(9):e2910].
The researchers evaluated 68 consecutive patients with follicular lymphoma; 16 had bone marrow involvement – 13 had biopsy-proven involvement and 3 had a negative biopsy with increased medullary uptake that normalized after treatment. BM FDG uptake was diffuse in 8 of them and focal in the other 8.
While focal increased uptake is indicative of bone marrow involvement, diffuse uptake can be associated with false-positive results, as it was in the case of 17 patients (32.7% of those with diffuse uptake). Overall, visual assessment of scan results had a negative predictive value of 100% and a positive predictive value (PPV) of 48.5%.
On a quantitative assessment, however, BM SUV mean was significantly higher in patients with bone marrow involvement (SUV mean of 3.7 [1.7-6] vs. 1.4 [0.4-2.65]; P less than .001). On the receiver operator curve (ROC) analysis, a BM SUV mean exceeding 2.7 had a positive predictive value of 100% for bone marrow involvement (sensitivity of 68%). A BM SUV mean less than 1.7 had an negative predictive value of 100% (specificity of 73%).
A mean standardized uptake value (BM SUV mean) below 1.7 may spare the need for bone marrow biopsy while a BM SUV mean above 2.7 is compatible with bone marrow involvement, although biopsy may still be recommended to exclude large cell transformation, the researchers concluded.
On Twitter @maryjodales
Quantifying bone marrow uptake of FDG (18fluorodeoxyglucose) improved the diagnostic accuracy of PET/CT for predicting bone marrow involvement in patients with follicular lymphoma, based on the results of a retrospective study.
Visual evidence of focal increased uptake on PET/CT indicates marrow involvement in follicular lymphoma; however, diffuse uptake is a nonspecific finding. Measuring the mean bone marrow standardized uptake value (BM SUV mean) improves PET/CT diagnostic accuracy, Dr. Chava Perry and his colleagues at Tel Aviv Sourasky Medical Center reported in Medicine [(Baltimore). 2016 Mar;95(9):e2910].
The researchers evaluated 68 consecutive patients with follicular lymphoma; 16 had bone marrow involvement – 13 had biopsy-proven involvement and 3 had a negative biopsy with increased medullary uptake that normalized after treatment. BM FDG uptake was diffuse in 8 of them and focal in the other 8.
While focal increased uptake is indicative of bone marrow involvement, diffuse uptake can be associated with false-positive results, as it was in the case of 17 patients (32.7% of those with diffuse uptake). Overall, visual assessment of scan results had a negative predictive value of 100% and a positive predictive value (PPV) of 48.5%.
On a quantitative assessment, however, BM SUV mean was significantly higher in patients with bone marrow involvement (SUV mean of 3.7 [1.7-6] vs. 1.4 [0.4-2.65]; P less than .001). On the receiver operator curve (ROC) analysis, a BM SUV mean exceeding 2.7 had a positive predictive value of 100% for bone marrow involvement (sensitivity of 68%). A BM SUV mean less than 1.7 had an negative predictive value of 100% (specificity of 73%).
A mean standardized uptake value (BM SUV mean) below 1.7 may spare the need for bone marrow biopsy while a BM SUV mean above 2.7 is compatible with bone marrow involvement, although biopsy may still be recommended to exclude large cell transformation, the researchers concluded.
On Twitter @maryjodales
Quantifying bone marrow uptake of FDG (18fluorodeoxyglucose) improved the diagnostic accuracy of PET/CT for predicting bone marrow involvement in patients with follicular lymphoma, based on the results of a retrospective study.
Visual evidence of focal increased uptake on PET/CT indicates marrow involvement in follicular lymphoma; however, diffuse uptake is a nonspecific finding. Measuring the mean bone marrow standardized uptake value (BM SUV mean) improves PET/CT diagnostic accuracy, Dr. Chava Perry and his colleagues at Tel Aviv Sourasky Medical Center reported in Medicine [(Baltimore). 2016 Mar;95(9):e2910].
The researchers evaluated 68 consecutive patients with follicular lymphoma; 16 had bone marrow involvement – 13 had biopsy-proven involvement and 3 had a negative biopsy with increased medullary uptake that normalized after treatment. BM FDG uptake was diffuse in 8 of them and focal in the other 8.
While focal increased uptake is indicative of bone marrow involvement, diffuse uptake can be associated with false-positive results, as it was in the case of 17 patients (32.7% of those with diffuse uptake). Overall, visual assessment of scan results had a negative predictive value of 100% and a positive predictive value (PPV) of 48.5%.
On a quantitative assessment, however, BM SUV mean was significantly higher in patients with bone marrow involvement (SUV mean of 3.7 [1.7-6] vs. 1.4 [0.4-2.65]; P less than .001). On the receiver operator curve (ROC) analysis, a BM SUV mean exceeding 2.7 had a positive predictive value of 100% for bone marrow involvement (sensitivity of 68%). A BM SUV mean less than 1.7 had an negative predictive value of 100% (specificity of 73%).
A mean standardized uptake value (BM SUV mean) below 1.7 may spare the need for bone marrow biopsy while a BM SUV mean above 2.7 is compatible with bone marrow involvement, although biopsy may still be recommended to exclude large cell transformation, the researchers concluded.
On Twitter @maryjodales
FROM MEDICINE
Key clinical point: Measuring the mean standardized uptake value of 18fluorodeoxyglucose in the bone marrow of patients with follicular lymphoma improves the diagnostic accuracy of PET/CT.
Major finding: In this study, diffuse uptake was associated with 17 (32.7%) false positive cases.
Data source: Retrospective study of 68 consecutive patients with follicular lymphoma.
Disclosures: The authors had no funding and conflicts of interest to disclose.
Generic sildenafil approved for erectile dysfunction
The first generic version of sildenafil citrate has been approved for the treatment of erectile dysfunction, the Food and Drug Administration has announced.
Teva Pharmaceutical Industries received approval to market generic sildenafil citrate tablets in 25-mg, 50-mg, and 100-mg doses and has been granted 180-day exclusivity, according to the FDA.
The first generic version of sildenafil citrate has been approved for the treatment of erectile dysfunction, the Food and Drug Administration has announced.
Teva Pharmaceutical Industries received approval to market generic sildenafil citrate tablets in 25-mg, 50-mg, and 100-mg doses and has been granted 180-day exclusivity, according to the FDA.
The first generic version of sildenafil citrate has been approved for the treatment of erectile dysfunction, the Food and Drug Administration has announced.
Teva Pharmaceutical Industries received approval to market generic sildenafil citrate tablets in 25-mg, 50-mg, and 100-mg doses and has been granted 180-day exclusivity, according to the FDA.
Ibrutinib approved as first-line therapy for all CLL patients
Ibrutinib (Imbruvica) has been approved as a first-line treatment for patients with chronic lymphocytic leukemia (CLL), irrespective of their treatment history, according to a statement issued by the manufacturer, AbbVie.
The approval is based on data from the randomized, multicenter, open-label phase III RESONATE-2 trial, which evaluated the use of ibrutinib versus chlorambucil in 269 treatment-naive patients with CLL or small lymphocytic lymphoma (SLL) aged 65 years or older. The RESONATE-2 data were presented at the 2015 annual meeting of the American Society of Hematology.
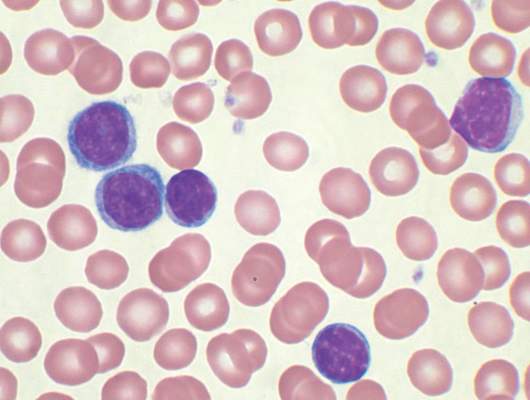
“The progression-free survival data seen in these previously untreated CLL patients are strong and encouraging,” Dr. Jan Burger of the University of Texas MD Anderson Cancer Center, Houston, and the lead investigator of RESONATE-2, said in the AbbVie statement. “This is especially important for first-line CLL patients, when considering the safety profile. This treatment represents another option for these patients.”
The National Comprehensive Cancer Network also recently published an update to its clinical practice guidelines for non-Hodgkin’s lymphomas, granting Imbruvica a category 1 recommendation for certain CLL patients, including as a first-line treatment option for frail CLL patients with significant comorbidities, as well as for CLL patients with or without del 17p or the genetic mutation TP53 who are 70 years or older, or younger patients with significant comorbidities, the AbbVie statement noted.
Imbruvica has been jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and by Janssen Biotech.
Ibrutinib (Imbruvica) has been approved as a first-line treatment for patients with chronic lymphocytic leukemia (CLL), irrespective of their treatment history, according to a statement issued by the manufacturer, AbbVie.
The approval is based on data from the randomized, multicenter, open-label phase III RESONATE-2 trial, which evaluated the use of ibrutinib versus chlorambucil in 269 treatment-naive patients with CLL or small lymphocytic lymphoma (SLL) aged 65 years or older. The RESONATE-2 data were presented at the 2015 annual meeting of the American Society of Hematology.
“The progression-free survival data seen in these previously untreated CLL patients are strong and encouraging,” Dr. Jan Burger of the University of Texas MD Anderson Cancer Center, Houston, and the lead investigator of RESONATE-2, said in the AbbVie statement. “This is especially important for first-line CLL patients, when considering the safety profile. This treatment represents another option for these patients.”
The National Comprehensive Cancer Network also recently published an update to its clinical practice guidelines for non-Hodgkin’s lymphomas, granting Imbruvica a category 1 recommendation for certain CLL patients, including as a first-line treatment option for frail CLL patients with significant comorbidities, as well as for CLL patients with or without del 17p or the genetic mutation TP53 who are 70 years or older, or younger patients with significant comorbidities, the AbbVie statement noted.
Imbruvica has been jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and by Janssen Biotech.
Ibrutinib (Imbruvica) has been approved as a first-line treatment for patients with chronic lymphocytic leukemia (CLL), irrespective of their treatment history, according to a statement issued by the manufacturer, AbbVie.
The approval is based on data from the randomized, multicenter, open-label phase III RESONATE-2 trial, which evaluated the use of ibrutinib versus chlorambucil in 269 treatment-naive patients with CLL or small lymphocytic lymphoma (SLL) aged 65 years or older. The RESONATE-2 data were presented at the 2015 annual meeting of the American Society of Hematology.
“The progression-free survival data seen in these previously untreated CLL patients are strong and encouraging,” Dr. Jan Burger of the University of Texas MD Anderson Cancer Center, Houston, and the lead investigator of RESONATE-2, said in the AbbVie statement. “This is especially important for first-line CLL patients, when considering the safety profile. This treatment represents another option for these patients.”
The National Comprehensive Cancer Network also recently published an update to its clinical practice guidelines for non-Hodgkin’s lymphomas, granting Imbruvica a category 1 recommendation for certain CLL patients, including as a first-line treatment option for frail CLL patients with significant comorbidities, as well as for CLL patients with or without del 17p or the genetic mutation TP53 who are 70 years or older, or younger patients with significant comorbidities, the AbbVie statement noted.
Imbruvica has been jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and by Janssen Biotech.
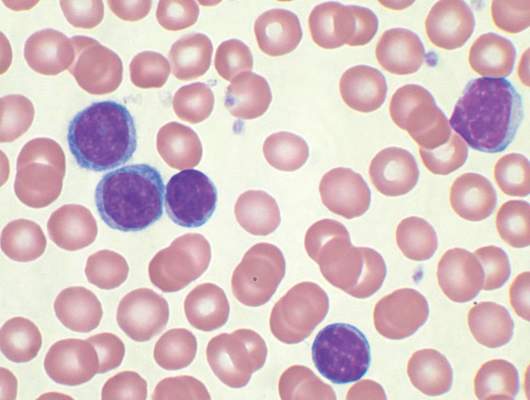
Novel test detects low levels of residual CML
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.

Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
FROM THE JOURNAL OF MOLECULAR DIAGNOSTICS
Key clinical point: A novel test may predict which CML patients in remission can safely stop TKI therapy.
Major finding: Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%).
Data source: DNA-based dPCR measures in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Disclosures: The researchers had no relevant disclosures.

Medicare to cover HSCT in approved clinical trials for myeloma, myelofibrosis, sickle cell disease
Medicare will cover allogeneic hematopoietic stem cell transplantation (HSCT) for beneficiaries with multiple myeloma, myelofibrosis, or sickle cell disease in the context of approved, prospective clinical trials, the Centers for Medicare & Medicaid Services announced in a final decision memo Jan. 27.
Approvable studies must examine whether Medicare beneficiaries who receive allogeneic HSCT have improved outcomes, compared with patients who do not receive allogeneic HSCT as measured by graft vs. host disease, other transplant-related adverse events, overall survival, and, optionally, quality of life measures.
In multiple myeloma, allogeneic HSCT will be covered only for Medicare beneficiaries who have Durie-Salmon Stage II or III multiple myeloma, or International Staging System (ISS) Stage II or Stage III multiple myeloma, and are participating in an approved prospective clinical study. Such studies must control for selection bias and potential confounding by age, duration of diagnosis, disease classification, International Myeloma Working Group (IMWG) classification, ISS staging, Durie-Salmon staging, comorbid conditions, type of preparative/conditioning regimen, graft vs. host disease (GVHD) prophylaxis, donor type, and cell source, the CMS said in its memo.
In myelofibrosis, allogeneic HSCT will be covered by Medicare in an approved prospective study only for beneficiaries with Dynamic International Prognostic Scoring System (DIPSS plus) intermediate-2 or high primary or secondary myelofibrosis. Studies must be controlled for selection bias and potential confounding by age, duration of diagnosis, disease classification, DIPSS plus score, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
In sickle cell disease, allogeneic HSCT will be covered by Medicare only for beneficiaries who have severe, symptomatic disease. Approvable studies must control for selection bias and potential confounding by age, duration of diagnosis, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
On Twitter @maryjodales
Dr. Navneet Majhail comments: I welcome the decision by CMS [the Centers for Medicare & Medicaid Services] to cover allogeneic hematopoietic stem cell transplantation (HSCT) for multiple myeloma, myelofibrosis, and sickle cell disease under the coverage with evidence development (CED) mechanism. This action will allow us to provide transplant as a treatment option for older patients with myeloma and myelofibrosis and for Medicare beneficiaries with sickle cell disease: Lack of coverage is a real challenge at present for this population and prevents us from offering a potentially curative treatment option to these high-risk patients.

|
Dr. Navneet Majhail |
The decision is the result of collective advocacy efforts of our transplant community, patients, and patient advocacy organizations, Be the Match, and the American Society for Blood and Marrow Transplantation.
The CED asks for a prospective clinical trial that mandates the presence of a control arm of comparable patients who do not receive allogeneic transplantation. I completely support provision of transplantation on a clinical trial for CED purposes; however, I believe it would have been better to allow hematology and transplant experts to determine the appropriate study design in consultation with CMS to fulfill the CED requirements. For example, on the basis of available evidence, it will be challenging to enroll patients with high-risk myelofibrosis to a nontransplant arm. Irrespective, this is a big win for patients with these life-threatening diseases and for physicians who treat them.
Dr. Navneet Majhail is the director of the Cleveland Clinic’s Blood & Marrow Transplant Program. He serves as a staff physician at the Taussig Cancer Institute and is a professor of medicine with the Cleveland Clinic Lerner College of Medicine.
Dr. Navneet Majhail comments: I welcome the decision by CMS [the Centers for Medicare & Medicaid Services] to cover allogeneic hematopoietic stem cell transplantation (HSCT) for multiple myeloma, myelofibrosis, and sickle cell disease under the coverage with evidence development (CED) mechanism. This action will allow us to provide transplant as a treatment option for older patients with myeloma and myelofibrosis and for Medicare beneficiaries with sickle cell disease: Lack of coverage is a real challenge at present for this population and prevents us from offering a potentially curative treatment option to these high-risk patients.
|
|
Dr. Navneet Majhail |
The decision is the result of collective advocacy efforts of our transplant community, patients, and patient advocacy organizations, Be the Match, and the American Society for Blood and Marrow Transplantation.
The CED asks for a prospective clinical trial that mandates the presence of a control arm of comparable patients who do not receive allogeneic transplantation. I completely support provision of transplantation on a clinical trial for CED purposes; however, I believe it would have been better to allow hematology and transplant experts to determine the appropriate study design in consultation with CMS to fulfill the CED requirements. For example, on the basis of available evidence, it will be challenging to enroll patients with high-risk myelofibrosis to a nontransplant arm. Irrespective, this is a big win for patients with these life-threatening diseases and for physicians who treat them.
Dr. Navneet Majhail is the director of the Cleveland Clinic’s Blood & Marrow Transplant Program. He serves as a staff physician at the Taussig Cancer Institute and is a professor of medicine with the Cleveland Clinic Lerner College of Medicine.
Dr. Navneet Majhail comments: I welcome the decision by CMS [the Centers for Medicare & Medicaid Services] to cover allogeneic hematopoietic stem cell transplantation (HSCT) for multiple myeloma, myelofibrosis, and sickle cell disease under the coverage with evidence development (CED) mechanism. This action will allow us to provide transplant as a treatment option for older patients with myeloma and myelofibrosis and for Medicare beneficiaries with sickle cell disease: Lack of coverage is a real challenge at present for this population and prevents us from offering a potentially curative treatment option to these high-risk patients.
|
|
Dr. Navneet Majhail |
The decision is the result of collective advocacy efforts of our transplant community, patients, and patient advocacy organizations, Be the Match, and the American Society for Blood and Marrow Transplantation.
The CED asks for a prospective clinical trial that mandates the presence of a control arm of comparable patients who do not receive allogeneic transplantation. I completely support provision of transplantation on a clinical trial for CED purposes; however, I believe it would have been better to allow hematology and transplant experts to determine the appropriate study design in consultation with CMS to fulfill the CED requirements. For example, on the basis of available evidence, it will be challenging to enroll patients with high-risk myelofibrosis to a nontransplant arm. Irrespective, this is a big win for patients with these life-threatening diseases and for physicians who treat them.
Dr. Navneet Majhail is the director of the Cleveland Clinic’s Blood & Marrow Transplant Program. He serves as a staff physician at the Taussig Cancer Institute and is a professor of medicine with the Cleveland Clinic Lerner College of Medicine.
Medicare will cover allogeneic hematopoietic stem cell transplantation (HSCT) for beneficiaries with multiple myeloma, myelofibrosis, or sickle cell disease in the context of approved, prospective clinical trials, the Centers for Medicare & Medicaid Services announced in a final decision memo Jan. 27.
Approvable studies must examine whether Medicare beneficiaries who receive allogeneic HSCT have improved outcomes, compared with patients who do not receive allogeneic HSCT as measured by graft vs. host disease, other transplant-related adverse events, overall survival, and, optionally, quality of life measures.
In multiple myeloma, allogeneic HSCT will be covered only for Medicare beneficiaries who have Durie-Salmon Stage II or III multiple myeloma, or International Staging System (ISS) Stage II or Stage III multiple myeloma, and are participating in an approved prospective clinical study. Such studies must control for selection bias and potential confounding by age, duration of diagnosis, disease classification, International Myeloma Working Group (IMWG) classification, ISS staging, Durie-Salmon staging, comorbid conditions, type of preparative/conditioning regimen, graft vs. host disease (GVHD) prophylaxis, donor type, and cell source, the CMS said in its memo.
In myelofibrosis, allogeneic HSCT will be covered by Medicare in an approved prospective study only for beneficiaries with Dynamic International Prognostic Scoring System (DIPSS plus) intermediate-2 or high primary or secondary myelofibrosis. Studies must be controlled for selection bias and potential confounding by age, duration of diagnosis, disease classification, DIPSS plus score, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
In sickle cell disease, allogeneic HSCT will be covered by Medicare only for beneficiaries who have severe, symptomatic disease. Approvable studies must control for selection bias and potential confounding by age, duration of diagnosis, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
On Twitter @maryjodales
Medicare will cover allogeneic hematopoietic stem cell transplantation (HSCT) for beneficiaries with multiple myeloma, myelofibrosis, or sickle cell disease in the context of approved, prospective clinical trials, the Centers for Medicare & Medicaid Services announced in a final decision memo Jan. 27.
Approvable studies must examine whether Medicare beneficiaries who receive allogeneic HSCT have improved outcomes, compared with patients who do not receive allogeneic HSCT as measured by graft vs. host disease, other transplant-related adverse events, overall survival, and, optionally, quality of life measures.
In multiple myeloma, allogeneic HSCT will be covered only for Medicare beneficiaries who have Durie-Salmon Stage II or III multiple myeloma, or International Staging System (ISS) Stage II or Stage III multiple myeloma, and are participating in an approved prospective clinical study. Such studies must control for selection bias and potential confounding by age, duration of diagnosis, disease classification, International Myeloma Working Group (IMWG) classification, ISS staging, Durie-Salmon staging, comorbid conditions, type of preparative/conditioning regimen, graft vs. host disease (GVHD) prophylaxis, donor type, and cell source, the CMS said in its memo.
In myelofibrosis, allogeneic HSCT will be covered by Medicare in an approved prospective study only for beneficiaries with Dynamic International Prognostic Scoring System (DIPSS plus) intermediate-2 or high primary or secondary myelofibrosis. Studies must be controlled for selection bias and potential confounding by age, duration of diagnosis, disease classification, DIPSS plus score, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
In sickle cell disease, allogeneic HSCT will be covered by Medicare only for beneficiaries who have severe, symptomatic disease. Approvable studies must control for selection bias and potential confounding by age, duration of diagnosis, comorbid conditions, type of preparative/conditioning regimen, GVHD prophylaxis, donor type, and cell source.
On Twitter @maryjodales

Consensus document issued on hematology research priorities for Europe
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.

|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.
|
|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
In a time of restricted federal budgets, research funding becomes somewhat of a luxury. Yet, research and innovation are the primary movers of change and progress, both of which are needed to drive growth to ease budget restrictions. In order to ensure that precious resources are allocated to the most promising endeavors, federal governments establish bureaucracies charged with the task of allocating funding to the “best” proposals. Unfortunately, the task of defining “best” is imprecise and largely subjective.
To address this systemic deficiency and to improve the efficient allocation of resources, the scientific community often provides guidance to funding agencies to help choose among competing proposals. In 2015, the American Society of Hematology (ASH) announced its “Agenda for Hematology Research.” The agenda included recommendations to prioritize funding to projects in the following domains: genomic profiling and chemical biology, immunologic treatments of hematologic malignancies, genome editing and gene therapy, stem cell biology and regenerative medicine, epigenetic mechanisms, and venous thromboembolic disease.
|
|
Dr. Matt Kalyacio |
More recently, the European Hematology Association has published its Roadmap for European Hematology Research. Ostensibly similar to the ASH Agenda, the EHA document is a much more detailed policy statement that calls out the most promising research opportunities across the nine major components of hematology: normal hematopoiesis, malignant lymphoid disorders, malignant myeloid disease, anemias and related diseases, platelet disorders, blood coagulation and hemostatic disorders, transfusion medicine, infections in hematology, and hematopoietic stem cell transplantation and other cell based therapies.
I find the two documents complementary in that the ASH Agenda is more accessible to grant reviewers and funding agencies, while the EHA Roadmap seems more directed to the scientific community. Whether a grant writer or a grant reviewer, these documents should help researchers focus their applications on preferred projects and help reviewers prioritize proposals.
While laudable in their goals globally, such consensus documents place much faith in the knowable future and less in the unknowable, disruptive future. Researchers with innovative ideas that do not fall into the prioritizations set forth by the community at large might find themselves struggling for resources. This unintended consequence of consensus building risks the loss of inspired science on the altar of groupthink. The “moonshot” championed by Vice-President Biden will be more likely to succeed when consensus science allows for novel approaches that have yet to be revealed.
Dr. Matt Kalaycio is the editor-in-chief of Hematology News and chairs the department of hematologic oncology and blood disorders at Cleveland Clinic Taussig Cancer Institute, Cleveland. Leave your comments on our website or write to Dr. Kalaycio at [email protected].
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.
A consensus document that summarizes the status of basic, translational, and clinical hematology research and identifies areas of unmet scientific and medical needs in Europe has been published in the February 2016 issue of Haematologica.
“For the first time, hematologists in Europe came together to develop a road map to guide hematology research in Europe,” Professor Andreas Engert, chair of the European Hematology Association’s Research Roadmap Task Force, said in a written statement. “Hematology ... must focus and collaborate to be efficient and remain successful in improving patient outcomes.”
Some 300 experts from over 20 countries in Europe helped to draft the road map. A wide variety of stakeholders, such as national hematology societies, patient organizations, hematology trial groups, and other European organizations, were consulted to comment on the final draft.
“The document reflects the views of the hematological research community in Europe, Professor Tony Green, president of the European Hematology Association (EHA), noted in the statement. “This is crucial if we want to convince policy makers to support the realization of this important research.”
“With an aging population, the slow recovery from the financial and Euro crises, costly medical breakthroughs and innovations – quite a few of which involve hematology researchers, Europe faces increased health expenditures while budgets are limited,” Professor Ulrich Jäger, chair of the EHA European Affairs Committee, said in the statement. “So it is our responsibility to provide the policy makers with the information and evidence they need to decide where their support impacts knowledge and health most efficiently, to the benefit of patients and society. ... Now it is up to the policy makers in the EU to deliver, too.”
You may find the full article in Haematologica 2016 Jan. doi: 10.3324/haematol.2015.136739.

Ofatumumab approved for extended treatment of CLL patients in complete or partial response
Ofatumumab has been approved for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive chronic lymphocytic leukemia (CLL), the U.S. Food and Drug Administration announced on Jan. 19.
Ofatumumab (Arzerra Injection, Novartis Pharmaceuticals) was previously approved for treatment-naive patients with CLL for whom fludarabine-based therapy was considered inappropriate and for patients with CLL refractory to fludarabine and alemtuzumab.

Approval of the new indication was based on the results of a randomized, open-label trial that found improved progression-free survival with ofatumumab as compared with observation in patients whose disease had a complete or partial response after at least two lines of prior therapy, the FDA said in a press release.
In the study, 238 patients were randomized to ofatumumab and 236 to observation. Patients in the ofatumumab arm had received a range of two to five prior therapies. The median progression-free survival was significantly longer with ofatumumab at 29.4 months (95% confidence interval, 26.2-34.2) than with observation at 15.2 months (95% CI, 11.8-18.8).
Of patients treated with ofatumumab, 33% reported serious adverse reactions. The most common were pneumonia, pyrexia, and neutropenia (including febrile neutropenia).
The recommended dose and schedule for ofatumumab therapy is 300 mg by intravenous infusion on day 1 followed by 1,000 mg on day 8, and then 7 weeks later, and then every 8 weeks thereafter for up to a maximum of 2 years.
Full prescribing information is available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125326s062lbl.pdf.
Ofatumumab has been approved for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive chronic lymphocytic leukemia (CLL), the U.S. Food and Drug Administration announced on Jan. 19.
Ofatumumab (Arzerra Injection, Novartis Pharmaceuticals) was previously approved for treatment-naive patients with CLL for whom fludarabine-based therapy was considered inappropriate and for patients with CLL refractory to fludarabine and alemtuzumab.
Approval of the new indication was based on the results of a randomized, open-label trial that found improved progression-free survival with ofatumumab as compared with observation in patients whose disease had a complete or partial response after at least two lines of prior therapy, the FDA said in a press release.
In the study, 238 patients were randomized to ofatumumab and 236 to observation. Patients in the ofatumumab arm had received a range of two to five prior therapies. The median progression-free survival was significantly longer with ofatumumab at 29.4 months (95% confidence interval, 26.2-34.2) than with observation at 15.2 months (95% CI, 11.8-18.8).
Of patients treated with ofatumumab, 33% reported serious adverse reactions. The most common were pneumonia, pyrexia, and neutropenia (including febrile neutropenia).
The recommended dose and schedule for ofatumumab therapy is 300 mg by intravenous infusion on day 1 followed by 1,000 mg on day 8, and then 7 weeks later, and then every 8 weeks thereafter for up to a maximum of 2 years.
Full prescribing information is available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125326s062lbl.pdf.
Ofatumumab has been approved for extended treatment of patients who are in complete or partial response after at least two lines of therapy for recurrent or progressive chronic lymphocytic leukemia (CLL), the U.S. Food and Drug Administration announced on Jan. 19.
Ofatumumab (Arzerra Injection, Novartis Pharmaceuticals) was previously approved for treatment-naive patients with CLL for whom fludarabine-based therapy was considered inappropriate and for patients with CLL refractory to fludarabine and alemtuzumab.
Approval of the new indication was based on the results of a randomized, open-label trial that found improved progression-free survival with ofatumumab as compared with observation in patients whose disease had a complete or partial response after at least two lines of prior therapy, the FDA said in a press release.
In the study, 238 patients were randomized to ofatumumab and 236 to observation. Patients in the ofatumumab arm had received a range of two to five prior therapies. The median progression-free survival was significantly longer with ofatumumab at 29.4 months (95% confidence interval, 26.2-34.2) than with observation at 15.2 months (95% CI, 11.8-18.8).
Of patients treated with ofatumumab, 33% reported serious adverse reactions. The most common were pneumonia, pyrexia, and neutropenia (including febrile neutropenia).
The recommended dose and schedule for ofatumumab therapy is 300 mg by intravenous infusion on day 1 followed by 1,000 mg on day 8, and then 7 weeks later, and then every 8 weeks thereafter for up to a maximum of 2 years.
Full prescribing information is available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125326s062lbl.pdf.

Venetoclax gets 79% overall response rate in high-risk CLL
ORLANDO – Venetoclax monotherapy achieved an overall response rate of 79% in a high-risk population of 107 patients with relapsed or refractory del(17p) chronic lymphocytic leukemia, Dr. Stephan Stilgenbauer reported in a late-breaking abstract at the annual meeting of the American Society of Hematology.
Of the 85 responders, the response was maintained at 1 year in 85%. Of the 45 patients assessed for minimal residual disease in the blood, 18 achieved MRD negativity. Ten of these 18 patients also had bone marrow assessments and six were MRD negative.
Dr. Stilgenbauer of the University of Ulm (Germany), discussed the implications of the phase II study findings in our exclusive interview at ASH, as well as phase III study plans and the use of venetoclax in combination therapies.
He receives honoraria or research funding from a wide range of companies, including AbbVie and Genentech, the companies collaborating on the development of venetoclax.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @maryjodales
ORLANDO – Venetoclax monotherapy achieved an overall response rate of 79% in a high-risk population of 107 patients with relapsed or refractory del(17p) chronic lymphocytic leukemia, Dr. Stephan Stilgenbauer reported in a late-breaking abstract at the annual meeting of the American Society of Hematology.
Of the 85 responders, the response was maintained at 1 year in 85%. Of the 45 patients assessed for minimal residual disease in the blood, 18 achieved MRD negativity. Ten of these 18 patients also had bone marrow assessments and six were MRD negative.
Dr. Stilgenbauer of the University of Ulm (Germany), discussed the implications of the phase II study findings in our exclusive interview at ASH, as well as phase III study plans and the use of venetoclax in combination therapies.
He receives honoraria or research funding from a wide range of companies, including AbbVie and Genentech, the companies collaborating on the development of venetoclax.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @maryjodales
ORLANDO – Venetoclax monotherapy achieved an overall response rate of 79% in a high-risk population of 107 patients with relapsed or refractory del(17p) chronic lymphocytic leukemia, Dr. Stephan Stilgenbauer reported in a late-breaking abstract at the annual meeting of the American Society of Hematology.
Of the 85 responders, the response was maintained at 1 year in 85%. Of the 45 patients assessed for minimal residual disease in the blood, 18 achieved MRD negativity. Ten of these 18 patients also had bone marrow assessments and six were MRD negative.
Dr. Stilgenbauer of the University of Ulm (Germany), discussed the implications of the phase II study findings in our exclusive interview at ASH, as well as phase III study plans and the use of venetoclax in combination therapies.
He receives honoraria or research funding from a wide range of companies, including AbbVie and Genentech, the companies collaborating on the development of venetoclax.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @maryjodales
AT ASH 2015
