User login
respectively, of pityriasis lichenoides, an uncommon clonal T-cell disorder. The cause is unknown, although associations with infections have been reported. Pityriasis lichenoides more commonly affects children or young adults, usually before age 30 years. The disease can occur in all races.
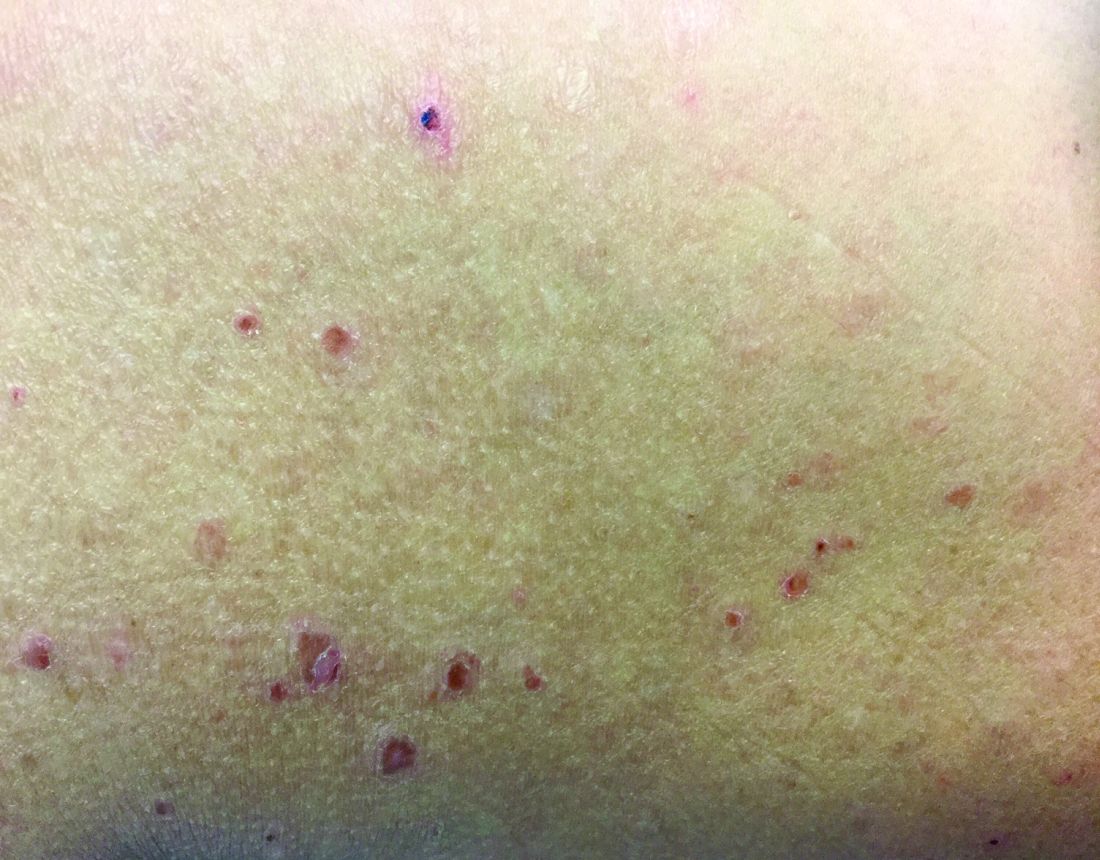
In PLEVA, erythematous to brown papules and macules, in various stages of evolution, appear suddenly and in crops. The trunk and flexural areas are most often affected, but lesions may become widespread and may be pruritic or painful. Lesions may crust, ulcerate, or become necrotic and can heal with scarring. In general, patients don’t have constitutional symptoms. Lesions tend to resolve spontaneously over 1-3 years.
Rarely, PLEVA may develop into a more severe form called febrile ulceronecrotic Mucha-Habermann disease, a dermatologic emergency. Patients (more commonly, young males) may present with high fever, malaise, and lymphadenopathy. Lesions become very painful, ulcerated, and necrotic, and extensive necrosis may be present. Changes in mental status, breathing difficulties, anemia, arthritis, abdominal pain, and sepsis may occur. Patients require hospitalization. There is a 25% mortality rate.
PLC is at the other end of this disease spectrum, representing the chronic, more mild stage of the disorder. Lesions present as indolent, asymptomatic, scaly macules and erythematous papules, favoring the trunk and proximal extremities. Lesions tend to be fewer in number than seen in PLEVA. They resolve over several months and may result in hypopigmentation, but usually don’t cause scarring. Patients may have long periods of remission between outbreaks. T-cell gene rearrangement may demonstrate monoclonality. PLC is generally considered a benign disease, although there are patients who have developed cutaneous T-cell lymphoma. For this reason, patients should be followed carefully for signs of malignant transformation.
Both forms share a common histologic picture. In PLEVA, focal parakeratosis and crusting is present. A dense, wedge-shaped infiltrate can be seen with prominent lymphocytic exocystosis in the epidermis. Necrotic keratinocytes are often seen. There may be spongiosis and intraepidermal vesicles. Extravasation of erythrocytes often occurs in the epidermis. PLC is histologically similar but far more subtle. There is less crusting, less spongiosis, fewer vesicles, and fewer necrotic keratinocytes. Generally, atypia of lymphocytes is absent.
Mucha-Habermann requires treatment with systemic steroids. Methotrexate, cyclosporine, or dapsone may be used as steroid-sparing agents. Upon treatment, lesions may resolve or revert back to more typical lesions of PLEVA. Treatment for PLEVA and PLC includes oral tetracycline or erythromycin, antihistamines (if pruritus is present), topical steroids, topical tacrolimus or pimecrolimus, or phototherapy. Low-dose weekly methotrexate may be helpful.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
respectively, of pityriasis lichenoides, an uncommon clonal T-cell disorder. The cause is unknown, although associations with infections have been reported. Pityriasis lichenoides more commonly affects children or young adults, usually before age 30 years. The disease can occur in all races.
In PLEVA, erythematous to brown papules and macules, in various stages of evolution, appear suddenly and in crops. The trunk and flexural areas are most often affected, but lesions may become widespread and may be pruritic or painful. Lesions may crust, ulcerate, or become necrotic and can heal with scarring. In general, patients don’t have constitutional symptoms. Lesions tend to resolve spontaneously over 1-3 years.
Rarely, PLEVA may develop into a more severe form called febrile ulceronecrotic Mucha-Habermann disease, a dermatologic emergency. Patients (more commonly, young males) may present with high fever, malaise, and lymphadenopathy. Lesions become very painful, ulcerated, and necrotic, and extensive necrosis may be present. Changes in mental status, breathing difficulties, anemia, arthritis, abdominal pain, and sepsis may occur. Patients require hospitalization. There is a 25% mortality rate.
PLC is at the other end of this disease spectrum, representing the chronic, more mild stage of the disorder. Lesions present as indolent, asymptomatic, scaly macules and erythematous papules, favoring the trunk and proximal extremities. Lesions tend to be fewer in number than seen in PLEVA. They resolve over several months and may result in hypopigmentation, but usually don’t cause scarring. Patients may have long periods of remission between outbreaks. T-cell gene rearrangement may demonstrate monoclonality. PLC is generally considered a benign disease, although there are patients who have developed cutaneous T-cell lymphoma. For this reason, patients should be followed carefully for signs of malignant transformation.
Both forms share a common histologic picture. In PLEVA, focal parakeratosis and crusting is present. A dense, wedge-shaped infiltrate can be seen with prominent lymphocytic exocystosis in the epidermis. Necrotic keratinocytes are often seen. There may be spongiosis and intraepidermal vesicles. Extravasation of erythrocytes often occurs in the epidermis. PLC is histologically similar but far more subtle. There is less crusting, less spongiosis, fewer vesicles, and fewer necrotic keratinocytes. Generally, atypia of lymphocytes is absent.
Mucha-Habermann requires treatment with systemic steroids. Methotrexate, cyclosporine, or dapsone may be used as steroid-sparing agents. Upon treatment, lesions may resolve or revert back to more typical lesions of PLEVA. Treatment for PLEVA and PLC includes oral tetracycline or erythromycin, antihistamines (if pruritus is present), topical steroids, topical tacrolimus or pimecrolimus, or phototherapy. Low-dose weekly methotrexate may be helpful.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
respectively, of pityriasis lichenoides, an uncommon clonal T-cell disorder. The cause is unknown, although associations with infections have been reported. Pityriasis lichenoides more commonly affects children or young adults, usually before age 30 years. The disease can occur in all races.
In PLEVA, erythematous to brown papules and macules, in various stages of evolution, appear suddenly and in crops. The trunk and flexural areas are most often affected, but lesions may become widespread and may be pruritic or painful. Lesions may crust, ulcerate, or become necrotic and can heal with scarring. In general, patients don’t have constitutional symptoms. Lesions tend to resolve spontaneously over 1-3 years.
Rarely, PLEVA may develop into a more severe form called febrile ulceronecrotic Mucha-Habermann disease, a dermatologic emergency. Patients (more commonly, young males) may present with high fever, malaise, and lymphadenopathy. Lesions become very painful, ulcerated, and necrotic, and extensive necrosis may be present. Changes in mental status, breathing difficulties, anemia, arthritis, abdominal pain, and sepsis may occur. Patients require hospitalization. There is a 25% mortality rate.
PLC is at the other end of this disease spectrum, representing the chronic, more mild stage of the disorder. Lesions present as indolent, asymptomatic, scaly macules and erythematous papules, favoring the trunk and proximal extremities. Lesions tend to be fewer in number than seen in PLEVA. They resolve over several months and may result in hypopigmentation, but usually don’t cause scarring. Patients may have long periods of remission between outbreaks. T-cell gene rearrangement may demonstrate monoclonality. PLC is generally considered a benign disease, although there are patients who have developed cutaneous T-cell lymphoma. For this reason, patients should be followed carefully for signs of malignant transformation.
Both forms share a common histologic picture. In PLEVA, focal parakeratosis and crusting is present. A dense, wedge-shaped infiltrate can be seen with prominent lymphocytic exocystosis in the epidermis. Necrotic keratinocytes are often seen. There may be spongiosis and intraepidermal vesicles. Extravasation of erythrocytes often occurs in the epidermis. PLC is histologically similar but far more subtle. There is less crusting, less spongiosis, fewer vesicles, and fewer necrotic keratinocytes. Generally, atypia of lymphocytes is absent.
Mucha-Habermann requires treatment with systemic steroids. Methotrexate, cyclosporine, or dapsone may be used as steroid-sparing agents. Upon treatment, lesions may resolve or revert back to more typical lesions of PLEVA. Treatment for PLEVA and PLC includes oral tetracycline or erythromycin, antihistamines (if pruritus is present), topical steroids, topical tacrolimus or pimecrolimus, or phototherapy. Low-dose weekly methotrexate may be helpful.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to [email protected].
A 28-year-old white female with no significant past medical history presents with a 10-year history of asymptomatic erythematous papules and scaly patches that come and go. She has used topical steroids in the past.