User login
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
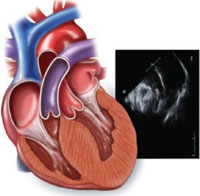
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment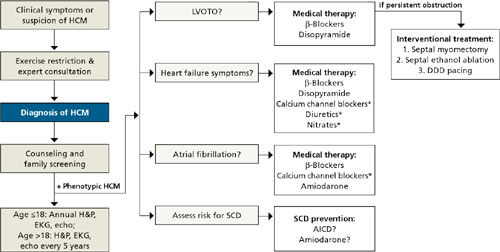
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
• Screen for hypertrophic cardiomyopathy (HCM) during preparticipation sports physicals. C
• Patients who have symptoms of, or are diagnosed with, HCM should be cleared by a cardiologist before being allowed to participate in organized sports or engaging in physical exercise. B
• Patients with HCM and a history of cardiac arrest should be given the opportunity to receive an automatic implantable cardioverter defibrillator (AICD). A
• Beta-blockers are first-line therapy for patients with HCM who have symptoms of heart failure. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Hypertrophic cardiomyopathy (HCM) is the most common cardiac genetic disorder.1 But it is best known for its tragic outcomes—the sudden cardiac death (SCD) of young athletes, many of whom are in high school or college.
In fact, HCM can present at any time from infancy through old age, and has a broad range of manifestations. Some individuals remain asymptomatic throughout their lives and enjoy a normal life span; others experience a range of symptoms, from chest pain and dyspnea on exertion to dizziness, light-headedness, palpitations, and syncope. Diagnosis is difficult, and predicting which patients will develop severe complications and face the greatest risk of premature SCD remains an inexact science.
What is clear is that in the United States, HCM causes SCD far more than any other condition.2 HCM has been positively identified in well over a third of cases (36%) of SCD in athletes under the age of 30, and cited as a possible cause in another 8%. Coronary artery anomalies is a distant second, responsible for 17% of sudden deaths in this patient population; myocarditis follows, at just 6%.2
We also know that HCM itself occurs much more frequently than previously believed. In the early 1980s, its prevalence was estimated at <1 in 100,000;3 today, HCM is believed to affect approximately 1 in 500 adults.2,4 Because this potentially fatal condition often remains undetected until an athlete collapses on the playing field, it is crucial that family physicians—who conduct many of the sports physicals typically required before students are permitted to play competitive sports—maintain a high degree of suspicion.
Knowing what signs and symptoms to look for and, perhaps more importantly, the key questions to ask, will help you identify young athletes at risk. This update, which begins with a review of the genetic basis and pathophysiology of HCM, will prepare you to take steps to protect your young patients.
Genetics and pathophysiology
HCM is associated with an autosomal-dominant family of disorders affecting any of 11 genes encoding for proteins in the myocardial sarcomere.5 Hundreds of mutations have been identified that can cause HCM disease. (You’ll find an updated list at http://genetics.med.harvard.edu/~seidman/cg3/index.html.)
The disease occurs equally in men and women,6 although far more males than females die on the playing field.3 Postulated reasons for the lower death rate in female athletes include differences in training regimen; lack of participation in high-risk sports such as football; and, until recently, fewer opportunities for young women to participate in high school and college athletic programs.3 HCM has no known racial predilection, although it may be underdiagnosed in women, minorities, and underserved populations.6
Myocardial tissue is disordered
The pathophysiology of HCM directly relates to the disordered myocardial tissue arising from the various gene mutations. Left ventricular myocytes appear hypertrophied and are chaotically arranged in bizarre structural patterns.1,7 This results in asymmetrically thickened and stiff ventricular walls, which can lead to diastolic dysfunction from poor left ventricle filling.
The abnormally thickened walls and septum also push the mitral valve anteriorly during systole. In some HCM patients, the anterior motion of the mitral valve, in conjunction with septal hypertrophy, result in obstruction of the subaortic left ventricular outflow tract. This obstruction—known as left ventricular outflow tract obstruction, or LVOTO—changes with physical position as well as physiological factors such as hydration level, heart rate, and vascular preload, making the murmur and abnormal pulses associated with it hard to detect during a physical exam.6
Ischemia and scarring result
The coronary arteries of patients with HCM are often tortuous, with thickened walls and narrow lumens. This leads to uneven perfusion and areas of relative myocardial ischemia, which result in patchy scarring. The ischemia, disorganized cellular architecture, and myocardial scarring create an environment that promotes the ventricular and atrial arrhythmias that commonly complicate HCM.1
While patients with HCM may develop heart failure at any age, only a minority ever experience severe heart failure—and more than 25% of patients with HCM live beyond the age of 75.8 Overall, adults with HCM have a 1% annual mortality rate, which is similar to that of the general adult population in the United States. For adults with HCM who have had an episode of cardiac arrest or have more than 1 major risk factor for SCD (TABLE 1), the annual mortality rate climbs to 6%.1
TABLE 1
HCM: Assessing risk of sudden cardiac death6,21,33
| Major risk factors Prior cardiac arrest* Unexplained syncope Family history of SCD Left ventricular wall thickness ≥30 mm Abnormal BP response to exercise Nonsustained spontaneous ventricular tachycardia |
| Possible risk factors LVOTO Late gadolinium enhancement on MRI Myocardial ischemia Specific troponin T and I mutations Intense physical exertion Atrial fibrillation |
| *Prior cardiac arrest is the most predictive of any major factor. |
| BP, blood pressure; HCM, hypertrophic cardiomyopathy; LVOTO, left ventricular outflow tract obstruction; MRI, magnetic resonance imaging; SCD, sudden cardiac death. |
The sports physical: Frustratingly “normal”
The mainstay of screening for HCM and other cardiac abnormalities associated with exertional SCD is the preparticipation physical examination, coupled with the medical history. The physical exam itself, however, is an insensitive screening tool for this condition. That’s because most patients with HCM have nonobstructive disease, meaning there is no murmur to be heard.1 Even among HCM patients with LVOTO, the murmur may be difficult to detect. Typically, it can be heard only when the patient stands or performs the Valsalva maneuver, movements that decrease preload. Clinicians who manage to detect the murmur generally describe it as a late-systolic ejection murmur best heard at the left sternal border radiating to the aortic and mitral areas, but not into the neck.9
Resting pulses, too, are typically normal in a patient with HCM/LVOTO, although “water hammer” and double-peak pulses may be present.10 At high levels of exertion, such patients may exhibit decreased peripheral pulses and an ominous decrease in systolic blood pressure.
It’s important to realize, however, that such findings are the exception. Because most patients with HCM have normal physical exams, the medical history plays a particularly important role in pinpointing patients at risk.
It’s time to tweak your preparticipation questionnaire
In recent years, efforts have been made to improve preparticipation questionnaires.11 Despite these efforts, only 17% of the preparticipation evaluation forms currently used by US high schools contain all the recommended screening elements.12 Validated screening questions are recommended by the American College of Cardiology (ACC), American Academy of Family Physicians, and other major organizations (TABLE 2). These 9 questions address symptoms triggered by exertion, such as chest pain, palpitations, syncope, or near syncope; history of heart murmur or need for an electrocardiogram (EKG); and family history of unexplained sudden death or premature heart disease.13 Further evaluation is critical if the answers indicate a suggestive patient or family history. (See “The sports physical: Should EKG be mandatory?”.)
TABLE 2
Screening for HCM: 9 questions you need to ask13
| 1. Have you ever passed out or nearly passed out during exercise? |
| 2. Have you ever passed out or nearly passed out after exercise? |
| 3. Have you ever had discomfort, pain, or pressure in your chest during exercise? |
| 4. Does your heart race or skip beats during exercise? |
| 5. Has a doctor ever told you that you have a heart murmur? |
| 6. Has a doctor ever ordered a test for your heart (for example, EKG, echocardiogram)? |
| 7. Has anyone in your family died for no apparent reason? |
| 8. Does anyone in your family have a heart problem? |
| 9. Has any family member or relative died of heart problems or of sudden death before age 50? |
| EKG, electrocardiogram; HCM, hypertrophic cardiomyopathy. |
Because both the physical exam and medical history are imperfect screening modalities, some clinicians have proposed the 12-lead EKG as an additional HCM screening tool. In the United States, the proposal is controversial, but the debate has intensified as a result of Italy’s experience: a 90% reduction in SCD following the implementation of a national EKG screening program for young athletes.37
Advocates in the United States cite the success of the Italian model and the lack of sensitivity in the standard history-physical HCM screening. Indeed, a retrospective study of US athletes who died suddenly showed that only 3% had been identified as having HCM during the traditional preparticipation screening, and none had been disqualified.38 Opponents of universal EKG screening point to the large number of potential candidates—approximately 12 million young people, ranging in age from adolescence through college, would need to be screened. Opponents also cite differences in the Italian and American populations; cost-benefit considerations; the large number of false-positive EKGs expected (10% to 15%); and most importantly, the lack of medical personnel to perform and interpret the EKGs.39 While advances in EKG technique may minimize false-positive readings and changes in the health care system may eventually create an environment more favorable to uniform screening procedures, current recommendations for preparticipation screening call for history and physical alone.
From suspicion to diagnosis
Patients with any abnormal findings or features suggestive of HCM should be referred to a cardiologist for further work-up.13 Accompany the referral with an order for complete exercise restriction until a more detailed analysis has been completed or HCM has been ruled out.
2-dimensional echocardiogram shows LV wall hypertrophy
Diagnosis is primarily made on the basis of 2-dimensional echocardiography showing asymmetric LV wall hypertrophy without chamber dilation (in the absence of another condition that might cause hypertrophy, such as hypertension, valvular disease, or amyloidosis). The anterior septum is commonly thickened; abnormal systolic anterior motion of the mitral valve may be evident, as well.1
Although increased LV wall thickness (≥13 mm) is the most common finding in HCM—and thicknesses up to 60 mm have been recorded—this is not a universal sign. Many people with genetic evidence of HCM have normal LV wall thickness. Conversely, some patients have increased LV wall thickness but do not have HCM.
HCM or “athlete’s heart”? Mild concentric LV hypertrophy (13-14 mm)—a level of thickening often referred to as the “athlete’s heart”—may be present in healthy individuals who exercise strenuously. In borderline cases, calculation of the LV mass distribution index by 3-dimensional echocardiography has been shown to have 100% specificity in distinguishing HCM from both athlete’s heart and hypertensive cardiomyopathy.14
Cardiac magnetic resonance imaging (MRI). With gadolinium as the contrast agent, cardiac MRI is another diagnostic tool. Imaging may reveal certain delayed enhancement patterns that are highly suggestive of HCM.15 Cardiac MRI can accurately quantify LV wall thickness and LV mass distribution index and identify subtle areas of patchy LV wall thickness that echocardiography may miss.16
Ensure that family members undergo screening
When physical exam, medical history, and follow-up tests are highly suggestive of HCM, clinical screening of asymptomatic first-degree relatives is recommended. In screening family members, it is important to remember that a normal physical exam, echocardiogram, and EKG do not definitively rule out HCM, as many people who have genetic mutations for this condition do not develop physical abnormalities until they reach adulthood.1 In such cases, genetic screening—the most definitive means of HCM diagnosis—may be considered, in consultation with a specialist.
Recognize the limits of genetic testing. Genetic testing is not universally recommended, however, for a number of reasons. Cost (about $3000) is a key factor. In addition, the test for HCM is not widely available. Nor is it highly sensitive, identifying only 50% to 60% of those with genetic mutations associated with HCM.4 What’s more, the presence of a genetic mutation does not guarantee that cardiac abnormalities will ever develop. Lifestyle, coexisting hypertension, and modifier genes may all play a role in determining whether an individual is affected.1,4
Provide follow-up. When genetic screening is not available, is declined, or is negative, stress the need for periodic clinical follow-up of family members. If the first-degree relative is an adolescent, he or she will need a history and physical examination, 12-lead EKG, and 2-dimensional echocardiography annually from the age of 12 to 18 years. If the relative is older than 18, he or she should be evaluated every 5 years.6
For patients themselves, SCD risk assessment is the next step
While family physicians may be involved in the care of a patient with HCM, an assessment of the individual’s risk for SCD is best done by a specialist. Risk stratification is typically based on the presence (or absence) of LVOTO, atrial fibrillation (AF), and heart failure.
LVOTO. Defined as a subaortic gradient of 30 mm Hg or more, LVOTO is present at rest in 25% of HCM patients.17 Because the obstruction is typically dynamic, treadmill or bicycle exercise testing in conjunction with Doppler echocardiography may be needed to identify it.6 LVOTO is a strong risk factor for death due to heart failure or stroke (relative risk [RR], 4.4, compared with HCM patients who do not have LVOTO) and death from any HCM-related cause (RR=2.0). Patients with LVOTO and left atrial enlargement are also at increased risk for bacterial infective endocarditis.18
AF, which occurs in approximately 25% of those with HCM and is more common in patients with LVOTO,19 often presents clinically as acute heart failure because of the reduced diastolic filling. Although AF is not as ominous as ventricular arrhythmia, it increases the risk for embolic stroke, the likelihood of severe functional disability, and the risk of death from HCM.19
Heart failure. This is the most common complication of HCM. In some cases, patients progress to a dilated cardiomyopathy that resembles classic systolic heart failure—and responds well to conventional treatments for systolic failure. More often, the condition more closely resembles diastolic failure and responds best to negative inotropic agents and the avoidance of volume depletion, both of which increase cardiac filling.20
Consensus guidelines weigh in on SCD risk
Age is another consideration in risk stratification for SCD, which primarily strikes those with HCM in adolescence or early adulthood. Consensus guidelines from the American College of Cardiology (ACC), American Heart Association (AHA), and European Society of Cardiology (ESC)21 (TABLE 1) offer additional considerations in assessing SCD risk.
Risk factors identified as “major” in the consensus guidelines include unexplained syncope, family history of premature cardiac death, left ventricular wall thickness ≥30 mm, abnormal blood pressure response to exercise, and nonsustained ventricular tachycardia, as well as a prior episode of cardiac arrest—the single most predictive risk factor.22 Because of the high risk of sudden death, exercise is absolutely contraindicated for many patients with certain HCM phenotypes and major risks.
The organizations also cite “possible” risk factors, and indicate in consensus statements that patients with more than 1 major or other possible risk factors are at higher risk for SCD.6,21 In cohort studies, however, other than a prior episode of cardiac arrest, no other risk factor has been found to predict SCD.
Cardiac MRI, discussed earlier for diagnostic purposes, may also have a role in stratifying risk. In small studies conducted recently, the presence of myocardial fibrosis (as demonstrated by delayed gadolinium enhancement) correlates with increased risk of nonsustained ventricular tachycardia (RR, 1.6, compared with HCM patients without myocardial fibrosis).23
HCM management: Pharmacologic and surgical options
There are no large-scale studies of medical treatments for HCM, and therapy is largely empiric and individualized based on complications, symptoms, and risk (FIGURE).24
FIGURE
HCM: A guide to screening, diagnosis, and treatment
*Relatively contraindicated in LVOTO patients.
AICD, automatic implantable cardioverter defibrillator; DDD, dual-chamber; echo, echocardiography; EKG, electrocardiography; HCM, hypertrophic cardiomyopathy; H&P, history and physical; LVOTO, left ventricular outflow tract obstruction; SCD, sudden cardiac death.
Adapted from: Soor GS, et al. J Clin Pathol.4
Which drugs for which patients?
For those with symptoms of heart failure, beta-blockers are first-line therapy. Additional therapeutic options for patients without LVOTO include calcium channel blockers, nitrates, and diuretics. But these additional therapies are relatively contraindicated in patients with LVOTO. For LVOTO patients, disopyramide can be added to the beta-blocker regimen, if needed for symptom control.4,24
Patients with LVOTO or abnormal mitral motion are at moderate risk of spontaneous bacterial endocarditis (SBE).18 But evolving evidence shows low baseline rates of SBE and increased complications in patients routinely receiving antimicrobial prophylaxis, so the most recent guidelines do not recommend such treatment for any HCM patient.25
Amiodarone is effective for atrial fibrillation in HCM when beta- or calcium channel blockers fail to provide sufficient rate control.4 Amiodarone can also be used to prevent SCD.26 Recent data show that an automatic implantable cardioverter defibrillator (AICD)—which we’ll discuss later—is superior to amiodarone in preventing SCD,27 but the drug may be used in addition to an AICD or given to patients who are not candidates for, or not interested in, an implantable device.22,24
Invasive treatments may be considered for patients with LVOTO who do not respond to medical management.
Septal myomectomy or ethanol ablation: Which is better?
Septal myomectomy is the gold standard for refractory LVOTO.28 This open-heart procedure, which involves the resection of a portion of the septum to remove the obstructing cardiac tissue, has an operative mortality rate <1%.28 Surgical complication rates are also low, but include aortic regurgitation, left bundle or complete heart block, and iatrogenic ventricular septal defect.
Septal ethanol ablation is a percutaneous alternative to surgical myomectomy. In this minimally invasive procedure, ethanol is injected into the first septal perforating branch of the left anterior descending (LAD) artery, resulting in myocardial necrosis and septal wall thinning, which relieves the obstruction. Complications include ablation of inappropriate myocardium, heart block, pericardial effusion, and LAD dissection.29 Maximum effects of the ablation are delayed, typically occurring 6 to 12 months after the procedure.
Although limited by a lack of randomized controlled trials, a recent meta-analysis found surgical myomectomy and ethanol ablation to be equally effective in decreasing the LV outflow gradient to <20 mm Hg. Notably, though, surgical myomectomy reduced the gradient to nearly 10 mm Hg, compared with an average of 18 mm Hg for ethanol ablation.28 What’s more, several studies have found a higher incidence of complete heart block in patients who had ethanol ablation compared with those who underwent myomectomy.30 For patients who cannot tolerate or are not interested in invasive surgery, ablation offers an effective option.
Dual-chamber (DDD) pacing can also be used to treat LVOTO, but studies comparing pacing with myomectomy and ablation have found mixed results.6 Despite recent data showing the benefits of pacing in HCM,31 DDD pacing is typically reserved for patients who are not candidates for either surgical myomectomy or ablation.4
AICDs for which patients? It’s not always clear
AICDs effectively prevent SCD in patients with HCM,1,6,22 but the substantial cost and high rate of complications (>36%) make the devices impractical and inadvisable for universal use. Adverse events include pneumothorax, pericardial effusion, device infection or malfunction, and physical and psychological sequelae from inappropriate shocks.32 In fact, several studies of AICDs in patients with HCM have found the yearly rate of inappropriate shocks to be higher than the rate of appropriate discharges.22,31 And, because HCM patients are typically decades younger than coronary disease patients when they undergo implantation, they have a significantly higher burden of complication.32
Consensus statements vs actual practice. The central question of HCM risk stratification is how to identify patients at risk of SCD, thereby making it possible for them to reap the benefits of an AICD and drug treatment while sparing low-risk candidates the morbidity and the expense. So far, that question has not been definitively answered. As noted earlier, consensus statements agree that patients with more than 1 major risk factor have a higher risk of SCD6,28 than those with only 1; yet many patients with a single risk factor (and no prior cardiac arrest) have received AICDs.33
Studies highlight limitations of consensus guidelines’ assessment of risk. In recent case studies of HCM patients with AICDs based on registry data, roughly 25% of those studied22 received AICDs for secondary prevention—that is, after surviving cardiac arrest; the rest received them for primary prevention based on clinical risk factors. Rates of appropriate AICD discharge were 3-fold higher in patients who had survived previous cardiac arrest than in those who had not,22,32 a finding that supports aggressive AICD implantation among these high-risk patients.
Among patients who had received AICDs for primary prevention, however, appropriate discharges occurred at statistically identical rates whether they had 1, 2, or 3 major risk factors. Further, there was no association between the number of risk factors and the likelihood of appropriate discharge. Given these results, the decision to use an AICD in an HCM patient for primary prevention should be made after careful consultation with the patient and an HCM disease specialist.
What to tell patients about sports activities
Just as there is no definitive means of deciding when, or whether, a patient who has never experienced cardiac arrest should receive an AICD, there is no clear, evidence-based consensus on exercise restriction. Recommendations, which are based on expert opinion, leave room for individualized decision-making.
For those with genetic mutations consistent with HCM but no associated cardiac abnormalities and no family history of sudden death, no objective data support exercise limitations.34 For such patients, education regarding warning signs and symptoms and annual follow-up should be sufficient.
For athletes with a probable or unequivocal diagnosis of HCM, the ACC recommends restriction from competitive sports, with the possible exception of low-intensity activities such as billiards, bowling, and golf.35 This recommendation is not dependent on the presence of LVOTO or on patient symptoms, medical or surgical therapy, or the placement of an AICD.
A consensus statement from the ESC also lists recreational doubles tennis and biking, lap swimming, and weight lifting as permissible activities for patients with probable or unequivocal HCM, with a cautionary note about avoiding the Valsalva maneuver.36 The society discourages those with HCM from engaging in any activity that provokes dyspnea. Primary care physicians, too, must be sure that young patients with this condition understand the importance of avoiding intense exertion—and immediately stopping any physical activity if they notice any signs or symptoms associated with HCM.
Disclosure
The views expressed here are those of the authors and do not represent the policy of the United States Air Force, United States Army, or Department of Defense.
CORRESPONDENCE Anthony Beutler, MD, FAAFP, Department of Family Medicine A-1038, Uniformed Services University, 4301 Jones Bridge Road, Bethesda, MD 20814; [email protected]
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.
1. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308-1320.
2. Maron BJ. Hypertrophic cardiomyopathy and other causes of sudden cardiac death in young competitive athletes, with considerations for preparticipation screening and criteria for disqualification. Cardiol Clin. 2007;25:399-414.
3. McKenna W, Deanfield J, Faruqui A, et al. Prognosis in hypertrophic cardiomyopathy: role of age and clinical, electrocardiographic and hemodynamic features. Am J Cardiol. 1981;47:532-538.
4. Soor GS, Luk A, Ahn E, et al. Hypertrophic cardiomyopathy: current understanding and treatment objectives. J Clin Pathol. 2009;62:226-235.
5. Watkins H, Ashrafian H, McKenna WJ. The genetics of hypertrophic cardiomyopathy: Teare redux. Heart. 2008;94:1264-1268.
6. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687-1713.
7. Teare D. Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958;2:1-8.
8. Maron BJ, Casey SA, Poliac LC, et al. Clinical course of hypertrophic cardiomyopathy in a regional United States cohort. JAMA. 1999;281:650-655.
9. Makenna W. Diseases of the Myocardium and Endocardium. Goldman L, Ausiello D, ed. Philadelphia: Saunders; 2008.
10. Giese EA, O’Connor FG, Brennan FH, et al. The athletic preparticipation evaluation: cardiovascular assessment. Am Fam Physician. 2007;75:1008-1114.
11. Glover DW, Maron BJ. Evolution in the process of screening United States high school student-athletes for cardiovascular disease. Am J Cardiol. 2007;100:1709-1712.
12. Rauch CM, Phillips GC. Adherence to guidelines for cardiovascular screening in current high school preparticipation evaluation forms. J Pediatr. 2009;155:584-586.
13. American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Osteopathic Society for Sports Medicine. The Preparticipation Physical Evaluation. 3rd ed. Minneapolis, Minn: McGrawHill; 2005:19–23.
14. Caselli S, Pelliccia A, Maron M, et al. Differentiation of hypertrophic cardiomyopathy from other forms of left ventricular hypertrophy by means of three-dimensional echocardiography. Am J Cardiol. 2008;102:616-620.
15. Olivotto I, Maron MS, Autore C, et al. Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;52:559-566.
16. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54:220-228.
17. Maron MS, Olivotto I, Betocchi S, et al. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med. 2003;348:295-303.
18. Spirito P, Rapezzi C, Bellone P, et al. Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation. 1999;99:2132-2137.
19. Olivotto I, Cecchi F, Casey SA, et al. Impact of atrial fibrillation on the clinical course of hypertrophic cardiomyopathy. Circulation. 2001;104:2517-2524.
20. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775-785.
21. Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur Heart J. 2006;27:2099-2140.
22. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298:405-412.
23. Nazarian S, Lima JA. Cardiovascular magnetic resonance for risk stratification of arrhythmia in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008;51:1375-1376.
24. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117:429-439.
25. Nishimura RA, Carabello BA, Faxon DP, et al. ACC/AHA 2008 Guideline update on valvular heart disease: focused update on infective endocarditis: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2008;52:676-685.
26. McKenna WJ, Oakley CM, Krikler DM, et al. Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J. 1985;53:412-416.
27. Maron BJ, Shen WK, Link MS, et al. Efficacy of implantable cardioverter-defibrillators for the prevention of sudden death in patients with hypertrophic cardiomyopathy. N Engl J Med. 2000;343:365-373.
28. Alam M, Dokainish H, Lakkis NM. Hypertrophic obstructive cardiomyopathy-alcohol septal ablation vs. myectomy: a meta-analysis. Eur Heart J. 2009;30:1080-1087.
29. Alam M, Dokainish H, Lakkis N. Alcohol septal ablation for hypertrophic obstructive cardiomyopathy: a systematic review of published studies. J Interv Cardiol. 2006;19:319-327.
30. Fernandes VL, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1:561-570.
31. Galve E, Sambola A, Saldana G, et al. Late benefits of dual-chamber pacing in obstructive hypertrophic cardiomyopathy. A 10-year follow-up study. Heart. 2009; May 28 [E-pub ahead of print.]
32. Lin G, Nishimura RA, Gersh BJ, et al. Device complications and inappropriate implantable cardioverter defibrillator shocks in patients with hypertrophic cardiomyopathy. Heart. 2009;95:709-714.
33. Elliott PM, Poloniecki J, Dickie S, et al. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212-2218.
34. Elliott P, Spirito P. Prevention of hypertrophic cardiomyopathy-related deaths: theory and practice. Heart. 2008;94:1269-1275.
35. Maron BJ, Ackerman MJ, Nishimura RA, et al. Task Force 4: HCM and other cardiomyopathies, mitral valve prolapse, myocarditis, and Marfan syndrome. J Am Coll Cardiol. 2005;45:1340-1345.
36. Pelliccia A, Fagard R, Bjornstad HH, et al. Recommendations for competitive sports participation in athletes with cardiovascular disease: a consensus document from the Study Group of Sports Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J. 2005;26:1422-1445.
37. Corrado D, Basso C, Pavei A, et al. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593-1601.
38. Maron BJ, Shirani J, Poliac LC, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199-204.
39. Maron BJ, Thompson PD, Ackerman MJ, et al. Recommendations and considerations related to preparticipation screening for cardiovascular abnormalities in competitive athletes: 2007 update: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism: endorsed by the American College of Cardiology Foundation. Circulation. 2007;115:1643-1655.