User login

Pathophysiology
HCL develops from activated, mature memory B-cells that, in most cases, have the acquired mutation in BRAF V600E, which is present in 80% to 90% of patients with classic HCL.1,3,5 BRAF is an integral part of the RAS-BRAF-MEK-ERK cellular pathway that transmits growth factor signals from the cell surface to the nucleus to regulate cell growth and proliferation.6 Mutated BRAF V600E continuously activates BRAF kinase and downstream signaling, resulting in enhanced HCL cell survival and unchecked proliferation.3
Variant HCL (HCLv) is a separate, more virulent disease that lacks BRAF V600E mutation and CD25 expression on flow cytometry.1,7-9 Patients with HCLv have a worse prognosis and poor responses to front-line purine analogs, and a higher proportion of these patients carry the unmutated immunoglobulin heavy chain variable (IGHV) gene (54% vs 17% in HCL).1,10,11 About 30% to 50% have wild-type BRAF and activating mutations in MAP2K1, which encodes aberrant MEK downstream of BRAF.10,12
Most patients with HCL have somatic mutations in the IGHV gene.3,13,14 Patients with unmutated IGHV4-34 and wildtype BRAF have an aggressive form of the disease, even if the HCL cells express CD25 as in classic HCL.1,15 HCL in patients with unmutated IGHV is often refractory to purine analogs and these patients have poor prognosis and rapid progression.16 Other identified mutations include CDKN1B in HCL and MAP2K1 and CCNC3 in HCLv.2
Signs and Symptoms
In many cases, HCL is asymptomatic, and diagnosed when pancytopenia, monocytopenia, and leukopenia are discovered on unrelated blood work.2,3,11 Monocytopenia is a specific presentation of HCL, but not HCLv.11 Typical systemic symptoms include unexplained weight loss and extreme fatigue (80%).1,3 Other symptoms can include fever, recurrent infections, night sweats, splenomegaly and related pain or abdominal fullness, hepatomegaly, and bleeding or bruising due to thrombocytopenia.1,3 Splenomegaly is associated with advanced disease.11
Up to 30% of patients may present with autoimmune disorders such as vasculitis or psoriasis. Although skin involvement is rare with HCL, 10% to 12% of patients will have dermatologic symptoms either due to recurrent infection or autoimmune reactions.1,2 Skin reactions include localized or generalized maculopapular rash, pyoderma gangrenosum (which may be severe), and recurrent bacterial or viral skin infections.17
Diagnosis
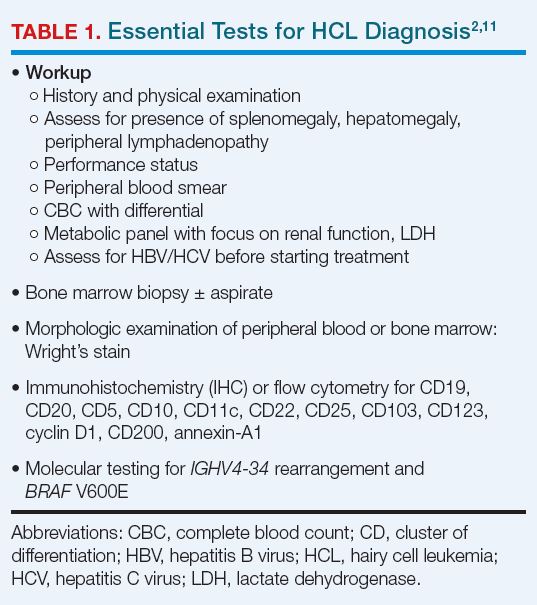
After complete history and physical examination, a diagnosis of HCL is usually made based on flow cytometry for immunophenotyping and molecular testing for BRAF V600E (Table 1).2,17
Disease-related fibrosis may impede bone marrow aspiration, and trephine biopsy should be done to make the diagnosis.11 On morphologic examination, HCL cells are small- to medium-sized, with round, oval, or indented, well-defined nuclei. Cytoplasm is pale blue, and cells have small cytoplasmic projections (Figure 1).2,18

On flow cytometry, HCL is positive for B-cell antigens (CD19, CD20, CD22), as well as antigens specific to the disease (CD11c, CD25, CD103, CD123), and by immunohistochemistry (IHC) for cyclin D1 and annexin-A1. CD20, CD123, and CD200 are bright in HCL. The presence of T-cell marker CD103 on B-cells indicates HCL.1-3 HCLv, in contrast, is positive for CD11c and CD103, but usually negative for CD25, CD123, and annexin-A1.2,19
BRAF V600E mutation can be identified using droplet digital polymerase chain reaction (PCR), next-generation molecular sequencing, or IHC with a VE1 stain.3,11 IHC for CD20, annexin-1, and VE1 establish the diagnosis, but also are useful in determining the extent to which leukemic cells have infiltrated bone marrow.11
Differential diagnosis of HCL includes HCL variants, splenic marginal zone lymphoma, and splenic diffuse red pulp small B-cell lymphoma.7,11
Indications for Treatment and Criteria for Response
Over time, about 90% of patients with HCL will require treatment. However, not all such patients will require urgent or immediate treatment, and some can be managed with observation and close monitoring.1,11 The indications for initiating treatment generally are systemic symptoms and significant pancytopenia (Table 2).2,11

The optimal response with treatment of HCL is complete response (CR) without minimal residual disease (MRD-free), which minimizes the risk for relapse.1,11 Hematologic and molecular response is assessed using peripheral blood samples; physical examination, ultrasound, computed tomography, or magnetic resonance imaging is used to determine response in lymph nodes, spleen, or liver.1 MRD-free is defined by the absence of HCL cells by the chosen method (IHC, flow cytometry, or PCR).20 Bone marrow aspirate flow cytometry is the most sensitive standard test for MRD detection.1 Table 3 summarizes response criteria for HCL.2,11

Initial Treatment of HCL
The purine nucleoside analogs (PNAs) cladribine (± rituximab) and pentostatin are widely recommended for initial treatment.1,2,11 As monotherapy, cladribine and pentostatin are considered similarly effective, with CR in 70% to 90% of patients and durations of response > 10 years.1 Adding the anti-CD20 monoclonal antibody rituximab in 8 weekly doses starting the first day of front-line cladribine (CDAR) improves remission, MRD-free rates, and duration of response (94% MRD-free at 96 months), with minimal added toxicity.21 Rituximab is often added 4 weeks after cladribine, which offers more convenience, an equally high CR rate of 100%, and a 76% MRD-free rate at 3 months.11 Bone marrow biopsy should be delayed for 4 to 6 months to allow a full response to develop with cladribine.1,11
Daily (intravenous or subcutaneous) and weekly cladribine are equally safe and effective.2,11 Pentostatin is administered intravenously every 2 weeks for 3 to 6 months, allowing time for hematologic recovery between doses.1,11 Patient factors to consider when choosing treatment include baseline neutropenia, patient preference, and comorbidities.
Toxicities of PNAs include neutropenia and fever, which typically occur during the first month of treatment and are more frequent in patients with baseline severe neutropenia; T-cell recovery may take years.1 CDAR is associated with higher transient thrombocytopenia, but faster platelet and neutrophil recovery at 4 weeks than cladribine alone.21 Both therapies are immunosuppressive. Patients should be evaluated for existing infections and watched for new infections during treatment. Control of active infection prior to treatment initiation is required.11,23
Patients with confirmed BRAF V600E mutation are candidates for vemurafenib if they are unable to tolerate a PNA, have an active infection, or would like effective vaccinations.2,23-25
Treatment at Relapse
At suspected HCL relapse, patients should be evaluated to determine whether cytopenia is due to recurrent disease or lingering effects from prior treatment. Use of successive flow cytometry over time can clarify whether symptoms are related to disease and need interventional treatment, or will resolve with additional time.1
Patients who have an HCL relapse after initial therapy with cladribine or pentostatin may be candidates for re-treatment with the same or alternate PNA plus rituximab.2 Rituximab
monotherapy has been used for patients unable to tolerate PNA but yields CR rates as low as 13%.26 Repeated courses of PNA therapy yield lower rates and durations of response with each course.1,2
For patients with primary refractory disease (less than CR with initial therapy) or relapse within 2 years of initial therapy, treatment with the BRAF V600E inhibitor vemurafenib off-label, with or without rituximab, is an option.2,5 In HCL, vemurafenib for patients with relapsed or refractory disease achieved CR in 35% and 42% in 2 small trials (N = 54). Relapse-free survival among people with CR was 19 months in 1 of the trials.27 Vemurafenib plus rituximab achieved CR in 87% of patients with relapsed or refractory HCL, and an MRD-free CR rate of 57%. Among patients with CR, 85% were relapse-free at a median follow-up of 34 months.5 Treatment with vemurafenib is not myelotoxic—an advantage for HCL patients. Adverse effects with vemurafenib are often manageable with dose reductions, if needed. A specific concern with vemurafenib is the potential development of secondary skin cancers.5,27,28
Novel Targeted Options and Recommended Use
Promising alternatives for patients with relapsed or refractory HCL include combined BRAF and MEK inhibitors and the Bruton tyrosine kinase (BTK) inhibitor ibrutinib. The concept of BRAF/MEK inhibition was validated in studies with BRAF-mutated melanoma, in which dabrafenib plus trametinib (the MEK inhibitor) improved overall survival (OS) with less toxicity and better quality of life than vemurafenib.1,29 In a phase 2 trial in HCL, dabrafenib monotherapy demonstrated an overall response rate (ORR) of 80%, including 30% CR.30 In a subsequent phase 2 trial, dabrafenib combined with trametinib was evaluated in refractory or late relapsed HCL. Among 55 enrolled patients, objective response rate was 89%, including 65.5% CR. Nine of 36 patients with CR were MRD-free. Among responding patients, duration of response was 97.7% at 24 months.31 The most common grade ≥ 3 toxicities were hyperglycemia, pyrexia, neutropenia, and pneumonia. Secondary skin cancers were seen in about 5% of patients.31
BRAF/MEK inhibitor combinations in HCL offer effective therapy with less myelosuppression than PNAs, making them useful for patients with or at risk for infection.23 Their use in HCL is off-label, as they currently are approved for treatment of BRAF-mutated melanoma and some other tumors.32 A study of encorafenib (a BRAF inhibitor) combined with binimetinib (a MEK inhibitor) is ongoing (Table 4).32

Ibrutinib interrupts B-cell receptor signaling to stop tumor cell growth. In a phase 2 trial, patients with relapsed or refractory HCL or HCLv were treated with once-daily oral ibrutinib. Best ORR was 54% (19% CR; 3% MRD-free). Despite the low CR rate, 3-year progression-free survival with ibrutinib was 73% and OS was 85%. Treatment was well-tolerated; cytopenia (including 22% grade ≥ 3 thrombocytopenia and neutropenia) and diarrhea were frequent toxicities.33
Moxetumomab pasudotox is a novel CD22-targeted antibody fused with protein toxin that interrupts protein synthesis in tumor cells.1 As treatment, it was studied in a phase 3 trial of relapsed HCL in heavily pretreated patients, and achieved a CR rate of 41%, including 36% durable CR.34 Although FDA-approved for relapsed or refractory HCL, the drug is being discontinued due to business decisions, not safety or efficacy concerns.2 It is notable that many types of B-cell lymphoma also express CD22.35
Enrollment in a clinical trial to study possible treatment advances is recommended by the National Comprehensive Cancer Network (NCCN) at first and subsequent relapses of HCL for appropriate patients.2 Figure 2 summarizes an approach to treatment choice and sequencing for patients with HCL.

Supportive Care
Patients being treated for HCL should have supportive care to manage adverse effects of their disease. Such care includes prophylaxis against herpes virus if CD4+ T cells < 200 cells/μL and other prophylactic vaccinations to hepatitis B virus, COVID-19 and Influenza. Patients with neutropeni may require broad-spectrum antibacterial prophylaxis or neutrophil growth factors if neutropenic fever develops. Blood product support is recommended if needed.2 Assessment of anti-COVID-19 antibodies is recommended to optimize immunity, particularly prior to beginning anti-CD20 antibody therapy like rituximab.23
Unmet Needs
Despite improvements in response and survival with newer therapies, not all patients with HCL benefit from these advances. Unmet needs are finding optimal treatment for patients with HCLv, despite some success with MEK inhibitors, and for patients with BRAF mutations other than V600E, who have few options beyond PNAs and rituximab.
- Kreitman RJ, Arons E. Diagnosis and treatment of hairy cell leukemia as the COVID-19 pandemic continues. Blood Rev. 2022;51:100888. doi:10.1016/j.blre.2021.100888
- National Comprehensive Cancer Network. NCCN clinical practice guideline in oncology: hairy cell leukemia. Version 1.2023. Published August 30, 2022. Accessed March 16, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hairy_cell.pdf
- Janus A, Robak T. Hairy cell leukemia. In: Li W, ed. Leukemia [Internet]. Brisbane: Exon Publications; 2022:chap3. Accessed February 16, 2023. doi:10.36255/exon-publications-leukemia-hairy-cell-leukemia
- Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol. 2015;28(4):175-179. doi:10.1016/j.beha.2015.10.014
- Tiacci E, De Carolis L, Simonetti E, et al. Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. N Engl J Med. 2021;384(19):1810-1823. doi:10.1056/NEJMoa20312986
- Falini B, Martelli MP, Tiacci E. BRAF V600E mutation in hairy cell leukemia: from bench to bedside. Blood. 2016;128(15):1918-1927. doi:10.1182/blood-2016-07-418434
- Matutes E. Diagnostic and therapeutic challenges in hairy cell leukemia-variant: where are we in 2021? Expert Rev Hematol. 2021;14(4):355-363. doi:10.1080/17474086.2021.1908121
- Cawley JC, Burns GF, Hayhoe FG. A chronic lymphoproliferative disorder with distinctive features: a distinct variant of hairy-cell leukaemia. Leuk Res. 1980;4(6):547-559. doi:10.1016/0145-2126(80)90066-1
- Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119(14):3330-3332. doi:10.1182/blood-2011-09-379339
- Durham BH, Getta B, Dietrich S, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130(14):1644-1648. doi:10.1182/blood-2017-01-76510711
- Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with hairy cell leukemia. Blood. 2017;129(5):553-560. doi:10.1182/blood-2016-01-689422
- Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46(1):8-10. doi:10.1038/ng.2828
- Arons E, Sunshine J, Suntum T, Kreitman RJ. Somatic hypermutation and VH gene usage in hairy cell leukaemia. Br J Haematol. 2006;133(5):504-512. doi:10.1111/j.1365-2141.2006.06066.x
- Arons E, Roth L, Sapolsky J, Suntum T, Stetler-Stevenson M, Kreitman RJ. Evidence of canonical somatic hypermutation in hairy cell leukemia. Blood. 2011;117(18):4844-4851. doi:10.1182/blood-2010-11-316737
- Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114(21):4687-4695. doi:10.1182/blood-2009-01-201731
- Forconi F, Sozzi E, Cencini E, et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood. 2009;114(21):4696-4702. doi:10.1182/blood-2009-03-212449
- Robak E, Jesionek-Kupnicka D, Robak T. Skin changes in hairy cell leukemia. Ann Hematol. 2021;100(3):615-625. doi:10.1007/s00277-020-04349-z
- Bouroncle BA. Thirty-five years in the progress of hairy cell leukemia. Leuk Lymphoma. 1994;14(suppl 1):1-12. https://pubmed.ncbi.nlm.nih.gov/7820038/
- Falini B, Tiacci E, Liso A, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet. 2004;363(9424): 1869-1870. doi:10.1016/S0140-6736(04)16356-3
- Robak T, Robak P. Measurable residual disease in hairy cell leukemia: technical considerations and clinical significance. Front Oncol. 2022;12:976374. doi:10.3389/fonc.2022.976374
- Chihara D, Arons E, Stetler-Stevenson M, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol. 2020;38(14):1527-1538. doi:10.1200/JCO.19.02250
- Chihara D, Kantarjian H, O’Brien S, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol. 2016;174(5):760-766. doi:10.1111/bjh.14129
- Grever M, Andritsos L, Banerji V, et al. Hairy cell leukemia and COVID-19 adaptation of treatment guidelines. Leukemia. 2021;35(7):1864-1872. doi:10.1038/s41375-021-01257-7
- Konrat J, Rösler W, Roiss M, et al. BRAF inhibitor treatment of classical hairy cell leukemia allows successful vaccination against SARS-CoV-2. Ann Hematol. 2023;102(2):403-406. doi:10.1007/s00277-022-05026-z
- Park JH, Shukla M, Salcedo JM, et al. First-line chemo-free therapy with the BRAF inhibitor vemurafenib combined with obinutuzumab is effective in patients with HCL. Blood. 2019;134(suppl 1):Abstract 3998. https://doi.org/10.1182/blood-2019-124478
- Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood. 2003;102(3):810-813. doi:10.1182/blood-2003-01-0014
- Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373(18):1733-1747. doi:10.1056/NEJMoa1506583
- Maitre E, Paillassa J, Troussard X. Novel targeted treatments in hairy cell leukemia and other hairy cell-like disorders. Front Oncol. 2022;12:1068981. doi:10.3389/fonc.2022.1068981
- Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015;16(13):1389-1398. doi:10.1016/S1470-2045(15)00087-X
- Tiacci E, De Carolis L, Simonetti E, et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: a pilot phase-2 clinical trial. Leukemia. 2021;35(11):3314-3318. doi:10.1038/s41375-021-01210-8
- Kreitman RJ, Moreau P, Ravandi F, et al. Dabrafenib plus trametinib in patients with relapsed/refractory BRAF V600E mutation-positive hairy cell leukemia. Blood. 2023;141(9):996-1006. doi:10.1182/blood.2021013658
- Adashek JJ, Menta AK, Reddy NK, Desai AP, Roszik J, Subbiah V. Tissue agnostic activity of BRAF plus MEK inhibitor in BRAF V600E-mutated tumors. Mol Cancer Ther. 2022;21(6):871-878. doi:10.1158/1535-7163.MCT-21-0950
- Rogers KA, Andritsos LA, Wei L, et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood. 2021;137(25):3473-3483. doi:10.1182/blood.2020009688
- Kreitman RJ, Dearden C, Zinzani PL, et al; Study 1053 investigators. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol. 2021;14(1):35. doi:10.1186/s13045-020-01004-y
- Leonard JP, Goldenberg DM. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene. 2007;26(25):3704-3713. doi:10.1038/sj.onc.1210370
Pathophysiology
HCL develops from activated, mature memory B-cells that, in most cases, have the acquired mutation in BRAF V600E, which is present in 80% to 90% of patients with classic HCL.1,3,5 BRAF is an integral part of the RAS-BRAF-MEK-ERK cellular pathway that transmits growth factor signals from the cell surface to the nucleus to regulate cell growth and proliferation.6 Mutated BRAF V600E continuously activates BRAF kinase and downstream signaling, resulting in enhanced HCL cell survival and unchecked proliferation.3
Variant HCL (HCLv) is a separate, more virulent disease that lacks BRAF V600E mutation and CD25 expression on flow cytometry.1,7-9 Patients with HCLv have a worse prognosis and poor responses to front-line purine analogs, and a higher proportion of these patients carry the unmutated immunoglobulin heavy chain variable (IGHV) gene (54% vs 17% in HCL).1,10,11 About 30% to 50% have wild-type BRAF and activating mutations in MAP2K1, which encodes aberrant MEK downstream of BRAF.10,12
Most patients with HCL have somatic mutations in the IGHV gene.3,13,14 Patients with unmutated IGHV4-34 and wildtype BRAF have an aggressive form of the disease, even if the HCL cells express CD25 as in classic HCL.1,15 HCL in patients with unmutated IGHV is often refractory to purine analogs and these patients have poor prognosis and rapid progression.16 Other identified mutations include CDKN1B in HCL and MAP2K1 and CCNC3 in HCLv.2
Signs and Symptoms
In many cases, HCL is asymptomatic, and diagnosed when pancytopenia, monocytopenia, and leukopenia are discovered on unrelated blood work.2,3,11 Monocytopenia is a specific presentation of HCL, but not HCLv.11 Typical systemic symptoms include unexplained weight loss and extreme fatigue (80%).1,3 Other symptoms can include fever, recurrent infections, night sweats, splenomegaly and related pain or abdominal fullness, hepatomegaly, and bleeding or bruising due to thrombocytopenia.1,3 Splenomegaly is associated with advanced disease.11
Up to 30% of patients may present with autoimmune disorders such as vasculitis or psoriasis. Although skin involvement is rare with HCL, 10% to 12% of patients will have dermatologic symptoms either due to recurrent infection or autoimmune reactions.1,2 Skin reactions include localized or generalized maculopapular rash, pyoderma gangrenosum (which may be severe), and recurrent bacterial or viral skin infections.17
Diagnosis
After complete history and physical examination, a diagnosis of HCL is usually made based on flow cytometry for immunophenotyping and molecular testing for BRAF V600E (Table 1).2,17
Disease-related fibrosis may impede bone marrow aspiration, and trephine biopsy should be done to make the diagnosis.11 On morphologic examination, HCL cells are small- to medium-sized, with round, oval, or indented, well-defined nuclei. Cytoplasm is pale blue, and cells have small cytoplasmic projections (Figure 1).2,18
On flow cytometry, HCL is positive for B-cell antigens (CD19, CD20, CD22), as well as antigens specific to the disease (CD11c, CD25, CD103, CD123), and by immunohistochemistry (IHC) for cyclin D1 and annexin-A1. CD20, CD123, and CD200 are bright in HCL. The presence of T-cell marker CD103 on B-cells indicates HCL.1-3 HCLv, in contrast, is positive for CD11c and CD103, but usually negative for CD25, CD123, and annexin-A1.2,19
BRAF V600E mutation can be identified using droplet digital polymerase chain reaction (PCR), next-generation molecular sequencing, or IHC with a VE1 stain.3,11 IHC for CD20, annexin-1, and VE1 establish the diagnosis, but also are useful in determining the extent to which leukemic cells have infiltrated bone marrow.11
Differential diagnosis of HCL includes HCL variants, splenic marginal zone lymphoma, and splenic diffuse red pulp small B-cell lymphoma.7,11
Indications for Treatment and Criteria for Response
Over time, about 90% of patients with HCL will require treatment. However, not all such patients will require urgent or immediate treatment, and some can be managed with observation and close monitoring.1,11 The indications for initiating treatment generally are systemic symptoms and significant pancytopenia (Table 2).2,11
The optimal response with treatment of HCL is complete response (CR) without minimal residual disease (MRD-free), which minimizes the risk for relapse.1,11 Hematologic and molecular response is assessed using peripheral blood samples; physical examination, ultrasound, computed tomography, or magnetic resonance imaging is used to determine response in lymph nodes, spleen, or liver.1 MRD-free is defined by the absence of HCL cells by the chosen method (IHC, flow cytometry, or PCR).20 Bone marrow aspirate flow cytometry is the most sensitive standard test for MRD detection.1 Table 3 summarizes response criteria for HCL.2,11
Initial Treatment of HCL
The purine nucleoside analogs (PNAs) cladribine (± rituximab) and pentostatin are widely recommended for initial treatment.1,2,11 As monotherapy, cladribine and pentostatin are considered similarly effective, with CR in 70% to 90% of patients and durations of response > 10 years.1 Adding the anti-CD20 monoclonal antibody rituximab in 8 weekly doses starting the first day of front-line cladribine (CDAR) improves remission, MRD-free rates, and duration of response (94% MRD-free at 96 months), with minimal added toxicity.21 Rituximab is often added 4 weeks after cladribine, which offers more convenience, an equally high CR rate of 100%, and a 76% MRD-free rate at 3 months.11 Bone marrow biopsy should be delayed for 4 to 6 months to allow a full response to develop with cladribine.1,11
Daily (intravenous or subcutaneous) and weekly cladribine are equally safe and effective.2,11 Pentostatin is administered intravenously every 2 weeks for 3 to 6 months, allowing time for hematologic recovery between doses.1,11 Patient factors to consider when choosing treatment include baseline neutropenia, patient preference, and comorbidities.
Toxicities of PNAs include neutropenia and fever, which typically occur during the first month of treatment and are more frequent in patients with baseline severe neutropenia; T-cell recovery may take years.1 CDAR is associated with higher transient thrombocytopenia, but faster platelet and neutrophil recovery at 4 weeks than cladribine alone.21 Both therapies are immunosuppressive. Patients should be evaluated for existing infections and watched for new infections during treatment. Control of active infection prior to treatment initiation is required.11,23
Patients with confirmed BRAF V600E mutation are candidates for vemurafenib if they are unable to tolerate a PNA, have an active infection, or would like effective vaccinations.2,23-25
Treatment at Relapse
At suspected HCL relapse, patients should be evaluated to determine whether cytopenia is due to recurrent disease or lingering effects from prior treatment. Use of successive flow cytometry over time can clarify whether symptoms are related to disease and need interventional treatment, or will resolve with additional time.1
Patients who have an HCL relapse after initial therapy with cladribine or pentostatin may be candidates for re-treatment with the same or alternate PNA plus rituximab.2 Rituximab
monotherapy has been used for patients unable to tolerate PNA but yields CR rates as low as 13%.26 Repeated courses of PNA therapy yield lower rates and durations of response with each course.1,2
For patients with primary refractory disease (less than CR with initial therapy) or relapse within 2 years of initial therapy, treatment with the BRAF V600E inhibitor vemurafenib off-label, with or without rituximab, is an option.2,5 In HCL, vemurafenib for patients with relapsed or refractory disease achieved CR in 35% and 42% in 2 small trials (N = 54). Relapse-free survival among people with CR was 19 months in 1 of the trials.27 Vemurafenib plus rituximab achieved CR in 87% of patients with relapsed or refractory HCL, and an MRD-free CR rate of 57%. Among patients with CR, 85% were relapse-free at a median follow-up of 34 months.5 Treatment with vemurafenib is not myelotoxic—an advantage for HCL patients. Adverse effects with vemurafenib are often manageable with dose reductions, if needed. A specific concern with vemurafenib is the potential development of secondary skin cancers.5,27,28
Novel Targeted Options and Recommended Use
Promising alternatives for patients with relapsed or refractory HCL include combined BRAF and MEK inhibitors and the Bruton tyrosine kinase (BTK) inhibitor ibrutinib. The concept of BRAF/MEK inhibition was validated in studies with BRAF-mutated melanoma, in which dabrafenib plus trametinib (the MEK inhibitor) improved overall survival (OS) with less toxicity and better quality of life than vemurafenib.1,29 In a phase 2 trial in HCL, dabrafenib monotherapy demonstrated an overall response rate (ORR) of 80%, including 30% CR.30 In a subsequent phase 2 trial, dabrafenib combined with trametinib was evaluated in refractory or late relapsed HCL. Among 55 enrolled patients, objective response rate was 89%, including 65.5% CR. Nine of 36 patients with CR were MRD-free. Among responding patients, duration of response was 97.7% at 24 months.31 The most common grade ≥ 3 toxicities were hyperglycemia, pyrexia, neutropenia, and pneumonia. Secondary skin cancers were seen in about 5% of patients.31
BRAF/MEK inhibitor combinations in HCL offer effective therapy with less myelosuppression than PNAs, making them useful for patients with or at risk for infection.23 Their use in HCL is off-label, as they currently are approved for treatment of BRAF-mutated melanoma and some other tumors.32 A study of encorafenib (a BRAF inhibitor) combined with binimetinib (a MEK inhibitor) is ongoing (Table 4).32
Ibrutinib interrupts B-cell receptor signaling to stop tumor cell growth. In a phase 2 trial, patients with relapsed or refractory HCL or HCLv were treated with once-daily oral ibrutinib. Best ORR was 54% (19% CR; 3% MRD-free). Despite the low CR rate, 3-year progression-free survival with ibrutinib was 73% and OS was 85%. Treatment was well-tolerated; cytopenia (including 22% grade ≥ 3 thrombocytopenia and neutropenia) and diarrhea were frequent toxicities.33
Moxetumomab pasudotox is a novel CD22-targeted antibody fused with protein toxin that interrupts protein synthesis in tumor cells.1 As treatment, it was studied in a phase 3 trial of relapsed HCL in heavily pretreated patients, and achieved a CR rate of 41%, including 36% durable CR.34 Although FDA-approved for relapsed or refractory HCL, the drug is being discontinued due to business decisions, not safety or efficacy concerns.2 It is notable that many types of B-cell lymphoma also express CD22.35
Enrollment in a clinical trial to study possible treatment advances is recommended by the National Comprehensive Cancer Network (NCCN) at first and subsequent relapses of HCL for appropriate patients.2 Figure 2 summarizes an approach to treatment choice and sequencing for patients with HCL.
Supportive Care
Patients being treated for HCL should have supportive care to manage adverse effects of their disease. Such care includes prophylaxis against herpes virus if CD4+ T cells < 200 cells/μL and other prophylactic vaccinations to hepatitis B virus, COVID-19 and Influenza. Patients with neutropeni may require broad-spectrum antibacterial prophylaxis or neutrophil growth factors if neutropenic fever develops. Blood product support is recommended if needed.2 Assessment of anti-COVID-19 antibodies is recommended to optimize immunity, particularly prior to beginning anti-CD20 antibody therapy like rituximab.23
Unmet Needs
Despite improvements in response and survival with newer therapies, not all patients with HCL benefit from these advances. Unmet needs are finding optimal treatment for patients with HCLv, despite some success with MEK inhibitors, and for patients with BRAF mutations other than V600E, who have few options beyond PNAs and rituximab.
Pathophysiology
HCL develops from activated, mature memory B-cells that, in most cases, have the acquired mutation in BRAF V600E, which is present in 80% to 90% of patients with classic HCL.1,3,5 BRAF is an integral part of the RAS-BRAF-MEK-ERK cellular pathway that transmits growth factor signals from the cell surface to the nucleus to regulate cell growth and proliferation.6 Mutated BRAF V600E continuously activates BRAF kinase and downstream signaling, resulting in enhanced HCL cell survival and unchecked proliferation.3
Variant HCL (HCLv) is a separate, more virulent disease that lacks BRAF V600E mutation and CD25 expression on flow cytometry.1,7-9 Patients with HCLv have a worse prognosis and poor responses to front-line purine analogs, and a higher proportion of these patients carry the unmutated immunoglobulin heavy chain variable (IGHV) gene (54% vs 17% in HCL).1,10,11 About 30% to 50% have wild-type BRAF and activating mutations in MAP2K1, which encodes aberrant MEK downstream of BRAF.10,12
Most patients with HCL have somatic mutations in the IGHV gene.3,13,14 Patients with unmutated IGHV4-34 and wildtype BRAF have an aggressive form of the disease, even if the HCL cells express CD25 as in classic HCL.1,15 HCL in patients with unmutated IGHV is often refractory to purine analogs and these patients have poor prognosis and rapid progression.16 Other identified mutations include CDKN1B in HCL and MAP2K1 and CCNC3 in HCLv.2
Signs and Symptoms
In many cases, HCL is asymptomatic, and diagnosed when pancytopenia, monocytopenia, and leukopenia are discovered on unrelated blood work.2,3,11 Monocytopenia is a specific presentation of HCL, but not HCLv.11 Typical systemic symptoms include unexplained weight loss and extreme fatigue (80%).1,3 Other symptoms can include fever, recurrent infections, night sweats, splenomegaly and related pain or abdominal fullness, hepatomegaly, and bleeding or bruising due to thrombocytopenia.1,3 Splenomegaly is associated with advanced disease.11
Up to 30% of patients may present with autoimmune disorders such as vasculitis or psoriasis. Although skin involvement is rare with HCL, 10% to 12% of patients will have dermatologic symptoms either due to recurrent infection or autoimmune reactions.1,2 Skin reactions include localized or generalized maculopapular rash, pyoderma gangrenosum (which may be severe), and recurrent bacterial or viral skin infections.17
Diagnosis
After complete history and physical examination, a diagnosis of HCL is usually made based on flow cytometry for immunophenotyping and molecular testing for BRAF V600E (Table 1).2,17
Disease-related fibrosis may impede bone marrow aspiration, and trephine biopsy should be done to make the diagnosis.11 On morphologic examination, HCL cells are small- to medium-sized, with round, oval, or indented, well-defined nuclei. Cytoplasm is pale blue, and cells have small cytoplasmic projections (Figure 1).2,18
On flow cytometry, HCL is positive for B-cell antigens (CD19, CD20, CD22), as well as antigens specific to the disease (CD11c, CD25, CD103, CD123), and by immunohistochemistry (IHC) for cyclin D1 and annexin-A1. CD20, CD123, and CD200 are bright in HCL. The presence of T-cell marker CD103 on B-cells indicates HCL.1-3 HCLv, in contrast, is positive for CD11c and CD103, but usually negative for CD25, CD123, and annexin-A1.2,19
BRAF V600E mutation can be identified using droplet digital polymerase chain reaction (PCR), next-generation molecular sequencing, or IHC with a VE1 stain.3,11 IHC for CD20, annexin-1, and VE1 establish the diagnosis, but also are useful in determining the extent to which leukemic cells have infiltrated bone marrow.11
Differential diagnosis of HCL includes HCL variants, splenic marginal zone lymphoma, and splenic diffuse red pulp small B-cell lymphoma.7,11
Indications for Treatment and Criteria for Response
Over time, about 90% of patients with HCL will require treatment. However, not all such patients will require urgent or immediate treatment, and some can be managed with observation and close monitoring.1,11 The indications for initiating treatment generally are systemic symptoms and significant pancytopenia (Table 2).2,11
The optimal response with treatment of HCL is complete response (CR) without minimal residual disease (MRD-free), which minimizes the risk for relapse.1,11 Hematologic and molecular response is assessed using peripheral blood samples; physical examination, ultrasound, computed tomography, or magnetic resonance imaging is used to determine response in lymph nodes, spleen, or liver.1 MRD-free is defined by the absence of HCL cells by the chosen method (IHC, flow cytometry, or PCR).20 Bone marrow aspirate flow cytometry is the most sensitive standard test for MRD detection.1 Table 3 summarizes response criteria for HCL.2,11
Initial Treatment of HCL
The purine nucleoside analogs (PNAs) cladribine (± rituximab) and pentostatin are widely recommended for initial treatment.1,2,11 As monotherapy, cladribine and pentostatin are considered similarly effective, with CR in 70% to 90% of patients and durations of response > 10 years.1 Adding the anti-CD20 monoclonal antibody rituximab in 8 weekly doses starting the first day of front-line cladribine (CDAR) improves remission, MRD-free rates, and duration of response (94% MRD-free at 96 months), with minimal added toxicity.21 Rituximab is often added 4 weeks after cladribine, which offers more convenience, an equally high CR rate of 100%, and a 76% MRD-free rate at 3 months.11 Bone marrow biopsy should be delayed for 4 to 6 months to allow a full response to develop with cladribine.1,11
Daily (intravenous or subcutaneous) and weekly cladribine are equally safe and effective.2,11 Pentostatin is administered intravenously every 2 weeks for 3 to 6 months, allowing time for hematologic recovery between doses.1,11 Patient factors to consider when choosing treatment include baseline neutropenia, patient preference, and comorbidities.
Toxicities of PNAs include neutropenia and fever, which typically occur during the first month of treatment and are more frequent in patients with baseline severe neutropenia; T-cell recovery may take years.1 CDAR is associated with higher transient thrombocytopenia, but faster platelet and neutrophil recovery at 4 weeks than cladribine alone.21 Both therapies are immunosuppressive. Patients should be evaluated for existing infections and watched for new infections during treatment. Control of active infection prior to treatment initiation is required.11,23
Patients with confirmed BRAF V600E mutation are candidates for vemurafenib if they are unable to tolerate a PNA, have an active infection, or would like effective vaccinations.2,23-25
Treatment at Relapse
At suspected HCL relapse, patients should be evaluated to determine whether cytopenia is due to recurrent disease or lingering effects from prior treatment. Use of successive flow cytometry over time can clarify whether symptoms are related to disease and need interventional treatment, or will resolve with additional time.1
Patients who have an HCL relapse after initial therapy with cladribine or pentostatin may be candidates for re-treatment with the same or alternate PNA plus rituximab.2 Rituximab
monotherapy has been used for patients unable to tolerate PNA but yields CR rates as low as 13%.26 Repeated courses of PNA therapy yield lower rates and durations of response with each course.1,2
For patients with primary refractory disease (less than CR with initial therapy) or relapse within 2 years of initial therapy, treatment with the BRAF V600E inhibitor vemurafenib off-label, with or without rituximab, is an option.2,5 In HCL, vemurafenib for patients with relapsed or refractory disease achieved CR in 35% and 42% in 2 small trials (N = 54). Relapse-free survival among people with CR was 19 months in 1 of the trials.27 Vemurafenib plus rituximab achieved CR in 87% of patients with relapsed or refractory HCL, and an MRD-free CR rate of 57%. Among patients with CR, 85% were relapse-free at a median follow-up of 34 months.5 Treatment with vemurafenib is not myelotoxic—an advantage for HCL patients. Adverse effects with vemurafenib are often manageable with dose reductions, if needed. A specific concern with vemurafenib is the potential development of secondary skin cancers.5,27,28
Novel Targeted Options and Recommended Use
Promising alternatives for patients with relapsed or refractory HCL include combined BRAF and MEK inhibitors and the Bruton tyrosine kinase (BTK) inhibitor ibrutinib. The concept of BRAF/MEK inhibition was validated in studies with BRAF-mutated melanoma, in which dabrafenib plus trametinib (the MEK inhibitor) improved overall survival (OS) with less toxicity and better quality of life than vemurafenib.1,29 In a phase 2 trial in HCL, dabrafenib monotherapy demonstrated an overall response rate (ORR) of 80%, including 30% CR.30 In a subsequent phase 2 trial, dabrafenib combined with trametinib was evaluated in refractory or late relapsed HCL. Among 55 enrolled patients, objective response rate was 89%, including 65.5% CR. Nine of 36 patients with CR were MRD-free. Among responding patients, duration of response was 97.7% at 24 months.31 The most common grade ≥ 3 toxicities were hyperglycemia, pyrexia, neutropenia, and pneumonia. Secondary skin cancers were seen in about 5% of patients.31
BRAF/MEK inhibitor combinations in HCL offer effective therapy with less myelosuppression than PNAs, making them useful for patients with or at risk for infection.23 Their use in HCL is off-label, as they currently are approved for treatment of BRAF-mutated melanoma and some other tumors.32 A study of encorafenib (a BRAF inhibitor) combined with binimetinib (a MEK inhibitor) is ongoing (Table 4).32
Ibrutinib interrupts B-cell receptor signaling to stop tumor cell growth. In a phase 2 trial, patients with relapsed or refractory HCL or HCLv were treated with once-daily oral ibrutinib. Best ORR was 54% (19% CR; 3% MRD-free). Despite the low CR rate, 3-year progression-free survival with ibrutinib was 73% and OS was 85%. Treatment was well-tolerated; cytopenia (including 22% grade ≥ 3 thrombocytopenia and neutropenia) and diarrhea were frequent toxicities.33
Moxetumomab pasudotox is a novel CD22-targeted antibody fused with protein toxin that interrupts protein synthesis in tumor cells.1 As treatment, it was studied in a phase 3 trial of relapsed HCL in heavily pretreated patients, and achieved a CR rate of 41%, including 36% durable CR.34 Although FDA-approved for relapsed or refractory HCL, the drug is being discontinued due to business decisions, not safety or efficacy concerns.2 It is notable that many types of B-cell lymphoma also express CD22.35
Enrollment in a clinical trial to study possible treatment advances is recommended by the National Comprehensive Cancer Network (NCCN) at first and subsequent relapses of HCL for appropriate patients.2 Figure 2 summarizes an approach to treatment choice and sequencing for patients with HCL.
Supportive Care
Patients being treated for HCL should have supportive care to manage adverse effects of their disease. Such care includes prophylaxis against herpes virus if CD4+ T cells < 200 cells/μL and other prophylactic vaccinations to hepatitis B virus, COVID-19 and Influenza. Patients with neutropeni may require broad-spectrum antibacterial prophylaxis or neutrophil growth factors if neutropenic fever develops. Blood product support is recommended if needed.2 Assessment of anti-COVID-19 antibodies is recommended to optimize immunity, particularly prior to beginning anti-CD20 antibody therapy like rituximab.23
Unmet Needs
Despite improvements in response and survival with newer therapies, not all patients with HCL benefit from these advances. Unmet needs are finding optimal treatment for patients with HCLv, despite some success with MEK inhibitors, and for patients with BRAF mutations other than V600E, who have few options beyond PNAs and rituximab.
- Kreitman RJ, Arons E. Diagnosis and treatment of hairy cell leukemia as the COVID-19 pandemic continues. Blood Rev. 2022;51:100888. doi:10.1016/j.blre.2021.100888
- National Comprehensive Cancer Network. NCCN clinical practice guideline in oncology: hairy cell leukemia. Version 1.2023. Published August 30, 2022. Accessed March 16, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hairy_cell.pdf
- Janus A, Robak T. Hairy cell leukemia. In: Li W, ed. Leukemia [Internet]. Brisbane: Exon Publications; 2022:chap3. Accessed February 16, 2023. doi:10.36255/exon-publications-leukemia-hairy-cell-leukemia
- Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol. 2015;28(4):175-179. doi:10.1016/j.beha.2015.10.014
- Tiacci E, De Carolis L, Simonetti E, et al. Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. N Engl J Med. 2021;384(19):1810-1823. doi:10.1056/NEJMoa20312986
- Falini B, Martelli MP, Tiacci E. BRAF V600E mutation in hairy cell leukemia: from bench to bedside. Blood. 2016;128(15):1918-1927. doi:10.1182/blood-2016-07-418434
- Matutes E. Diagnostic and therapeutic challenges in hairy cell leukemia-variant: where are we in 2021? Expert Rev Hematol. 2021;14(4):355-363. doi:10.1080/17474086.2021.1908121
- Cawley JC, Burns GF, Hayhoe FG. A chronic lymphoproliferative disorder with distinctive features: a distinct variant of hairy-cell leukaemia. Leuk Res. 1980;4(6):547-559. doi:10.1016/0145-2126(80)90066-1
- Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119(14):3330-3332. doi:10.1182/blood-2011-09-379339
- Durham BH, Getta B, Dietrich S, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130(14):1644-1648. doi:10.1182/blood-2017-01-76510711
- Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with hairy cell leukemia. Blood. 2017;129(5):553-560. doi:10.1182/blood-2016-01-689422
- Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46(1):8-10. doi:10.1038/ng.2828
- Arons E, Sunshine J, Suntum T, Kreitman RJ. Somatic hypermutation and VH gene usage in hairy cell leukaemia. Br J Haematol. 2006;133(5):504-512. doi:10.1111/j.1365-2141.2006.06066.x
- Arons E, Roth L, Sapolsky J, Suntum T, Stetler-Stevenson M, Kreitman RJ. Evidence of canonical somatic hypermutation in hairy cell leukemia. Blood. 2011;117(18):4844-4851. doi:10.1182/blood-2010-11-316737
- Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114(21):4687-4695. doi:10.1182/blood-2009-01-201731
- Forconi F, Sozzi E, Cencini E, et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood. 2009;114(21):4696-4702. doi:10.1182/blood-2009-03-212449
- Robak E, Jesionek-Kupnicka D, Robak T. Skin changes in hairy cell leukemia. Ann Hematol. 2021;100(3):615-625. doi:10.1007/s00277-020-04349-z
- Bouroncle BA. Thirty-five years in the progress of hairy cell leukemia. Leuk Lymphoma. 1994;14(suppl 1):1-12. https://pubmed.ncbi.nlm.nih.gov/7820038/
- Falini B, Tiacci E, Liso A, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet. 2004;363(9424): 1869-1870. doi:10.1016/S0140-6736(04)16356-3
- Robak T, Robak P. Measurable residual disease in hairy cell leukemia: technical considerations and clinical significance. Front Oncol. 2022;12:976374. doi:10.3389/fonc.2022.976374
- Chihara D, Arons E, Stetler-Stevenson M, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol. 2020;38(14):1527-1538. doi:10.1200/JCO.19.02250
- Chihara D, Kantarjian H, O’Brien S, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol. 2016;174(5):760-766. doi:10.1111/bjh.14129
- Grever M, Andritsos L, Banerji V, et al. Hairy cell leukemia and COVID-19 adaptation of treatment guidelines. Leukemia. 2021;35(7):1864-1872. doi:10.1038/s41375-021-01257-7
- Konrat J, Rösler W, Roiss M, et al. BRAF inhibitor treatment of classical hairy cell leukemia allows successful vaccination against SARS-CoV-2. Ann Hematol. 2023;102(2):403-406. doi:10.1007/s00277-022-05026-z
- Park JH, Shukla M, Salcedo JM, et al. First-line chemo-free therapy with the BRAF inhibitor vemurafenib combined with obinutuzumab is effective in patients with HCL. Blood. 2019;134(suppl 1):Abstract 3998. https://doi.org/10.1182/blood-2019-124478
- Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood. 2003;102(3):810-813. doi:10.1182/blood-2003-01-0014
- Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373(18):1733-1747. doi:10.1056/NEJMoa1506583
- Maitre E, Paillassa J, Troussard X. Novel targeted treatments in hairy cell leukemia and other hairy cell-like disorders. Front Oncol. 2022;12:1068981. doi:10.3389/fonc.2022.1068981
- Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015;16(13):1389-1398. doi:10.1016/S1470-2045(15)00087-X
- Tiacci E, De Carolis L, Simonetti E, et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: a pilot phase-2 clinical trial. Leukemia. 2021;35(11):3314-3318. doi:10.1038/s41375-021-01210-8
- Kreitman RJ, Moreau P, Ravandi F, et al. Dabrafenib plus trametinib in patients with relapsed/refractory BRAF V600E mutation-positive hairy cell leukemia. Blood. 2023;141(9):996-1006. doi:10.1182/blood.2021013658
- Adashek JJ, Menta AK, Reddy NK, Desai AP, Roszik J, Subbiah V. Tissue agnostic activity of BRAF plus MEK inhibitor in BRAF V600E-mutated tumors. Mol Cancer Ther. 2022;21(6):871-878. doi:10.1158/1535-7163.MCT-21-0950
- Rogers KA, Andritsos LA, Wei L, et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood. 2021;137(25):3473-3483. doi:10.1182/blood.2020009688
- Kreitman RJ, Dearden C, Zinzani PL, et al; Study 1053 investigators. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol. 2021;14(1):35. doi:10.1186/s13045-020-01004-y
- Leonard JP, Goldenberg DM. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene. 2007;26(25):3704-3713. doi:10.1038/sj.onc.1210370
- Kreitman RJ, Arons E. Diagnosis and treatment of hairy cell leukemia as the COVID-19 pandemic continues. Blood Rev. 2022;51:100888. doi:10.1016/j.blre.2021.100888
- National Comprehensive Cancer Network. NCCN clinical practice guideline in oncology: hairy cell leukemia. Version 1.2023. Published August 30, 2022. Accessed March 16, 2023. https://www.nccn.org/professionals/physician_gls/pdf/hairy_cell.pdf
- Janus A, Robak T. Hairy cell leukemia. In: Li W, ed. Leukemia [Internet]. Brisbane: Exon Publications; 2022:chap3. Accessed February 16, 2023. doi:10.36255/exon-publications-leukemia-hairy-cell-leukemia
- Tadmor T, Polliack A. Epidemiology and environmental risk in hairy cell leukemia. Best Pract Res Clin Haematol. 2015;28(4):175-179. doi:10.1016/j.beha.2015.10.014
- Tiacci E, De Carolis L, Simonetti E, et al. Vemurafenib plus rituximab in refractory or relapsed hairy-cell leukemia. N Engl J Med. 2021;384(19):1810-1823. doi:10.1056/NEJMoa20312986
- Falini B, Martelli MP, Tiacci E. BRAF V600E mutation in hairy cell leukemia: from bench to bedside. Blood. 2016;128(15):1918-1927. doi:10.1182/blood-2016-07-418434
- Matutes E. Diagnostic and therapeutic challenges in hairy cell leukemia-variant: where are we in 2021? Expert Rev Hematol. 2021;14(4):355-363. doi:10.1080/17474086.2021.1908121
- Cawley JC, Burns GF, Hayhoe FG. A chronic lymphoproliferative disorder with distinctive features: a distinct variant of hairy-cell leukaemia. Leuk Res. 1980;4(6):547-559. doi:10.1016/0145-2126(80)90066-1
- Xi L, Arons E, Navarro W, et al. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood. 2012;119(14):3330-3332. doi:10.1182/blood-2011-09-379339
- Durham BH, Getta B, Dietrich S, et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood. 2017;130(14):1644-1648. doi:10.1182/blood-2017-01-76510711
- Grever MR, Abdel-Wahab O, Andritsos LA, et al. Consensus guidelines for the diagnosis and management of patients with hairy cell leukemia. Blood. 2017;129(5):553-560. doi:10.1182/blood-2016-01-689422
- Waterfall JJ, Arons E, Walker RL, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46(1):8-10. doi:10.1038/ng.2828
- Arons E, Sunshine J, Suntum T, Kreitman RJ. Somatic hypermutation and VH gene usage in hairy cell leukaemia. Br J Haematol. 2006;133(5):504-512. doi:10.1111/j.1365-2141.2006.06066.x
- Arons E, Roth L, Sapolsky J, Suntum T, Stetler-Stevenson M, Kreitman RJ. Evidence of canonical somatic hypermutation in hairy cell leukemia. Blood. 2011;117(18):4844-4851. doi:10.1182/blood-2010-11-316737
- Arons E, Suntum T, Stetler-Stevenson M, Kreitman RJ. VH4-34+ hairy cell leukemia, a new variant with poor prognosis despite standard therapy. Blood. 2009;114(21):4687-4695. doi:10.1182/blood-2009-01-201731
- Forconi F, Sozzi E, Cencini E, et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood. 2009;114(21):4696-4702. doi:10.1182/blood-2009-03-212449
- Robak E, Jesionek-Kupnicka D, Robak T. Skin changes in hairy cell leukemia. Ann Hematol. 2021;100(3):615-625. doi:10.1007/s00277-020-04349-z
- Bouroncle BA. Thirty-five years in the progress of hairy cell leukemia. Leuk Lymphoma. 1994;14(suppl 1):1-12. https://pubmed.ncbi.nlm.nih.gov/7820038/
- Falini B, Tiacci E, Liso A, et al. Simple diagnostic assay for hairy cell leukaemia by immunocytochemical detection of annexin A1 (ANXA1). Lancet. 2004;363(9424): 1869-1870. doi:10.1016/S0140-6736(04)16356-3
- Robak T, Robak P. Measurable residual disease in hairy cell leukemia: technical considerations and clinical significance. Front Oncol. 2022;12:976374. doi:10.3389/fonc.2022.976374
- Chihara D, Arons E, Stetler-Stevenson M, et al. Randomized phase II study of first-line cladribine with concurrent or delayed rituximab in patients with hairy cell leukemia. J Clin Oncol. 2020;38(14):1527-1538. doi:10.1200/JCO.19.02250
- Chihara D, Kantarjian H, O’Brien S, et al. Long-term durable remission by cladribine followed by rituximab in patients with hairy cell leukaemia: update of a phase II trial. Br J Haematol. 2016;174(5):760-766. doi:10.1111/bjh.14129
- Grever M, Andritsos L, Banerji V, et al. Hairy cell leukemia and COVID-19 adaptation of treatment guidelines. Leukemia. 2021;35(7):1864-1872. doi:10.1038/s41375-021-01257-7
- Konrat J, Rösler W, Roiss M, et al. BRAF inhibitor treatment of classical hairy cell leukemia allows successful vaccination against SARS-CoV-2. Ann Hematol. 2023;102(2):403-406. doi:10.1007/s00277-022-05026-z
- Park JH, Shukla M, Salcedo JM, et al. First-line chemo-free therapy with the BRAF inhibitor vemurafenib combined with obinutuzumab is effective in patients with HCL. Blood. 2019;134(suppl 1):Abstract 3998. https://doi.org/10.1182/blood-2019-124478
- Nieva J, Bethel K, Saven A. Phase 2 study of rituximab in the treatment of cladribine-failed patients with hairy cell leukemia. Blood. 2003;102(3):810-813. doi:10.1182/blood-2003-01-0014
- Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373(18):1733-1747. doi:10.1056/NEJMoa1506583
- Maitre E, Paillassa J, Troussard X. Novel targeted treatments in hairy cell leukemia and other hairy cell-like disorders. Front Oncol. 2022;12:1068981. doi:10.3389/fonc.2022.1068981
- Grob JJ, Amonkar MM, Karaszewska B, et al. Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial. Lancet Oncol. 2015;16(13):1389-1398. doi:10.1016/S1470-2045(15)00087-X
- Tiacci E, De Carolis L, Simonetti E, et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: a pilot phase-2 clinical trial. Leukemia. 2021;35(11):3314-3318. doi:10.1038/s41375-021-01210-8
- Kreitman RJ, Moreau P, Ravandi F, et al. Dabrafenib plus trametinib in patients with relapsed/refractory BRAF V600E mutation-positive hairy cell leukemia. Blood. 2023;141(9):996-1006. doi:10.1182/blood.2021013658
- Adashek JJ, Menta AK, Reddy NK, Desai AP, Roszik J, Subbiah V. Tissue agnostic activity of BRAF plus MEK inhibitor in BRAF V600E-mutated tumors. Mol Cancer Ther. 2022;21(6):871-878. doi:10.1158/1535-7163.MCT-21-0950
- Rogers KA, Andritsos LA, Wei L, et al. Phase 2 study of ibrutinib in classic and variant hairy cell leukemia. Blood. 2021;137(25):3473-3483. doi:10.1182/blood.2020009688
- Kreitman RJ, Dearden C, Zinzani PL, et al; Study 1053 investigators. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol. 2021;14(1):35. doi:10.1186/s13045-020-01004-y
- Leonard JP, Goldenberg DM. Preclinical and clinical evaluation of epratuzumab (anti-CD22 IgG) in B-cell malignancies. Oncogene. 2007;26(25):3704-3713. doi:10.1038/sj.onc.1210370