User login
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
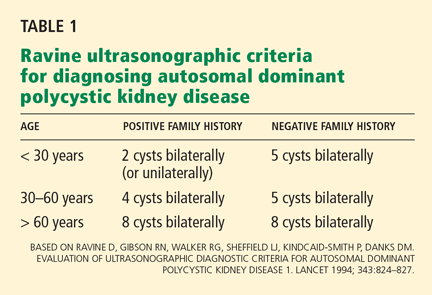
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
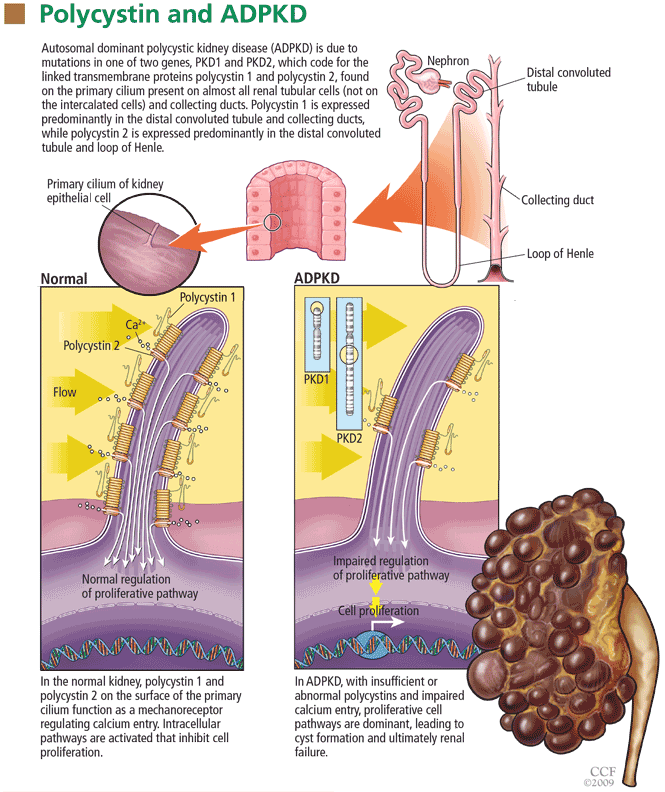
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
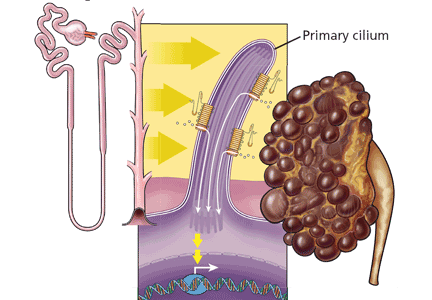
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
A 25-year-old married white woman presented to a clinic because of pelvic pain. A computed tomographic scan of her abdomen and pelvis without intravenous contrast showed two definite cysts in the right kidney (the larger measuring 2.5 cm) and a 1.5-cm cyst in the left kidney. It also showed several smaller (< 1 cm) areas of low density in both kidneys that suggested cysts. Renal ultrasonography also showed two cysts in the left kidney and one in the right kidney. The kidneys were normal-sized—the right one measured 12.5 cm and the left one 12.7 cm.
She had no family history of autosomal dominant polycystic kidney disease (ADPKD), and renal ultrasonography of her parents showed no cystic disease. She had no history of headache or heart murmur, and her blood pressure was normal. Her kidneys were barely palpable, her liver was not enlarged, and she had no cardiac murmur or click. She was not taking any medications. Her serum creatinine level was 0.7 mg/dL, hemoglobin 14.0 g/dL, and urinalysis normal.
Does this patient have ADPKD? Based on the studies done so far, would genetic testing be useful? If the genetic analysis does show a mutation, what additional information can be derived from the location of that mutation? Can she do anything to improve her prognosis?
ADPKD ACCOUNTS FOR ABOUT 3% OF END-STAGE RENAL DISEASE
ADPKD is the most common of all inherited renal diseases, with 600,000 to 700,000 cases in the United States and about 12.5 million cases worldwide. About 5,000 to 6,000 new cases are diagnosed yearly in the United States, about 40% of them by age 45. Typically, patients with ADPKD have a family history of the disease, but about 5% to 10% do not. In about 50% of cases, ADPKD progresses to end-stage renal disease by age 60, and it accounts for about 3% of cases of end-stage renal disease in the United States.1
CYSTS IN KIDNEYS AND OTHER ORGANS, AND NONCYSTIC FEATURES
In ADPKD, cysts in the kidneys increase in number and size over time, ultimately destroying normal renal tissue. However, renal function remains steady over many years until the kidneys have approximately quadrupled in volume to 1,500 cm3 (normal combined kidney volume is about 250 to 400 cm3), which defines a tipping point beyond which renal function can rapidly decline.2,3 Ultimately, the patient will need renal replacement therapy, ie, dialysis or renal transplantation.
The cysts (kidney and liver) cause discomfort and pain by putting pressure on the abdominal wall, flanks, and back, by impinging on neighboring organs, by bleeding into the cysts, and by the development of kidney stones or infected cysts (which are uncommon, though urinary tract infections themselves are more frequent). Kidney stones occur in about 20% of patients with ADPKD, and uric acid stones are almost as common as calcium oxalate stones. Compression of the iliac vein and inferior vena cava with possible thrombus formation and pulmonary embolism can be caused by enormous enlargement of the cystic kidneys, particularly the right.4 Interestingly, the patients at greatest risk of pulmonary embolism after renal transplantation are those with ADPKD.5
Cysts can also develop in other organs. Liver cysts develop in about 80% of patients. Usually, the cysts do not affect liver function, but because they are substantially estrogen-dependent they can be more of a clinical problem in women. About 10% of patients have cysts in the pancreas, but these are functionally insignificant. Other locations of cysts include the spleen, arachnoid membranes, and seminal vesicles in men.
Intracranial aneurysms are a key noncystic feature, and these are strongly influenced by family history. A patient with ADPKD who has a family member with ADPKD as well as an intracranial aneurysm or subarachnoid hemorrhage has about a 20% chance of having an intracranial aneurysm. A key clinical warning is a “sentinel” or “thunderclap” headache, which patients typically rate as at least a 10 on a scale of 10 in severity. In a patient with ADPKD, this type of headache can signal a leaking aneurysm causing irritation and edema of the surrounding brain tissue that temporarily tamponades the bleeding before the aneurysm actually ruptures. This is a critical period when a patient should immediately obtain emergency care.
Cardiac valve abnormalities occur in about one-third of patients. Most common is mitral valve prolapse, which is usually mild. Abnormalities can also occur in the aortic valve and the left ventricular outflow tract.
Hernias are the third general noncystic feature of ADPKD. Patients with ADPKD have an increased prevalence of umbilical, hiatal, and inguinal hernias, as well as diverticulae of the colon.
DOES THIS PATIENT HAVE ADPKD?
The Ravine ultrasonographic criteria for the diagnosis of ADPKD are based on the patient’s age, family history, and number of cysts (Table 1).6,7 Alternatively, Torres (Vincent E. Torres, personal communication, March 2008) recommends that, in the absence of a family history of ADPKD or other findings to suggest other cystic disease, the diagnosis of ADPKD can be made if the patient has a total of at least 20 renal cysts.
Our patient had only three definite cysts, was 25 years old, and had no family history of ADPKD and so did not technically meet the Ravine criteria of five cysts at this age, or the Torres criteria, for having ADPKD. Nevertheless, because she was concerned about overt disease possibly developing later and about passing on a genetic defect to her future offspring, she decided to undergo genetic testing.
CLINICAL GENETICS OF ADPKD: TWO MAJOR TYPES
There are two major genetic forms of ADPKD, caused by mutations in the genes PKD1 and PKD2.
PKD1 has been mapped to the short arm of the 16th chromosome. Its gene product is polycystin 1. Mutations in PKD1 account for about 85% of all cases of polycystic kidney disease. The cysts appear when patients are in their 20s, and the disease progresses relatively rapidly, so that most patients enter end-stage renal disease when they are in their 50s.
PKD2 has been mapped to the long arm of the fourth chromosome. Its product is polycystin 2. PKD2 mutations account for about 15% of all cases of ADPKD, and the disease progresses more slowly, usually with end-stage disease developing when the patients usually are in their 70s.
Screening for mutations by direct DNA sequencing in ADPKD
Genetic testing for PKD1 and PKD2 mutations is available (www.athenadiagnostics.com).8 The Human Gene Mutation Database lists at least 270 different PKD1 mutations and 70 different PKD2 mutations.8 Most are unique to a single family.
Our patient was tested for mutations of the PKD1 and PKD2 genes by polymerase chain reaction amplification and direct DNA sequencing. She was found to possess a DNA sequence variant at a nucleotide position in the PKD1 gene previously reported as a disease-associated mutation. She is therefore likely to be affected with or predisposed to developing ADPKD.
Furthermore, the position of her mutation means she has a worse prognosis. Rossetti et al,9 in a study of 324 PKD1 patients, found that only 19% of those who had mutations in the 5′ region of the gene (ie, at positions below 7,812) still had adequate renal function at 60 years of age, compared with 40% of those with mutations in the 3′ region (P = .025).
Other risk factors for more rapid kidney failure in ADPKD include male sex, onset of hypertension before age 35, gross hematuria before age 30 in men, and, in women, having had three or more pregnancies.
THE ‘TWO-HIT’ HYPOTHESIS
The time of onset and the rate of progression of ADPKD can vary from patient to patient, even in the same family. Besides the factors mentioned above, another reason may be that second mutations (“second hits”) have to occur before the cysts develop.
The first mutation exists in all the kidney tubular cells and is the germline mutation in the PKD gene inherited from the affected parent. This is necessary but not sufficient for cyst formation.
The second hit is a somatic mutation in an individual tubular cell that inactivates to varying degrees the unaffected gene from the normal parent. It is these second hits that allow abnormal focal (monoclonal) proliferation of renal tubular cells and cyst formation (reviewed by Arnaout10 and by Pei11). There is no way to predict these second hits, and their identity is unknown.
Other genetic variations may occur, such as transheterozygous mutations, in which a person may have a mutation of PKD1 as well as PKD2.
Germline mutations of PKD1 or PKD2 combined with somatic mutations of the normal paired chromosome depress levels of their normal gene products (polycystin 1 and polycystin 2) to the point that cysts develop.
The timing and frequency of these second hits blur the distinction between the time course for the progression of PKD1 and PKD2 disease, and can accelerate the course of both.
BASIC RESEARCH POINTS THE WAY TO TREATMENTS FOR ADPKD
Polycystin 1 and polycystin 2 are the normal gene products of the genes which, when mutated, are responsible for PKD1 and PKD2, respectively. Research into the structure and function of the polycystin 1 and polycystin 2 proteins—and what goes wrong when they are not produced in sufficient quantity or accurately—is pointing the way to possible treatments for ADPKD.
When the polycystins are not functioning, as in ADPKD, these proliferative pathways are unopposed. However, proliferation can be countered in other ways. One of the prime movers of cell proliferation, acting through adenylyl cyclase and cAMP, is vasopressin. In genetically produced polycystic animals, two antagonists of the vasopressin V2 receptor (VPV2R), OPC31260 and OPC41061 (tolvaptan), decreased cAMP and ERK, prevented or reduced renal cysts, and preserved renal function.15,16 Not surprisingly, simply increasing water intake decreases vasopressin production and the development of polycystic kidney disease in rats.17 Definitive proof of the role of vasopressin in causing cyst formation was achieved by crossing PCK rats (genetically destined to develop polycystic kidneys) with Brattleboro rats (totally lacking vasopressin) in order to generate rats with polycystic kidneys and varying amounts of vasopressin.18 PCK animals with no vasopressin had virtually no cAMP or renal cysts, whereas PCK animals with increasing amounts of vasopressin had progressively larger kidneys with more numerous cysts. Administration of synthetic vasopressin to PCK rats that totally lacked vasopressin re-created the full cystic disease.
Normally, cAMP is broken down by phosphodiesterases. Caffeine and methylxanthine products such as theophylline interfere with phosphodiesterase activity, raise cAMP in epithelial cell cultures from patients with ADPKD,19 and increase cyst formation in canine kidney cell cultures.20 One could infer that caffeine-containing drinks and foods would be undesirable for ADPKD patients.
The absence of polycystin permits excessive kinase activity in the mTOR pathway and the development of renal cysts.14 The mTOR system can be blocked by rapamycin (sirolimus, Rapamune). Wahl et al21 found that inhibition of mTOR with rapamycin slows PKD progression in rats. In a prospective study in humans, rapamycin reduced polycystic liver volumes in ADPKD renal transplant recipients.22
Rapamycin, however, can have significant side effects that include hypertriglyceridemia, hypercholesterolemia, thrombocytopenia, anemia, leukopenia, oral ulcers, impaired wound healing, proteinuria, thrombotic thrombocytopenic purpura, interstitial pneumonia, infection, and venous thrombosis. Many of these appear to be dose-related and can generally be reversed by stopping or reducing the dose. However, this drug is not approved by the US Food and Drug Administration for the treatment of ADPKD, and we absolutely do not advocate using it “off-label.”
What does this mean for our patient?
Although these results were derived primarily from animal experiments, they do provide a substantial rationale for advising our patient to:
Drink approximately 3 L of water throughout the day right up to bedtime in order to suppress vasopressin secretion and the stimulation of cAMP. This should be done under a doctor’s direction and with regular monitoring.15,17,18,23
Avoid caffeine and methylxanthines because they block phosphodiesterase, thereby leaving more cAMP to stimulate cyst formation.19,20
Follow a low-sodium diet (< 2,300 mg/day), which, while helping to control hypertension and kidney stone formation, may also help to maintain smaller cysts and kidneys. Keith et al,24 in an experiment in rats, found that the greater the sodium content of the rats’ diet, the greater the cyst sizes and kidney volumes by the end of 3 months.
Consider participating in a study. Several clinical treatment studies in ADPKD are currently enrolling patients who qualify:
- The Halt Progression of Polycystic Kidney Disease (HALT PKD) study, funded by the National Institutes of Health, is comparing the combination of an angiotensin-converting enzyme (ACE) inhibitor and an angiotensin receptor blocker (ARB) vs an ACE inhibitor plus placebo. Participating centers are Beth Israel Deaconess Medical Center, Cleveland Clinic, Emory University, Mayo Clinic, Tufts-New England Medical Center, University of Colorado Health Sciences Center, and University of Kansas Medical Center. This study involves approximately 1,020 patients nationwide.
- The Tolvaptan Efficacy and Safety in Management of Polycystic Disease and its Outcomes (TEMPO) study plans to enroll approximately 1,500 patients.
- Rapamycin is being studied in a pilot study at Cleveland Clinic and in another study in Zurich, Switzerland.
- A study of everolimus, a shorter-acting mTOR inhibitor, is beginning.
- A study of somatostatin is under way in Italy.
HYPERTENSION AND ADPKD
Uncontrolled hypertension is a key factor in the rate of progression of kidney disease in general and ADPKD in particular. It needs to be effectively treated. The target blood pressure should be in the range of 110 to 130 mm Hg systolic and 70 to 80 mm Hg diastolic.
Hypertension develops at least in part because the renin-angiotensin-aldosterone system (RAAS) is up-regulated in ADPKD due to renal cysts compressing and stretching blood vessels.25 Synthesis of immunoreactive renin, which normally takes place in the juxtaglomerular apparatus, shifts to the walls of the arterioles. There is also ectopic renin synthesis in the epithelium of dilated tubules and cysts. Greater renin production causes increases in angiotensin II and vasoconstriction, in aldosterone and sodium retention, and both angiotensin II and aldosterone can cause fibrosis and mitogenesis, which enhance cyst formation.
ACE inhibitors partially reverse the decrease in renal blood flow, renal vascular resistance, and the increase in filtration fraction. However, because some angiotensin II is also produced by an ACE-independent pathway via a chymase-like enzyme, ARBs may have a broader role in treating ADPKD.
In experimental rats with polycystic kidney disease, Keith et al24 found that blood pressure, kidney weight, plasma creatinine, and histology score (reflecting the volume of cysts as a percentage of the cortex) were all lower in animals receiving the ACE inhibitor enalapril (Vasotec) or the ARB losartan (Cozaar) than in controls or those receiving hydralazine. They also reported that the number of cysts and the size of the kidneys increased as the amount of sodium in the animals’ drinking water increased.
The potential benefits of giving ACE inhibitors or ARBs to interrupt the RAAS in polycystic disease include reduced intraglomerular pressure, reduced renal vasoconstriction (and consequently, increased renal blood flow), less proteinuria, and decreased production of transforming growth factor beta with less fibrosis. In addition, Schrier et al26 found that “rigorous blood pressure control” (goal < 120/80 mm Hg) led to a greater reduction in left ventricular mass index over time than did standard blood pressure control (goal 135–140/85–90 mm Hg) in patients with ADPKD, and that treatment with enalapril led to a greater reduction than with amlodipine (Norvasc), a calcium channel blocker.
The renal risks of ACE inhibitors include ischemia from further reduction in renal blood flow (which is already compromised by expanding cysts), hyperkalemia, and reversible renal failure that can typically be avoided by judicious dosing and monitoring.27 In addition, these drugs have the well-known side effects of cough and angioedema, and they should be avoided in pregnancy.
If diuretics are used, hypokalemia should be avoided because of both clinical and experimental evidence that it promotes cyst development. In patients who have hyperaldosteronism and hypokalemia, the degree of cyst formation in their kidneys is much greater than in other forms of hypertension. Hypokalemia has also been shown to increase cyst formation in rat models.
What does this mean for our patient?
When hypertension develops in an ADPKD patient, it would probably be best treated with an ACE inhibitor or an ARB. However, should our patient become pregnant, these drugs are to be avoided. Children of a parent with ADPKD have a 50:50 chance of having ADPKD. Genetic counseling may be advisable.
Chapman et al28 found that pregnant women with ADPKD have a significantly higher frequency of maternal complications (particularly hypertension, edema, and preeclampsia) than patients without ADPKD (35% vs 19%, P < .001). Normotensive women with ADPKD and serum creatinine levels of 1.2 mg/dL or less typically had successful, uncomplicated pregnancies. However, 16% of normotensive ADPKD women developed new-onset hypertension in pregnancy and 11% developed preeclampsia; these patients were more likely to develop chronic hypertension. Preeclampsia developed in 7 (54%) of 13 hypertensive women with ADPKD vs 13 (8%) of 157 normotensive ADPKD women. Moreover, 4 (80%) of 5 women with ADPKD who had prepregnancy serum creatinine levels higher than 1.2 mg/dL developed end-stage renal disease 15 years earlier than the general ADPKD population. Overall fetal complication rates were similar in those with or without ADPKD (32.6% vs 26.2%), but fetal prematurity due to preeclampsia was increased significantly (28% vs 10%, P < .01).28
The authors concluded that hypertensive ADPKD women are at high risk of fetal and maternal complications and measures should be taken to prevent the development of preeclampsia in these women.
In conclusion, the patient with ADPKD can present many therapeutic challenges. Fortunately, new treatment approaches combined with established ones should begin to have a favorable impact on outcomes.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
- US Renal Data Services. Table A.1, Incident counts of reported ESRD: all patients. USRDS 2008 Annual Data Report, Vol. 3, page 7.
- Grantham JJ, Torres VE, Chapman AB, et al; CRISP Investigators. Volume progression in polycystic kidney disease. N Engl J Med 2006; 354:2122–2130.
- Grantham JJ, Cook LT, Torres VE, et al. Determinants of renal volume in autosomal-dominant polycystic kidney disease. Kidney Int 2008; 73:108–116.
- O’Sullivan DA, Torres VE, Heit JA, Liggett S, King BF. Compression of the inferior vena cava by right renal cysts: an unusual cause of IVC and/or iliofemoral thrombosis with pulmonary embolism in autosomal dominant polycystic kidney disease. Clin Nephrol 1998; 49:332–334.
- Tveit DP, Hypolite I, Bucci J, et al. Risk factors for hospitalizations resulting from pulmonary embolism after renal transplantation in the United States. J Nephrol 2001; 14:361–368.
- Ravine D, Gibson RN, Walker RG, Sheffield LJ, Kincaid-Smith P, Danks DM. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994; 343:824–827.
- Rizk D, Chapman AB. Cystic and inherited kidney disease. Am J Kidney Dis 2004; 42:1305–1317.
- Rossetti S, Consugar MB, Chapman AB, et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2007; 18:2143–2160.
- Rossetti S, Burton S, Strmecki L, et al. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol 2002; 13:1230–1237.
- Arnaout MA. Molecular genetics and pathogenesis of autosomal dominant polycystic kidney disease. Annu Rev Med 2001; 52:93–123.
- Pei Y. A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 2001; 7:151–156.
- Nauli S, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 2003; 33:129–137.
- Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 2004; 279:40419–40430.
- Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 2006; 103:5466–5471.
- Wang X, Gattone V, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 2005; 16:846–851.
- Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 2003; 9:1323–1326.
- Nagao S, Nishii K, Katsuvama M, et al. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 2006; 17:2220–2227.
- Wang W, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 2008; 19:102–108.
- Belibi FA, Wallace DP, Yamaguchi T, Christensen M, Reif G, Grantham JJ. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 13:2723–2729.
- Mangoo-Karim R, Uchich M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A 1989; 86:6007–6011.
- Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant 2006; 21:598–604.
- Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol 2008; 19:631–638.
- Grantham JJ. Therapy for polycystic kidney disease? It’s water, stupid! J Am Soc Nephrol 2008: 12:1–2.
- Keith DS, Torres VE, Johnson CM, Holley KE. Effect of sodium chloride, enalapril, and losartan on the development of polycystic kidney disease in Han:SPRD rats. Am J Kidney Dis 1994; 24:491–498.
- Ecder T, Schrier RW. Hypertension in autosomal dominant polycystic kidney disease: early occurrence and unique aspects. J Am Soc Nephrol 2001; 12:194–200.
- Schrier R, McFann K, Johnson A, et al. Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: results of a seven-year prospective randomized study. J Am Soc Nephrol 2002; 13:1733–1739.
- Chapman AB, Gabow PA, Schrier RW. Reversible renal failure associated with angiotensin-converting enzyme inhibitors in polycystic kidney disease. Ann Intern Med 1991; 115:769–773.
- Chapman AB, Johnson AM, Gabow PA. Pregnancy outcome and its relationship to progression of renal failure in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1994; 5:1178–1185.
KEY POINTS
- In ADPKD the expanding cysts destroy normally functioning kidney tissue, causing hypertension, pain, and other complications, but renal function remains relatively stable until kidney volumes reach a critical size.
- Testing for genetic defects that cause ADPKD is available. The specific mutation involved (PKD1 or PKD2) affects the age of onset and therefore the rate of disease progression as well as the likelihood of cardiovascular complications. Other factors include somatic mutations (“second hits”) of the normal paired chromosome.
- Intracranial aneurysms are a key noncystic feature and may present with a very severe (“sentinel” or “thunderclap”) headache requiring immediate medical attention. Their occurrence is strongly influenced by family history.
- Basic research indicates that patients may be advised to increase their water intake, limit their sodium intake, and avoid caffeine and methylxanthine derivatives.