User login
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
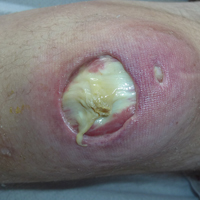
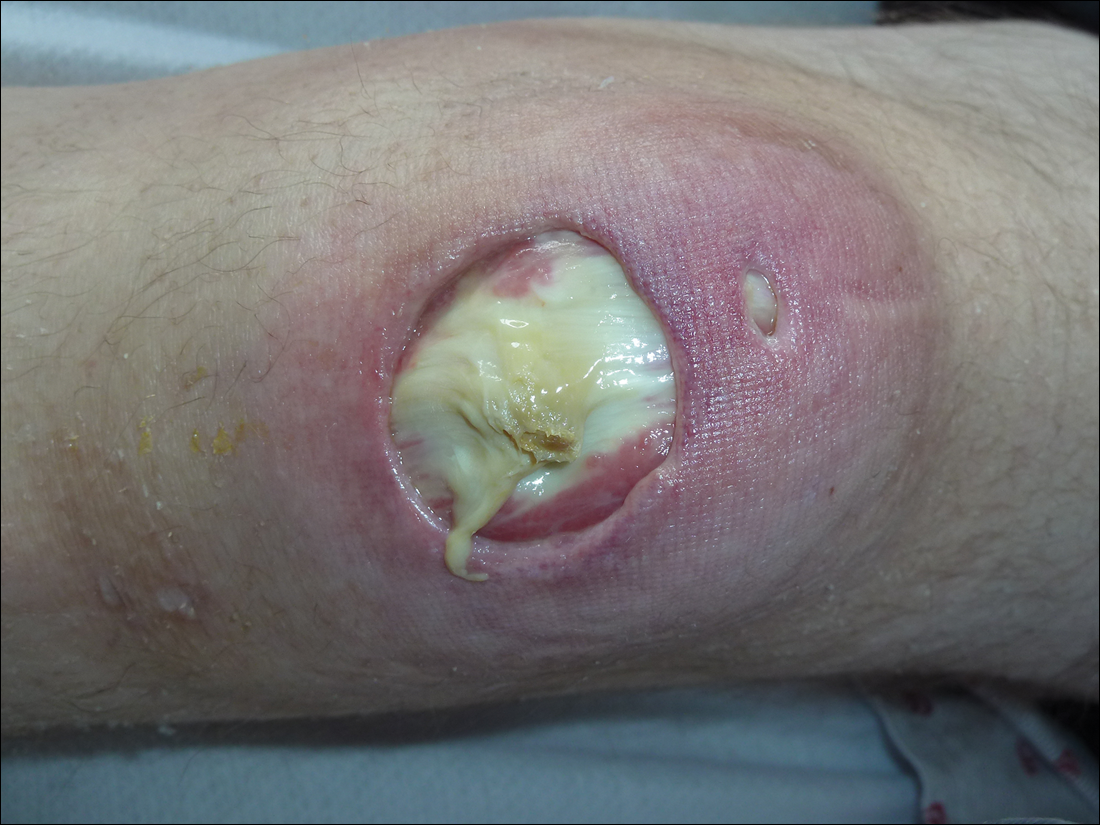
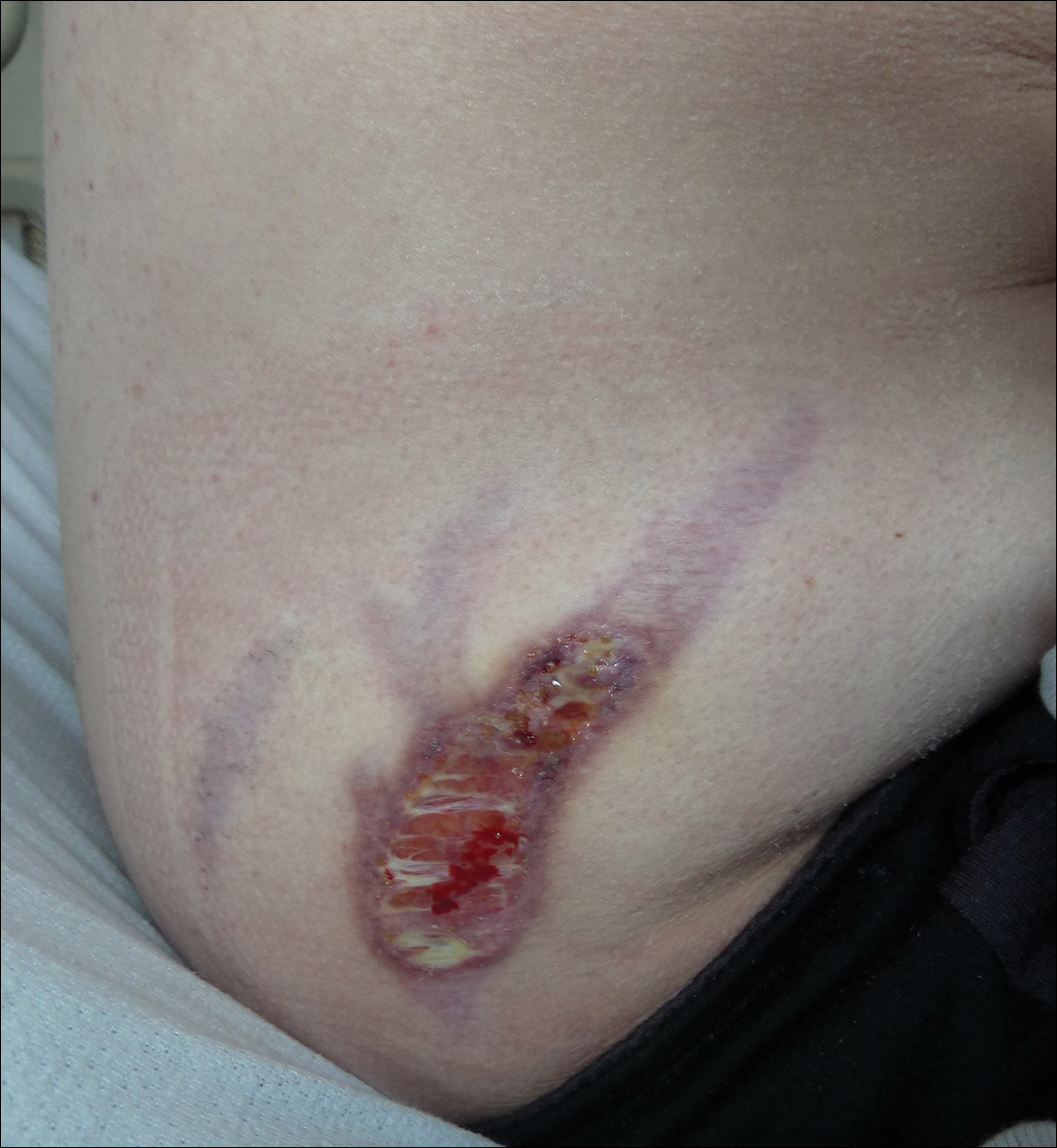
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
The process of wound healing has been well characterized. Immediately following injury, neutrophils arrive at the site in response to chemotactic factors produced by the coagulation cascade. Monocytes follow 24 to 36 hours later; transform into macrophages; and begin to phagocytose tissue debris, organisms, and any remaining neutrophils. In turn, macrophages release chemotactic factors such as basic fibroblast growth factor to attract fibroblasts to the wound, which then begin the process of synthesizing collagen and ground substance. Fibroblasts then take over as the dominant cell type, with collagen synthesis continuing for approximately 6 weeks. Keratinocytes and endothelial cells also proliferate during this time. After approximately 6 weeks, collagen remodeling begins. Tensile strength of the wound may continue to increase up to one year after the injury.1,2
Corticosteroids inhibit wound healing in several ways. Notably, they decrease the number of circulating monocytes, leading to fewer macrophages in the tissue at the site of injury, which then leads to impaired phagocytosis and reduced release of chemotactic factors that attract fibroblasts. Additionally, corticosteroids can inhibit collagen synthesis and remodeling, leading to delayed wound healing and decreased tensile strength of the wound as well as impacting capillary proliferation.3
The subtypes of EDS were reclassified in 1998 by Beighton et al,4 and the benign hypermobility type (EDS-BHT)(formerly type III) is considered the least severe. There is some controversy as to whether this subtype constitutes a separate diagnosis from the benign familial joint hypermobility syndrome. It is characterized by hypermobility of the joints (objectively measured with the Beighton scale) and mild hyperextensibility of the skin, and patients often have a history of joint subluxations and dislocations with resultant degenerative joint disease and chronic pain. Manifestations of fragile skin and soft tissue (eg, abnormal wound healing or scarring; spontaneous tearing of the skin, ligaments, tendons, or organs) are notably absent from the findings in this syndrome.5 The genetic basis for EDS is unknown in the majority of patients, although a deficiency in tenascin X (secondary to defects in the tenascin XB gene [TNXB]) has been identified in a small subset (<5%) of patients, leading to elastic fiber abnormalities, reduced collagen deposition, and impaired cross-linking of collagen.6,7 Inheritance usually is autosomal dominant but also can be autosomal recessive. In contrast, the classic type of EDS (formerly types I and II) is associated with atrophic scarring and tissue fragility, in addition to joint hypermobility and skin hyperextensibility. Type V collagen mutations are found in more than half of patients with this disorder.8
We present the case of a patient with EDS-BHT who developed large nonhealing cutaneous ulcerations with initiation of high-dose systemic corticosteroids for treatment of dermatomyositis. This case provides a dramatic illustration of the effects of the use of chronic systemic corticosteroids on skin fragility and wound healing in patients with an underlying inherited defect in collagen or connective tissue.
Case Report
A 23-year-old man with an unremarkable medical history was admitted to our inpatient cardiology service with palpitations attributable to new-onset atrial fibrillation. Dermatology was consulted to evaluate a rash of approximately 4 months’ duration that started on the dorsal aspect of the hands, then progressed to involve the extensor elbows and knees. The rash also was associated with fatigue, arthralgia, and proximal muscle weakness. A taper of prednisone that was prescribed approximately 2 months prior to admission by a rheumatologist for presumed dermatomyositis improved his symptoms, but they recurred with discontinuation of the medication.
Physical examination revealed reddish, violaceous and hyperpigmented patches on the dorsal aspect of the hands and digits and the extensor aspect of the knees and elbows. A skin biopsy from the right elbow showed a mild interface reaction and nonspecific direct immunofluorescence, consistent with a diagnosis of dermatomyositis. Autoimmune serologies were negative, including antinuclear, anti–Jo-1, anti–Mi-2, anti–Sjögren syndrome antigen A, anti–Sjögren syndrome antigen B, anti-Smith, and antiribonucleoprotein antibodies. Creatine kinase and rheumatoid factor levels were within reference range. Electromyogram was supportive of the diagnosis of dermatomyositis, showing an irritable myopathy. Cardiac magnetic resonance imaging showed an acute inflammatory process of the myocardium, and a transthoracic echocardiogram revealed a depressed left ventricular ejection fraction of 35% to 40% (reference range, 55%–70%). His cardiac disease also was attributed to dermatomyositis, and he was managed by cardiology with anangiotensin-converting enzyme inhibitor and antiarrhythmic therapy. Rheumatology was consulted and prednisone 60 mg once daily was started, with the patient reporting improvement in his muscle weakness and the rash.
Interestingly, the patient also noted a history of joint hypermobility, and a genetics consultation was obtained during the current hospitalization. He denied a history of abnormal scarring or skin problems, but he did note dislocation of the patella on 2 occasions and an umbilical hernia repair at 3 years of age. A paternal uncle had a history of similar joint hypermobility. His Beighton score was noted to be 8/8 (bending at the waist was unable to be tested due to recent lumbar puncture obtained during this hospitalization). The patient was diagnosed with EDS-BHT, and no further workup was recommended.
Subsequent to his hospitalization for several days, the patient’s prednisone was slowly tapered down from 60 mg once daily to 12.5 mg once daily, and azathioprine was started and titrated up to 150 mg once daily. Approximately 6 months after his initial hospitalization, he was readmitted due to increased pain of the right knee with concern for osteomyelitis. Dermatology was again consulted, and at this time, the patient reported a 4-month history of nonhealing ulcers to the knees and elbows (Figure 1). He stated that the ulcers were initially about the size of a pencil eraser and had started approximately 2 months after the prednisone was started, with subsequent slow enlargement. He noted a stinging sensation with enlargement of the ulcers, but otherwise they were not painful. He denied major trauma to the areas. He noted that his prior rash from the dermatomyositis seemed to have resolved, along with his muscle weakness, and he reported weight gain and improvement in his energy levels. Physical examination at this time revealed several stigmata of chronic systemic corticosteroids, including fatty deposits in the face (moon facies) and between the shoulders (buffalo hump), facial acne, and numerous erythematous striae on the trunk and proximal extremities (Figure 2). Multiple noninflammatory ulcers with punched-out borders ranging in size from 0.5 to 6 cm were seen at sites overlying bony prominences, including the bilateral extensor elbows and knees and the right plantar foot. Similar ulcers were noted on the trunk within the striae. Some of the ulcers were covered with a thick hyperkeratotic crust. A biopsy from the edge of an ulcer on the right side of the flank showed only dermal fibrosis. Workup by orthopedic surgery was felt to be inconsistent with osteomyelitis, and plastic surgery was consulted to consider surgical options for repair. Consequently, the patient was taken to the operating room for primary closure of the ulcers to the bilateral knees and right elbow. He has been followed closely by plastic surgery, with the use of joint immobilization to promote wound healing.
Comment
This case represents a dramatic illustration of the effects of chronic systemic corticosteroids on skin fragility and wound healing in a patient with an underlying genetic defect in the connective tissue. The ulcers were all located within striae or overlying bony prominences where the skin was subjected to increased tension; however, the patient reported no problems with wound healing or scarring at these sites prior to the initiation of corticosteroids, suggesting that the addition of this medication was disruptive to the cutaneous wound healing mechanisms. This case is unique because abnormal wound healing in an EDS patient was so clearly linked to the initiation of systemic steroids.
The exact pathogenesis of the patient’s ulcers is unclear. The diagnosis of EDS was primarily clinical, and without genetic testing, we cannot state with certainty the underlying molecular problem in this patient. Although tenascin X deficiency has been found in a few patients, a genetic defect remains uncharacterized in most patients with EDS-BHT, and in most situations, EDS-BHT remains a clinical diagnosis. In 2001, Schalkwijk et al9 first described the association of tenascin X deficiency and EDS in 5 patients, and they noted delayed wound healing in 1 patient who had received systemic corticosteroids for congenital adrenal hyperplasia. The authors remarked that it was not clear whether the abnormality was linked to the patient’s EDS or to his treatment with systemic corticosteroids.9 Furthermore, it is possible that our patient in fact has a milder variant of classic type EDS and that the manifestations of tissue fragility remained subclinical until the addition of systemic corticosteroids. It also is interesting to note that muscle weakness can be a symptom of EDS, both classic and BHT of EDS, but our patient’s muscle weakness improved with immunosuppression, supporting an underlying autoimmune disease as the cause for it.10 Skin ulcerations have been reported as a rare manifestation of dermatomyositis, but it is remarkable that his ulcers progressed as his other dermatomyositis symptoms improved with therapy, suggesting that his autoimmune disease was not the underlying cause for the ulcers.11-13 This case points to the need to thoughtfully consider the adverse effects of corticosteroids on wound healing in patients with an inherited disorder of collagen or connective tissue such as EDS.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
- Bolognia JL, Jorizzo JL, Rapini RP, et al. Dermatology. 2nd ed. Philadelphia, PA: Mosby Elsevier; 2008.
- Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314-321.
- Poetker DM, Reh DD. A comprehensive review of the adverse effects of systemic corticosteroids. Otolaryng Clin N Am. 2010;43:753-768.
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31-37.
- Levy HP. Ehlers-Danlos syndrome, hypermobility type. In: Pagon RA, Bird TD, Dolan CR, et al, es. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1279/. Accessed August 5, 2015.
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214-217.
- Brellier F, Tucker RP, Chiquet-Ehrismann R. Tenascins and their implications in diseases and tissue mechanics. Scand J Med Sci Spor. 2009;19:511-519.
- Malfait F, Wenstrup R, De Paepe A. Ehlers-Danlos syndrome, classic type. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle,WA: University of Washington, Seattle; 1993-2015. http://www.ncbi.nlm.nih.gov/books/NBK1244/. Accessed August 5, 2015.
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin X deficiency. N Engl J Med. 2001;345:1167-1175.
- Voermans NC, Alfen NV, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65:687-697.
- Scheinfeld NS. Ulcerative paraneoplastic dermatomyositis secondary to metastatic breast cancer. Skinmed. 2006;5:94-96.
- Tomb R, Stephan F. Perforating skin ulcers occurring in an adult with dermatomyositis [in French]. Ann Dermatol Venerol. 2002;129:1383-1385.
- Yosipovitch G, Feinmesser M, David M. Adult dermatomyositis with livedo reticularis and multiple skin ulcers. J Eur Acad Dermatol. 1998;11:48-50.
Practice Points
- Chronic corticosteroids have profound effects on the wound-healing process, and their detrimental effects may be amplified in patients with underlying connective tissue defects.
- Although genetic testing is available, the diagnosis of Ehlers-Danlos syndrome benign hypermobility type usually is made clinically.