User login
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
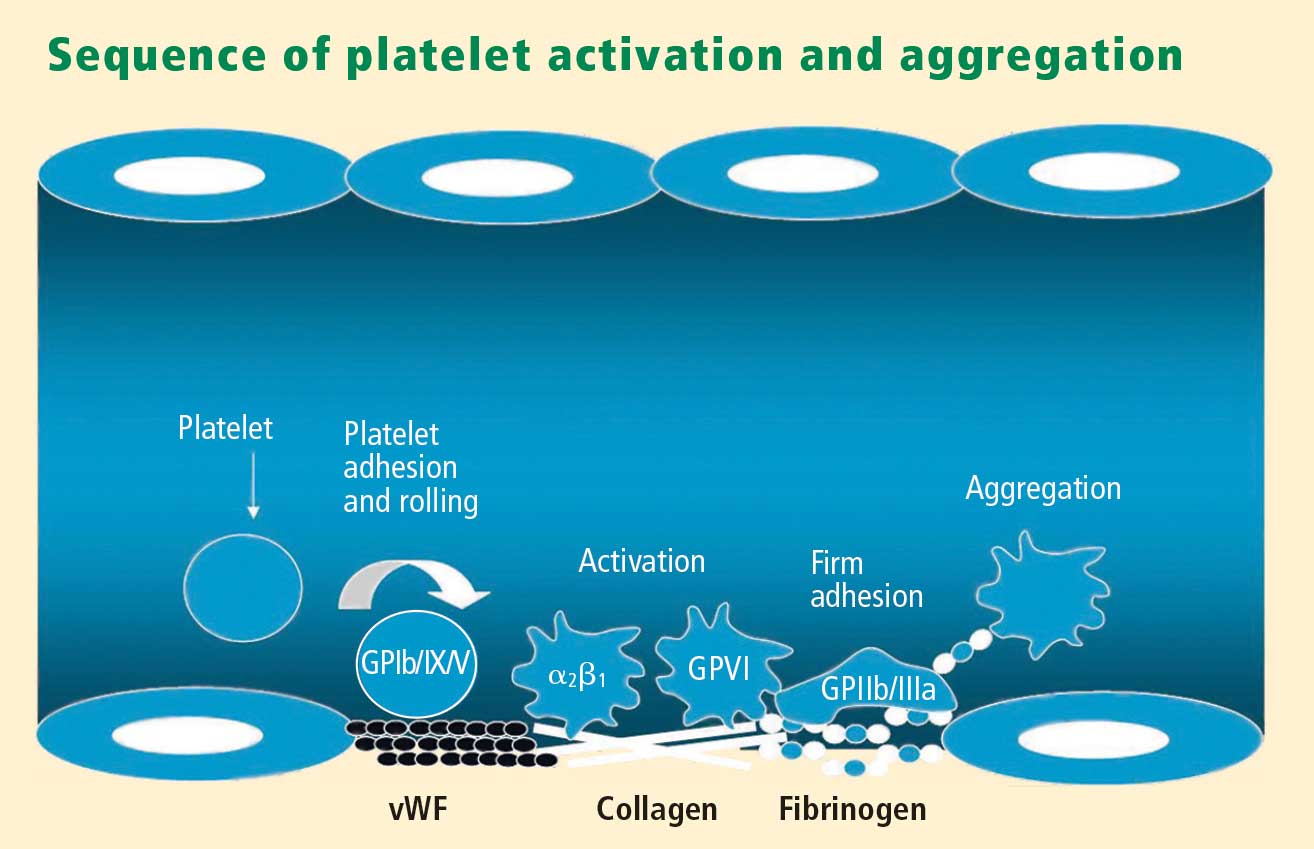
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
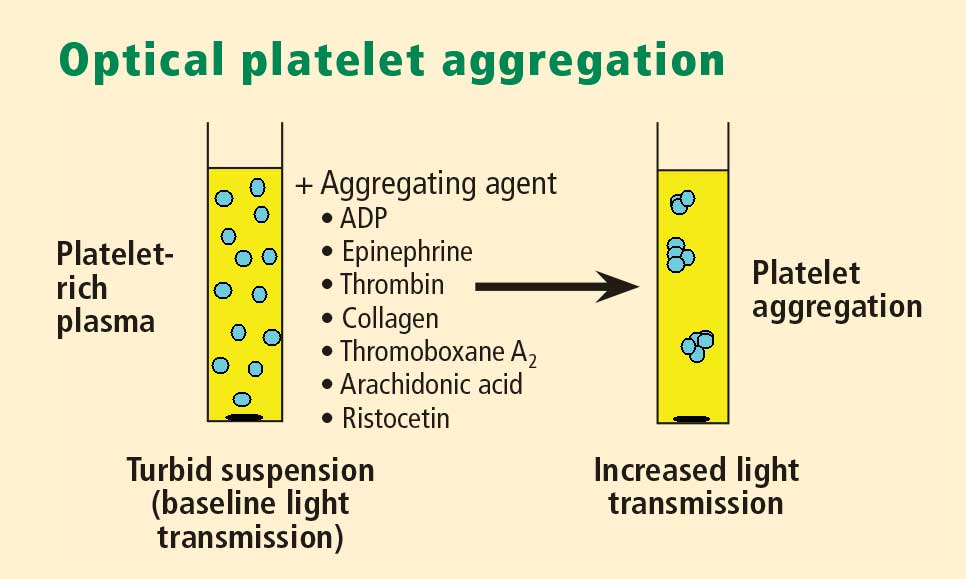
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
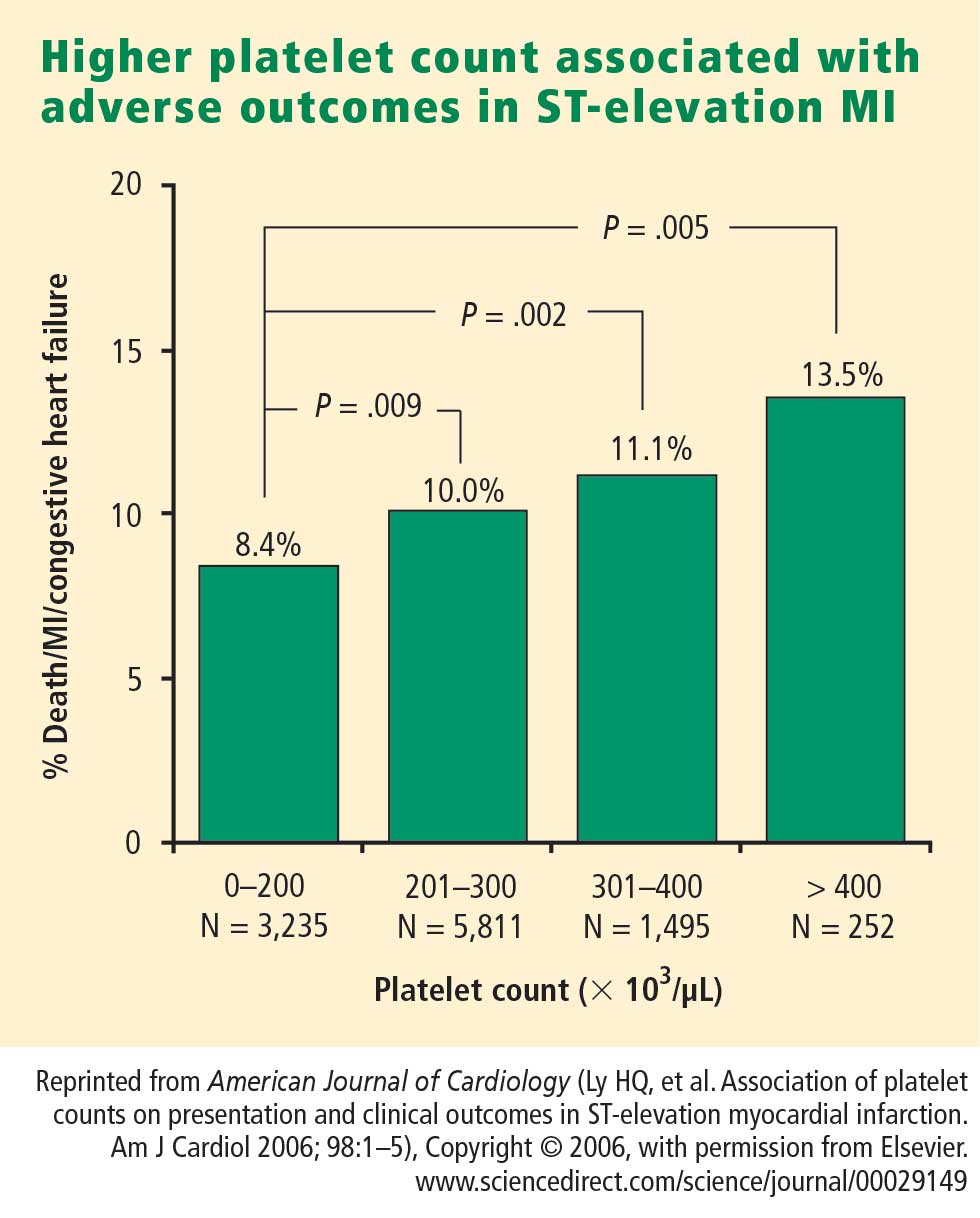
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
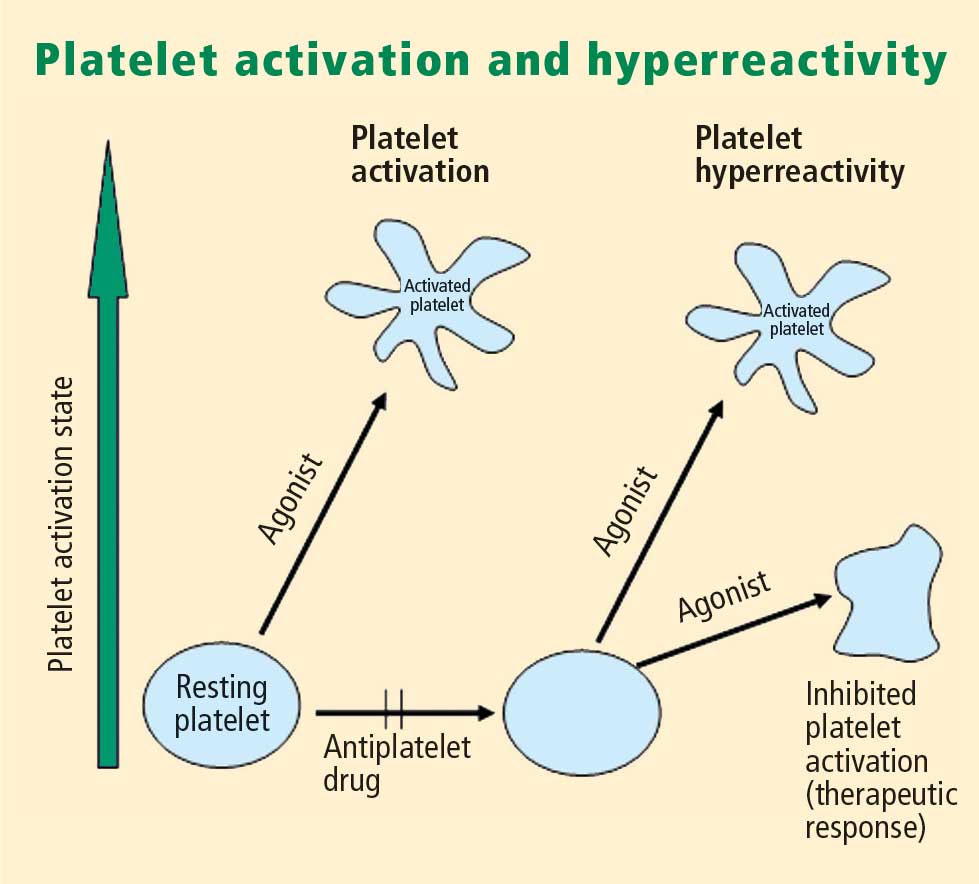
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
PLATELET FUNCTION
Platelets are non-nucleated cells produced by megakaryocytes, which are very large cells (50 to 100 μm in diameter) found in bone marrow. The megakaryocyte surface membrane forms protoplatelet extensions from which platelets “bud off” and are emitted into the circulation, where they number approximately 200,000 to 400,000 per microliter of blood.
Platelet activation
Platelets play a crucial role in the vascular response to injury, and activation of platelets has long been recognized as an important step. Platelets release dense granules that contain the nucleotide adenosine diphosphate (ADP), which activates other platelets. They also possess alpha granules, which contain proteins and protein mediators (eg, platelet-derived growth factor, platelet factor 4) that are involved in inflammatory processes. The platelet surface is coated with hundreds of thousands of receptors for other cells, including activated vascular wall cells and extracellular matrix proteins. Platelets possess an affinity for adherence, especially to injured vessel walls, where they release their granule contents and then aggregate. These properties promote platelets’ involvement in many vascular processes, including ACS, as will be explored below.
Platelets exist in a nonactivated state and are drawn passively into areas of vascular injury. Initially, they adhere to proteins such as von Willebrand factor, which is a large extracellular matrix protein produced by endothelial cells. The platelet glycoprotein Ib/IX/V binds to von Willebrand factor, forming a loose association that results in platelets rolling on the surface of the vessel wall. As a multimer, von Willebrand factor exists in one subunit that is dimerized and then polymerized, making it an ideal substrate for platelets because of the multiple substrates to which platelets can adhere.
Platelet fibrinogen receptor
The platelet fibrinogen receptor (glycoprotein IIb/IIIa receptor) is an αIIbβ3 integrin that binds to arginine-glycine-aspartic acid (RGD) epitopes of proteins, such as fibrinogen. Fibrinogen has a two-dimensional symmetry, with RGD groups on both ends of the molecule, which makes it an ideal molecule for linking platelet to platelet.
von Willebrand factor has RGD groups, as do both fibronectin and glycoprotein IIb/IIIa vitronectin, and can therefore bind to many plasma and extracellular matrix proteins. The glycoprotein IIb/IIIa receptor is inactive in resting platelets. It becomes activated during the platelet activation process and binds to fibrinogen, which bridges to other platelets, causing aggregation.
ADP receptors
Various receptors on platelet surfaces are responsible for platelet activation. One is a family of receptors for ADP. As ADP is released from platelets, it can then activate other platelets by binding to the receptors. The ADP receptor P2Y12 signals through G protein pathways and is coupled to adenylate cyclase, an enzyme that catalyzes the conversion of adenosine triphosphate to cyclic adenosine monophosphate (cAMP). High levels of cAMP inhibit platelet function; ADP binding to P2Y12 shuts down adenylate cyclase, which leads to phosphoinositide 3-kinase activation and accelerated aggregation and platelet release.
A final notable factor in the mediation of platelet activation and aggregation is phospholipase A2, which liberates arachidonic acid from the platelet membrane, metabolizing it through cyclooxygenase and thromboxane synthase to generate thromoboxane A2, which leads to release of platelet granule contents and aggregation of other platelets.
PLATELET FUNCTION TESTS
Platelet function assays are inherently variable because they measure cell function rather than a single analyte. Several new platelet testing devices have come to market with the goal of ease of use; many can now be used at the bedside to measure platelet function.
Platelet count
In my view, the platelet count remains one of the best tests for assessing bleeding risk, as a low platelet count is one of the most common causes of bleeding. However, the platelet count is not a functional assay because it does not evaluate other platelet functions.
Screening tests
Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of many different aspects of platelet function, such as adhesion, aggregation and granule release.
Bleeding time. The bleeding time is an archaic test because of the poor correlation between bleeding time and bleeding disorders or thrombotic disorders. Its utility in measuring platelet function is therefore highly limited.
PFA-100. The PFA-100 Platelet Function Analyzer system (PFA-100) is one example of a global platelet function assay that measures multiple platelet functions, including platelet adhesion and aggregation. The instrument, which is about the size of a bread box, uses a citrate-anticoagulated whole blood specimen to measure platelet reaction in a high-shear environment. Blood travels at high shear rates through membranes coated with either collagen and ADP or collagen and epinephrine (epinephrine receptors exist on platelet surfaces). Platelets adhere to the membranes and then activate, aggregate, and occlude a small aperture in the center of each membrane, yielding a measurable closure time.
Since the PFA-100 was developed before the availability of the thienopyridine antiplatelet drugs, its utility lies not in monitoring the effects of those agents but in its ability to detect aspirin-induced platelet dysfunction or intrinsic platelet function disorders. An abnormal epinephrine cartridge closure time in the presence of a normal ADP cartridge closure time indicates aspirin-induced platelet dysfunction. An abnormal closure time on both measures is indicative of von Willebrand disease or a platelet defect such as Glanzmann thrombasthenia or Bernard-Soulier syndrome.
Specific functional tests
Whole blood platelet aggregation is typically a high-complexity laboratory test. Recently, self-contained assay platforms that can measure whole blood aggregation have been developed. These are applicable for smaller hospitals and near-patient settings. One such rapid platelet function analyzer, known commercially as VerifyNow, offers point-of-care assessment of platelet function. The instrument, which is the size of a telephone answering machine, operates by a principle similar to that of optical platelet aggregation: platelet function is measured by the rate and extent of change in light transmittance in response to the introduction of agonists specific to various antiplatelet medications. Low light transmittance indicates a blood sample with inhibited platelet function; high light transmittance indicates normal platelet function.
Measurement of VASP phosphorylation. Vasodilator-stimulated phosphoprotein (VASP) is an intracellular platelet protein that is nonphosphorylated in basal state. The phosphorylation of VASP depends on the level of activation of the P2Y12 receptor, a target of thienopyridine drugs. Thus, measuring VASP phosphorylation by flow cytometry using citrated whole blood can be a highly specific indicator of the action and efficacy of clopidogrel and other thienopyridine drugs.
A flow cytometry assay that measures VASP phosphorylation requires a whole blood sample that is incubated with ADP to measure what is called the platelet reactivity index. Adding ADP to whole blood stimulates adenylate cyclase, lowering cAMP and shutting off protein kinase, which results in low levels of VASP phosphorylation. Thus, if VASP is phosphorylated, the platelets are inhibited; if VASP is not phosphorylated, the platelets are activated. A satisfactory therapeutic response to clopidogrel or another thienopyridine drug produces a low platelet reactivity index, reflecting platelet inhibition.
ROLE OF PLATELETS IN ATHEROSCLEROSIS
Platelets serve major functions in three key aspects of atherosclerosis: atherogenesis, inflammation, and atherothrombosis.
Atherogenesis
Platelets play a pivotal role in atherogenesis.1 They release matrix metalloproteinases that are involved in degrading the matrix in atherosclerotic plaques. Moreover, they contain and release chemokines and growth factors, including:
- RANTES, a chemokine that stimulates monocytes and T cells to increase the production of monocyte inflammatory mediators
- Platelet-derived growth factor, which stimulates the migration and proliferation of smooth muscle cells
- Transforming growth factor–β, which also stimulates proliferation of smooth muscle cells.
Inflammation
Activated platelets release inflammatory mediators and thereby change the adhesive and chemotactic properties of endothelial cells. Likewise, mediators derived from inflammatory cells (neutrophils) can affect platelet function.
Platelet-derived mediators include the following:
- Pro‑interleukin (IL)-β, which triggers the synthesis of E-selectin that enables endothelial cells to interact with leukocytes
- Thromboxane A2, which increases neutrophil adhesion to facilitate platelet aggregation
- Platelet-derived growth factor and platelet factor 4, which increase neutrophil chemotaxis (the ability of neutrophils to infiltrate atherosclerotic plaque)
- CD40 ligand, a protein expressed on platelets that induces inflammatory responses in the endothelium
- P-selectin, a cell adhesion molecule expressed on activated platelets that enhances the adhesion of monocytes on activated endothelial cells.
Among the neutrophil-derived mediators, some—such as superoxide and leukotrienes—enhance platelet activation, whereas elastases inhibit platelet activation.
Overall, once inflammation begins in an atherosclerotic plaque, much reciprocal platelet activation can occur, so that the inflammatory process can become a feed-forward loop to eventually promote atherothrombosis.
Atherothrombosis
In the last stage of the atherosclerotic process, platelet enzymes that degrade the matrix may make plaques vulnerable to rupture by creating fissures in the fibrous plaque cap. This exposes the lipid-rich core, which contains a significant amount of thromboplastin. Exposure to the extracellular matrix can lead to further platelet adhesion, activation, and aggregation. The development of a platelet thrombus is usually one of the ultimate steps in atherothrombosis leading to ACS, including MI.
ROLE OF PLATELETS IN ACUTE CORONARY SYNDROMES: WHAT IS THE EVIDENCE?
How predictive is an elevated platelet count?
However, another study conducted in a slightly different population—1,616 patients with non‑ST-segment-elevation MI/unstable angina—found no correlation between platelet count (by quintiles) and death at 60 months.3 The lowest mortality was observed in patients with a platelet count in the second-lowest quintile, although the highest mortality was indeed observed in the quintile of patients with the lowest platelet counts.3
The differing results in the above two studies suggest that additional platelet factors, beyond platelet count, contribute to the risk of adverse outcomes following ACS.
Platelet hyperreactivity and outcomes in ACS
Platelet hyperreactivity—ie, residual platelet activity despite antiplatelet therapy—appears to be involved in the spectrum of ACS. A recent study evaluated the association between hyperreactivity of platelets to ADP and outcomes in 600 patients with stable cardiovascular disease who were on aspirin therapy.4 Hyperreactivity was defined as a collagen/ADP closure time of less than 90 seconds on the PFA-100 system (short collagen/ADP closure time). On receiver operating characteristic (ROC) curve analysis, a short collagen/ADP closure time served as a significant predictor of recurrent events (relative risk [RR] = 3.65; 95% CI, 1.76–7.57) and death (RR = 6.56; 95% CI, 1.93–22.35) compared with a closure time of 90 seconds or greater. The authors concluded that there appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major adverse events associated with platelet hyperreactivity.4
An earlier study by Harrison et al assessed platelet function using the PFA-100 in 78 patients presenting with acute chest pain classified as MI, unstable angina, or nonspecific chest pain.5 Using the PFA-100, they found shorter collagen/ADP closure times and higher levels of von Willebrand factor in subjects with MI compared with those who had unstable angina or nonspecific chest pain.5 Fuchs et al reported a similar association between von Willebrand factor and outcomes in 208 patients with ACS,6 raising the possibility that von Willebrand factor, through its association with increased platelet adhesion and activation, may be a major contributor to risk in ACS.
Similarly, an association between platelet hyperreactivity and cardiovascular events has been suggested in patients with type 2 diabetes. In a 2007 study of 173 patients with type 2 diabetes and coronary artery disease receiving dual antiplatelet therapy (aspirin plus clopidogrel), the 2-year risk of major cardiovascular events was significantly higher in those in the highest quartile of platelet aggregation compared with those in the lower three quartiles (hazard ratio = 3.35; 95% CI, 1.68–6.66).7 In a separate study, Serebruany et al measured platelet activity by five different testing methods in 822 patients with coronary artery disease and found significantly higher platelet hyperreactivity by all methods in those patients who had diabetes (n = 257) than in those who did not (n = 565).8
Marcucci et al recently examined the relationship between clinical characteristics and residual platelet activity in 386 patients with ACS on dual antiplatelet therapy (aspirin plus clopidogrel).9 The presence of residual platelet activity (determined by platelet aggregation in response to the agonists arachidonic acid and ADP, as well as by the PFA-100) was associated with significantly higher inflammatory status, as determined by leukocyte count and erythrocyte sedimentation rate. The same association was observed among a subset of patients in this study undergoing percutaneous coronary intervention (PCI) who were receiving dual antiplatelet therapy; additionally, residual platelet activity was associated with a significantly higher incidence of diabetes and a significantly lower ejection fraction in this subset.9
Platelet hyperreactivity while on dual antiplatelet therapy (aspirin plus clopidogrel) was also found to be predictive of clinical outcome in a study of 195 patients with non-ST-elevation MI undergoing PCI.10 Hyporesponse to antiplatelet therapy, as measured by a high VASP platelet reactivity index (PRI), predicted an increased risk of recurrent ischemic events within 30 days of PCI. Using ROC curve analysis, the investigators found that a VASP PRI cutoff value of 53% (ie, a high PRI [> 53%] indicates residual platelet activity despite clopidogrel) had a sensitivity of 93%, a specificity of 50%, a positive predictive value of 12%, and a negative predictive value of 99% for ischemic events.10 Similarly, among 144 patients undergoing PCI assessed for decreased platelet reactivity to a loading dose of clopidogrel, Bonello et al also found that a VASP PRI greater than 50% was optimal for predicting major adverse cardiovascular events: all 21 events in the study occurred among patients whose VASP PRI was in the highest four quintiles.11
CONCLUSIONS AND GENERAL ASSESSMENT OF PLATELET FUNCTION TESTS
Platelets clearly are involved in the pathogenesis of atherothrombosis. Accumulating evidence suggests that both an elevated platelet count and platelet hyperreactivity (residual platelet activity despite dual antiplatelet therapy) may be associated with adverse cardiovascular events in patients with ACS.
Platelet function can be measured using several different assays and measures of platelet activation. The best assays for measuring residual platelet activity in the setting of antiplatelet therapy are still being defined, as are their predictive values. Platelet aggregation remains the gold standard. The PFA-100 may detect overall platelet hyperreactivity despite the use of antiplatelet therapy, and is attracting increasing use for this purpose. VASP phosphorylation may be a good assay for detecting P2Y12 inhibition but is limited to thienopyridines in terms of detecting platelet hyperreactivity. For predicting adverse cardiac events, ROC curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrarily defined cutoff values.
Moving forward, standard testing protocols for platelet aggregation clearly are needed to achieve consistency among studies.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
- Davì G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357:2482–2494.
- Ly HQ, Kirtane AJ, Murphy SA, et al. Association of platelet counts on presentation and clinical outcomes in ST-elevation myocardial infarction (from the TIMI Trials). Am J Cardiol 2006; 98:1–5.
- Mueller C, Neumann FJ, Hochholzer W, et al. The impact of platelet count on mortality in unstable angina/non-ST-segment elevation myocardial infarction. Am Heart J 2006; 151:1214.e1–7.
- Christie DJ, Kottke-Marchant K, Gorman RT. Hypersensitivity of platelets to adenosine diphosphate in patients with stable cardiovascular disease predicts major adverse events despite antiplatelet therapy. Platelets 2008; 19:104–110.
- Harrison P, Mackie I, Mathur A, et al. Platelet hyperfunction in acute coronary syndromes. Blood Coagul Fibrinolysis 2005; 16:557–562.
- Fuchs I, Frossard M, Spiel A, Riedmüller E, Laggner AN, Jilma B. Platelet function in patients with acute coronary syndrome (ACS) predicts recurrent ACS. J Thromb Haemost 2006; 4:2547–2552.
- Angiolillo DJ, Bernardo E, Sabaté M, et al. Impact of platelet reactivity on cardiovascular outcomes in patients with type 2 diabetes mellitus and coronary artery disease. J Am Coll Cardiol 2007; 50:1541–1547.
- Serebruany V, Pokov I, Kuliczkowski W, Chesebro J, Badimon J. Baseline platelet activity and response after clopidogrel in 257 diabetics among 822 patients with coronary artery disease. Thromb Haemost 2008; 100:76–82.
- Marcucci R, Gori AM, Paniccia R, et al. Residual platelet reactivity is associated with clinical and laboratory characteristics in patients with ischemic heart disease undergoing PCI on dual antiplatelet therapy. Atherosclerosis 2007; 195:e217–e223.
- Frere C, Cuisset T, Quilici J, et al. ADP-induced platelet aggregation and platelet reactivity index VASP are good predictive markers for clinical outcomes in non-ST elevation acute coronary syndrome. Thromb Haemost 2007; 98:838–843.
- Bonello L, Paganelli F, Arpin-Bornet M, et al. Vasodilator-stimulated phosphoprotein phosphorylation analysis prior to percutaneous coronary intervention for exclusion of postprocedural major adverse cardiovascular events. J Thromb Haemost 2007; 5:1630–1636.
KEY POINTS
- Platelet function assays are inherently variable because they measure cell function rather than a single analyte.
- Screening tests, or global tests for platelet function, do not identify specific causes of platelet dysfunction but combine measurement of different aspects of platelet function.
- There appears to be a subgroup of patients with stable cardiovascular disease who have an increased risk of major cardiac events associated with platelet hyperreactivity.
- For predicting cardiac events, receiver operating characteristic (ROC) curve analysis should be used to objectively define cutoff values for platelet hyperreactivity as opposed to reliance on arbitrary cutoff values.