User login
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
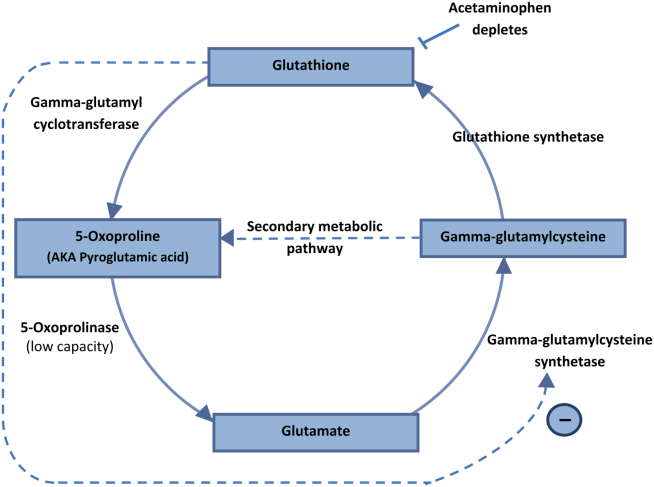
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
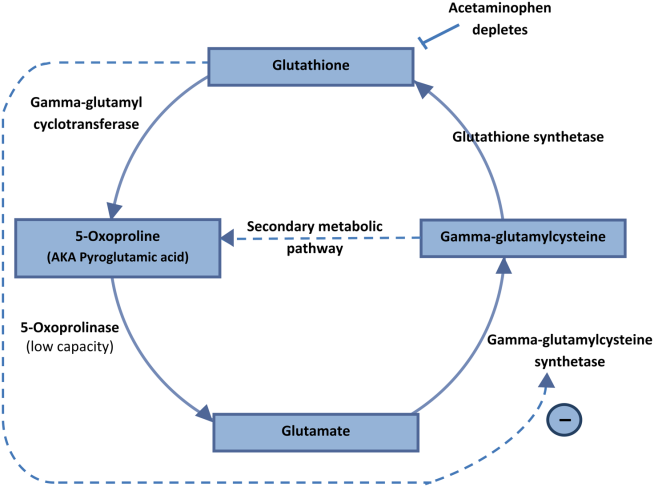
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2 20 mm Hg, PaO2 127 mm Hg, and HCO3 5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of 5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal 120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.