User login
Blood smear analysis in babesiosis, ehrlichiosis, relapsing fever, malaria, and Chagas disease
Blood smear analysis, while commonly used to evaluate hematologic conditions, is infrequently used to diagnose infectious diseases. This is because of the rarity of diseases for which blood smear analysis is indicated. Consequently, such testing is often overlooked when it is diagnostically important.
Nonspecific changes may include morphologic changes in leukocytes and erythrocytes (eg, toxic granulations, macrocytosis).1 And with certain pathogens, identifying organisms in a peripheral blood smear allows for a rapid diagnosis.
This paper discusses the epidemiology, clinical manifestations, laboratory findings, and management of five infectious diseases in which direct visualization of the organism in the blood plays a major diagnostic role. Our intent is to summarize the clinical findings that should prompt blood smear analysis so that these uncommon conditions are not overlooked.
BABESIOSIS
Babesiosis, a tick-borne protozoal disease, occurs principally in the United States and Europe. Of the more than 100 species of Babesia, two account for almost all human disease: B microti and B divergens.
Both species are transmitted by Ixodes ticks, although patients often do not recall being bitten. The disease may rarely complicate blood transfusion. Most cases occur from May to September, when tick exposure is highest. The incubation period varies from 1 to 4 weeks.
Common in the Northeast, usually asymptomatic
B microti infection occurs predominantly in the United States. Rodents, especially the white-footed mouse, are the principal reservoir.2,3 Endemic areas, where seropositivity rates range from 4% to 21%, include the coastal areas and islands off of Massachusetts, particularly Cape Cod, Nantucket, and Martha’s Vineyard; the islands near New York City, especially Long Island, Shelter Island, and Fire Island; Block Island, off the coast of Rhode Island; and certain areas in Connecticut.4 WA-1, a species that is morphologically identical to B microti, is emerging in California and Washington.5,6
Infection with B microti is usually asymptomatic. Elderly and immunosuppressed people, especially those without a spleen or with impaired cellular immunity, are more likely to become ill. Symptoms, including fever, malaise, headache, nausea, and generalized aching, may last weeks to months.
About one-fourth of patients with babesiosis are coinfected with the Lyme disease bacterium (Borrelia burgdorferi) and often have more severe illness.3
Hepatomegaly, splenomegaly, jaundice, and dark urine are common findings in patients with symptoms. Severe hemolysis, often accompanied by thrombocytopenia, leukopenia, and atypical lymphocytosis, is more common in high-risk patients. Hepatic transaminases may be elevated. Urinalysis may show proteinuria and hemoglobinuria. Acute respiratory distress syndrome has been reported in severe cases.7–10
B divergens infection: A serious but rare disease seen in Europe
B divergens is found mainly in Europe. Altogether, fewer than 50 cases of infection have been reported in France, Spain, Germany, Great Britain, Ireland, Yugoslavia, and the former Soviet Union.11,12 Cattle are the principal reservoir of infection.
Infection with B divergens causes a rare but devastating disease mainly in asplenic people, usually resulting in coma and death. No cases of subclinical infection have been reported. The clinical course is fulminant, and hemolytic anemia is common.2,3
Suspect babesiosis in endemic areas in cases of prolonged ‘flu’
Babesiosis should be considered when a patient residing in or traveling from an endemic area presents with a prolonged flu-like illness and hemolysis, with or without organomegaly and jaundice.
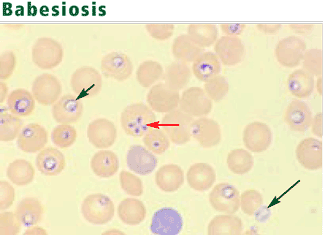
The protozoa may resemble the rings of malaria parasites. Distinguishing traits include exoerythrocytic organisms; the absence of pigmented granules in infected red blood cells; and a “maltese cross,” a rare pattern produced by tetrads of Babesia merozoites.13 An infected erythrocyte may contain up to eight parasites.
Serologic and polymerase chain reaction tests are useful when the organism is not visible.14,15
Treat patients with severe disease
Most patients with B microti infection have a mild illness that resolves without treatment. Treatment is recommended for those with severe infection and in those with high-level parasitemia. Agents with consistent activity against B microti include clindamycin (Cleocin), azithromycin (Zithromax), ato-vaquone (Mepron), doxycycline (Vibramycin), and quinine (Quinamm). Combination therapy with either clindamycin and quinine or azithromycin and atovaquone is recommended. B divergens infection has been successfully treated with a combination of clindamycin, quinine, and exchange transfusion.16
EHRLICHIOSIS
Ehrlichiosis, nicknamed Rocky Mountain “spotless” fever, is a seasonal, tick-borne disease caused by obligate intracellular bacteria. Bacteria of the genus Ehrlichia grow within the cytoplasmic vacuoles of leukocytes and cause mainly zoonotic infections. Several species, especially Ehrlichia chaffeensis and Anaplasma phagocytophilum, are recognized as human pathogens.17,18E chaffeensis infects mononuclear cells, causing a condition known as human monocytic ehrlichiosis (HME). A phagocytophilum infects neutrophils, producing a condition called human granulocytic anaplasmosis (HGA).
Deer are the principal reservoir for E chaffeensis19; white-footed mice, other rodents, and deer are the principal reservoirs for A phagocytophilum. HME is transmitted by Dermacentor and Ixodes ticks, and HGA by Ixodes ticks. Human infection usually occurs in the spring and summer, when tick exposure is greatest. Co-infection of ticks with the organisms causing Lyme disease or babesiosis may result in simultaneous transmission of these diseases.
More than 1,000 cases of HME have been reported in the southeastern, south-central, and mid-Atlantic regions of the United States.20 The prevalence of HME in the United States appears to follow that of Rocky Mountain spotted fever. Some cases have been described in New England and in the Pacific Northwest. The more than 600 reported cases of HGA have come from Wisconsin, Minnesota, Connecticut, New York, Massachusetts, California, Florida, and Western Europe.21,22 The distribution of HGA follows that of Lyme disease, because the two diseases share the same tick vector.
Acute onset of fever and myalgias
HME and HGA have an incubation period of 1 to 2 weeks. The symptoms are similar and are usually acute, ranging from mild to severe. Most patients have fever, chills, malaise, headache, and myalgias. Many also have nausea, vomiting, cough, and arthralgias. Symptoms are similar to those in Rocky Mountain spotted fever (caused by Rickettsia rickettsii), except that rash is uncommon in HME (seen in approximately a third of patients) and rare in HGA.23–25 Neurologic findings, such as altered sensorium and neck stiffness, may be accompanied by lymphocytic pleocytosis and elevated protein levels in the cerebrospinal fluid.26 Subclinical and subacute presentations (eg, a fever lasting up to 2 months) are uncommon. No chronic cases have been reported.
The estimated death rate is 1% to 10%, and hospitalization rates are as high as 60%. Most deaths occur in the elderly, often following such complications as congestive heart failure,27 cardiac tamponade, respiratory or renal failure, seizures, and coma. Patients with human immunodeficiency virus infection also have a poor prognosis. Convalescence may be prolonged.
Laboratory abnormalities include leukopenia, thrombocytopenia, and elevated hepatic transaminase levels. Leukopenia may be associated with lymphopenia or neutropenia. Lymphopenia occurs early in the course of illness and is usually followed by an atypical lymphocytosis. Prolonged symptoms are associated with a decreased total neutrophil count and an increased band neutrophil count.28
Suspect ehrlichiosis in endemic areas in patients with fever, leukopenia, or thrombocytopenia
Ehrlichiosis should be suspected when a febrile patient with leukopenia or thrombocytopenia has been exposed to ticks in an endemic area. Even patients whose cell counts and liver enzyme levels are normal should be evaluated if the clinical and epidemiologic situations suggest this disease.
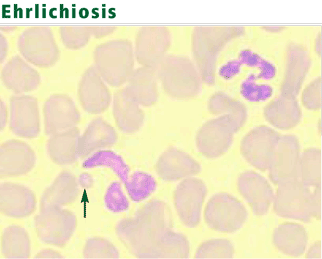
Other diagnostic tests include polymerase chain reaction and serologic assays, which are highly sensitive and specific.30,31 Because the organisms are difficult to culture in vitro, blood cultures are not useful diagnostically.
Treatment
Doxycycline 100 mg twice daily for 7 to 10 days is the treatment of choice for both HME and HGA. No role has been defined for fluoroquinolones for treating these diseases. Avoiding ticks and removing ticks promptly are the best preventive strategies.
RELAPSING FEVER
Relapsing fever is an acute febrile illness caused by spirochetes of the genus Borrelia. The disease has two forms: tick-borne, in which human infection is zoonotic, and louse-borne, in which humans are the only known reservoir of infection.32
Few tick-borne cases in the United States
Tick-borne disease is caused by many species of Borrelia. Those found in the United States occur in the western mountains and high deserts and plains of the Southwest.33 Fewer than 30 cases of tick-borne relapsing fever are diagnosed in the United States annually.
Tick-borne relapsing fever is transmitted by soft-bodied argasid ticks (Ornithodoros genus), which feed for less than an hour (usually at night) and can survive for years without a blood meal. They stay close to human and animal habitations. Exposure often occurs in cabins, under buildings, in caves, near woodpiles, and in rooms shared with animals. Rodents are the primary animal reservoir. In contrast, most other tick-borne diseases—babesiosis, ehrlichiosis, Lyme disease, Rocky Mountain spotted fever, Colorado tick fever—are transmitted by hard-bodied ixodid ticks, which live in brush and forested areas and attach to passersby, on whom they feed for days if not removed.34–36
Louse-borne disease is endemic in Africa
Louse-borne relapsing fever is caused by a single species, B recurrentis, endemic in Ethiopia and Sudan. It may occur sporadically or in epidemics. War, famine, and mass migrations predispose to epidemics with death rates ranging from 30% to 70% if untreated.37,38 Disease is spread between humans by the human body louse (Pediculus humanus).
Relapsing high fevers
The clinical manifestations of tick-borne and louse-borne relapsing fever are similar, although louse-borne relapsing fever often has a longer incubation period and a longer duration of illness. Bacteremia is heralded by the acute onset of high fever (usually above 39°C [102.2°F]), accompanied by headache, nausea, myalgias, and arthralgias. On average, clinical illness remits in 3 days in tick-borne relapsing fever, but may take 5 to 6 days in louse-borne relapsing fever. Physical findings may include altered sensorium, petechiae, hepatosplenomegaly, and conjunctival suffusion. The fever culminates in a “crisis,” characterized by rigors and a precipitous rise in temperature, pulse, and blood pressure. This is followed by defervescence, diaphoresis, and hypotension. The risk of death is highest during this period and immediately afterwards.
With resolution of the bacteremia, an afebrile period ensues, lasting 4 to 14 days. Fever then recurs, although usually milder, again associated with bacteremia. On average, people with tick-borne relapsing fever have three febrile relapses; those with louse-borne relapsing fever have one.39 Relapse occurs because of antigenic variation, in which a major surface antigen of the spirochete is changed to evade the host’s immune system.40–42
Borrelia may invade organs and the nervous system
With each episode of bacteremia, spirochetes may penetrate the brain, heart, liver, eye, or inner ear. Involvement of the central nervous system is more common with tick-borne than with louse-borne relapsing fever. Nervous system involvement may include facial palsy, myelitis, radiculopathy, aphasia, hemiplegia, stupor, or coma.43,44 Myocarditis, common in both forms of relapsing fever, portends a poor prognosis.45 Invasion of the eye or ear may result in visual impairment or dizziness. Bleeding disorders, manifested by epistaxis, petechiae, and ecchymoses, are typical of louse-borne disease and may be associated with low-grade disseminated intravascular coagulation.46 Splenomegaly is more common in louse-borne than in tick-borne disease.
Auxiliary test findings
Laboratory findings include normocytic anemia, leukocytosis, and thrombocytopenia. Liver enzyme levels may be elevated and coagulation tests may be prolonged. Patients with cardiac involvement may have a prolonged QTc interval. Cardiomegaly and pulmonary edema may be seen on chest radiography. The cerebrospinal fluid in patients with neurologic involvement has a mononuclear pleocytosis and a mildly elevated protein concentration.
Suspect if recurrent fever in endemic areas
Treatment
Relapsing fever can be successfully treated with tetracycline, penicillin, or erythromycin.47 The preferred regimen for a nonpregnant adult with louse-borne relapsing fever is a single 0.5-g dose of tetracycline. In tick-borne relapsing fever, 0.5 g of tetracycline four times daily for 5 to 10 days is recommended. Meningitis or encephalitis is usually treated with parenteral penicillin or ceftriaxone (Rocephin). The death rate in treated disease is usually less than 5%.39 Treatment can induce a Jarisch-Herxheimer reaction (rigors and hypotension, resembling a febrile crisis), and patients must be watched closely for the first 4 hours after the antibiotic is given. Avoiding ticks and practicing good personal hygiene to prevent acquiring lice are the major preventive strategies.
MALARIA
Malaria is caused by protozoa of the genus Plasmodium. Injected during a mosquito bite, sporozoites enter the bloodstream and travel to the liver. During the asymptomatic hepatic stage, the sporozoites multiply within hepatocytes to form mature schizonts. Within 1 to 2 weeks, the schizonts rupture, releasing thousands of merozoites into the bloodstream. The merozoites enter red blood cells, where they mature through the trophozoite stage to become schizonts, which release more merozoites into the blood that may infect other red blood cells. Symptoms occur during this erythrocytic stage, usually 1 to 4 weeks after a mosquito bite.
Four Plasmodia species regularly cause human disease.48 All can digest hemoglobin and cause hemolysis. P falciparum is uniquely dangerous, in part because it can alter the erythrocyte surface and obstruct the microcirculation.49,50 Further, P falciparum can cause high levels of parasitemia because it can infect red blood cells in all stages of their maturation. In contrast, P vivax and P ovale invade only reticulocytes, so levels of parasitemia are low (< 1%). P vivax and P ovale produce dormant liver forms (hypnozoites), which may cause relapses, usually within 3 years of exposure. Falciparum malaria does not have a dormant form, so relapses do not occur. P malariae infects only mature red blood cells, producing low levels of parasitemia. A fifth malaria species once thought to be confined to monkeys, P knowlesi, is a rare cause of severe human disease.51
Up to a half-billion cases of malaria occur worldwide each year.52 Although the disease predominates among residents of endemic areas, about 30,000 travelers from industrialized countries are infected each year. Fewer than 1,000 cases are reported in the United States annually, which were usually acquired during travel to endemic areas.53,54 The risk of transmission varies from region to region, with highest rates (listed in descending order) occurring in Oceania, sub-Saharan Africa, the Indian subcontinent, Southeast Asia, South America, and Central America.55 More than 3 million people die of malaria annually, most of them in sub-Saharan Africa.
Malaria is transmitted principally by the bite of the female Anopheles mosquito but can also be transmitted by blood transfusion, by the use of contaminated needles, congenitally, and through organ transplantation. Because immigrants to the United States from endemic areas may harbor the parasites for months to years, malaria may rarely be acquired by autochthonous transmission, in which a parasitized individual infects competent vectors, which then bite uninfected persons.55 The disease may also be transmitted from parasitized mosquitoes that arrive on an aircraft (“airport malaria”). There is no animal reservoir for human malaria parasites.
Recurring fever
Fever is universally present, irrespective of the species causing infection. Common symptoms include malaise, chills, headache, myalgias, abdominal pain, night sweats, nausea, and diarrhea. Febrile episodes, initially frequent and irregular, may become regular, producing temperature spikes every second or third day depending on the species. The severity of the paroxysms usually diminishes over time. Eventually, anemia, thrombocytopenia, jaundice, splenomegaly, and hepatomegaly occur.
Falciparum malaria may result in pulmonary edema, renal failure, gastroenteritis, bleeding, or hypoglycemia. Cerebral disease, presenting as altered sensorium or seizures, can be fatal.56 Death, common in untreated patients, correlates with the degree of parasitemia.57 Severe disease often occurs in children, pregnant women, asplenic patients, and nonimmune adults.
Infection with P vivax or P ovale does not produce the microvascular complications of falciparum malaria, and thrombocytopenia is less common. P malariae infection may lead to immune complex deposition and nephrotic syndrome. Symptoms, although mild, are often prolonged.
Consider malaria in travelers with recurring fever
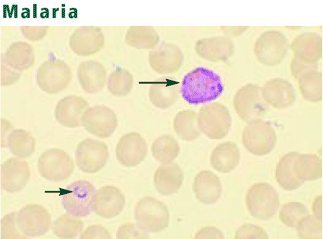
Antigen-capture test kits are useful for rapid diagnosis. These portable devices detect parasitic antigens from a drop of blood in just 15 minutes.61 Polymerase chain reaction techniques offer high sensitivity and specificity but are more expensive and less available.62
The management of patients with suspected malaria should be done by experienced health care providers. Recommendations about malaria treatment are beyond the scope of this review. The choice of antimalarial drugs requires knowledge of the regional distribution of drug resistance and the adverse effects of the agents. Patients with non-falciparum malaria rarely require hospitalization.
Malaria is prevented by mosquito avoidance and chemoprophylaxis.
AMERICAN TRYPANOSOMIASIS (CHAGAS DISEASE)
American trypanosomiasis is a zoonotic, protozoal disease caused by Trypanosoma cruzi.
The parasite is transmitted by bloodsucking triatomine insects, or “kissing bugs.”63 These insects favor mud-brick and clay houses, where they live in wall cracks, under furniture, and behind pictures. The insects acquire the organism by feeding on infected animals or on humans that have circulating trypomastigotes. The organisms then multiply in the gut of the insects and are transmitted to a second vertebrate host as the insect defecates following a blood meal. The parasite then enters the body through the skin, conjunctivae, or mucous membranes. After entering the body, the parasite disseminates through the bloodstream, invading many cell types, especially muscle and nerve.
American trypanosomiasis occurs in Central and South America, Mexico, and the southern United States. Between 16 and 18 million people are infected with the parasite, and nearly 50,000 die annually, usually from cardiac complications. Once confined to rural areas, the disease is now common in cities. The majority of reported cases come from Brazil. Only a few cases have been reported in the United States, but an estimated 50,000 to 100,000 immigrants are thought to be infected.64–68
Transmission of the parasite may also occur with blood transfusion, with organ transplantation, and congenitally.63,69,70 An increase in transfusion-related cases is expected in the United States because of an influx of migrant workers from Mexico and Central America.
Acute phase ranges from asymptomatic to multiorgan involvement
Most infected people are asymptomatic carriers. Only 1% become acutely ill, but up to a third of those infected may develop chronic symptoms decades later. Acute Chagas disease, usually seen in children, is characterized by fever, malaise, and anorexia, often accompanied by vomiting, diarrhea, and rash.64,71,72 The heart, liver, spleen, and lymph nodes become enlarged. A red and indurated nodule or furuncle (chagoma) appears at the inoculation site. If inoculation occurs across the conjunctivae, painless edema of the palpebrae and periocular tissues may be observed (Romaña sign). Generalized lymphadenopathy and hepatosplenomegaly may also be seen.
Myocarditis and meningoencephalitis occur in some cases of acute Chagas disease. Myocardial inflammation may extend to the pericardium, causing pericardial effusion, and to the endocardium, causing thrombus formation. All cardiac chambers become enlarged and the conduction system is disrupted.73 Brain damage from meningoencephalitis usually occurs in infants and young children, and may result in death.
This acute phase lasts 4 to 8 weeks and is characterized by profound parasitemia, tissue invasion, and inflammation. Following the acute phase, an asymptomatic latent or indeterminate phase lasting 10 to 40 years occurs.74 Less than half of those in clinical latency enter a chronic phase of disease.
Severe chronic phase may occur decades later
The chronic phase, which occurs years to decades after the initial infection, is characterized by cardiac, esophageal, and colonic enlargement. Cardiac involvement is associated with congestive heart failure, arrhythmias, and cardiac arrest. Intracardiac thrombi may embolize, causing systemic and pulmonary infarctions.71–73,75 Enlargement of the esophagus is associated with dysphagia, chest pain, weight loss, and sometimes perforation or aspiration-related pneumonitis. Colonic enlargement may result in constipation, abdominal distention, and intestinal obstruction. Sometimes, the small bowel, ureters, and bronchi become dilated as well. These findings are the result of low-grade parasitemia, tissue inflammation, and immune-mediated disruption of the microvasculature.71,76
Diagnosis: Protozoa evident in acute phase
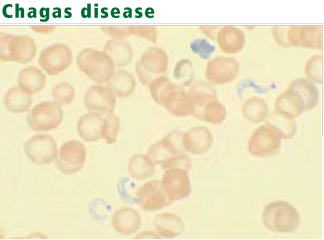
Serologic tests are most useful for diagnosing chronic Chagas disease.77 The organism can be identified by polymerase chain reaction, but the sensitivity of this test is highly variable.
Treatment reduces symptoms
Two drugs are used to treat American trypanosomiasis. Nifurtimox (Lampit) reduces symptom duration and severity, as well as mortality rates, in acute and congenital Chagas disease; however, fewer than 75% of patients have a parasitologic cure, and adverse effects limit tolerability.78 Benznidazole (Rochagan, Radanil) has an efficacy similar to that of nifurtimox and is considered the drug of choice in Latin America. These drugs are not commercially available in the United States. Therapy may help during the indeterminate phase but is rarely effective for chronic Chagas disease. Treatment of Chagas disease with triazoles is under evaluation.
- Kroft SH. Infectious diseases manifested in the peripheral blood. Clin Lab Med 2002; 22:253–277.
- Meldrum SC, Birkhead GS, White DJ, Benach JL, Morse DL. Human babesiosis in New York State: an epidemiological description of 136 cases. Clin Infect Dis 1992; 15:1019–1023.
- Pruthi RK, Marshall WF, Wiltsie JC, Persing DH. Human babesiosis. Mayo Clin Proc 1995; 70:853–862.
- Eskow ES, Krause PJ, Spielman A, Freeman K, Aslanzadeh J. Southern extension of the range of human babesiosis in the eastern United States. J Clin Microbiol 1999; 37:2051–2052.
- Quick RE, Herwaldt BL, Thomford JW, et al. Babesiosis in Washington State: a new species of Babesia? Ann Intern Med 1993; 119:284–290.
- Persing DH, Herwaldt BL, Glaser C, et al. Infection with a babesia-like organism in northern California. N Engl J Med 1995; 332:298–303.
- Gorenflot A, Moubri K, Precigout E, Carcy B, Schetters TP. Human babesiosis. Ann Trop Med Parasitol 1998; 92:489–501.
- Villar BF, White DJ, Benach JL. Human babesiosis. Prog Clin Parasitol 1991; 2:129–143.
- Sun T, Tenenbaum MJ, Greenspan J, et al. Morphologic and clinical observations in human infection with Babesia microti. J Infect Dis 1983; 148:239–248.
- Benach JL, Habicht GS. Clinical characteristics of human babesiosis. J Infect Dis 1981; 144:481.
- Boustani MR, Gelfand JA. Babesiosis. Clin Infect Dis 1996; 22:611–615.
- Meer-Scherrer L, Adelson M, Mordechai E, Lottaz B, Tilton R. Babesia microti infection in Europe. Curr Microbiol 2004; 48:435–437.
- Krause PJ. Babesiosis diagnosis and treatment. Vector Borne Zoonotic Dis 2003: 3:45–51.
- Persing DH, Mathiesen D, Marshall WF, et al. Detection of Babesia microti by polymerase chain reaction. J Clin Microbiol 1992; 30:2097–2103.
- Chisholm ES, Sulzer AJ, Ruebush TK. Indirect immunofluorescence test for human Babesia microti infection: antigen specificity. Am J Trop Med Hyg 1986; 35:921–925.
- Berry A, Morassin B, Kamar N, Magnaval JF. Clinical picture: human babesiosis. Lancet 2001; 357:341.
- Maeda K, Markowitz N, Hawley R, Ristic M, Cox D, McDade JE. Human infection with Ehrlichia canis, a leukocytic rickettsia. N Engl J Med 1987; 316:853–856.
- Chen SM, Dumler JS, Bakken JS, Walker DH. Identification of a granulocytotrophic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol 1994; 32:589–595.
- Lockhart JM, Davidson WR, Stallknecht DE, Dawson JE, Howerth EW. Isolation of Ehrlichia chaffeensis from wild white-tailed deer (Odocoileus virginianus) confirms their role as natural reservoir hosts. J Clin Microbiol 1997; 35:1681–1686.
- Stone JH, Dierberg K, Aram G, Dumler JS. Human monocytic erhlichiosis. JAMA 2004; 292:2263–2270.
- Bakken JS, Dumler JS. Human granulocytic ehrlichiosis. Clin Infect Dis 2000; 31:554–560.
- Dumler JS, Bakken JS. Ehrlichial diseases of humans: emerging tick-borne infections. Clin Infect Dis 1995; 20:1102–1110.
- Everett ED, Evans KA, Henry RB, McDonald G. Human ehrlichiosis in adults after tick exposure. Diagnosis using polymerase chain reaction. Ann Intern Med 1994; 120:730–735.
- Fishbein DB, Dawson JE, Robinson LE. Human ehrlichiosis in the United States, 1985 to 1990. Ann Intern Med 1994; 120:736–743.
- Bakken JS, Krueth J, Wilson-Nordskog C, Tilden RL, Asanovich K, Dumler JS. Clinical and laboratory characteristics of human granulocytic ehrlichiosis. JAMA 1996; 275:199–205.
- Ratnasamy N, Everett ED, Roland WE, McDonald G, Caldwell CW. Central nervous system manifestations of human ehrlichiosis. Clin Infect Dis 1996; 23:314–319.
- Vanek NN, Kazi S, Cepero NM, Tang S, Rex JH. Human ehrlichiosis causing left ventricular dilation and dysfunction. Clin Infect Dis 1996; 22:386–387.
- Bakken JS, Aguero-Rosenfeld ME, Tilden RL, et al. Serial measurements of hematologic counts during the active phase of human granulocytic ehrlichiosis. Clin Infect Dis 2001; 32:862–870.
- Hamilton KS, Standaert SM, Kinney MC. Characteristic peripheral blood findings in human ehrlichiosis. Mod Path 2004; 17:512–517.
- Dawson JE, Fishbein DB, Eng TR, Redus MA, Green NR. Diagnosis of human ehrlichiosis with the indirect fluorescent antibody test: kinetics and specificity. J Infect Dis 1990; 162:91–95.
- Walls JJ, Caturegli P, Bakken JS, Asanovich KM, Dumler JS. Improved sensitivity of PCR for diagnosis of human granulocytic ehrlichiosis using epank1 genes of Ehrlichia phagocytophila-group ehrlichiae. J Clin Microbiol 2000; 38:354–356.
- Barbour AG. Relapsing fever. In:Goodman JL, Dennis DT, Sonenshine DE, editors. Tick-Borne Diseases of Humans. Washington, DC: ASM Press; 2005:268.
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev 1986; 50:381–400.
- Hoogstraal H. Ticks and spirochetes. Acta Trop 1979; 36:133–136.
- Sonenshine D. Biology of Ticks. New York, NY: Oxford University Press; 1991.
- Felsenfeld O. Borrelia; Strains, Vectors, Human, and Animal Borreliosis. St Louis, MO: Warren H Greene, Inc; 1971.
- Bryceson AD, Parry EH, Perine PL, Warrell DA, Vukotich D, Leithead CS. Louse-borne relapsing fever. Q J Med 1970; 39:129–170.
- Judge DM, Samuel I, Perine PL, Vukotic D, Ababa A. Louse-borne relapsing fever in man. Arch Pathol 1974; 97:136–170.
- Southern P, Sanford J. Relapsing fever. A clinical and microbiological review. Medicine 1969; 48:129–149.
- Barbour AG. Antigenic variation of a relapsing fever Borrelia species. Annu Rev Microbiol 1990; 44:155–171.
- Stoenner HG, Dodd T, Larsen C. Antigenic variation of Borrelia hermsii. J Exp Med 1982; 156:1297–1311.
- Barbour AG. Immunobiology of relapsing fever. Contrib Microbiol Immunol 1987; 8:125–137.
- Scott R. Neurological complications of relapsing fever. Lancet 1944; 247:436–438.
- Cadavid D, Barbour AG. Neuroborreliosis during relapsing fever: review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin Infect Dis 1998; 26:151–164.
- Wengrower D, Knobler H, Gillis S, Chajek-Shaul T. Myocarditis in tick-borne relapsing fever. J Infect Dis 1984; 149:1033.
- Perine PL, Parry EH, Vukotich D, Warrell DA, Bryceson AD. Bleeding in louse-borne relapsing fever. I. Clinical studies in 37 patients. Trans R Soc Trop Med Hyg 1971; 65:776–781.
- Rhee KY, Johnson WD. Borrelia species (relapsing fever). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:2795–2798.
- Greenwood BM, Bojang K, Whitty CJ, Targett GA. Malaria. Lancet 2005; 365:1487–1498.
- Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol 1999; 29:927–937.
- Oh SS, Chishti AH, Palek J, Liu SC. Erythrocyte membrane alterations in Plasmodium falciparum malaria sequestration. Curr Opin Hematol 1997; 4:148–154.
- Singh B, Kim Sung L, Matusop A, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004; 363:1017–1024.
- Fairhurst RM, Wellems TE. Plasmodium species (malaria). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3121,3122.
- Filler S, Causer LM, Newman RD, et al Centers for Disease Control and Prevention (CDC). . Malaria surveillance—United States, 2001. MMWR Surveill Summ 2003; 52:1–14.
- Olliaro P, Cattani J, Wirth D. Malaria, the submerged disease. JAMA 1996; 275:230–233.
- Kain KC, Keystone JS. Malaria in travelers. Epidemiology, disease, and prevention. Infect Dis Clin North Am 1998; 12:267–284.
- World Health Organization, Division of Control of Tropical Diseases. Severe and complicated malaria. Trans R Soc Trop Med Hyg 1990; 84(suppl 2):1–65.
- Field JW. Blood examination and prognosis in acute falciparum malaria. Trans R Soc Trop Med Hyg 1949; 43:33–48.
- Microscopic procedures for diagnosing malaria. Appendix MMWR Surveill Summ 2003; 52:15–16.
- Lema OE, Carter JY, Nagelkerke N, et al. Comparison of five methods of malaria detection in the outpatient setting. Am J Trop Med Hyg 1999; 60:177–182.
- White NJ. The treatment of malaria. N Engl J Med 1996; 335:800–806.
- Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev 2002; 15:66–78.
- Makler MT, Palmer CJ, Ager AL. A review of practical techniques for the diagnosis of malaria. Ann Trop Med Parasitol 1998; 92:419–433.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3157–3158.
- Barrett MP, Burchmore RJ, Stich A, et al. The trypanosomiases. Lancet 2003; 362:1469–1480.
- Chagas disease. Interruption of transmission. Wkly Epidemiol Rec 1997; 71:1
- Control of Chagas disease. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 1991; 811:1–95.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). In:Guerrant R, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens and Practice; vol. 1. Philadelphia, PA: Churchill Livingstone; 1999:785.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). Gastroenterol Clin North Am 1996; 25:517–533.
- Wanderley DM, Corrêa FM. Epidemiology of Chagas’ heart disease. Sao Paul Med J 1995; 113:742–749.
- Schmuñis GA. Trypanosoma cruzi, the etiologic agent of Chagas’ disease: status in the blood supply in endemic and nonendemic countries. Transfusion 1991; 31:547–557.
- Köberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol 1968; 6:63–116.
- Dias E, Laranja FS, Miranda A, Nobrega G. Chagas’ disease; a clinical, epidemiologic, and pathologic study. Circulation 1956; 14:1035–1060.
- Prata A, Andrade Z, Guimaraes AC. Chagas’ heart disease. In:Shaper AG, Hutt MS, Fejfar Z, editors. Cardiovascular disease in the tropics. London, England: British Medical Association; 1974:264.
- Dias JC. The indeterminate form of human chronic Chagas’ disease. A clinical epidemiological study. Rev Soc Bras Med Trop 1989; 22:147–156.
- Lopes ER, Tafuri WL. Involvement of the autonomic nervous system in Chagas’ heart disease. Rev Soc Bras Med Trop 1983; 16:206.
- Meneghelli UG. Chagas’ disease: a model of denervation in the study of digestive tract motility. Braz J Med Biol Res 1985; 18:255–264.
- Pirard M, Iihoshi N, Boelaert M, Basanta P, Lopez F, Van der Stuyft P. The validity of serologic tests for Trypanosoma cruzi and the effectiveness of transfusional screening strategies in a hyperendemic region. Transfusion 2005; 45:554–561.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3162–3163.
Blood smear analysis, while commonly used to evaluate hematologic conditions, is infrequently used to diagnose infectious diseases. This is because of the rarity of diseases for which blood smear analysis is indicated. Consequently, such testing is often overlooked when it is diagnostically important.
Nonspecific changes may include morphologic changes in leukocytes and erythrocytes (eg, toxic granulations, macrocytosis).1 And with certain pathogens, identifying organisms in a peripheral blood smear allows for a rapid diagnosis.
This paper discusses the epidemiology, clinical manifestations, laboratory findings, and management of five infectious diseases in which direct visualization of the organism in the blood plays a major diagnostic role. Our intent is to summarize the clinical findings that should prompt blood smear analysis so that these uncommon conditions are not overlooked.
BABESIOSIS
Babesiosis, a tick-borne protozoal disease, occurs principally in the United States and Europe. Of the more than 100 species of Babesia, two account for almost all human disease: B microti and B divergens.
Both species are transmitted by Ixodes ticks, although patients often do not recall being bitten. The disease may rarely complicate blood transfusion. Most cases occur from May to September, when tick exposure is highest. The incubation period varies from 1 to 4 weeks.
Common in the Northeast, usually asymptomatic
B microti infection occurs predominantly in the United States. Rodents, especially the white-footed mouse, are the principal reservoir.2,3 Endemic areas, where seropositivity rates range from 4% to 21%, include the coastal areas and islands off of Massachusetts, particularly Cape Cod, Nantucket, and Martha’s Vineyard; the islands near New York City, especially Long Island, Shelter Island, and Fire Island; Block Island, off the coast of Rhode Island; and certain areas in Connecticut.4 WA-1, a species that is morphologically identical to B microti, is emerging in California and Washington.5,6
Infection with B microti is usually asymptomatic. Elderly and immunosuppressed people, especially those without a spleen or with impaired cellular immunity, are more likely to become ill. Symptoms, including fever, malaise, headache, nausea, and generalized aching, may last weeks to months.
About one-fourth of patients with babesiosis are coinfected with the Lyme disease bacterium (Borrelia burgdorferi) and often have more severe illness.3
Hepatomegaly, splenomegaly, jaundice, and dark urine are common findings in patients with symptoms. Severe hemolysis, often accompanied by thrombocytopenia, leukopenia, and atypical lymphocytosis, is more common in high-risk patients. Hepatic transaminases may be elevated. Urinalysis may show proteinuria and hemoglobinuria. Acute respiratory distress syndrome has been reported in severe cases.7–10
B divergens infection: A serious but rare disease seen in Europe
B divergens is found mainly in Europe. Altogether, fewer than 50 cases of infection have been reported in France, Spain, Germany, Great Britain, Ireland, Yugoslavia, and the former Soviet Union.11,12 Cattle are the principal reservoir of infection.
Infection with B divergens causes a rare but devastating disease mainly in asplenic people, usually resulting in coma and death. No cases of subclinical infection have been reported. The clinical course is fulminant, and hemolytic anemia is common.2,3
Suspect babesiosis in endemic areas in cases of prolonged ‘flu’
Babesiosis should be considered when a patient residing in or traveling from an endemic area presents with a prolonged flu-like illness and hemolysis, with or without organomegaly and jaundice.
The protozoa may resemble the rings of malaria parasites. Distinguishing traits include exoerythrocytic organisms; the absence of pigmented granules in infected red blood cells; and a “maltese cross,” a rare pattern produced by tetrads of Babesia merozoites.13 An infected erythrocyte may contain up to eight parasites.
Serologic and polymerase chain reaction tests are useful when the organism is not visible.14,15
Treat patients with severe disease
Most patients with B microti infection have a mild illness that resolves without treatment. Treatment is recommended for those with severe infection and in those with high-level parasitemia. Agents with consistent activity against B microti include clindamycin (Cleocin), azithromycin (Zithromax), ato-vaquone (Mepron), doxycycline (Vibramycin), and quinine (Quinamm). Combination therapy with either clindamycin and quinine or azithromycin and atovaquone is recommended. B divergens infection has been successfully treated with a combination of clindamycin, quinine, and exchange transfusion.16
EHRLICHIOSIS
Ehrlichiosis, nicknamed Rocky Mountain “spotless” fever, is a seasonal, tick-borne disease caused by obligate intracellular bacteria. Bacteria of the genus Ehrlichia grow within the cytoplasmic vacuoles of leukocytes and cause mainly zoonotic infections. Several species, especially Ehrlichia chaffeensis and Anaplasma phagocytophilum, are recognized as human pathogens.17,18E chaffeensis infects mononuclear cells, causing a condition known as human monocytic ehrlichiosis (HME). A phagocytophilum infects neutrophils, producing a condition called human granulocytic anaplasmosis (HGA).
Deer are the principal reservoir for E chaffeensis19; white-footed mice, other rodents, and deer are the principal reservoirs for A phagocytophilum. HME is transmitted by Dermacentor and Ixodes ticks, and HGA by Ixodes ticks. Human infection usually occurs in the spring and summer, when tick exposure is greatest. Co-infection of ticks with the organisms causing Lyme disease or babesiosis may result in simultaneous transmission of these diseases.
More than 1,000 cases of HME have been reported in the southeastern, south-central, and mid-Atlantic regions of the United States.20 The prevalence of HME in the United States appears to follow that of Rocky Mountain spotted fever. Some cases have been described in New England and in the Pacific Northwest. The more than 600 reported cases of HGA have come from Wisconsin, Minnesota, Connecticut, New York, Massachusetts, California, Florida, and Western Europe.21,22 The distribution of HGA follows that of Lyme disease, because the two diseases share the same tick vector.
Acute onset of fever and myalgias
HME and HGA have an incubation period of 1 to 2 weeks. The symptoms are similar and are usually acute, ranging from mild to severe. Most patients have fever, chills, malaise, headache, and myalgias. Many also have nausea, vomiting, cough, and arthralgias. Symptoms are similar to those in Rocky Mountain spotted fever (caused by Rickettsia rickettsii), except that rash is uncommon in HME (seen in approximately a third of patients) and rare in HGA.23–25 Neurologic findings, such as altered sensorium and neck stiffness, may be accompanied by lymphocytic pleocytosis and elevated protein levels in the cerebrospinal fluid.26 Subclinical and subacute presentations (eg, a fever lasting up to 2 months) are uncommon. No chronic cases have been reported.
The estimated death rate is 1% to 10%, and hospitalization rates are as high as 60%. Most deaths occur in the elderly, often following such complications as congestive heart failure,27 cardiac tamponade, respiratory or renal failure, seizures, and coma. Patients with human immunodeficiency virus infection also have a poor prognosis. Convalescence may be prolonged.
Laboratory abnormalities include leukopenia, thrombocytopenia, and elevated hepatic transaminase levels. Leukopenia may be associated with lymphopenia or neutropenia. Lymphopenia occurs early in the course of illness and is usually followed by an atypical lymphocytosis. Prolonged symptoms are associated with a decreased total neutrophil count and an increased band neutrophil count.28
Suspect ehrlichiosis in endemic areas in patients with fever, leukopenia, or thrombocytopenia
Ehrlichiosis should be suspected when a febrile patient with leukopenia or thrombocytopenia has been exposed to ticks in an endemic area. Even patients whose cell counts and liver enzyme levels are normal should be evaluated if the clinical and epidemiologic situations suggest this disease.
Other diagnostic tests include polymerase chain reaction and serologic assays, which are highly sensitive and specific.30,31 Because the organisms are difficult to culture in vitro, blood cultures are not useful diagnostically.
Treatment
Doxycycline 100 mg twice daily for 7 to 10 days is the treatment of choice for both HME and HGA. No role has been defined for fluoroquinolones for treating these diseases. Avoiding ticks and removing ticks promptly are the best preventive strategies.
RELAPSING FEVER
Relapsing fever is an acute febrile illness caused by spirochetes of the genus Borrelia. The disease has two forms: tick-borne, in which human infection is zoonotic, and louse-borne, in which humans are the only known reservoir of infection.32
Few tick-borne cases in the United States
Tick-borne disease is caused by many species of Borrelia. Those found in the United States occur in the western mountains and high deserts and plains of the Southwest.33 Fewer than 30 cases of tick-borne relapsing fever are diagnosed in the United States annually.
Tick-borne relapsing fever is transmitted by soft-bodied argasid ticks (Ornithodoros genus), which feed for less than an hour (usually at night) and can survive for years without a blood meal. They stay close to human and animal habitations. Exposure often occurs in cabins, under buildings, in caves, near woodpiles, and in rooms shared with animals. Rodents are the primary animal reservoir. In contrast, most other tick-borne diseases—babesiosis, ehrlichiosis, Lyme disease, Rocky Mountain spotted fever, Colorado tick fever—are transmitted by hard-bodied ixodid ticks, which live in brush and forested areas and attach to passersby, on whom they feed for days if not removed.34–36
Louse-borne disease is endemic in Africa
Louse-borne relapsing fever is caused by a single species, B recurrentis, endemic in Ethiopia and Sudan. It may occur sporadically or in epidemics. War, famine, and mass migrations predispose to epidemics with death rates ranging from 30% to 70% if untreated.37,38 Disease is spread between humans by the human body louse (Pediculus humanus).
Relapsing high fevers
The clinical manifestations of tick-borne and louse-borne relapsing fever are similar, although louse-borne relapsing fever often has a longer incubation period and a longer duration of illness. Bacteremia is heralded by the acute onset of high fever (usually above 39°C [102.2°F]), accompanied by headache, nausea, myalgias, and arthralgias. On average, clinical illness remits in 3 days in tick-borne relapsing fever, but may take 5 to 6 days in louse-borne relapsing fever. Physical findings may include altered sensorium, petechiae, hepatosplenomegaly, and conjunctival suffusion. The fever culminates in a “crisis,” characterized by rigors and a precipitous rise in temperature, pulse, and blood pressure. This is followed by defervescence, diaphoresis, and hypotension. The risk of death is highest during this period and immediately afterwards.
With resolution of the bacteremia, an afebrile period ensues, lasting 4 to 14 days. Fever then recurs, although usually milder, again associated with bacteremia. On average, people with tick-borne relapsing fever have three febrile relapses; those with louse-borne relapsing fever have one.39 Relapse occurs because of antigenic variation, in which a major surface antigen of the spirochete is changed to evade the host’s immune system.40–42
Borrelia may invade organs and the nervous system
With each episode of bacteremia, spirochetes may penetrate the brain, heart, liver, eye, or inner ear. Involvement of the central nervous system is more common with tick-borne than with louse-borne relapsing fever. Nervous system involvement may include facial palsy, myelitis, radiculopathy, aphasia, hemiplegia, stupor, or coma.43,44 Myocarditis, common in both forms of relapsing fever, portends a poor prognosis.45 Invasion of the eye or ear may result in visual impairment or dizziness. Bleeding disorders, manifested by epistaxis, petechiae, and ecchymoses, are typical of louse-borne disease and may be associated with low-grade disseminated intravascular coagulation.46 Splenomegaly is more common in louse-borne than in tick-borne disease.
Auxiliary test findings
Laboratory findings include normocytic anemia, leukocytosis, and thrombocytopenia. Liver enzyme levels may be elevated and coagulation tests may be prolonged. Patients with cardiac involvement may have a prolonged QTc interval. Cardiomegaly and pulmonary edema may be seen on chest radiography. The cerebrospinal fluid in patients with neurologic involvement has a mononuclear pleocytosis and a mildly elevated protein concentration.
Suspect if recurrent fever in endemic areas
Treatment
Relapsing fever can be successfully treated with tetracycline, penicillin, or erythromycin.47 The preferred regimen for a nonpregnant adult with louse-borne relapsing fever is a single 0.5-g dose of tetracycline. In tick-borne relapsing fever, 0.5 g of tetracycline four times daily for 5 to 10 days is recommended. Meningitis or encephalitis is usually treated with parenteral penicillin or ceftriaxone (Rocephin). The death rate in treated disease is usually less than 5%.39 Treatment can induce a Jarisch-Herxheimer reaction (rigors and hypotension, resembling a febrile crisis), and patients must be watched closely for the first 4 hours after the antibiotic is given. Avoiding ticks and practicing good personal hygiene to prevent acquiring lice are the major preventive strategies.
MALARIA
Malaria is caused by protozoa of the genus Plasmodium. Injected during a mosquito bite, sporozoites enter the bloodstream and travel to the liver. During the asymptomatic hepatic stage, the sporozoites multiply within hepatocytes to form mature schizonts. Within 1 to 2 weeks, the schizonts rupture, releasing thousands of merozoites into the bloodstream. The merozoites enter red blood cells, where they mature through the trophozoite stage to become schizonts, which release more merozoites into the blood that may infect other red blood cells. Symptoms occur during this erythrocytic stage, usually 1 to 4 weeks after a mosquito bite.
Four Plasmodia species regularly cause human disease.48 All can digest hemoglobin and cause hemolysis. P falciparum is uniquely dangerous, in part because it can alter the erythrocyte surface and obstruct the microcirculation.49,50 Further, P falciparum can cause high levels of parasitemia because it can infect red blood cells in all stages of their maturation. In contrast, P vivax and P ovale invade only reticulocytes, so levels of parasitemia are low (< 1%). P vivax and P ovale produce dormant liver forms (hypnozoites), which may cause relapses, usually within 3 years of exposure. Falciparum malaria does not have a dormant form, so relapses do not occur. P malariae infects only mature red blood cells, producing low levels of parasitemia. A fifth malaria species once thought to be confined to monkeys, P knowlesi, is a rare cause of severe human disease.51
Up to a half-billion cases of malaria occur worldwide each year.52 Although the disease predominates among residents of endemic areas, about 30,000 travelers from industrialized countries are infected each year. Fewer than 1,000 cases are reported in the United States annually, which were usually acquired during travel to endemic areas.53,54 The risk of transmission varies from region to region, with highest rates (listed in descending order) occurring in Oceania, sub-Saharan Africa, the Indian subcontinent, Southeast Asia, South America, and Central America.55 More than 3 million people die of malaria annually, most of them in sub-Saharan Africa.
Malaria is transmitted principally by the bite of the female Anopheles mosquito but can also be transmitted by blood transfusion, by the use of contaminated needles, congenitally, and through organ transplantation. Because immigrants to the United States from endemic areas may harbor the parasites for months to years, malaria may rarely be acquired by autochthonous transmission, in which a parasitized individual infects competent vectors, which then bite uninfected persons.55 The disease may also be transmitted from parasitized mosquitoes that arrive on an aircraft (“airport malaria”). There is no animal reservoir for human malaria parasites.
Recurring fever
Fever is universally present, irrespective of the species causing infection. Common symptoms include malaise, chills, headache, myalgias, abdominal pain, night sweats, nausea, and diarrhea. Febrile episodes, initially frequent and irregular, may become regular, producing temperature spikes every second or third day depending on the species. The severity of the paroxysms usually diminishes over time. Eventually, anemia, thrombocytopenia, jaundice, splenomegaly, and hepatomegaly occur.
Falciparum malaria may result in pulmonary edema, renal failure, gastroenteritis, bleeding, or hypoglycemia. Cerebral disease, presenting as altered sensorium or seizures, can be fatal.56 Death, common in untreated patients, correlates with the degree of parasitemia.57 Severe disease often occurs in children, pregnant women, asplenic patients, and nonimmune adults.
Infection with P vivax or P ovale does not produce the microvascular complications of falciparum malaria, and thrombocytopenia is less common. P malariae infection may lead to immune complex deposition and nephrotic syndrome. Symptoms, although mild, are often prolonged.
Consider malaria in travelers with recurring fever
Antigen-capture test kits are useful for rapid diagnosis. These portable devices detect parasitic antigens from a drop of blood in just 15 minutes.61 Polymerase chain reaction techniques offer high sensitivity and specificity but are more expensive and less available.62
The management of patients with suspected malaria should be done by experienced health care providers. Recommendations about malaria treatment are beyond the scope of this review. The choice of antimalarial drugs requires knowledge of the regional distribution of drug resistance and the adverse effects of the agents. Patients with non-falciparum malaria rarely require hospitalization.
Malaria is prevented by mosquito avoidance and chemoprophylaxis.
AMERICAN TRYPANOSOMIASIS (CHAGAS DISEASE)
American trypanosomiasis is a zoonotic, protozoal disease caused by Trypanosoma cruzi.
The parasite is transmitted by bloodsucking triatomine insects, or “kissing bugs.”63 These insects favor mud-brick and clay houses, where they live in wall cracks, under furniture, and behind pictures. The insects acquire the organism by feeding on infected animals or on humans that have circulating trypomastigotes. The organisms then multiply in the gut of the insects and are transmitted to a second vertebrate host as the insect defecates following a blood meal. The parasite then enters the body through the skin, conjunctivae, or mucous membranes. After entering the body, the parasite disseminates through the bloodstream, invading many cell types, especially muscle and nerve.
American trypanosomiasis occurs in Central and South America, Mexico, and the southern United States. Between 16 and 18 million people are infected with the parasite, and nearly 50,000 die annually, usually from cardiac complications. Once confined to rural areas, the disease is now common in cities. The majority of reported cases come from Brazil. Only a few cases have been reported in the United States, but an estimated 50,000 to 100,000 immigrants are thought to be infected.64–68
Transmission of the parasite may also occur with blood transfusion, with organ transplantation, and congenitally.63,69,70 An increase in transfusion-related cases is expected in the United States because of an influx of migrant workers from Mexico and Central America.
Acute phase ranges from asymptomatic to multiorgan involvement
Most infected people are asymptomatic carriers. Only 1% become acutely ill, but up to a third of those infected may develop chronic symptoms decades later. Acute Chagas disease, usually seen in children, is characterized by fever, malaise, and anorexia, often accompanied by vomiting, diarrhea, and rash.64,71,72 The heart, liver, spleen, and lymph nodes become enlarged. A red and indurated nodule or furuncle (chagoma) appears at the inoculation site. If inoculation occurs across the conjunctivae, painless edema of the palpebrae and periocular tissues may be observed (Romaña sign). Generalized lymphadenopathy and hepatosplenomegaly may also be seen.
Myocarditis and meningoencephalitis occur in some cases of acute Chagas disease. Myocardial inflammation may extend to the pericardium, causing pericardial effusion, and to the endocardium, causing thrombus formation. All cardiac chambers become enlarged and the conduction system is disrupted.73 Brain damage from meningoencephalitis usually occurs in infants and young children, and may result in death.
This acute phase lasts 4 to 8 weeks and is characterized by profound parasitemia, tissue invasion, and inflammation. Following the acute phase, an asymptomatic latent or indeterminate phase lasting 10 to 40 years occurs.74 Less than half of those in clinical latency enter a chronic phase of disease.
Severe chronic phase may occur decades later
The chronic phase, which occurs years to decades after the initial infection, is characterized by cardiac, esophageal, and colonic enlargement. Cardiac involvement is associated with congestive heart failure, arrhythmias, and cardiac arrest. Intracardiac thrombi may embolize, causing systemic and pulmonary infarctions.71–73,75 Enlargement of the esophagus is associated with dysphagia, chest pain, weight loss, and sometimes perforation or aspiration-related pneumonitis. Colonic enlargement may result in constipation, abdominal distention, and intestinal obstruction. Sometimes, the small bowel, ureters, and bronchi become dilated as well. These findings are the result of low-grade parasitemia, tissue inflammation, and immune-mediated disruption of the microvasculature.71,76
Diagnosis: Protozoa evident in acute phase
Serologic tests are most useful for diagnosing chronic Chagas disease.77 The organism can be identified by polymerase chain reaction, but the sensitivity of this test is highly variable.
Treatment reduces symptoms
Two drugs are used to treat American trypanosomiasis. Nifurtimox (Lampit) reduces symptom duration and severity, as well as mortality rates, in acute and congenital Chagas disease; however, fewer than 75% of patients have a parasitologic cure, and adverse effects limit tolerability.78 Benznidazole (Rochagan, Radanil) has an efficacy similar to that of nifurtimox and is considered the drug of choice in Latin America. These drugs are not commercially available in the United States. Therapy may help during the indeterminate phase but is rarely effective for chronic Chagas disease. Treatment of Chagas disease with triazoles is under evaluation.
Blood smear analysis, while commonly used to evaluate hematologic conditions, is infrequently used to diagnose infectious diseases. This is because of the rarity of diseases for which blood smear analysis is indicated. Consequently, such testing is often overlooked when it is diagnostically important.
Nonspecific changes may include morphologic changes in leukocytes and erythrocytes (eg, toxic granulations, macrocytosis).1 And with certain pathogens, identifying organisms in a peripheral blood smear allows for a rapid diagnosis.
This paper discusses the epidemiology, clinical manifestations, laboratory findings, and management of five infectious diseases in which direct visualization of the organism in the blood plays a major diagnostic role. Our intent is to summarize the clinical findings that should prompt blood smear analysis so that these uncommon conditions are not overlooked.
BABESIOSIS
Babesiosis, a tick-borne protozoal disease, occurs principally in the United States and Europe. Of the more than 100 species of Babesia, two account for almost all human disease: B microti and B divergens.
Both species are transmitted by Ixodes ticks, although patients often do not recall being bitten. The disease may rarely complicate blood transfusion. Most cases occur from May to September, when tick exposure is highest. The incubation period varies from 1 to 4 weeks.
Common in the Northeast, usually asymptomatic
B microti infection occurs predominantly in the United States. Rodents, especially the white-footed mouse, are the principal reservoir.2,3 Endemic areas, where seropositivity rates range from 4% to 21%, include the coastal areas and islands off of Massachusetts, particularly Cape Cod, Nantucket, and Martha’s Vineyard; the islands near New York City, especially Long Island, Shelter Island, and Fire Island; Block Island, off the coast of Rhode Island; and certain areas in Connecticut.4 WA-1, a species that is morphologically identical to B microti, is emerging in California and Washington.5,6
Infection with B microti is usually asymptomatic. Elderly and immunosuppressed people, especially those without a spleen or with impaired cellular immunity, are more likely to become ill. Symptoms, including fever, malaise, headache, nausea, and generalized aching, may last weeks to months.
About one-fourth of patients with babesiosis are coinfected with the Lyme disease bacterium (Borrelia burgdorferi) and often have more severe illness.3
Hepatomegaly, splenomegaly, jaundice, and dark urine are common findings in patients with symptoms. Severe hemolysis, often accompanied by thrombocytopenia, leukopenia, and atypical lymphocytosis, is more common in high-risk patients. Hepatic transaminases may be elevated. Urinalysis may show proteinuria and hemoglobinuria. Acute respiratory distress syndrome has been reported in severe cases.7–10
B divergens infection: A serious but rare disease seen in Europe
B divergens is found mainly in Europe. Altogether, fewer than 50 cases of infection have been reported in France, Spain, Germany, Great Britain, Ireland, Yugoslavia, and the former Soviet Union.11,12 Cattle are the principal reservoir of infection.
Infection with B divergens causes a rare but devastating disease mainly in asplenic people, usually resulting in coma and death. No cases of subclinical infection have been reported. The clinical course is fulminant, and hemolytic anemia is common.2,3
Suspect babesiosis in endemic areas in cases of prolonged ‘flu’
Babesiosis should be considered when a patient residing in or traveling from an endemic area presents with a prolonged flu-like illness and hemolysis, with or without organomegaly and jaundice.
The protozoa may resemble the rings of malaria parasites. Distinguishing traits include exoerythrocytic organisms; the absence of pigmented granules in infected red blood cells; and a “maltese cross,” a rare pattern produced by tetrads of Babesia merozoites.13 An infected erythrocyte may contain up to eight parasites.
Serologic and polymerase chain reaction tests are useful when the organism is not visible.14,15
Treat patients with severe disease
Most patients with B microti infection have a mild illness that resolves without treatment. Treatment is recommended for those with severe infection and in those with high-level parasitemia. Agents with consistent activity against B microti include clindamycin (Cleocin), azithromycin (Zithromax), ato-vaquone (Mepron), doxycycline (Vibramycin), and quinine (Quinamm). Combination therapy with either clindamycin and quinine or azithromycin and atovaquone is recommended. B divergens infection has been successfully treated with a combination of clindamycin, quinine, and exchange transfusion.16
EHRLICHIOSIS
Ehrlichiosis, nicknamed Rocky Mountain “spotless” fever, is a seasonal, tick-borne disease caused by obligate intracellular bacteria. Bacteria of the genus Ehrlichia grow within the cytoplasmic vacuoles of leukocytes and cause mainly zoonotic infections. Several species, especially Ehrlichia chaffeensis and Anaplasma phagocytophilum, are recognized as human pathogens.17,18E chaffeensis infects mononuclear cells, causing a condition known as human monocytic ehrlichiosis (HME). A phagocytophilum infects neutrophils, producing a condition called human granulocytic anaplasmosis (HGA).
Deer are the principal reservoir for E chaffeensis19; white-footed mice, other rodents, and deer are the principal reservoirs for A phagocytophilum. HME is transmitted by Dermacentor and Ixodes ticks, and HGA by Ixodes ticks. Human infection usually occurs in the spring and summer, when tick exposure is greatest. Co-infection of ticks with the organisms causing Lyme disease or babesiosis may result in simultaneous transmission of these diseases.
More than 1,000 cases of HME have been reported in the southeastern, south-central, and mid-Atlantic regions of the United States.20 The prevalence of HME in the United States appears to follow that of Rocky Mountain spotted fever. Some cases have been described in New England and in the Pacific Northwest. The more than 600 reported cases of HGA have come from Wisconsin, Minnesota, Connecticut, New York, Massachusetts, California, Florida, and Western Europe.21,22 The distribution of HGA follows that of Lyme disease, because the two diseases share the same tick vector.
Acute onset of fever and myalgias
HME and HGA have an incubation period of 1 to 2 weeks. The symptoms are similar and are usually acute, ranging from mild to severe. Most patients have fever, chills, malaise, headache, and myalgias. Many also have nausea, vomiting, cough, and arthralgias. Symptoms are similar to those in Rocky Mountain spotted fever (caused by Rickettsia rickettsii), except that rash is uncommon in HME (seen in approximately a third of patients) and rare in HGA.23–25 Neurologic findings, such as altered sensorium and neck stiffness, may be accompanied by lymphocytic pleocytosis and elevated protein levels in the cerebrospinal fluid.26 Subclinical and subacute presentations (eg, a fever lasting up to 2 months) are uncommon. No chronic cases have been reported.
The estimated death rate is 1% to 10%, and hospitalization rates are as high as 60%. Most deaths occur in the elderly, often following such complications as congestive heart failure,27 cardiac tamponade, respiratory or renal failure, seizures, and coma. Patients with human immunodeficiency virus infection also have a poor prognosis. Convalescence may be prolonged.
Laboratory abnormalities include leukopenia, thrombocytopenia, and elevated hepatic transaminase levels. Leukopenia may be associated with lymphopenia or neutropenia. Lymphopenia occurs early in the course of illness and is usually followed by an atypical lymphocytosis. Prolonged symptoms are associated with a decreased total neutrophil count and an increased band neutrophil count.28
Suspect ehrlichiosis in endemic areas in patients with fever, leukopenia, or thrombocytopenia
Ehrlichiosis should be suspected when a febrile patient with leukopenia or thrombocytopenia has been exposed to ticks in an endemic area. Even patients whose cell counts and liver enzyme levels are normal should be evaluated if the clinical and epidemiologic situations suggest this disease.
Other diagnostic tests include polymerase chain reaction and serologic assays, which are highly sensitive and specific.30,31 Because the organisms are difficult to culture in vitro, blood cultures are not useful diagnostically.
Treatment
Doxycycline 100 mg twice daily for 7 to 10 days is the treatment of choice for both HME and HGA. No role has been defined for fluoroquinolones for treating these diseases. Avoiding ticks and removing ticks promptly are the best preventive strategies.
RELAPSING FEVER
Relapsing fever is an acute febrile illness caused by spirochetes of the genus Borrelia. The disease has two forms: tick-borne, in which human infection is zoonotic, and louse-borne, in which humans are the only known reservoir of infection.32
Few tick-borne cases in the United States
Tick-borne disease is caused by many species of Borrelia. Those found in the United States occur in the western mountains and high deserts and plains of the Southwest.33 Fewer than 30 cases of tick-borne relapsing fever are diagnosed in the United States annually.
Tick-borne relapsing fever is transmitted by soft-bodied argasid ticks (Ornithodoros genus), which feed for less than an hour (usually at night) and can survive for years without a blood meal. They stay close to human and animal habitations. Exposure often occurs in cabins, under buildings, in caves, near woodpiles, and in rooms shared with animals. Rodents are the primary animal reservoir. In contrast, most other tick-borne diseases—babesiosis, ehrlichiosis, Lyme disease, Rocky Mountain spotted fever, Colorado tick fever—are transmitted by hard-bodied ixodid ticks, which live in brush and forested areas and attach to passersby, on whom they feed for days if not removed.34–36
Louse-borne disease is endemic in Africa
Louse-borne relapsing fever is caused by a single species, B recurrentis, endemic in Ethiopia and Sudan. It may occur sporadically or in epidemics. War, famine, and mass migrations predispose to epidemics with death rates ranging from 30% to 70% if untreated.37,38 Disease is spread between humans by the human body louse (Pediculus humanus).
Relapsing high fevers
The clinical manifestations of tick-borne and louse-borne relapsing fever are similar, although louse-borne relapsing fever often has a longer incubation period and a longer duration of illness. Bacteremia is heralded by the acute onset of high fever (usually above 39°C [102.2°F]), accompanied by headache, nausea, myalgias, and arthralgias. On average, clinical illness remits in 3 days in tick-borne relapsing fever, but may take 5 to 6 days in louse-borne relapsing fever. Physical findings may include altered sensorium, petechiae, hepatosplenomegaly, and conjunctival suffusion. The fever culminates in a “crisis,” characterized by rigors and a precipitous rise in temperature, pulse, and blood pressure. This is followed by defervescence, diaphoresis, and hypotension. The risk of death is highest during this period and immediately afterwards.
With resolution of the bacteremia, an afebrile period ensues, lasting 4 to 14 days. Fever then recurs, although usually milder, again associated with bacteremia. On average, people with tick-borne relapsing fever have three febrile relapses; those with louse-borne relapsing fever have one.39 Relapse occurs because of antigenic variation, in which a major surface antigen of the spirochete is changed to evade the host’s immune system.40–42
Borrelia may invade organs and the nervous system
With each episode of bacteremia, spirochetes may penetrate the brain, heart, liver, eye, or inner ear. Involvement of the central nervous system is more common with tick-borne than with louse-borne relapsing fever. Nervous system involvement may include facial palsy, myelitis, radiculopathy, aphasia, hemiplegia, stupor, or coma.43,44 Myocarditis, common in both forms of relapsing fever, portends a poor prognosis.45 Invasion of the eye or ear may result in visual impairment or dizziness. Bleeding disorders, manifested by epistaxis, petechiae, and ecchymoses, are typical of louse-borne disease and may be associated with low-grade disseminated intravascular coagulation.46 Splenomegaly is more common in louse-borne than in tick-borne disease.
Auxiliary test findings
Laboratory findings include normocytic anemia, leukocytosis, and thrombocytopenia. Liver enzyme levels may be elevated and coagulation tests may be prolonged. Patients with cardiac involvement may have a prolonged QTc interval. Cardiomegaly and pulmonary edema may be seen on chest radiography. The cerebrospinal fluid in patients with neurologic involvement has a mononuclear pleocytosis and a mildly elevated protein concentration.
Suspect if recurrent fever in endemic areas
Treatment
Relapsing fever can be successfully treated with tetracycline, penicillin, or erythromycin.47 The preferred regimen for a nonpregnant adult with louse-borne relapsing fever is a single 0.5-g dose of tetracycline. In tick-borne relapsing fever, 0.5 g of tetracycline four times daily for 5 to 10 days is recommended. Meningitis or encephalitis is usually treated with parenteral penicillin or ceftriaxone (Rocephin). The death rate in treated disease is usually less than 5%.39 Treatment can induce a Jarisch-Herxheimer reaction (rigors and hypotension, resembling a febrile crisis), and patients must be watched closely for the first 4 hours after the antibiotic is given. Avoiding ticks and practicing good personal hygiene to prevent acquiring lice are the major preventive strategies.
MALARIA
Malaria is caused by protozoa of the genus Plasmodium. Injected during a mosquito bite, sporozoites enter the bloodstream and travel to the liver. During the asymptomatic hepatic stage, the sporozoites multiply within hepatocytes to form mature schizonts. Within 1 to 2 weeks, the schizonts rupture, releasing thousands of merozoites into the bloodstream. The merozoites enter red blood cells, where they mature through the trophozoite stage to become schizonts, which release more merozoites into the blood that may infect other red blood cells. Symptoms occur during this erythrocytic stage, usually 1 to 4 weeks after a mosquito bite.
Four Plasmodia species regularly cause human disease.48 All can digest hemoglobin and cause hemolysis. P falciparum is uniquely dangerous, in part because it can alter the erythrocyte surface and obstruct the microcirculation.49,50 Further, P falciparum can cause high levels of parasitemia because it can infect red blood cells in all stages of their maturation. In contrast, P vivax and P ovale invade only reticulocytes, so levels of parasitemia are low (< 1%). P vivax and P ovale produce dormant liver forms (hypnozoites), which may cause relapses, usually within 3 years of exposure. Falciparum malaria does not have a dormant form, so relapses do not occur. P malariae infects only mature red blood cells, producing low levels of parasitemia. A fifth malaria species once thought to be confined to monkeys, P knowlesi, is a rare cause of severe human disease.51
Up to a half-billion cases of malaria occur worldwide each year.52 Although the disease predominates among residents of endemic areas, about 30,000 travelers from industrialized countries are infected each year. Fewer than 1,000 cases are reported in the United States annually, which were usually acquired during travel to endemic areas.53,54 The risk of transmission varies from region to region, with highest rates (listed in descending order) occurring in Oceania, sub-Saharan Africa, the Indian subcontinent, Southeast Asia, South America, and Central America.55 More than 3 million people die of malaria annually, most of them in sub-Saharan Africa.
Malaria is transmitted principally by the bite of the female Anopheles mosquito but can also be transmitted by blood transfusion, by the use of contaminated needles, congenitally, and through organ transplantation. Because immigrants to the United States from endemic areas may harbor the parasites for months to years, malaria may rarely be acquired by autochthonous transmission, in which a parasitized individual infects competent vectors, which then bite uninfected persons.55 The disease may also be transmitted from parasitized mosquitoes that arrive on an aircraft (“airport malaria”). There is no animal reservoir for human malaria parasites.
Recurring fever
Fever is universally present, irrespective of the species causing infection. Common symptoms include malaise, chills, headache, myalgias, abdominal pain, night sweats, nausea, and diarrhea. Febrile episodes, initially frequent and irregular, may become regular, producing temperature spikes every second or third day depending on the species. The severity of the paroxysms usually diminishes over time. Eventually, anemia, thrombocytopenia, jaundice, splenomegaly, and hepatomegaly occur.
Falciparum malaria may result in pulmonary edema, renal failure, gastroenteritis, bleeding, or hypoglycemia. Cerebral disease, presenting as altered sensorium or seizures, can be fatal.56 Death, common in untreated patients, correlates with the degree of parasitemia.57 Severe disease often occurs in children, pregnant women, asplenic patients, and nonimmune adults.
Infection with P vivax or P ovale does not produce the microvascular complications of falciparum malaria, and thrombocytopenia is less common. P malariae infection may lead to immune complex deposition and nephrotic syndrome. Symptoms, although mild, are often prolonged.
Consider malaria in travelers with recurring fever
Antigen-capture test kits are useful for rapid diagnosis. These portable devices detect parasitic antigens from a drop of blood in just 15 minutes.61 Polymerase chain reaction techniques offer high sensitivity and specificity but are more expensive and less available.62
The management of patients with suspected malaria should be done by experienced health care providers. Recommendations about malaria treatment are beyond the scope of this review. The choice of antimalarial drugs requires knowledge of the regional distribution of drug resistance and the adverse effects of the agents. Patients with non-falciparum malaria rarely require hospitalization.
Malaria is prevented by mosquito avoidance and chemoprophylaxis.
AMERICAN TRYPANOSOMIASIS (CHAGAS DISEASE)
American trypanosomiasis is a zoonotic, protozoal disease caused by Trypanosoma cruzi.
The parasite is transmitted by bloodsucking triatomine insects, or “kissing bugs.”63 These insects favor mud-brick and clay houses, where they live in wall cracks, under furniture, and behind pictures. The insects acquire the organism by feeding on infected animals or on humans that have circulating trypomastigotes. The organisms then multiply in the gut of the insects and are transmitted to a second vertebrate host as the insect defecates following a blood meal. The parasite then enters the body through the skin, conjunctivae, or mucous membranes. After entering the body, the parasite disseminates through the bloodstream, invading many cell types, especially muscle and nerve.
American trypanosomiasis occurs in Central and South America, Mexico, and the southern United States. Between 16 and 18 million people are infected with the parasite, and nearly 50,000 die annually, usually from cardiac complications. Once confined to rural areas, the disease is now common in cities. The majority of reported cases come from Brazil. Only a few cases have been reported in the United States, but an estimated 50,000 to 100,000 immigrants are thought to be infected.64–68
Transmission of the parasite may also occur with blood transfusion, with organ transplantation, and congenitally.63,69,70 An increase in transfusion-related cases is expected in the United States because of an influx of migrant workers from Mexico and Central America.
Acute phase ranges from asymptomatic to multiorgan involvement
Most infected people are asymptomatic carriers. Only 1% become acutely ill, but up to a third of those infected may develop chronic symptoms decades later. Acute Chagas disease, usually seen in children, is characterized by fever, malaise, and anorexia, often accompanied by vomiting, diarrhea, and rash.64,71,72 The heart, liver, spleen, and lymph nodes become enlarged. A red and indurated nodule or furuncle (chagoma) appears at the inoculation site. If inoculation occurs across the conjunctivae, painless edema of the palpebrae and periocular tissues may be observed (Romaña sign). Generalized lymphadenopathy and hepatosplenomegaly may also be seen.
Myocarditis and meningoencephalitis occur in some cases of acute Chagas disease. Myocardial inflammation may extend to the pericardium, causing pericardial effusion, and to the endocardium, causing thrombus formation. All cardiac chambers become enlarged and the conduction system is disrupted.73 Brain damage from meningoencephalitis usually occurs in infants and young children, and may result in death.
This acute phase lasts 4 to 8 weeks and is characterized by profound parasitemia, tissue invasion, and inflammation. Following the acute phase, an asymptomatic latent or indeterminate phase lasting 10 to 40 years occurs.74 Less than half of those in clinical latency enter a chronic phase of disease.
Severe chronic phase may occur decades later
The chronic phase, which occurs years to decades after the initial infection, is characterized by cardiac, esophageal, and colonic enlargement. Cardiac involvement is associated with congestive heart failure, arrhythmias, and cardiac arrest. Intracardiac thrombi may embolize, causing systemic and pulmonary infarctions.71–73,75 Enlargement of the esophagus is associated with dysphagia, chest pain, weight loss, and sometimes perforation or aspiration-related pneumonitis. Colonic enlargement may result in constipation, abdominal distention, and intestinal obstruction. Sometimes, the small bowel, ureters, and bronchi become dilated as well. These findings are the result of low-grade parasitemia, tissue inflammation, and immune-mediated disruption of the microvasculature.71,76
Diagnosis: Protozoa evident in acute phase
Serologic tests are most useful for diagnosing chronic Chagas disease.77 The organism can be identified by polymerase chain reaction, but the sensitivity of this test is highly variable.
Treatment reduces symptoms
Two drugs are used to treat American trypanosomiasis. Nifurtimox (Lampit) reduces symptom duration and severity, as well as mortality rates, in acute and congenital Chagas disease; however, fewer than 75% of patients have a parasitologic cure, and adverse effects limit tolerability.78 Benznidazole (Rochagan, Radanil) has an efficacy similar to that of nifurtimox and is considered the drug of choice in Latin America. These drugs are not commercially available in the United States. Therapy may help during the indeterminate phase but is rarely effective for chronic Chagas disease. Treatment of Chagas disease with triazoles is under evaluation.
- Kroft SH. Infectious diseases manifested in the peripheral blood. Clin Lab Med 2002; 22:253–277.
- Meldrum SC, Birkhead GS, White DJ, Benach JL, Morse DL. Human babesiosis in New York State: an epidemiological description of 136 cases. Clin Infect Dis 1992; 15:1019–1023.
- Pruthi RK, Marshall WF, Wiltsie JC, Persing DH. Human babesiosis. Mayo Clin Proc 1995; 70:853–862.
- Eskow ES, Krause PJ, Spielman A, Freeman K, Aslanzadeh J. Southern extension of the range of human babesiosis in the eastern United States. J Clin Microbiol 1999; 37:2051–2052.
- Quick RE, Herwaldt BL, Thomford JW, et al. Babesiosis in Washington State: a new species of Babesia? Ann Intern Med 1993; 119:284–290.
- Persing DH, Herwaldt BL, Glaser C, et al. Infection with a babesia-like organism in northern California. N Engl J Med 1995; 332:298–303.
- Gorenflot A, Moubri K, Precigout E, Carcy B, Schetters TP. Human babesiosis. Ann Trop Med Parasitol 1998; 92:489–501.
- Villar BF, White DJ, Benach JL. Human babesiosis. Prog Clin Parasitol 1991; 2:129–143.
- Sun T, Tenenbaum MJ, Greenspan J, et al. Morphologic and clinical observations in human infection with Babesia microti. J Infect Dis 1983; 148:239–248.
- Benach JL, Habicht GS. Clinical characteristics of human babesiosis. J Infect Dis 1981; 144:481.
- Boustani MR, Gelfand JA. Babesiosis. Clin Infect Dis 1996; 22:611–615.
- Meer-Scherrer L, Adelson M, Mordechai E, Lottaz B, Tilton R. Babesia microti infection in Europe. Curr Microbiol 2004; 48:435–437.
- Krause PJ. Babesiosis diagnosis and treatment. Vector Borne Zoonotic Dis 2003: 3:45–51.
- Persing DH, Mathiesen D, Marshall WF, et al. Detection of Babesia microti by polymerase chain reaction. J Clin Microbiol 1992; 30:2097–2103.
- Chisholm ES, Sulzer AJ, Ruebush TK. Indirect immunofluorescence test for human Babesia microti infection: antigen specificity. Am J Trop Med Hyg 1986; 35:921–925.
- Berry A, Morassin B, Kamar N, Magnaval JF. Clinical picture: human babesiosis. Lancet 2001; 357:341.
- Maeda K, Markowitz N, Hawley R, Ristic M, Cox D, McDade JE. Human infection with Ehrlichia canis, a leukocytic rickettsia. N Engl J Med 1987; 316:853–856.
- Chen SM, Dumler JS, Bakken JS, Walker DH. Identification of a granulocytotrophic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol 1994; 32:589–595.
- Lockhart JM, Davidson WR, Stallknecht DE, Dawson JE, Howerth EW. Isolation of Ehrlichia chaffeensis from wild white-tailed deer (Odocoileus virginianus) confirms their role as natural reservoir hosts. J Clin Microbiol 1997; 35:1681–1686.
- Stone JH, Dierberg K, Aram G, Dumler JS. Human monocytic erhlichiosis. JAMA 2004; 292:2263–2270.
- Bakken JS, Dumler JS. Human granulocytic ehrlichiosis. Clin Infect Dis 2000; 31:554–560.
- Dumler JS, Bakken JS. Ehrlichial diseases of humans: emerging tick-borne infections. Clin Infect Dis 1995; 20:1102–1110.
- Everett ED, Evans KA, Henry RB, McDonald G. Human ehrlichiosis in adults after tick exposure. Diagnosis using polymerase chain reaction. Ann Intern Med 1994; 120:730–735.
- Fishbein DB, Dawson JE, Robinson LE. Human ehrlichiosis in the United States, 1985 to 1990. Ann Intern Med 1994; 120:736–743.
- Bakken JS, Krueth J, Wilson-Nordskog C, Tilden RL, Asanovich K, Dumler JS. Clinical and laboratory characteristics of human granulocytic ehrlichiosis. JAMA 1996; 275:199–205.
- Ratnasamy N, Everett ED, Roland WE, McDonald G, Caldwell CW. Central nervous system manifestations of human ehrlichiosis. Clin Infect Dis 1996; 23:314–319.
- Vanek NN, Kazi S, Cepero NM, Tang S, Rex JH. Human ehrlichiosis causing left ventricular dilation and dysfunction. Clin Infect Dis 1996; 22:386–387.
- Bakken JS, Aguero-Rosenfeld ME, Tilden RL, et al. Serial measurements of hematologic counts during the active phase of human granulocytic ehrlichiosis. Clin Infect Dis 2001; 32:862–870.
- Hamilton KS, Standaert SM, Kinney MC. Characteristic peripheral blood findings in human ehrlichiosis. Mod Path 2004; 17:512–517.
- Dawson JE, Fishbein DB, Eng TR, Redus MA, Green NR. Diagnosis of human ehrlichiosis with the indirect fluorescent antibody test: kinetics and specificity. J Infect Dis 1990; 162:91–95.
- Walls JJ, Caturegli P, Bakken JS, Asanovich KM, Dumler JS. Improved sensitivity of PCR for diagnosis of human granulocytic ehrlichiosis using epank1 genes of Ehrlichia phagocytophila-group ehrlichiae. J Clin Microbiol 2000; 38:354–356.
- Barbour AG. Relapsing fever. In:Goodman JL, Dennis DT, Sonenshine DE, editors. Tick-Borne Diseases of Humans. Washington, DC: ASM Press; 2005:268.
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev 1986; 50:381–400.
- Hoogstraal H. Ticks and spirochetes. Acta Trop 1979; 36:133–136.
- Sonenshine D. Biology of Ticks. New York, NY: Oxford University Press; 1991.
- Felsenfeld O. Borrelia; Strains, Vectors, Human, and Animal Borreliosis. St Louis, MO: Warren H Greene, Inc; 1971.
- Bryceson AD, Parry EH, Perine PL, Warrell DA, Vukotich D, Leithead CS. Louse-borne relapsing fever. Q J Med 1970; 39:129–170.
- Judge DM, Samuel I, Perine PL, Vukotic D, Ababa A. Louse-borne relapsing fever in man. Arch Pathol 1974; 97:136–170.
- Southern P, Sanford J. Relapsing fever. A clinical and microbiological review. Medicine 1969; 48:129–149.
- Barbour AG. Antigenic variation of a relapsing fever Borrelia species. Annu Rev Microbiol 1990; 44:155–171.
- Stoenner HG, Dodd T, Larsen C. Antigenic variation of Borrelia hermsii. J Exp Med 1982; 156:1297–1311.
- Barbour AG. Immunobiology of relapsing fever. Contrib Microbiol Immunol 1987; 8:125–137.
- Scott R. Neurological complications of relapsing fever. Lancet 1944; 247:436–438.
- Cadavid D, Barbour AG. Neuroborreliosis during relapsing fever: review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin Infect Dis 1998; 26:151–164.
- Wengrower D, Knobler H, Gillis S, Chajek-Shaul T. Myocarditis in tick-borne relapsing fever. J Infect Dis 1984; 149:1033.
- Perine PL, Parry EH, Vukotich D, Warrell DA, Bryceson AD. Bleeding in louse-borne relapsing fever. I. Clinical studies in 37 patients. Trans R Soc Trop Med Hyg 1971; 65:776–781.
- Rhee KY, Johnson WD. Borrelia species (relapsing fever). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:2795–2798.
- Greenwood BM, Bojang K, Whitty CJ, Targett GA. Malaria. Lancet 2005; 365:1487–1498.
- Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol 1999; 29:927–937.
- Oh SS, Chishti AH, Palek J, Liu SC. Erythrocyte membrane alterations in Plasmodium falciparum malaria sequestration. Curr Opin Hematol 1997; 4:148–154.
- Singh B, Kim Sung L, Matusop A, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004; 363:1017–1024.
- Fairhurst RM, Wellems TE. Plasmodium species (malaria). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3121,3122.
- Filler S, Causer LM, Newman RD, et al Centers for Disease Control and Prevention (CDC). . Malaria surveillance—United States, 2001. MMWR Surveill Summ 2003; 52:1–14.
- Olliaro P, Cattani J, Wirth D. Malaria, the submerged disease. JAMA 1996; 275:230–233.
- Kain KC, Keystone JS. Malaria in travelers. Epidemiology, disease, and prevention. Infect Dis Clin North Am 1998; 12:267–284.
- World Health Organization, Division of Control of Tropical Diseases. Severe and complicated malaria. Trans R Soc Trop Med Hyg 1990; 84(suppl 2):1–65.
- Field JW. Blood examination and prognosis in acute falciparum malaria. Trans R Soc Trop Med Hyg 1949; 43:33–48.
- Microscopic procedures for diagnosing malaria. Appendix MMWR Surveill Summ 2003; 52:15–16.
- Lema OE, Carter JY, Nagelkerke N, et al. Comparison of five methods of malaria detection in the outpatient setting. Am J Trop Med Hyg 1999; 60:177–182.
- White NJ. The treatment of malaria. N Engl J Med 1996; 335:800–806.
- Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev 2002; 15:66–78.
- Makler MT, Palmer CJ, Ager AL. A review of practical techniques for the diagnosis of malaria. Ann Trop Med Parasitol 1998; 92:419–433.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3157–3158.
- Barrett MP, Burchmore RJ, Stich A, et al. The trypanosomiases. Lancet 2003; 362:1469–1480.
- Chagas disease. Interruption of transmission. Wkly Epidemiol Rec 1997; 71:1
- Control of Chagas disease. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 1991; 811:1–95.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). In:Guerrant R, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens and Practice; vol. 1. Philadelphia, PA: Churchill Livingstone; 1999:785.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). Gastroenterol Clin North Am 1996; 25:517–533.
- Wanderley DM, Corrêa FM. Epidemiology of Chagas’ heart disease. Sao Paul Med J 1995; 113:742–749.
- Schmuñis GA. Trypanosoma cruzi, the etiologic agent of Chagas’ disease: status in the blood supply in endemic and nonendemic countries. Transfusion 1991; 31:547–557.
- Köberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol 1968; 6:63–116.
- Dias E, Laranja FS, Miranda A, Nobrega G. Chagas’ disease; a clinical, epidemiologic, and pathologic study. Circulation 1956; 14:1035–1060.
- Prata A, Andrade Z, Guimaraes AC. Chagas’ heart disease. In:Shaper AG, Hutt MS, Fejfar Z, editors. Cardiovascular disease in the tropics. London, England: British Medical Association; 1974:264.
- Dias JC. The indeterminate form of human chronic Chagas’ disease. A clinical epidemiological study. Rev Soc Bras Med Trop 1989; 22:147–156.
- Lopes ER, Tafuri WL. Involvement of the autonomic nervous system in Chagas’ heart disease. Rev Soc Bras Med Trop 1983; 16:206.
- Meneghelli UG. Chagas’ disease: a model of denervation in the study of digestive tract motility. Braz J Med Biol Res 1985; 18:255–264.
- Pirard M, Iihoshi N, Boelaert M, Basanta P, Lopez F, Van der Stuyft P. The validity of serologic tests for Trypanosoma cruzi and the effectiveness of transfusional screening strategies in a hyperendemic region. Transfusion 2005; 45:554–561.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3162–3163.
- Kroft SH. Infectious diseases manifested in the peripheral blood. Clin Lab Med 2002; 22:253–277.
- Meldrum SC, Birkhead GS, White DJ, Benach JL, Morse DL. Human babesiosis in New York State: an epidemiological description of 136 cases. Clin Infect Dis 1992; 15:1019–1023.
- Pruthi RK, Marshall WF, Wiltsie JC, Persing DH. Human babesiosis. Mayo Clin Proc 1995; 70:853–862.
- Eskow ES, Krause PJ, Spielman A, Freeman K, Aslanzadeh J. Southern extension of the range of human babesiosis in the eastern United States. J Clin Microbiol 1999; 37:2051–2052.
- Quick RE, Herwaldt BL, Thomford JW, et al. Babesiosis in Washington State: a new species of Babesia? Ann Intern Med 1993; 119:284–290.
- Persing DH, Herwaldt BL, Glaser C, et al. Infection with a babesia-like organism in northern California. N Engl J Med 1995; 332:298–303.
- Gorenflot A, Moubri K, Precigout E, Carcy B, Schetters TP. Human babesiosis. Ann Trop Med Parasitol 1998; 92:489–501.
- Villar BF, White DJ, Benach JL. Human babesiosis. Prog Clin Parasitol 1991; 2:129–143.
- Sun T, Tenenbaum MJ, Greenspan J, et al. Morphologic and clinical observations in human infection with Babesia microti. J Infect Dis 1983; 148:239–248.
- Benach JL, Habicht GS. Clinical characteristics of human babesiosis. J Infect Dis 1981; 144:481.
- Boustani MR, Gelfand JA. Babesiosis. Clin Infect Dis 1996; 22:611–615.
- Meer-Scherrer L, Adelson M, Mordechai E, Lottaz B, Tilton R. Babesia microti infection in Europe. Curr Microbiol 2004; 48:435–437.
- Krause PJ. Babesiosis diagnosis and treatment. Vector Borne Zoonotic Dis 2003: 3:45–51.
- Persing DH, Mathiesen D, Marshall WF, et al. Detection of Babesia microti by polymerase chain reaction. J Clin Microbiol 1992; 30:2097–2103.
- Chisholm ES, Sulzer AJ, Ruebush TK. Indirect immunofluorescence test for human Babesia microti infection: antigen specificity. Am J Trop Med Hyg 1986; 35:921–925.
- Berry A, Morassin B, Kamar N, Magnaval JF. Clinical picture: human babesiosis. Lancet 2001; 357:341.
- Maeda K, Markowitz N, Hawley R, Ristic M, Cox D, McDade JE. Human infection with Ehrlichia canis, a leukocytic rickettsia. N Engl J Med 1987; 316:853–856.
- Chen SM, Dumler JS, Bakken JS, Walker DH. Identification of a granulocytotrophic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol 1994; 32:589–595.
- Lockhart JM, Davidson WR, Stallknecht DE, Dawson JE, Howerth EW. Isolation of Ehrlichia chaffeensis from wild white-tailed deer (Odocoileus virginianus) confirms their role as natural reservoir hosts. J Clin Microbiol 1997; 35:1681–1686.
- Stone JH, Dierberg K, Aram G, Dumler JS. Human monocytic erhlichiosis. JAMA 2004; 292:2263–2270.
- Bakken JS, Dumler JS. Human granulocytic ehrlichiosis. Clin Infect Dis 2000; 31:554–560.
- Dumler JS, Bakken JS. Ehrlichial diseases of humans: emerging tick-borne infections. Clin Infect Dis 1995; 20:1102–1110.
- Everett ED, Evans KA, Henry RB, McDonald G. Human ehrlichiosis in adults after tick exposure. Diagnosis using polymerase chain reaction. Ann Intern Med 1994; 120:730–735.
- Fishbein DB, Dawson JE, Robinson LE. Human ehrlichiosis in the United States, 1985 to 1990. Ann Intern Med 1994; 120:736–743.
- Bakken JS, Krueth J, Wilson-Nordskog C, Tilden RL, Asanovich K, Dumler JS. Clinical and laboratory characteristics of human granulocytic ehrlichiosis. JAMA 1996; 275:199–205.
- Ratnasamy N, Everett ED, Roland WE, McDonald G, Caldwell CW. Central nervous system manifestations of human ehrlichiosis. Clin Infect Dis 1996; 23:314–319.
- Vanek NN, Kazi S, Cepero NM, Tang S, Rex JH. Human ehrlichiosis causing left ventricular dilation and dysfunction. Clin Infect Dis 1996; 22:386–387.
- Bakken JS, Aguero-Rosenfeld ME, Tilden RL, et al. Serial measurements of hematologic counts during the active phase of human granulocytic ehrlichiosis. Clin Infect Dis 2001; 32:862–870.
- Hamilton KS, Standaert SM, Kinney MC. Characteristic peripheral blood findings in human ehrlichiosis. Mod Path 2004; 17:512–517.
- Dawson JE, Fishbein DB, Eng TR, Redus MA, Green NR. Diagnosis of human ehrlichiosis with the indirect fluorescent antibody test: kinetics and specificity. J Infect Dis 1990; 162:91–95.
- Walls JJ, Caturegli P, Bakken JS, Asanovich KM, Dumler JS. Improved sensitivity of PCR for diagnosis of human granulocytic ehrlichiosis using epank1 genes of Ehrlichia phagocytophila-group ehrlichiae. J Clin Microbiol 2000; 38:354–356.
- Barbour AG. Relapsing fever. In:Goodman JL, Dennis DT, Sonenshine DE, editors. Tick-Borne Diseases of Humans. Washington, DC: ASM Press; 2005:268.
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev 1986; 50:381–400.
- Hoogstraal H. Ticks and spirochetes. Acta Trop 1979; 36:133–136.
- Sonenshine D. Biology of Ticks. New York, NY: Oxford University Press; 1991.
- Felsenfeld O. Borrelia; Strains, Vectors, Human, and Animal Borreliosis. St Louis, MO: Warren H Greene, Inc; 1971.
- Bryceson AD, Parry EH, Perine PL, Warrell DA, Vukotich D, Leithead CS. Louse-borne relapsing fever. Q J Med 1970; 39:129–170.
- Judge DM, Samuel I, Perine PL, Vukotic D, Ababa A. Louse-borne relapsing fever in man. Arch Pathol 1974; 97:136–170.
- Southern P, Sanford J. Relapsing fever. A clinical and microbiological review. Medicine 1969; 48:129–149.
- Barbour AG. Antigenic variation of a relapsing fever Borrelia species. Annu Rev Microbiol 1990; 44:155–171.
- Stoenner HG, Dodd T, Larsen C. Antigenic variation of Borrelia hermsii. J Exp Med 1982; 156:1297–1311.
- Barbour AG. Immunobiology of relapsing fever. Contrib Microbiol Immunol 1987; 8:125–137.
- Scott R. Neurological complications of relapsing fever. Lancet 1944; 247:436–438.
- Cadavid D, Barbour AG. Neuroborreliosis during relapsing fever: review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin Infect Dis 1998; 26:151–164.
- Wengrower D, Knobler H, Gillis S, Chajek-Shaul T. Myocarditis in tick-borne relapsing fever. J Infect Dis 1984; 149:1033.
- Perine PL, Parry EH, Vukotich D, Warrell DA, Bryceson AD. Bleeding in louse-borne relapsing fever. I. Clinical studies in 37 patients. Trans R Soc Trop Med Hyg 1971; 65:776–781.
- Rhee KY, Johnson WD. Borrelia species (relapsing fever). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:2795–2798.
- Greenwood BM, Bojang K, Whitty CJ, Targett GA. Malaria. Lancet 2005; 365:1487–1498.
- Newbold C, Craig A, Kyes S, Rowe A, Fernandez-Reyes D, Fagan T. Cytoadherence, pathogenesis and the infected red cell surface in Plasmodium falciparum. Int J Parasitol 1999; 29:927–937.
- Oh SS, Chishti AH, Palek J, Liu SC. Erythrocyte membrane alterations in Plasmodium falciparum malaria sequestration. Curr Opin Hematol 1997; 4:148–154.
- Singh B, Kim Sung L, Matusop A, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 2004; 363:1017–1024.
- Fairhurst RM, Wellems TE. Plasmodium species (malaria). In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3121,3122.
- Filler S, Causer LM, Newman RD, et al Centers for Disease Control and Prevention (CDC). . Malaria surveillance—United States, 2001. MMWR Surveill Summ 2003; 52:1–14.
- Olliaro P, Cattani J, Wirth D. Malaria, the submerged disease. JAMA 1996; 275:230–233.
- Kain KC, Keystone JS. Malaria in travelers. Epidemiology, disease, and prevention. Infect Dis Clin North Am 1998; 12:267–284.
- World Health Organization, Division of Control of Tropical Diseases. Severe and complicated malaria. Trans R Soc Trop Med Hyg 1990; 84(suppl 2):1–65.
- Field JW. Blood examination and prognosis in acute falciparum malaria. Trans R Soc Trop Med Hyg 1949; 43:33–48.
- Microscopic procedures for diagnosing malaria. Appendix MMWR Surveill Summ 2003; 52:15–16.
- Lema OE, Carter JY, Nagelkerke N, et al. Comparison of five methods of malaria detection in the outpatient setting. Am J Trop Med Hyg 1999; 60:177–182.
- White NJ. The treatment of malaria. N Engl J Med 1996; 335:800–806.
- Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev 2002; 15:66–78.
- Makler MT, Palmer CJ, Ager AL. A review of practical techniques for the diagnosis of malaria. Ann Trop Med Parasitol 1998; 92:419–433.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3157–3158.
- Barrett MP, Burchmore RJ, Stich A, et al. The trypanosomiases. Lancet 2003; 362:1469–1480.
- Chagas disease. Interruption of transmission. Wkly Epidemiol Rec 1997; 71:1
- Control of Chagas disease. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser 1991; 811:1–95.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). In:Guerrant R, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens and Practice; vol. 1. Philadelphia, PA: Churchill Livingstone; 1999:785.
- Kirchhoff LV. American trypanosomiasis (Chagas’ disease). Gastroenterol Clin North Am 1996; 25:517–533.
- Wanderley DM, Corrêa FM. Epidemiology of Chagas’ heart disease. Sao Paul Med J 1995; 113:742–749.
- Schmuñis GA. Trypanosoma cruzi, the etiologic agent of Chagas’ disease: status in the blood supply in endemic and nonendemic countries. Transfusion 1991; 31:547–557.
- Köberle F. Chagas’ disease and Chagas’ syndromes: the pathology of American trypanosomiasis. Adv Parasitol 1968; 6:63–116.
- Dias E, Laranja FS, Miranda A, Nobrega G. Chagas’ disease; a clinical, epidemiologic, and pathologic study. Circulation 1956; 14:1035–1060.
- Prata A, Andrade Z, Guimaraes AC. Chagas’ heart disease. In:Shaper AG, Hutt MS, Fejfar Z, editors. Cardiovascular disease in the tropics. London, England: British Medical Association; 1974:264.
- Dias JC. The indeterminate form of human chronic Chagas’ disease. A clinical epidemiological study. Rev Soc Bras Med Trop 1989; 22:147–156.
- Lopes ER, Tafuri WL. Involvement of the autonomic nervous system in Chagas’ heart disease. Rev Soc Bras Med Trop 1983; 16:206.
- Meneghelli UG. Chagas’ disease: a model of denervation in the study of digestive tract motility. Braz J Med Biol Res 1985; 18:255–264.
- Pirard M, Iihoshi N, Boelaert M, Basanta P, Lopez F, Van der Stuyft P. The validity of serologic tests for Trypanosoma cruzi and the effectiveness of transfusional screening strategies in a hyperendemic region. Transfusion 2005; 45:554–561.
- Kirchhoff LV. Trypanosoma species (American trypanosomiasis, Chagas’ disease): Biology of trypanosomes. In:Mandell GL, Bennett JE, Dolin R, editors. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 6th ed. Philadelphia, PA: Elsevier Churchill Livingstone; 2005:3162–3163.
KEY POINTS
- In the United States, malaria and American trypanosomiasis principally affect travelers from the developing world.
- Babesiosis, ehrlichiosis, and relapsing fever are transmitted by ticks and may produce thrombocytopenia and elevation of liver enzyme levels.
- Malaria and babesiosis cause hemolytic anemia and may be associated with hepatomegaly and splenomegaly.
- Recurring fever is typical of malaria and Borrelia infection.