User login
From the Department of Neurology, Oregon Health & Science University, Portland, OR.
Abstract
- Objective: To review the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms (NPS) in Parkinson’s disease (PD).
- Methods: Literature review.
- Results: PD has traditionally been considered a disease of impaired motor function. However, neuropsychiatric complications, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, frequently complicate the course of the illness. Although the development of new medication options in recent years has had a positive benefit on the management of these troublesome symptoms, responses are frequently suboptimal. The development of valid instruments to measure neuropsychiatric symptoms has been vital in research efforts to bridge the gaps in our understanding. Further elucidation of neuropsychiatric pathophysiologies will help to define treatment targets and has the potential to expand our therapeutic armamentarium.
- Conclusion: While NPS affect patients with established disease, recent investigations have demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of NPS should be the goal of practitioners treating the entire continuum of PD.
Parkinson’s disease (PD) has traditionally been considered a disease of impaired motor function, but increased recognition of nonmotor symptoms and in particular neuropsychiatric symptoms, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, offer new opportunities for better care of patients. While neuropsychiatric symptoms affect patients with established disease, recent investigations have clearly demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of neuropsychiatric symptoms should be the goal of practitioners treating the entire continuum of PD. This review will focus on the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms in PD.
Impulse Control Disorders
The recognition that dopaminergic drugs were successful at treating many symptoms of PD was followed by the disturbing realization that impulse control disorders could be an unfortunate side effect in a substantial minority. Impulse control disorders as defined by DSM-IV [1] are disinhibited behaviors that are maladaptive and recurrent, causing personal and relationship consequences. The impulse control disorders that became associated with PD and medication intake, particularly dopamine agonist use, included gambling, hobbyism, punding (stereotyped, seemingly purposeless behaviors), excessive sexual behavior, shopping, hoarding, and less commonly, compulsive eating. The prevalence estimates of these behavioral disturbances range from 6% to 15.5%, compared with < 2% in the general population [2,3]. The addiction-like dopamine dysregulation syndrome, whereby patients self-medicate with high doses of levodopa and short-acting dopamine agonists beyond what is needed for motor control, can lead to significant impairment of the therapeutic alliance in addition to other patient personal relations. With the advent of surgical options to treat PD and its medication complications, it was observed that stimulation of the subthalamic nucleus could be associated with the spectrum of impulse control disorders [4].
Epidemiology/Risk Factors
In a recent systematic review of the literature of impulse control disorders in PD [5], the authors determined that dopaminergic therapy caused compulsive or impulsive behaviors in approximately 10% of PD patients in the course of their treatment, with pathologic gambling and hypersexuality most frequently experienced. Multiple impulse control disorders are not uncommon and may coexist in one-quarter of patients with compulsivity. There appeared to be more disordered behavior with higher comparable doses of agonists. The authors concluded that impulse control disorder symptoms tended to occur with initiation or dose increases of direct D2/D3 agonists, such as pramipexole and ropinirole. Importantly, impulse control disorder behavior improved if not resolved with discontinuation or reduction of dosage of the agonist, even if a compensatory levodopa dosage is added or increased. Perhaps not surprisingly, it was observed that if patients had a preexisting impulse control disorder prior to PD or the initiation of treatment, there was a high likelihood of worsening of symptoms. This small subgroup is estimated at about 1% of PD subjects, which corresponds to the prevalence of impulse control disorders in the general population. Other identified potential risk factors for impulse control disorder development include male gender, young age at onset, a personal or family history of addiction, novelty or risk seeking personality, and a concurrent diagnosis of depression [3]. In a recent study of early PD patients, the risk of developing an impulse control disorder became important once treatment with dopaminergic drugs began and continued for a year or more [6].
Pathogenesis
The pathogenesis is not fully understood, however, mesolimbic dopamine alterations are strongly suspected. It has been long speculated that the high doses of dopamine needed to replete the relatively depleted dorsal striatum overdose the “intact” ventral striatum and cause this neuropsychiatric disorder [7–9]. The additional cognitive impairments in PD, which can include problems with attention, working memory, planning, forethought and decision-making, are faculties that can markedly increase susceptibility to impulse control disorder [8].
The role of serotonin deficiency in the PD brain and its part in inhibiting the patient’s ability to delay rewards adds to the complexity of impulse control disorder pathogenesis. Dorsal raphe nuclei disease in PD results in loss of serotonin innervation to substantial portions of the prefrontal and motor cortices in addition to basal ganglia substructures like the striatum, pallidum and subthalamic nucleus [10]. Together with dopamine, serotonin may work to regulate risk-sensitive decision making, response inhibition, waiting for future rewards, and overall impulse control. Its relative loss therefore also likely contributes to tipping the balance towards impulse dyscontrol [11,12]. The role of other neurotransmitters such as opiate systems involved in the process of acquisition and maintenance of addictive behaviors like dopamine dysregulation syndrome remains to be fully understood.
Treatment
The most successful strategy to address this problem is to reduce or eliminate the offending medication, usually the dopamine agonist. This may be associated with worsening apathy, anxiety or depression; however, substituting levodopa can be a successful strategy in many cases [13]. Zonisamide was described to be possibly effective in a trial of 15 subjects; however, the open label nature of this evidence must be considered as with other case reports using valproate, donepezil, and selective serotonin reuptake inhibitors (SSRIs) [14–16].
Fatigue
An easy to understand operational definition of fatigue is that it is a state of extreme tiredness, weakness, or exhaustion, either physical or mental or both. Fatigue is not uncommon in the general population [17] but is increasingly recognized to occur in numerous disease conditions and is frequently encountered in PD and multiple sclerosis. The latter is of special significance in the consideration of the neurotransmission of fatigue, as it is not thought to be a disease of dopamine deficiency. The pathophysiology remains unclear, and it may differ depending on whether the fatigue is experienced as more physical or mental, or rather motor versus nonmotor as some authors propose.
Fatigue has been conceptualized as central or peripheral in character. Peripheral fatigue is best understood as muscular fatigue caused by repetitive muscular contraction or reduced force generation [18]. Central fatigue however, is divided into mental or physical fatigue. Mental fatigue can occur after sustained attentive or emotional activity. It may alternatively be provoked after boring repetitive tasks or lack of intellectually stimulating activity. Physical fatigue is the sense of body exhaustion or energy to perform physical tasks even though the ability to carry them out exists.
Epidemiology
As recognition of the problem of fatigue increased in the last 2 decades, the realization that one-third to one-half of patients experience it at some point has improved opportunities for recognition and treatment [19]. Fatigue may be the presenting symptom in one-third of patients prior to actual motor symptom onset [20]. Half of untreated PD patients in a biomarker cohort study reported fatigue [6]. Unfortunately, it is also described by patients as one of the most disabling symptoms, causing significant impact on quality of life [19]. Fatigue in PD is associated with higher rates of depressive symptoms, but occurs with higher prevalence in nondepressed patients [21]. Poor ability to initiate and sustain activity due to fatigue is different from depression, excessive sleepiness, or impaired motor function [22,23].
Pathophysiology
The pathophysiology of fatigue remains somewhat unclear, though physical fatigue is likely a significant part of the problem and related to dopamine deficiency based on studies of time and force generation of keyboard strikes in PD subjects before and after L-dopa administration. These subjects had declines in force and increased physical fatigue which improved after L-dopa [24]. In other studies using transcranial magnetic stimulation to study changes in cortical excitability, the degree of physical fatigue correlated with abnormalities in motor evoked potentials during fatiguing exercising. These studies also support the hypothesis that fatigue is a motor symptom [25,26]. In the ELLDOPA study, fatigue worsened more in PD subjects treated with placebo [27]. Other imaging studies have suggested suggested nondopaminergic mechanisms including serotonergic pathway abnormalities [28], thus the question behind the etiology and solution for all cases of fatigue remains to be settled.
Diagnosis
The diagnosis is fatigue may be challenging as it may mask as depression or apathy. There are a number of fatigue rating scales available; however, the validated Parkinson’s Fatigue Scale (PFS) supersedes many of the problems of using a generic scale which could overlap motor questions and potentially be confounding [29,30].
Treatment
Most important is awareness and vigilance for the symptoms of fatigue, depression, and apathy and effort to distinguish between them. It may require structured interviews or assessment tools to properly diagnose the problem. Treatment is less clear in that few studies have clearly indicated the best treatment options. In placebo-controlled trials, methylphenidate did improve fatigue as did levodopa [31]. Modafinil, a hypocretin modulator and a drug first approved by the FDA for treatment of narcolepsy, has demonstrated mixed results in recent years. It may reduce physical fatigue and reduce excessive daytime sleepiness but likely does not reduce subjective symptoms of fatigue [32]. L-dopa can significantly reduce fatigue in many patients, which would argue that it often is a motor symptom [33,24]. In a post-hoc analysis of the ADAGIO delayed start study, patients taking rasagiline 1 mg/day and 2 mg/day (the latter dose exceeds the usual clinical dosing) showed significantly less worsening of symptoms on the PFS compared to placebo over time [34]. It is important to realize that once motor symptoms are optimally treated with dopaminergic medications, while many patients will feel significant relief from fatigue some patients will continue to feel symptomatic.
Apathy
The definition of apathy has become more complicated and refined, incorporating findings from the study of brain disease and behavioral analysis. Marin’s classic elaboration of apathy as lack of motivation not attributable to diminished level of consciousness, cognitive impairment, or emotional distress has been built upon by Levy and Dubois [35–37]. They suggest apathy may be better thought of as an observable behavioral syndrome characterized by a quantitative reduction of self-generated voluntary and purposeful behaviors. They suggest 3 apathetic subtypes: emotional, cognitive, and auto-activational, which reflect different disease states accounting for failure of normal goal-directed behavior.
Epidemiology
Prevalence estimates for apathy in PD vary. This is likely due to the varying recruitment criteria among studies, with some including patients with comorbid depression and dementia and others containing only “pure apathy.” Other reports may have had referral bias issues, as community-based studies report lower prevalences in general. In a group of newly diagnosed PD patients, using more restrictive criteria (apathy subscale of the neuropsychiatric inventory and the diagnostic consensus criteria for apathy validated in PD), Pedersen reported a prevalence of apathy of 14.3% [38]. In a 4-year prospective longitudinal cohort study, an annual incidence rate of 12.3% was reported, with apathy developing in 60% of the cohort by the study’s conclusion [39].
Apathy has been associated with longer disease duration, male gender [40], higher daily levodopa doses [41], more severe parkinsonism [38], and lower education status, though the latter feature remains under debate. Early cognitive deficits appear to be a risk factor for development of apathy [42]. The patterns of cognitive dysfunction and apathy remain unsettled in the literature.
Pathology
The pathology of atrophy remains unexplained and is unlikely to be reduced to a simple atrophy of one nucleus or the tone of one circuit. However, in a small neuroimaging study, severity of apathy correlated with atrophy of the bilateral nucleus accumbens [43], and it is notable that one major input to the nucleus accumbens is the amygdala. According to Braak staging, by stage 4 significant involvement of the amygdala by Lewy bodies has occurred. Others have found changes in grey matter density that could correlate with deficits of the prefrontal-basal ganglia circuitry to produce dysfunction of segregated frontal-subcortical loops. These may correlate with the “autoactivation” deficit pattern of apathy in which patients have a lack of self-initiated actions, even thoughts, though appear more normal when giving externally prompted responses [37,44].
Assessment
Clinically, the relationship between apathy and depression can be hard to disentangle, especially since many studies have found an association between them, especially with regards to apathy and anhedonia. Depression may feature negative self thoughts and sadness while apathy is notable for lack of initiation and effort. Viewed over a longer period of time, apathy tended to worsen in a linear fashion, where depression tended to fluctuate with improvements and exacerbations.
The Movement Disorders Society task force has recommended the Lille Apathy Rating Scale (LARS) for assessment of apathy; English and French versions have been validated in PD patients. It uses a semi-structured interview format assessing 4 dimensions of apathy: self awareness, intellectual curiosity, emotion, and action initiation [45–47].
The impact of apathy cannot be underestimated as this poor show of motivation or effort leads to lack of engagement in old activities or interest in new ones. Spouses may misinterpret this change in behavior as laziness or deliberate social withdrawal, or perhaps entitlement. It is not surprising that apathy routinely shows up on quality of life (QoL) questionnaires as highly impacting patients and families. In one study, apathy was the nonmotor symptom most likely to cause caregiver distress in PD [40,48–50].
Treatment
No approved drugs exist for treatment of apathy. However, clinical experience often confirms that dopaminergic modulation can be helpful in the treatment of apathy as indirect evidence suggests. A meta-analysis of controlled trials using pramipexole and Part I of the Unified Parkinson's disease rating scale (UPDRS) (secondary measure) showed the medication improved scores on this measure of motivation and mood in non-depressed subjects [51] with PD. Rare patients undergoing subthalamic deep brain stimulation have been reported to experience new and sometimes severe apathy after surgery [52]. This was posited at least in part to be the result of reduction of dopaminergic medication due to surgery.
Nondopaminergic pharmacotherapy of apathy is in its infancy. A recent controlled trial of rivastigmine in 31 French subjects with moderate to severe apathy based on LARS showed that 6 months of treatment at 9.5 mg/day improved average scores from –11.5 to –20 compared with placebo. While quality of life did not improve, caregiver burden did. The investigators found in this group of subjects that apathy was a possible herald for early dementia in PD [53].
A post-hoc analysis of the ADAGIO study (rasagiline or placebo in PD patients taking antidepressants) found that rasagiline use was associated with a nonsignificant slowing of apathy development during the trial [54].
Psychosis
Psychotic symptoms are a common occurrence in drug-treated patients, with visual hallucinations occurring in up to 30%, though over a 20-year period up to three-quarters of patients may develop visual hallucinations.After visual, the most common type of hallucination is auditory, followed by the other affected senses such as tactile, olfactory, or even taste [57]. Delusions, which tend to be paranoid in nature, occur in about 5% of patients [55–57]. The presence of psychotic symptoms is associated with poorer quality of life [58].
Symptomatology
The visual hallucinations of PD are usually quite stereotyped, and have been described as “minor” and “non-minor”[59]. Minor hallucinations refer to transient peripheral field stimuli that disappear when brought into central focus, “something flashed by,” a sense of a living being nearby, “a presence in the room,” or illusions whereby objects are transformed, eg, a bush in the yard is a deer.
Auditory hallucinations tend to be vague or indistinct sounds, like music in another room as opposed to voices speaking directly to the patient as might be experienced in a primary psychotic disorder. Tactile forms often involve insects or other animals crawling on the skin. Olfactory hallucinations may take the form of smelling perfume, toxic odors from room vents, etc.
Early in the experience, the visual hallucinations may be amusing in that they consistently remain nonthreatening, similar day to day, and sometimes oddly provide an aspect of comfort or companionship to the patient. More commonly, the hallucinations are bothersome to the patient because the experience indicates to the patient that there is something wrong with their mind. Visual hallucinations often begin in low-stimulus environments, often in the evening or other low-light conditions, but as the problem advances they can occur at any time of day. While visual hallucinations may initially occur for only seconds at a time many days apart, the frequency and duration can increase until they occur hours at a time every day and are accompanied by multiple other visual hallucinations, delusions, and confusion [60].
Delusions tend to be more distressing to patients and caregivers because they are often paranoid in nature. The patient is more likely to act out due to the anxiety the paranoia creates. For example, she may change passwords to online accounts due to a belief that unknown assailants are after her finances. He may go to great lengths trying to prove his wife is cheating.
Risk Factors
While the primary risk factor for psychotic symptom development is dementia [57], it occurs in nondemented patients. Other associations include reduced visual acuity [56], visual processing impairment [61–65], use of dopamine agonists, REM behavior disorder, duration of PD, axial rigidity subtype of disease [61,66–68]. The pathophysiology of psychosis in PD is likely complex and remains currently unexplained. The role of excess dopamine has been described above, but there is also data suggesting cholinergic deficits in the cortex may also contribute. Excess serotonin (increased 5HT2A receptor subtypes) in the temporal lobe within the visual processing pathway has been postulated to be of significance [69,70]. Hypometabolism in visual association areas of the brain in subjects with visual hallucinations has been demonstrated in PET and functional MRI studies [64,71]. This is similar to findings in patients with dementia with Lewy bodies [72].
This review focuses on the primary forms of PD-related psychosis, which occur with a clear sensorium and generally longer exposure to dopaminergic medication. It is important to distinguish 2 other common scenarios in which hallucinations or delusions may occur. In the common toxic-metabolic delirium, a clouded sensorium with attention deficits may be the only clue to the etiology of new onset confusion with visual hallucinations. It is highly likely that resolution of the underlying medical problem will lead to resolution of the new onset psychosis and encephalopathy. In a second scenario, hallucinations precede or occur very shortly after the onset of initiation of dopaminergic medication. This differs from the classic syndrome described earlier, in particular when visual hallucinations precede any initiation of medication, and likely represents the distinction between a diagnosis of Lewy body disease and PD [60].
Treatment
Management of psychosis is approachable, but often the outcome is unsatisfactory and associated with trade-offs in motor control. It is unfortunately true that psychotic symptoms are often associated with increased caregiver burden and are a cause of increased nursing home placements [73]. When considering the workup of psychotic symptoms, the differential diagnosis includes delirium, dream enactment (REM behavior disorder), or less commonly, Bonnet syndrome.
A delirium may be precipitated by a difficult to diagnose infection; new-onset confusion and psychotic symptoms may be the heralding presentation. Urinary tract or upper respiratory tract are common vulnerable sources of infection. Once infection is ruled out, the next practical step is to review the patient’s medication list and manage centrally acting drugs that could be contributing to the altered sensorium. A recent prescription of opioids for a dental treatment or a new muscle relaxant may be a culprit, though it is not that usual. A bladder anticholinergic could be suspect and is worth eliminating especially if its addition coincided with the appearance of the psychotic symptoms. Once the non-dopaminergic medications have been reduced/eliminated, then the PD medications should be considered. The general approach is to eliminate the medications that provide the least benefit while being more likely to contribute to psychotic symptoms. Anticholinergic medications, dopamine agonists, selegiline should all be uppermost in that consideration until one is left with L-dopa and COMT inhibitors (the latter function to increase levodopa availability). Then COMT inhibitors and levodopa can be reduced; however, at any point motor control can suffer with the loss of symptomatic therapies [74].
Clozapine is effective against psychotic symptoms in PD, at doses much lower than used in schizophrenia (300-600mg/day). The average dose in the US randomized controlled clinical trial was 25 mg/day, with no associated motor worsening. Patients in the United States are required by the FDA to be placed in a computer-based registry and monitored for agranulocytosis for the duration of clozapine therapy. This rare adverse event is not dose related. Orthostasis can occur at these low doses however. Fortunately the metabolic syndrome is not associated with this range of administration [75,76].
Quetiapine was not found to be effective in 3 blinded randomized controlled trials despite its rather common use for this purpose. It was not associated with motor worsening, however.
Other neuroleptic medications have not resulted in widespread use, because trials have been open label, or outcomes demonstrated motor worsening. Cholinesterase inhibitors have been the subject of a few positive case series, however results appear to be sporadic, the effect size is relatively small, and side effects of this medication class are common [77–79]. It is clear that there is an unmet need for a medication for psychotic symptoms. Clozapine is effective but onerous in its monitoring requirements. Practically speaking, there are relatively few PD patients who take advantage of it because of its feasibility challenges. Yet the problem of psychotic symptoms is a significant one that imposes important challenges to the patient and caregiver, and may limit the number of medications that the patient needs in order to optimize quality of life.
Pimavanserin, a novel medication which acts as a selective serotonin inverse agonist, is in the early application stages for FDA approval for treatment of psychotic symptoms in PD [80]. In its pivotal phase III controlled trial, the drug reduced not only positive symptoms (hallucinations/delusions) without causing motoric worsening, but also reduced caregiver burden. Pimavanserin improved certain sleep features without causing daytime sedation. If this drug meets final approval, it may present an exciting option for many patients for whom treatment was previously limited.
Depression
A study of early PD suggested that depression is often unrecognized and frequently untreated [1]. Indeed it is not unusual for depression to predate the diagnosis of PD by an average of 4 to 6 years [81]. Expanding to the larger PD population, it is generally accepted that about 30% to 50% of PD patients experience clinically significant depression, and once diagnosed may have a long term course, or may recur [82,83]. This is important as untreated depression is an important cause of poor quality of life in early PD. In addition depression can exacerbate motor disability, lead to earlier motor treatment with medication, and increase caregiver stress [83–85].
Diagnosis
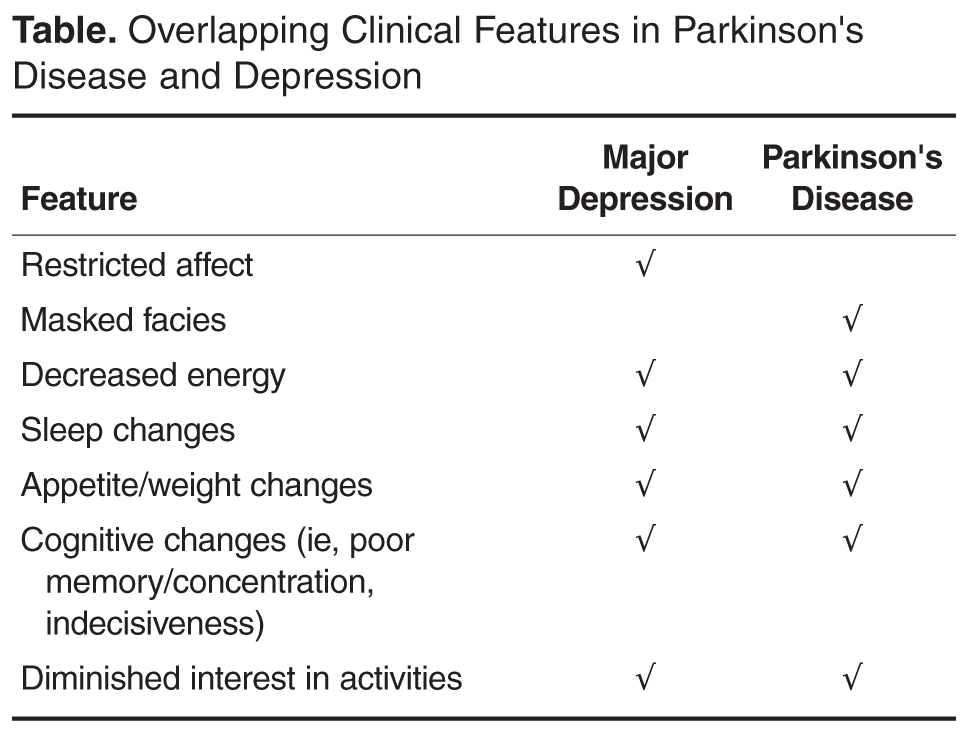
A number of clinimetric rating scales for depression have been used and their advantages have been largely related to their objective nature (quantifiable); thus, they tend to be most useful in epidemiologic research studies or for larger scale screening purposes. Examples include the the Beck Depression Inventory, the Geriatric Depression Scale, and the Hamilton Depression Scale, all of which have been shown to be valid tools in PD (with the exception of the UPDRS Depression). It is important to note that they do not substitute for a diagnostic clinical interview [89].
Suicide is not common in PD, however suicidal ideation is estimated at about 11% in PD patients [90], and while there was concern initially after deep brain stimulation procedures began that suicide incidence was increased, evidence does not support this [91].
Pathophysiology
The pathophysiology of depression in PD is largely unknown however is thought to be less causally due to psychosocial factors and more etiologically driven by brainstem monoamine and serotonergic dysfunction [92]. Nonetheless, similar to other chronic conditions, PD patients can certainly develop fear of disability, guilt about impact on others, or other reactive mood changes. Overall, rates of depression are higher in PD compared with patients with similar conditions matched for disability [93].
Treatment
First, the clinician must determine if depression is a result of short-term fluctuations, chronic undertreatment of motor disease, or longer-term mood phenomenon. One important pattern to recognize are mood fluctuations, which can parallel motor OFF-ON cycling. It can be valuable to distinguish this as “subsyndromic” depression or anxiety (sometimes referred to as “OFF dysphoria”), as it can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations[94–96]. Similarly, elevating chronic motor undertreatment to goal therapy can result in mood normalization.
If symptoms persist despite optimization of motor/nonmotor fluctuations or chronic undertreatment and are severe enough to warrant treatment, then therapies used can range from nonpharmacologic education, support, and mental health referrals, as well as pharmacologic support in the form of medications.
A frequent but uncontrolled observation was that when undertreatment of motor disease was finally redressed, mood often improved. A multicenter randomized controlled trial of pramipexole in PD patients without motor fluctuations but with mild to moderate depressive symptoms showed the drug improved scores on the Beck Depression Inventory over a 12-week period. The improvement in mood was 6 points overall, but by 2 points over placebo, illustrating the importance of the size of the placebo effect [97]. Given the potential side effect profile of dopamine agonists, it may be useful to weigh the antidepressant effects only when their motor benefits are already being employed.
Controlled trials have demonstrated efficacy of both selective serotonin reuptake inhibitors (SSRI) and selective norepinephrine reuptake inhibitors (SNRI) antidepressants in PD. Clinical trials have demonstrated efficacy against placebo or with other antidepressant comparators. Examples of drugs with demonstrated efficacy include citalopram, paroxetine, venlafaxine, and nortriptyline. Results have attempted to illuminate the small unique differences between classes of antidepressants or dynamic properties between drugs within a class. For example, desipramine may nudge scores on a mood scale a few weeks sooner than a purer SSRI. Paroxetine (SSRI) versus venlafaxine (SNRI) improved mood scores comparably in a multicenter trial with a placebo comparator. In general, all have all been demonstrated to be effective and with a relatively low side-effect profile, comparable to the general population[98–102]. While case reports exist in the literature, the interaction of monoamine oxidase B inhibitors and SSRIs has not caused significant hypertensive crises or risk of serotonin syndrome [103,104]. Electroconvulsive therapy (ECT) can be used for severe refractory depression in PD as for non-PD patients, with case reports of very effective results. Due to the rarity of use, systematic evidence for its use is lacking [105,106].
Other novel agents and techniques such as omega-3 fatty acids [107] and repetitive transcranial magnetic stimulation [108] have been reported with promising early results. Cognitive behavioral therapy (CBT), which may involve stress management techniques, sleep hygiene, and caregiver support, additionally almost always provided improvement in measured outcomes, whether the trial was controlled or open label in design. In one RCT of CBT in PD of 14 weeks’ duration, there were significantly more treatment responders in the CBT group, with a number needed to treat of only 2 [109].
Anxiety
Anxiety is also common in PD, at least as common as depression considering that prevalence estimates suggest up to 50% of patients experience it [110–112]. Manifestations of anxiety may include panic attacks, generalized anxiety disorder, social anxiety, or other phobias [113]. Anxiety has an important negative impact on health-related quality of life and is often underrecognized by clinicians [114]. While reliable and valid scales to measure anxiety have been lacking in PD, a new effort has yielded the “Parkinson Anxiety Scale” though full clinimetric properties of the scale remain to be demonstrated (sensitivity to change) [115].
Anxiety that parallels the timing of motor OFF-ON cycling is important to recognize. This “subsyndromic” anxiety or anxiety disorder not otherwise specified (ie, the anxiety does not meet DSM-IV criteria) can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations [116,117]. Indeed, the presence of motor fluctuations is the principle marker of anxiety in many studies [118–120]. In an analogous manner, anxiety can predate PD by years and be part of the nonmotor amalgam of features heralding the disease [6,121].
Treatment
Systematic controlled trials of anxiolytic treatment for PD are lacking; therefore, SSRIs are prescribed for this purpose as in non-PD patients. Until SSRIs are demonstrated to be of benefit in anxiety, they are likely safer than use of benzodiazepines, which are associated with risk for falling, cognitive dysfunction, or autonomic dysregulation in PD patients when used during waking hours. Psychotherapy and other nonpharmacologic approaches are likely to be of benefit. A small study of neuromuscular (massage) therapy demonstrated improvement on the Beck Anxiety Inventory in PD [122]. A case report of ECT for severe anxiety has been published [123].
Conclusion
Neuropsychiatric symptoms are common in PD and new knowledge about clinical features, epidemiology, and treatment options has been gained in the last decade, though much remains to be discovered. The development of valid instruments to measure neuropsychiatric symptoms has been vital in these research efforts to bridge the gaps in our understanding. Further elucidation of the pathophysiologies of neuropsychiatric symptoms will help to define treatment targets and likely fuel drug development and the discovery of drugs with more potent benefit and fewer side effects.
Corresponding author: Kathryn A. Chung, MD, Department of Neurology, Oregon Health & Science University, Portland, OR, [email protected].
Financial disclosures: None.
1. American Psychiatric Association. DSM-IV. 2000.
2. Voon V, Hassan K, Zurowski M, et al. Prevalence of behaviors in Parkinson disease Neurology 2006;67;1254–7.
3. D. Weintraub D, J. Koester J, M. N. Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67589–95.
4. Hälbig TD, Tse W, Frisina PG, et al. Subthalamic deep brain stimulation and impulse control in Parkinson’s disease. Eur J Neuro 2009;16:493–7.
5. Callesen MB, Scheel-Krüger J, Kringelbach ML, Møller A. A systematic review of impulse control disorders in Parkinson’s disease. J Parkinsons Dis 2013;3:105–38.
6. De Riva P, Smith K, Xie SX, Weintraub D. Course of psychiatric symptoms and global cognition in early course of psychiatric symptoms and global cognition in early Parkinson disease Neurology 2014;83:1–8.
7. Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev 2006;30:1–23.
8. Rowe JB, Hughes L, Ghosh BCP, et al. Parkinson’s disease and dopaminergic therapy – Differential effects on movement, reward and cognition. Brain 2008;131:2094–105.
9. Cools R, Frank MJ, Gibbs SE, et al. Striatal dopamine predicts outcome-specific reversal learning and its sensitivity to dopaminergic drug administration. J Neurosci 2009;29:1538–43.
10. Di Matteo V, Pierucci M, Esposito E, et al. Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson’s disease and other motor disorders. Prog Brain Res 2008;172:423–463.
11. Campbell-Meiklejohn D, Wakeley J, Herbert V, et al. Serotonin and dopamine play complementary roles in gambling to recover losses. Neuropsychopharmacology 2011;36:402–10.
12. Macoveanu J, Rowe JB, Hornboll B, et al. Playing it safe but losing anyway-Serotonergic signaling of negative outcomes in dorsomedial prefrontal cortex in the context of risk-aversion. Eur Neuropsychopharmacol 2013;23:919–30.
13. Mamikonyan E, Siderowf AD, Duda JE, et al. NIH Public Access 2009;23:75–80.
14. Ivanco LS, Bohnen NI. Effects of donepezil on compulsive hypersexual behavior in Parkinson disease: a single case study. Am J Ther 2005;12:467–8.
15. Hicks CW, Pandya MM, Itin I, Fernandez HH. Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease. Parkinsonism Relat Disord 2011;5:379–81.
16. Bermejo PE, Ruiz-Huete C, Anciones B. Zonisamide in managing impulse control disorders in Parkinson’s disease. J Neurol 2010;257:1682–5.
17. Pawlikowska T, Chalder T, Hirsch SR, et al. Population based study of fatigue and psychological distress. BMJ 1994;308:763–6.
18. Lou JS, Kearns G, Oken B, et al. Exacerbated physical fatigue and mental fatigue in Parkinson’s disease. Mov Disord 2001;16:190–6.
19. Herlofson K, Larsen JP. The influence of fatigue on health-related quality of life in patients with Parkinson’s disease. Acta Neurol Scand 2003;107:1–6.
20. Hagell P, Brundin L. Towards an understanding of fatigue in Parkinson disease. J Neurol Neurosurg Psychiatry 2009;80:489–92.
21. Alves G, Wentzel-Larsen T, Larsen JP. Is fatigue an independent and persistent symptom in patients with Parkinson disease? Neurology 2004;63:1908–11.
22. Martinez-Martin P, Catalan MJ, Benito-Leon J, et al. Impact of fatigue in Parkinson’s disease: The fatigue impact scale for daily use (D-FIS). Qual Life Res 2006:15:597–606.
23. Havlikova E, Rosenberger J, Nagyova I, et al. Clinical and psychosocial factors associated with fatigue in patients with Parkinson’s disease. Parkinsonism Relat Disord 2008;14:187–92.
24. Lou JS, Kearns G, Benice T, et al. Levodopa improves physical fatigue in Parkinson’s disease: A double-blind, placebo controlled, crossover study. Mov Disord 2003;18:1108–14.
25. Khedr EM, Galal O, Said A, et al. Lack of post-exercise depression of corticospinal excitability in patients with Parkinson’s disease. Eur J Neurol 2007;14:793–6.
26. Lou JS, Benice T, Kearns G, et al. Levodopa normalizes exercise related cortico-motoneuron excitability abnormalities in Parkinson’s disease. Clin. Neurophysiol 2003;114:930–7.
27. Schifitto G, Friedman JH, Oakes D, et al. Fatigue in levodopa-naive subjects with Parkinson disease. Neurology 2008;71:481–5.
28. Pavese N, Metta V, Bose SK, et al. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010;133:3434–43.
29. Brown RG, Dittner A, Findley L, Wessely SC. The Parkinson fatigue scale. Park Relat Disord 2005;11:49–55.
30. Grace J, Mendelsohn A, Friedman JH. A comparison of fatigue measures in Parkinson’s disease. Parkinsonism Relat Disord 2007;13:443–5.
31. Mendonça DA, Menezes K, Jog MS. Methylphenidate improves fatigue scores in Parkinson disease: a randomized controlled trial. Mov Disord 2007;22:2070–6.
32. Lou JS, Dimitrova DM, Park BS, et al. Using modafinil to treat fatigue in Parkinson disease: a double-blind, placebo-controlled pilot study. Clin Neuropharmacol 2009;32:305–10.
33. Ziv I, Avraham M, Michaelov Y, et al. Enhanced fatigue during motor performance in patients with Parkinson’s disease. Neurology 1998;51:1583–6.
34. Stocchi F. Benefits of treatment with rasagiline for fatigue symptoms in patients with early Parkinson’s disease. Eur J Neurol 2014;21:357–60.
35. Marin RS. Apathy: a neuropsychiatric syndrome. J. Neuropsychiatry Clin Neurosci 1991;3:243–4.
36. Marin RS. Apathy: concept, syndrome, neural mechanisms, and treatment. Semin Clin Neuropsychiatry 1996:1:304–14.
37. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cerebral Cortex 2006;16:916–28.
38. Pedersen KF, Larsen JP, Alves G, Aarsland D. Prevalence and clinical correlates of apathy in Parkinson’s disease: a community-based study. Parkinsonism Relat Disord 2009;15:295–9.
39. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
40. Pedersen KF, Alves G, Brønnick K, et al. Apathy in drug-naïve patients with incident Parkinson’s disease: The Norwegian ParkWest study. J Neurol 2010;257:217–23.
41. Ziropadja L, Stefanova E, Petrovic M, et al. Apathy and depression in Parkinson’s disease: The Belgrade PD study report. Parkinsonism Relat Disord 2012;18:339–42.
42. Dujardin K, Sockeel P, Devos D, et al. Characteristics of apathy in Parkinson’s disease. Mov Disord 2007;22:778–84.
43. Carriere N, Besson P, Dujardin K, et al. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov Disord 2014;29;897–903.
44. Reijnders JSAM, Scholtissen B, Weber WEJ, et al. Neuroanatomical correlates of apathy in Parkinson’s disease: a magnetic resonance imaging study using voxel-based morphometry. Mov Disord 2010;25:2318–25.
45. Leroi I, Harbishettar V, Andrews M, et al. Carer burden in apathy and impulse control disorders in Parkinson’s disease. Int J Geriatr Psychiatry 2012;27:160–6.
46. Schiehser DM, Liu L, Lessig SL, et al. Predictors of discrepancies in Parkinson’s disease patient and caregiver ratings of apathy, disinhibition, and executive dysfunction before and after diagnosis. J Int Neuropsychol Soc 2013;19:295–304.
47. Benito-León J, Cubo E, Coronell C, et al. Impact of apathy on health-related quality of life in recently diagnosed Parkinson’s disease: The ANIMO study. Mov Disord 2012;27:211–8.
48. Sockeel P, Dujardin K, Devos D, et al. The Lille apathy rating scale (LARS), a new instrument for detecting and quantifying apathy: validation in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2006;77:579–84.
49. Zahodne LB, Young S, Kirsch-Darrow L, et al. Examination of the Lille Apathy Rating Scale in Parkinson disease. Mov Disord 2009;24:677–83.
50. Drijgers RL, Dujardin K, Reijnders JSAM, et al. Validation of diagnostic criteria for apathy in Parkinson’s disease. Parkinsonism Relat Disord 2010;16:656–60.
51. Leentjens AFG, Koester J, Fruh B, et al. The effect of pramipexole on mood and motivational symptoms in parkinson’s disease: A meta-analysis of placebo-controlled studies. Clin Ther 2009;31:89–98.
52. Thobois S, Ardouin C, Lhommée E, et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: Predictors and underlying mesolimbic denervation. Brain 2010;133:1111–27.
53. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry 2014;85:668–74.
54. Smith KM, Eyal E, Weintraub D; ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 2015;72:88–95.
55. Forsaa EB, Larsen JP, Wentzel-Larsen T, et al. A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol 2010;67:996–1001.
56. Holroyd S, Currie L, Wooten GF. Prospective study of hallucinations and delusions in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2001;70:734–8.
57. Fénelon G, Alves G. Epidemiology of psychosis in Parkinson’s disease. J Neurol Sci 2010;289:12–7.
58. Mack J, Rabins P, Anderson K, et al. Prevalence of psychotic symptoms in a community-based Parkinson disease sample. Am J Geriatr Psychiatry 2012;20:123–32.
59. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: Report of an NINDS, NIMH Work Group. Mov Disord 2007;22:1061–8.
60. Diederich NJ, Fénelon G, Stebbins G, Goetz CG. Hallucinations in Parkinson disease. Nat Rev Neurol 2009;5:331–42.
61. Gallagher DA, Parkkinen L, O’Sullivan SS, et al. Testing an aetiological model of visual hallucinations in Parkinson’s disease. Brain 2011;134:3299–309.
62. Barnes J, Boubert L, Harris J, et al. Reality monitoring and visual hallucinations in Parkinson’s disease. Neuropsychologia 2003:41:565–74.
63. Kurita A, Murakami M, Takagi S, et al. Visual hallucinations and altered visual information processing in Parkinson disease and dementia with Lewy bodies. Mov Disord 2010;25:167–71.
64. Stebbins GT, Goetz CG, Carrillo MC, et al. Altered cortical visual processing in PD with hallucinations: an fMRI study. Neurology 2004;63:1409–16.
65. Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: A retrospective autopsy study. Lancet Neurol 2005:4:605–10.
66. Biglan KM, Holloway RG, McDermott MP, Richard IH. Risk factors for somnolence, edema, and hallucinations in early Parkinson disease. Neurology 2007;69:187–95.
67. Kiziltan G, Ozekmekci S, Ertan S, et al. Relationship between age and subtypes of psychotic symptoms in Parkinson’s disease. J Neurol 2007;254:448–52.
68. Ecker D, Unrath A, Kassubek J, Sabolek M. Dopamine agonists and their risk to induce psychotic episodes in Parkinson’s disease: a case-control study. BMC Neurol 2009;9:23.
69. Huot P, Johnston TH, Darr T, et al. Increased 5-HT2A receptors in the temporal cortex of Parkinsonian patients with visual hallucinations. Mov Disord 2010;25:1399–408.
70. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch. Neurol 2010;67:416–21.
71. Boecker H, Ceballos-Baumann O, Volk D, et al. Metabolic alterations in patients with Parkinson disease and visual hallucinations. Arch Neurol 2007;64:984–8.
72. Sanchez-Castaneda C, Rene R, Ramirez-Ruiz B, et al. Frontal and associative visual areas related to visual hallucinations in dementia with Lewy bodies and Parkinson’s disease with dementia. Mov Disord 2010;25:615–22.
73. Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993;43:2227–9.
74. Friedman JH. Parkinson disease psychosis: update. Behav.Neurol 2013;27:469–77.
75. Pollak P, Tison F, Rascol O, et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004;75:689–95.
76. Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis. N Engl J Med 1999;340:757–63.
77. Sobow T. Parkinson’s disease-related visual hallucinations unresponsive to atypical antipsychotics treated with cholinesterase inhibitors: a case series. Neurol Neurochir Pol 2007;41:276–9.
78. Fabbrini G, Barbanti P, Aurilia C, et al. Donepezil in the treatment of hallucinations and delusions in Parkinson’s disease. Neurol Sci 2002;23:41–3.
79. Bullock R, Cameron A, Hospital WH. Rivastigmine for the treatment of dementia and visual hallucinations associated with Parkinson’s disease: a case series. Curr Med Res Opin 2002;18,:258–64.
80. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383:533–40.
81. L. Ishihara L, and C. Brayne C. A systematic review of depression and mental illness preceding Parkinson’s disease. Acta Neurol Scand 2006;113:211–20.
82. Reijnders JSAM, Ehrt U, Weber WEJ, et al. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord 2008;23:183–9.
83. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992:55:377–82.
84. Müller B, Assmus J, Herlofson K, et al. Importance of motor vs. non-motor symptoms for health-related quality of life in early Parkinson’s disease. Parkinsonism Relat Disord 2013;19:1027–32.
85. Ravina B, Camicioli R, Como PG, et al. The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
86. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148–58.
87. Marsh L. Depression and Parkinson’s disease: current knowledge. Curr. Neurol. Neurosci. Rep 2013; 13:409.
88. Starkstein SE, Merello M, Jorge R, et al. A validation study of depressive syndromes in Parkinson’s disease. Mov Disord 2008;23,:538–46.
89. Williams JR, Hirsch ES, Anderson K, et al. A comparison of nine scales to detect depression in Parkinson disease: which scale to use? Neurology 2012;78:998–1006.
90. Nazem S, Siderowf AD, Duda JE, et al. Suicidal and death ideation in Parkinson’s disease. Mov Disord 2008;23:1573–9.
91. Weintraub D, Duda JE, Carlson K, et al. Suicide ideation and behaviours after STN and GPi DBS surgery for Parkinson’s disease: results from a randomised, controlled trial. J Neurol Neurosurg Psychiatry 2013;84:1113–8.
92. Aarsland D, Påhlhagen S, Ballard CG, et al. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol 2011;8:35–47.
93. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson’s disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
94. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology 2002:59:408–13.
95. Nissenbaum H, Quinn NP, Brown RG, et al. Mood swings associated with the ‘on-off’ phenomenon in Parkinson’s disease. Psychol Med 1987;17:899–904.
96. Maricle RA, Nutt JG, Carter JH. Mood and anxiety fluctuation in Parkinson’s disease associated with levodopa infusion: preliminary findings. Mov Disord 1995;10:329–32.
97. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
98. Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008;23:850–7.
99. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
100. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
101. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord 2011;26 Suppl 3:S42–80.
102. Skapinakis P, Bakola E, Salanti G, et al. Efficacy and acceptability of selective serotonin reuptake inhibitors for the treatment of depression in Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. BMC Neurol 2010;10:49.
103. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Parkinson Study Group. Neurology 1997;48:1070–7.
104. Montgomery EB, Panisset M. Retrospective statistical analysis of the incidence of serotonin toxicity in patients taking rasagiline and anti-depressants in clinical trials. Mov Disord 2009;24:359.
105. Bailine S, Kremen N, Kohen I, et al. Bitemporal electroconvulsive therapy for depression in a Parkinson disease patient with a deep-brain stimulator. J ECT 2008;24:171–2.
106. Faber R, Trimble MR. Electroconvulsive therapy in Parkinson’s disease and other movement disorders. Mov Disord 1991;6:293–303.
107. da Silva TM, Munhoz RP, Alvarez C, et al. Depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled pilot study of omega-3 fatty-acid supplementation. J Affect Disord 2008;111:351–9.
108. Pal E, Nagy F, Aschermann Z, et al. The impact of left prefrontal repetitive transcranial magnetic stimulation on depression in Parkinson’s disease: a randomized, double-blind, placebo-controlled study. Mov Disord 2010;25:2311–7.
109. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry 2011;168:1066–74.
110. Stein MB, Heuser IJ, Juncos JL, Uhde TW. Anxiety disorders in patients with parkinson’s disease. Am J Psychiatry 1990;147:217–20.
111. Nuti A, Ceravolo R, Piccinni A, et al. Psychiatric comorbidity in a population of Parkinson’s disease patients. Eur J Neurol 2004;11:315–20.
112. Marinus J, Leentjens AFG, Visser M, et al. Evaluation of the Hospital Anxiety and Depression Scale in patients With Parkinson’s disease. Clin Neuropharmacol 2002:25:318–24.
113. Dissanayaka NNW, White E, O’Sullivan JD, et al. The clinical spectrum of anxiety in Parkinson’s disease. Mov Disord 2014;29:967–75.
114. Dissanayaka NNW, Sellbach A, Matheson S, et al. Anxiety disorders in Parkinson’s disease: prevalence and risk factors. Mov Disord 2010;25:838–45.
115. Leentjens AFG, Dujardin K, Pontone GM, et al. The Parkinson anxiety scale (PAS): Development and validation of a new anxiety scale. Mov Disord 2014;29:1035–43.
116. Maricle RA, Nutt JG, Valentine RJ, Carter JH. Dose-response relationship of levodopa with mood and anxiety in fluctuating Parkinson’s disease: a double-blind, placebo-controlled study. Neurology 1995;45:1757–60.
117. Erdal KJ. Depression and anxiety in persons with Parkinson’s disease with and without ‘on–off’ phenomena. J Clin Psychol Med Settings 2001:8:293–9.
118. Leentjens AFG, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson’s disease: a cross-sectional study. Mov. Disord 2011;26:484–92.
119. Solla P, Cannas A, Floris GL, et al. Behavioral, neuropsychiatric and cognitive disorders in Parkinson’s disease patients with and without motor complications. Prog Neuropsychopharmacol Biol Psychiatry 2011;35:1009–13.
120. E. Stefanova E, L. Ziropadja L, M. Petrović M, et al. Screening for anxiety symptoms in Parkinson disease: a cross-sectional study. J Geriatr Psychiatry Neurol 2013;26:34–40.
121. Jacob EL, Gatto NM, Thompson A, et al. Occurrence of depression and anxiety prior to Parkinson’s disease. Parkinsonism Relat Disord 2010;16:576–81.
122. Craig LH, Svircev A, Haber M, Juncos JL. Controlled pilot study of the effects of neuromuscular therapy in patients with Parkinson’s disease. Mov Disord 2006;21:2127–33.
123. Marino L, Friedman JH. Letter to the editor: Successful use of electroconvulsive therapy for refractory anxiety in Parkinson’s disease. Int J Neurosci 2013;123:70–1.
From the Department of Neurology, Oregon Health & Science University, Portland, OR.
Abstract
- Objective: To review the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms (NPS) in Parkinson’s disease (PD).
- Methods: Literature review.
- Results: PD has traditionally been considered a disease of impaired motor function. However, neuropsychiatric complications, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, frequently complicate the course of the illness. Although the development of new medication options in recent years has had a positive benefit on the management of these troublesome symptoms, responses are frequently suboptimal. The development of valid instruments to measure neuropsychiatric symptoms has been vital in research efforts to bridge the gaps in our understanding. Further elucidation of neuropsychiatric pathophysiologies will help to define treatment targets and has the potential to expand our therapeutic armamentarium.
- Conclusion: While NPS affect patients with established disease, recent investigations have demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of NPS should be the goal of practitioners treating the entire continuum of PD.
Parkinson’s disease (PD) has traditionally been considered a disease of impaired motor function, but increased recognition of nonmotor symptoms and in particular neuropsychiatric symptoms, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, offer new opportunities for better care of patients. While neuropsychiatric symptoms affect patients with established disease, recent investigations have clearly demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of neuropsychiatric symptoms should be the goal of practitioners treating the entire continuum of PD. This review will focus on the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms in PD.
Impulse Control Disorders
The recognition that dopaminergic drugs were successful at treating many symptoms of PD was followed by the disturbing realization that impulse control disorders could be an unfortunate side effect in a substantial minority. Impulse control disorders as defined by DSM-IV [1] are disinhibited behaviors that are maladaptive and recurrent, causing personal and relationship consequences. The impulse control disorders that became associated with PD and medication intake, particularly dopamine agonist use, included gambling, hobbyism, punding (stereotyped, seemingly purposeless behaviors), excessive sexual behavior, shopping, hoarding, and less commonly, compulsive eating. The prevalence estimates of these behavioral disturbances range from 6% to 15.5%, compared with < 2% in the general population [2,3]. The addiction-like dopamine dysregulation syndrome, whereby patients self-medicate with high doses of levodopa and short-acting dopamine agonists beyond what is needed for motor control, can lead to significant impairment of the therapeutic alliance in addition to other patient personal relations. With the advent of surgical options to treat PD and its medication complications, it was observed that stimulation of the subthalamic nucleus could be associated with the spectrum of impulse control disorders [4].
Epidemiology/Risk Factors
In a recent systematic review of the literature of impulse control disorders in PD [5], the authors determined that dopaminergic therapy caused compulsive or impulsive behaviors in approximately 10% of PD patients in the course of their treatment, with pathologic gambling and hypersexuality most frequently experienced. Multiple impulse control disorders are not uncommon and may coexist in one-quarter of patients with compulsivity. There appeared to be more disordered behavior with higher comparable doses of agonists. The authors concluded that impulse control disorder symptoms tended to occur with initiation or dose increases of direct D2/D3 agonists, such as pramipexole and ropinirole. Importantly, impulse control disorder behavior improved if not resolved with discontinuation or reduction of dosage of the agonist, even if a compensatory levodopa dosage is added or increased. Perhaps not surprisingly, it was observed that if patients had a preexisting impulse control disorder prior to PD or the initiation of treatment, there was a high likelihood of worsening of symptoms. This small subgroup is estimated at about 1% of PD subjects, which corresponds to the prevalence of impulse control disorders in the general population. Other identified potential risk factors for impulse control disorder development include male gender, young age at onset, a personal or family history of addiction, novelty or risk seeking personality, and a concurrent diagnosis of depression [3]. In a recent study of early PD patients, the risk of developing an impulse control disorder became important once treatment with dopaminergic drugs began and continued for a year or more [6].
Pathogenesis
The pathogenesis is not fully understood, however, mesolimbic dopamine alterations are strongly suspected. It has been long speculated that the high doses of dopamine needed to replete the relatively depleted dorsal striatum overdose the “intact” ventral striatum and cause this neuropsychiatric disorder [7–9]. The additional cognitive impairments in PD, which can include problems with attention, working memory, planning, forethought and decision-making, are faculties that can markedly increase susceptibility to impulse control disorder [8].
The role of serotonin deficiency in the PD brain and its part in inhibiting the patient’s ability to delay rewards adds to the complexity of impulse control disorder pathogenesis. Dorsal raphe nuclei disease in PD results in loss of serotonin innervation to substantial portions of the prefrontal and motor cortices in addition to basal ganglia substructures like the striatum, pallidum and subthalamic nucleus [10]. Together with dopamine, serotonin may work to regulate risk-sensitive decision making, response inhibition, waiting for future rewards, and overall impulse control. Its relative loss therefore also likely contributes to tipping the balance towards impulse dyscontrol [11,12]. The role of other neurotransmitters such as opiate systems involved in the process of acquisition and maintenance of addictive behaviors like dopamine dysregulation syndrome remains to be fully understood.
Treatment
The most successful strategy to address this problem is to reduce or eliminate the offending medication, usually the dopamine agonist. This may be associated with worsening apathy, anxiety or depression; however, substituting levodopa can be a successful strategy in many cases [13]. Zonisamide was described to be possibly effective in a trial of 15 subjects; however, the open label nature of this evidence must be considered as with other case reports using valproate, donepezil, and selective serotonin reuptake inhibitors (SSRIs) [14–16].
Fatigue
An easy to understand operational definition of fatigue is that it is a state of extreme tiredness, weakness, or exhaustion, either physical or mental or both. Fatigue is not uncommon in the general population [17] but is increasingly recognized to occur in numerous disease conditions and is frequently encountered in PD and multiple sclerosis. The latter is of special significance in the consideration of the neurotransmission of fatigue, as it is not thought to be a disease of dopamine deficiency. The pathophysiology remains unclear, and it may differ depending on whether the fatigue is experienced as more physical or mental, or rather motor versus nonmotor as some authors propose.
Fatigue has been conceptualized as central or peripheral in character. Peripheral fatigue is best understood as muscular fatigue caused by repetitive muscular contraction or reduced force generation [18]. Central fatigue however, is divided into mental or physical fatigue. Mental fatigue can occur after sustained attentive or emotional activity. It may alternatively be provoked after boring repetitive tasks or lack of intellectually stimulating activity. Physical fatigue is the sense of body exhaustion or energy to perform physical tasks even though the ability to carry them out exists.
Epidemiology
As recognition of the problem of fatigue increased in the last 2 decades, the realization that one-third to one-half of patients experience it at some point has improved opportunities for recognition and treatment [19]. Fatigue may be the presenting symptom in one-third of patients prior to actual motor symptom onset [20]. Half of untreated PD patients in a biomarker cohort study reported fatigue [6]. Unfortunately, it is also described by patients as one of the most disabling symptoms, causing significant impact on quality of life [19]. Fatigue in PD is associated with higher rates of depressive symptoms, but occurs with higher prevalence in nondepressed patients [21]. Poor ability to initiate and sustain activity due to fatigue is different from depression, excessive sleepiness, or impaired motor function [22,23].
Pathophysiology
The pathophysiology of fatigue remains somewhat unclear, though physical fatigue is likely a significant part of the problem and related to dopamine deficiency based on studies of time and force generation of keyboard strikes in PD subjects before and after L-dopa administration. These subjects had declines in force and increased physical fatigue which improved after L-dopa [24]. In other studies using transcranial magnetic stimulation to study changes in cortical excitability, the degree of physical fatigue correlated with abnormalities in motor evoked potentials during fatiguing exercising. These studies also support the hypothesis that fatigue is a motor symptom [25,26]. In the ELLDOPA study, fatigue worsened more in PD subjects treated with placebo [27]. Other imaging studies have suggested suggested nondopaminergic mechanisms including serotonergic pathway abnormalities [28], thus the question behind the etiology and solution for all cases of fatigue remains to be settled.
Diagnosis
The diagnosis is fatigue may be challenging as it may mask as depression or apathy. There are a number of fatigue rating scales available; however, the validated Parkinson’s Fatigue Scale (PFS) supersedes many of the problems of using a generic scale which could overlap motor questions and potentially be confounding [29,30].
Treatment
Most important is awareness and vigilance for the symptoms of fatigue, depression, and apathy and effort to distinguish between them. It may require structured interviews or assessment tools to properly diagnose the problem. Treatment is less clear in that few studies have clearly indicated the best treatment options. In placebo-controlled trials, methylphenidate did improve fatigue as did levodopa [31]. Modafinil, a hypocretin modulator and a drug first approved by the FDA for treatment of narcolepsy, has demonstrated mixed results in recent years. It may reduce physical fatigue and reduce excessive daytime sleepiness but likely does not reduce subjective symptoms of fatigue [32]. L-dopa can significantly reduce fatigue in many patients, which would argue that it often is a motor symptom [33,24]. In a post-hoc analysis of the ADAGIO delayed start study, patients taking rasagiline 1 mg/day and 2 mg/day (the latter dose exceeds the usual clinical dosing) showed significantly less worsening of symptoms on the PFS compared to placebo over time [34]. It is important to realize that once motor symptoms are optimally treated with dopaminergic medications, while many patients will feel significant relief from fatigue some patients will continue to feel symptomatic.
Apathy
The definition of apathy has become more complicated and refined, incorporating findings from the study of brain disease and behavioral analysis. Marin’s classic elaboration of apathy as lack of motivation not attributable to diminished level of consciousness, cognitive impairment, or emotional distress has been built upon by Levy and Dubois [35–37]. They suggest apathy may be better thought of as an observable behavioral syndrome characterized by a quantitative reduction of self-generated voluntary and purposeful behaviors. They suggest 3 apathetic subtypes: emotional, cognitive, and auto-activational, which reflect different disease states accounting for failure of normal goal-directed behavior.
Epidemiology
Prevalence estimates for apathy in PD vary. This is likely due to the varying recruitment criteria among studies, with some including patients with comorbid depression and dementia and others containing only “pure apathy.” Other reports may have had referral bias issues, as community-based studies report lower prevalences in general. In a group of newly diagnosed PD patients, using more restrictive criteria (apathy subscale of the neuropsychiatric inventory and the diagnostic consensus criteria for apathy validated in PD), Pedersen reported a prevalence of apathy of 14.3% [38]. In a 4-year prospective longitudinal cohort study, an annual incidence rate of 12.3% was reported, with apathy developing in 60% of the cohort by the study’s conclusion [39].
Apathy has been associated with longer disease duration, male gender [40], higher daily levodopa doses [41], more severe parkinsonism [38], and lower education status, though the latter feature remains under debate. Early cognitive deficits appear to be a risk factor for development of apathy [42]. The patterns of cognitive dysfunction and apathy remain unsettled in the literature.
Pathology
The pathology of atrophy remains unexplained and is unlikely to be reduced to a simple atrophy of one nucleus or the tone of one circuit. However, in a small neuroimaging study, severity of apathy correlated with atrophy of the bilateral nucleus accumbens [43], and it is notable that one major input to the nucleus accumbens is the amygdala. According to Braak staging, by stage 4 significant involvement of the amygdala by Lewy bodies has occurred. Others have found changes in grey matter density that could correlate with deficits of the prefrontal-basal ganglia circuitry to produce dysfunction of segregated frontal-subcortical loops. These may correlate with the “autoactivation” deficit pattern of apathy in which patients have a lack of self-initiated actions, even thoughts, though appear more normal when giving externally prompted responses [37,44].
Assessment
Clinically, the relationship between apathy and depression can be hard to disentangle, especially since many studies have found an association between them, especially with regards to apathy and anhedonia. Depression may feature negative self thoughts and sadness while apathy is notable for lack of initiation and effort. Viewed over a longer period of time, apathy tended to worsen in a linear fashion, where depression tended to fluctuate with improvements and exacerbations.
The Movement Disorders Society task force has recommended the Lille Apathy Rating Scale (LARS) for assessment of apathy; English and French versions have been validated in PD patients. It uses a semi-structured interview format assessing 4 dimensions of apathy: self awareness, intellectual curiosity, emotion, and action initiation [45–47].
The impact of apathy cannot be underestimated as this poor show of motivation or effort leads to lack of engagement in old activities or interest in new ones. Spouses may misinterpret this change in behavior as laziness or deliberate social withdrawal, or perhaps entitlement. It is not surprising that apathy routinely shows up on quality of life (QoL) questionnaires as highly impacting patients and families. In one study, apathy was the nonmotor symptom most likely to cause caregiver distress in PD [40,48–50].
Treatment
No approved drugs exist for treatment of apathy. However, clinical experience often confirms that dopaminergic modulation can be helpful in the treatment of apathy as indirect evidence suggests. A meta-analysis of controlled trials using pramipexole and Part I of the Unified Parkinson's disease rating scale (UPDRS) (secondary measure) showed the medication improved scores on this measure of motivation and mood in non-depressed subjects [51] with PD. Rare patients undergoing subthalamic deep brain stimulation have been reported to experience new and sometimes severe apathy after surgery [52]. This was posited at least in part to be the result of reduction of dopaminergic medication due to surgery.
Nondopaminergic pharmacotherapy of apathy is in its infancy. A recent controlled trial of rivastigmine in 31 French subjects with moderate to severe apathy based on LARS showed that 6 months of treatment at 9.5 mg/day improved average scores from –11.5 to –20 compared with placebo. While quality of life did not improve, caregiver burden did. The investigators found in this group of subjects that apathy was a possible herald for early dementia in PD [53].
A post-hoc analysis of the ADAGIO study (rasagiline or placebo in PD patients taking antidepressants) found that rasagiline use was associated with a nonsignificant slowing of apathy development during the trial [54].
Psychosis
Psychotic symptoms are a common occurrence in drug-treated patients, with visual hallucinations occurring in up to 30%, though over a 20-year period up to three-quarters of patients may develop visual hallucinations.After visual, the most common type of hallucination is auditory, followed by the other affected senses such as tactile, olfactory, or even taste [57]. Delusions, which tend to be paranoid in nature, occur in about 5% of patients [55–57]. The presence of psychotic symptoms is associated with poorer quality of life [58].
Symptomatology
The visual hallucinations of PD are usually quite stereotyped, and have been described as “minor” and “non-minor”[59]. Minor hallucinations refer to transient peripheral field stimuli that disappear when brought into central focus, “something flashed by,” a sense of a living being nearby, “a presence in the room,” or illusions whereby objects are transformed, eg, a bush in the yard is a deer.
Auditory hallucinations tend to be vague or indistinct sounds, like music in another room as opposed to voices speaking directly to the patient as might be experienced in a primary psychotic disorder. Tactile forms often involve insects or other animals crawling on the skin. Olfactory hallucinations may take the form of smelling perfume, toxic odors from room vents, etc.
Early in the experience, the visual hallucinations may be amusing in that they consistently remain nonthreatening, similar day to day, and sometimes oddly provide an aspect of comfort or companionship to the patient. More commonly, the hallucinations are bothersome to the patient because the experience indicates to the patient that there is something wrong with their mind. Visual hallucinations often begin in low-stimulus environments, often in the evening or other low-light conditions, but as the problem advances they can occur at any time of day. While visual hallucinations may initially occur for only seconds at a time many days apart, the frequency and duration can increase until they occur hours at a time every day and are accompanied by multiple other visual hallucinations, delusions, and confusion [60].
Delusions tend to be more distressing to patients and caregivers because they are often paranoid in nature. The patient is more likely to act out due to the anxiety the paranoia creates. For example, she may change passwords to online accounts due to a belief that unknown assailants are after her finances. He may go to great lengths trying to prove his wife is cheating.
Risk Factors
While the primary risk factor for psychotic symptom development is dementia [57], it occurs in nondemented patients. Other associations include reduced visual acuity [56], visual processing impairment [61–65], use of dopamine agonists, REM behavior disorder, duration of PD, axial rigidity subtype of disease [61,66–68]. The pathophysiology of psychosis in PD is likely complex and remains currently unexplained. The role of excess dopamine has been described above, but there is also data suggesting cholinergic deficits in the cortex may also contribute. Excess serotonin (increased 5HT2A receptor subtypes) in the temporal lobe within the visual processing pathway has been postulated to be of significance [69,70]. Hypometabolism in visual association areas of the brain in subjects with visual hallucinations has been demonstrated in PET and functional MRI studies [64,71]. This is similar to findings in patients with dementia with Lewy bodies [72].
This review focuses on the primary forms of PD-related psychosis, which occur with a clear sensorium and generally longer exposure to dopaminergic medication. It is important to distinguish 2 other common scenarios in which hallucinations or delusions may occur. In the common toxic-metabolic delirium, a clouded sensorium with attention deficits may be the only clue to the etiology of new onset confusion with visual hallucinations. It is highly likely that resolution of the underlying medical problem will lead to resolution of the new onset psychosis and encephalopathy. In a second scenario, hallucinations precede or occur very shortly after the onset of initiation of dopaminergic medication. This differs from the classic syndrome described earlier, in particular when visual hallucinations precede any initiation of medication, and likely represents the distinction between a diagnosis of Lewy body disease and PD [60].
Treatment
Management of psychosis is approachable, but often the outcome is unsatisfactory and associated with trade-offs in motor control. It is unfortunately true that psychotic symptoms are often associated with increased caregiver burden and are a cause of increased nursing home placements [73]. When considering the workup of psychotic symptoms, the differential diagnosis includes delirium, dream enactment (REM behavior disorder), or less commonly, Bonnet syndrome.
A delirium may be precipitated by a difficult to diagnose infection; new-onset confusion and psychotic symptoms may be the heralding presentation. Urinary tract or upper respiratory tract are common vulnerable sources of infection. Once infection is ruled out, the next practical step is to review the patient’s medication list and manage centrally acting drugs that could be contributing to the altered sensorium. A recent prescription of opioids for a dental treatment or a new muscle relaxant may be a culprit, though it is not that usual. A bladder anticholinergic could be suspect and is worth eliminating especially if its addition coincided with the appearance of the psychotic symptoms. Once the non-dopaminergic medications have been reduced/eliminated, then the PD medications should be considered. The general approach is to eliminate the medications that provide the least benefit while being more likely to contribute to psychotic symptoms. Anticholinergic medications, dopamine agonists, selegiline should all be uppermost in that consideration until one is left with L-dopa and COMT inhibitors (the latter function to increase levodopa availability). Then COMT inhibitors and levodopa can be reduced; however, at any point motor control can suffer with the loss of symptomatic therapies [74].
Clozapine is effective against psychotic symptoms in PD, at doses much lower than used in schizophrenia (300-600mg/day). The average dose in the US randomized controlled clinical trial was 25 mg/day, with no associated motor worsening. Patients in the United States are required by the FDA to be placed in a computer-based registry and monitored for agranulocytosis for the duration of clozapine therapy. This rare adverse event is not dose related. Orthostasis can occur at these low doses however. Fortunately the metabolic syndrome is not associated with this range of administration [75,76].
Quetiapine was not found to be effective in 3 blinded randomized controlled trials despite its rather common use for this purpose. It was not associated with motor worsening, however.
Other neuroleptic medications have not resulted in widespread use, because trials have been open label, or outcomes demonstrated motor worsening. Cholinesterase inhibitors have been the subject of a few positive case series, however results appear to be sporadic, the effect size is relatively small, and side effects of this medication class are common [77–79]. It is clear that there is an unmet need for a medication for psychotic symptoms. Clozapine is effective but onerous in its monitoring requirements. Practically speaking, there are relatively few PD patients who take advantage of it because of its feasibility challenges. Yet the problem of psychotic symptoms is a significant one that imposes important challenges to the patient and caregiver, and may limit the number of medications that the patient needs in order to optimize quality of life.
Pimavanserin, a novel medication which acts as a selective serotonin inverse agonist, is in the early application stages for FDA approval for treatment of psychotic symptoms in PD [80]. In its pivotal phase III controlled trial, the drug reduced not only positive symptoms (hallucinations/delusions) without causing motoric worsening, but also reduced caregiver burden. Pimavanserin improved certain sleep features without causing daytime sedation. If this drug meets final approval, it may present an exciting option for many patients for whom treatment was previously limited.
Depression
A study of early PD suggested that depression is often unrecognized and frequently untreated [1]. Indeed it is not unusual for depression to predate the diagnosis of PD by an average of 4 to 6 years [81]. Expanding to the larger PD population, it is generally accepted that about 30% to 50% of PD patients experience clinically significant depression, and once diagnosed may have a long term course, or may recur [82,83]. This is important as untreated depression is an important cause of poor quality of life in early PD. In addition depression can exacerbate motor disability, lead to earlier motor treatment with medication, and increase caregiver stress [83–85].
Diagnosis
A number of clinimetric rating scales for depression have been used and their advantages have been largely related to their objective nature (quantifiable); thus, they tend to be most useful in epidemiologic research studies or for larger scale screening purposes. Examples include the the Beck Depression Inventory, the Geriatric Depression Scale, and the Hamilton Depression Scale, all of which have been shown to be valid tools in PD (with the exception of the UPDRS Depression). It is important to note that they do not substitute for a diagnostic clinical interview [89].
Suicide is not common in PD, however suicidal ideation is estimated at about 11% in PD patients [90], and while there was concern initially after deep brain stimulation procedures began that suicide incidence was increased, evidence does not support this [91].
Pathophysiology
The pathophysiology of depression in PD is largely unknown however is thought to be less causally due to psychosocial factors and more etiologically driven by brainstem monoamine and serotonergic dysfunction [92]. Nonetheless, similar to other chronic conditions, PD patients can certainly develop fear of disability, guilt about impact on others, or other reactive mood changes. Overall, rates of depression are higher in PD compared with patients with similar conditions matched for disability [93].
Treatment
First, the clinician must determine if depression is a result of short-term fluctuations, chronic undertreatment of motor disease, or longer-term mood phenomenon. One important pattern to recognize are mood fluctuations, which can parallel motor OFF-ON cycling. It can be valuable to distinguish this as “subsyndromic” depression or anxiety (sometimes referred to as “OFF dysphoria”), as it can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations[94–96]. Similarly, elevating chronic motor undertreatment to goal therapy can result in mood normalization.
If symptoms persist despite optimization of motor/nonmotor fluctuations or chronic undertreatment and are severe enough to warrant treatment, then therapies used can range from nonpharmacologic education, support, and mental health referrals, as well as pharmacologic support in the form of medications.
A frequent but uncontrolled observation was that when undertreatment of motor disease was finally redressed, mood often improved. A multicenter randomized controlled trial of pramipexole in PD patients without motor fluctuations but with mild to moderate depressive symptoms showed the drug improved scores on the Beck Depression Inventory over a 12-week period. The improvement in mood was 6 points overall, but by 2 points over placebo, illustrating the importance of the size of the placebo effect [97]. Given the potential side effect profile of dopamine agonists, it may be useful to weigh the antidepressant effects only when their motor benefits are already being employed.
Controlled trials have demonstrated efficacy of both selective serotonin reuptake inhibitors (SSRI) and selective norepinephrine reuptake inhibitors (SNRI) antidepressants in PD. Clinical trials have demonstrated efficacy against placebo or with other antidepressant comparators. Examples of drugs with demonstrated efficacy include citalopram, paroxetine, venlafaxine, and nortriptyline. Results have attempted to illuminate the small unique differences between classes of antidepressants or dynamic properties between drugs within a class. For example, desipramine may nudge scores on a mood scale a few weeks sooner than a purer SSRI. Paroxetine (SSRI) versus venlafaxine (SNRI) improved mood scores comparably in a multicenter trial with a placebo comparator. In general, all have all been demonstrated to be effective and with a relatively low side-effect profile, comparable to the general population[98–102]. While case reports exist in the literature, the interaction of monoamine oxidase B inhibitors and SSRIs has not caused significant hypertensive crises or risk of serotonin syndrome [103,104]. Electroconvulsive therapy (ECT) can be used for severe refractory depression in PD as for non-PD patients, with case reports of very effective results. Due to the rarity of use, systematic evidence for its use is lacking [105,106].
Other novel agents and techniques such as omega-3 fatty acids [107] and repetitive transcranial magnetic stimulation [108] have been reported with promising early results. Cognitive behavioral therapy (CBT), which may involve stress management techniques, sleep hygiene, and caregiver support, additionally almost always provided improvement in measured outcomes, whether the trial was controlled or open label in design. In one RCT of CBT in PD of 14 weeks’ duration, there were significantly more treatment responders in the CBT group, with a number needed to treat of only 2 [109].
Anxiety
Anxiety is also common in PD, at least as common as depression considering that prevalence estimates suggest up to 50% of patients experience it [110–112]. Manifestations of anxiety may include panic attacks, generalized anxiety disorder, social anxiety, or other phobias [113]. Anxiety has an important negative impact on health-related quality of life and is often underrecognized by clinicians [114]. While reliable and valid scales to measure anxiety have been lacking in PD, a new effort has yielded the “Parkinson Anxiety Scale” though full clinimetric properties of the scale remain to be demonstrated (sensitivity to change) [115].
Anxiety that parallels the timing of motor OFF-ON cycling is important to recognize. This “subsyndromic” anxiety or anxiety disorder not otherwise specified (ie, the anxiety does not meet DSM-IV criteria) can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations [116,117]. Indeed, the presence of motor fluctuations is the principle marker of anxiety in many studies [118–120]. In an analogous manner, anxiety can predate PD by years and be part of the nonmotor amalgam of features heralding the disease [6,121].
Treatment
Systematic controlled trials of anxiolytic treatment for PD are lacking; therefore, SSRIs are prescribed for this purpose as in non-PD patients. Until SSRIs are demonstrated to be of benefit in anxiety, they are likely safer than use of benzodiazepines, which are associated with risk for falling, cognitive dysfunction, or autonomic dysregulation in PD patients when used during waking hours. Psychotherapy and other nonpharmacologic approaches are likely to be of benefit. A small study of neuromuscular (massage) therapy demonstrated improvement on the Beck Anxiety Inventory in PD [122]. A case report of ECT for severe anxiety has been published [123].
Conclusion
Neuropsychiatric symptoms are common in PD and new knowledge about clinical features, epidemiology, and treatment options has been gained in the last decade, though much remains to be discovered. The development of valid instruments to measure neuropsychiatric symptoms has been vital in these research efforts to bridge the gaps in our understanding. Further elucidation of the pathophysiologies of neuropsychiatric symptoms will help to define treatment targets and likely fuel drug development and the discovery of drugs with more potent benefit and fewer side effects.
Corresponding author: Kathryn A. Chung, MD, Department of Neurology, Oregon Health & Science University, Portland, OR, [email protected].
Financial disclosures: None.
From the Department of Neurology, Oregon Health & Science University, Portland, OR.
Abstract
- Objective: To review the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms (NPS) in Parkinson’s disease (PD).
- Methods: Literature review.
- Results: PD has traditionally been considered a disease of impaired motor function. However, neuropsychiatric complications, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, frequently complicate the course of the illness. Although the development of new medication options in recent years has had a positive benefit on the management of these troublesome symptoms, responses are frequently suboptimal. The development of valid instruments to measure neuropsychiatric symptoms has been vital in research efforts to bridge the gaps in our understanding. Further elucidation of neuropsychiatric pathophysiologies will help to define treatment targets and has the potential to expand our therapeutic armamentarium.
- Conclusion: While NPS affect patients with established disease, recent investigations have demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of NPS should be the goal of practitioners treating the entire continuum of PD.
Parkinson’s disease (PD) has traditionally been considered a disease of impaired motor function, but increased recognition of nonmotor symptoms and in particular neuropsychiatric symptoms, such as fatigue, depression, anxiety, psychosis, impulse control disorders, and apathy, offer new opportunities for better care of patients. While neuropsychiatric symptoms affect patients with established disease, recent investigations have clearly demonstrated risk of symptoms in those with early untreated stages of PD; therefore, better understanding of neuropsychiatric symptoms should be the goal of practitioners treating the entire continuum of PD. This review will focus on the clinical characteristics, epidemiology, and management of the most common neuropsychiatric symptoms in PD.
Impulse Control Disorders
The recognition that dopaminergic drugs were successful at treating many symptoms of PD was followed by the disturbing realization that impulse control disorders could be an unfortunate side effect in a substantial minority. Impulse control disorders as defined by DSM-IV [1] are disinhibited behaviors that are maladaptive and recurrent, causing personal and relationship consequences. The impulse control disorders that became associated with PD and medication intake, particularly dopamine agonist use, included gambling, hobbyism, punding (stereotyped, seemingly purposeless behaviors), excessive sexual behavior, shopping, hoarding, and less commonly, compulsive eating. The prevalence estimates of these behavioral disturbances range from 6% to 15.5%, compared with < 2% in the general population [2,3]. The addiction-like dopamine dysregulation syndrome, whereby patients self-medicate with high doses of levodopa and short-acting dopamine agonists beyond what is needed for motor control, can lead to significant impairment of the therapeutic alliance in addition to other patient personal relations. With the advent of surgical options to treat PD and its medication complications, it was observed that stimulation of the subthalamic nucleus could be associated with the spectrum of impulse control disorders [4].
Epidemiology/Risk Factors
In a recent systematic review of the literature of impulse control disorders in PD [5], the authors determined that dopaminergic therapy caused compulsive or impulsive behaviors in approximately 10% of PD patients in the course of their treatment, with pathologic gambling and hypersexuality most frequently experienced. Multiple impulse control disorders are not uncommon and may coexist in one-quarter of patients with compulsivity. There appeared to be more disordered behavior with higher comparable doses of agonists. The authors concluded that impulse control disorder symptoms tended to occur with initiation or dose increases of direct D2/D3 agonists, such as pramipexole and ropinirole. Importantly, impulse control disorder behavior improved if not resolved with discontinuation or reduction of dosage of the agonist, even if a compensatory levodopa dosage is added or increased. Perhaps not surprisingly, it was observed that if patients had a preexisting impulse control disorder prior to PD or the initiation of treatment, there was a high likelihood of worsening of symptoms. This small subgroup is estimated at about 1% of PD subjects, which corresponds to the prevalence of impulse control disorders in the general population. Other identified potential risk factors for impulse control disorder development include male gender, young age at onset, a personal or family history of addiction, novelty or risk seeking personality, and a concurrent diagnosis of depression [3]. In a recent study of early PD patients, the risk of developing an impulse control disorder became important once treatment with dopaminergic drugs began and continued for a year or more [6].
Pathogenesis
The pathogenesis is not fully understood, however, mesolimbic dopamine alterations are strongly suspected. It has been long speculated that the high doses of dopamine needed to replete the relatively depleted dorsal striatum overdose the “intact” ventral striatum and cause this neuropsychiatric disorder [7–9]. The additional cognitive impairments in PD, which can include problems with attention, working memory, planning, forethought and decision-making, are faculties that can markedly increase susceptibility to impulse control disorder [8].
The role of serotonin deficiency in the PD brain and its part in inhibiting the patient’s ability to delay rewards adds to the complexity of impulse control disorder pathogenesis. Dorsal raphe nuclei disease in PD results in loss of serotonin innervation to substantial portions of the prefrontal and motor cortices in addition to basal ganglia substructures like the striatum, pallidum and subthalamic nucleus [10]. Together with dopamine, serotonin may work to regulate risk-sensitive decision making, response inhibition, waiting for future rewards, and overall impulse control. Its relative loss therefore also likely contributes to tipping the balance towards impulse dyscontrol [11,12]. The role of other neurotransmitters such as opiate systems involved in the process of acquisition and maintenance of addictive behaviors like dopamine dysregulation syndrome remains to be fully understood.
Treatment
The most successful strategy to address this problem is to reduce or eliminate the offending medication, usually the dopamine agonist. This may be associated with worsening apathy, anxiety or depression; however, substituting levodopa can be a successful strategy in many cases [13]. Zonisamide was described to be possibly effective in a trial of 15 subjects; however, the open label nature of this evidence must be considered as with other case reports using valproate, donepezil, and selective serotonin reuptake inhibitors (SSRIs) [14–16].
Fatigue
An easy to understand operational definition of fatigue is that it is a state of extreme tiredness, weakness, or exhaustion, either physical or mental or both. Fatigue is not uncommon in the general population [17] but is increasingly recognized to occur in numerous disease conditions and is frequently encountered in PD and multiple sclerosis. The latter is of special significance in the consideration of the neurotransmission of fatigue, as it is not thought to be a disease of dopamine deficiency. The pathophysiology remains unclear, and it may differ depending on whether the fatigue is experienced as more physical or mental, or rather motor versus nonmotor as some authors propose.
Fatigue has been conceptualized as central or peripheral in character. Peripheral fatigue is best understood as muscular fatigue caused by repetitive muscular contraction or reduced force generation [18]. Central fatigue however, is divided into mental or physical fatigue. Mental fatigue can occur after sustained attentive or emotional activity. It may alternatively be provoked after boring repetitive tasks or lack of intellectually stimulating activity. Physical fatigue is the sense of body exhaustion or energy to perform physical tasks even though the ability to carry them out exists.
Epidemiology
As recognition of the problem of fatigue increased in the last 2 decades, the realization that one-third to one-half of patients experience it at some point has improved opportunities for recognition and treatment [19]. Fatigue may be the presenting symptom in one-third of patients prior to actual motor symptom onset [20]. Half of untreated PD patients in a biomarker cohort study reported fatigue [6]. Unfortunately, it is also described by patients as one of the most disabling symptoms, causing significant impact on quality of life [19]. Fatigue in PD is associated with higher rates of depressive symptoms, but occurs with higher prevalence in nondepressed patients [21]. Poor ability to initiate and sustain activity due to fatigue is different from depression, excessive sleepiness, or impaired motor function [22,23].
Pathophysiology
The pathophysiology of fatigue remains somewhat unclear, though physical fatigue is likely a significant part of the problem and related to dopamine deficiency based on studies of time and force generation of keyboard strikes in PD subjects before and after L-dopa administration. These subjects had declines in force and increased physical fatigue which improved after L-dopa [24]. In other studies using transcranial magnetic stimulation to study changes in cortical excitability, the degree of physical fatigue correlated with abnormalities in motor evoked potentials during fatiguing exercising. These studies also support the hypothesis that fatigue is a motor symptom [25,26]. In the ELLDOPA study, fatigue worsened more in PD subjects treated with placebo [27]. Other imaging studies have suggested suggested nondopaminergic mechanisms including serotonergic pathway abnormalities [28], thus the question behind the etiology and solution for all cases of fatigue remains to be settled.
Diagnosis
The diagnosis is fatigue may be challenging as it may mask as depression or apathy. There are a number of fatigue rating scales available; however, the validated Parkinson’s Fatigue Scale (PFS) supersedes many of the problems of using a generic scale which could overlap motor questions and potentially be confounding [29,30].
Treatment
Most important is awareness and vigilance for the symptoms of fatigue, depression, and apathy and effort to distinguish between them. It may require structured interviews or assessment tools to properly diagnose the problem. Treatment is less clear in that few studies have clearly indicated the best treatment options. In placebo-controlled trials, methylphenidate did improve fatigue as did levodopa [31]. Modafinil, a hypocretin modulator and a drug first approved by the FDA for treatment of narcolepsy, has demonstrated mixed results in recent years. It may reduce physical fatigue and reduce excessive daytime sleepiness but likely does not reduce subjective symptoms of fatigue [32]. L-dopa can significantly reduce fatigue in many patients, which would argue that it often is a motor symptom [33,24]. In a post-hoc analysis of the ADAGIO delayed start study, patients taking rasagiline 1 mg/day and 2 mg/day (the latter dose exceeds the usual clinical dosing) showed significantly less worsening of symptoms on the PFS compared to placebo over time [34]. It is important to realize that once motor symptoms are optimally treated with dopaminergic medications, while many patients will feel significant relief from fatigue some patients will continue to feel symptomatic.
Apathy
The definition of apathy has become more complicated and refined, incorporating findings from the study of brain disease and behavioral analysis. Marin’s classic elaboration of apathy as lack of motivation not attributable to diminished level of consciousness, cognitive impairment, or emotional distress has been built upon by Levy and Dubois [35–37]. They suggest apathy may be better thought of as an observable behavioral syndrome characterized by a quantitative reduction of self-generated voluntary and purposeful behaviors. They suggest 3 apathetic subtypes: emotional, cognitive, and auto-activational, which reflect different disease states accounting for failure of normal goal-directed behavior.
Epidemiology
Prevalence estimates for apathy in PD vary. This is likely due to the varying recruitment criteria among studies, with some including patients with comorbid depression and dementia and others containing only “pure apathy.” Other reports may have had referral bias issues, as community-based studies report lower prevalences in general. In a group of newly diagnosed PD patients, using more restrictive criteria (apathy subscale of the neuropsychiatric inventory and the diagnostic consensus criteria for apathy validated in PD), Pedersen reported a prevalence of apathy of 14.3% [38]. In a 4-year prospective longitudinal cohort study, an annual incidence rate of 12.3% was reported, with apathy developing in 60% of the cohort by the study’s conclusion [39].
Apathy has been associated with longer disease duration, male gender [40], higher daily levodopa doses [41], more severe parkinsonism [38], and lower education status, though the latter feature remains under debate. Early cognitive deficits appear to be a risk factor for development of apathy [42]. The patterns of cognitive dysfunction and apathy remain unsettled in the literature.
Pathology
The pathology of atrophy remains unexplained and is unlikely to be reduced to a simple atrophy of one nucleus or the tone of one circuit. However, in a small neuroimaging study, severity of apathy correlated with atrophy of the bilateral nucleus accumbens [43], and it is notable that one major input to the nucleus accumbens is the amygdala. According to Braak staging, by stage 4 significant involvement of the amygdala by Lewy bodies has occurred. Others have found changes in grey matter density that could correlate with deficits of the prefrontal-basal ganglia circuitry to produce dysfunction of segregated frontal-subcortical loops. These may correlate with the “autoactivation” deficit pattern of apathy in which patients have a lack of self-initiated actions, even thoughts, though appear more normal when giving externally prompted responses [37,44].
Assessment
Clinically, the relationship between apathy and depression can be hard to disentangle, especially since many studies have found an association between them, especially with regards to apathy and anhedonia. Depression may feature negative self thoughts and sadness while apathy is notable for lack of initiation and effort. Viewed over a longer period of time, apathy tended to worsen in a linear fashion, where depression tended to fluctuate with improvements and exacerbations.
The Movement Disorders Society task force has recommended the Lille Apathy Rating Scale (LARS) for assessment of apathy; English and French versions have been validated in PD patients. It uses a semi-structured interview format assessing 4 dimensions of apathy: self awareness, intellectual curiosity, emotion, and action initiation [45–47].
The impact of apathy cannot be underestimated as this poor show of motivation or effort leads to lack of engagement in old activities or interest in new ones. Spouses may misinterpret this change in behavior as laziness or deliberate social withdrawal, or perhaps entitlement. It is not surprising that apathy routinely shows up on quality of life (QoL) questionnaires as highly impacting patients and families. In one study, apathy was the nonmotor symptom most likely to cause caregiver distress in PD [40,48–50].
Treatment
No approved drugs exist for treatment of apathy. However, clinical experience often confirms that dopaminergic modulation can be helpful in the treatment of apathy as indirect evidence suggests. A meta-analysis of controlled trials using pramipexole and Part I of the Unified Parkinson's disease rating scale (UPDRS) (secondary measure) showed the medication improved scores on this measure of motivation and mood in non-depressed subjects [51] with PD. Rare patients undergoing subthalamic deep brain stimulation have been reported to experience new and sometimes severe apathy after surgery [52]. This was posited at least in part to be the result of reduction of dopaminergic medication due to surgery.
Nondopaminergic pharmacotherapy of apathy is in its infancy. A recent controlled trial of rivastigmine in 31 French subjects with moderate to severe apathy based on LARS showed that 6 months of treatment at 9.5 mg/day improved average scores from –11.5 to –20 compared with placebo. While quality of life did not improve, caregiver burden did. The investigators found in this group of subjects that apathy was a possible herald for early dementia in PD [53].
A post-hoc analysis of the ADAGIO study (rasagiline or placebo in PD patients taking antidepressants) found that rasagiline use was associated with a nonsignificant slowing of apathy development during the trial [54].
Psychosis
Psychotic symptoms are a common occurrence in drug-treated patients, with visual hallucinations occurring in up to 30%, though over a 20-year period up to three-quarters of patients may develop visual hallucinations.After visual, the most common type of hallucination is auditory, followed by the other affected senses such as tactile, olfactory, or even taste [57]. Delusions, which tend to be paranoid in nature, occur in about 5% of patients [55–57]. The presence of psychotic symptoms is associated with poorer quality of life [58].
Symptomatology
The visual hallucinations of PD are usually quite stereotyped, and have been described as “minor” and “non-minor”[59]. Minor hallucinations refer to transient peripheral field stimuli that disappear when brought into central focus, “something flashed by,” a sense of a living being nearby, “a presence in the room,” or illusions whereby objects are transformed, eg, a bush in the yard is a deer.
Auditory hallucinations tend to be vague or indistinct sounds, like music in another room as opposed to voices speaking directly to the patient as might be experienced in a primary psychotic disorder. Tactile forms often involve insects or other animals crawling on the skin. Olfactory hallucinations may take the form of smelling perfume, toxic odors from room vents, etc.
Early in the experience, the visual hallucinations may be amusing in that they consistently remain nonthreatening, similar day to day, and sometimes oddly provide an aspect of comfort or companionship to the patient. More commonly, the hallucinations are bothersome to the patient because the experience indicates to the patient that there is something wrong with their mind. Visual hallucinations often begin in low-stimulus environments, often in the evening or other low-light conditions, but as the problem advances they can occur at any time of day. While visual hallucinations may initially occur for only seconds at a time many days apart, the frequency and duration can increase until they occur hours at a time every day and are accompanied by multiple other visual hallucinations, delusions, and confusion [60].
Delusions tend to be more distressing to patients and caregivers because they are often paranoid in nature. The patient is more likely to act out due to the anxiety the paranoia creates. For example, she may change passwords to online accounts due to a belief that unknown assailants are after her finances. He may go to great lengths trying to prove his wife is cheating.
Risk Factors
While the primary risk factor for psychotic symptom development is dementia [57], it occurs in nondemented patients. Other associations include reduced visual acuity [56], visual processing impairment [61–65], use of dopamine agonists, REM behavior disorder, duration of PD, axial rigidity subtype of disease [61,66–68]. The pathophysiology of psychosis in PD is likely complex and remains currently unexplained. The role of excess dopamine has been described above, but there is also data suggesting cholinergic deficits in the cortex may also contribute. Excess serotonin (increased 5HT2A receptor subtypes) in the temporal lobe within the visual processing pathway has been postulated to be of significance [69,70]. Hypometabolism in visual association areas of the brain in subjects with visual hallucinations has been demonstrated in PET and functional MRI studies [64,71]. This is similar to findings in patients with dementia with Lewy bodies [72].
This review focuses on the primary forms of PD-related psychosis, which occur with a clear sensorium and generally longer exposure to dopaminergic medication. It is important to distinguish 2 other common scenarios in which hallucinations or delusions may occur. In the common toxic-metabolic delirium, a clouded sensorium with attention deficits may be the only clue to the etiology of new onset confusion with visual hallucinations. It is highly likely that resolution of the underlying medical problem will lead to resolution of the new onset psychosis and encephalopathy. In a second scenario, hallucinations precede or occur very shortly after the onset of initiation of dopaminergic medication. This differs from the classic syndrome described earlier, in particular when visual hallucinations precede any initiation of medication, and likely represents the distinction between a diagnosis of Lewy body disease and PD [60].
Treatment
Management of psychosis is approachable, but often the outcome is unsatisfactory and associated with trade-offs in motor control. It is unfortunately true that psychotic symptoms are often associated with increased caregiver burden and are a cause of increased nursing home placements [73]. When considering the workup of psychotic symptoms, the differential diagnosis includes delirium, dream enactment (REM behavior disorder), or less commonly, Bonnet syndrome.
A delirium may be precipitated by a difficult to diagnose infection; new-onset confusion and psychotic symptoms may be the heralding presentation. Urinary tract or upper respiratory tract are common vulnerable sources of infection. Once infection is ruled out, the next practical step is to review the patient’s medication list and manage centrally acting drugs that could be contributing to the altered sensorium. A recent prescription of opioids for a dental treatment or a new muscle relaxant may be a culprit, though it is not that usual. A bladder anticholinergic could be suspect and is worth eliminating especially if its addition coincided with the appearance of the psychotic symptoms. Once the non-dopaminergic medications have been reduced/eliminated, then the PD medications should be considered. The general approach is to eliminate the medications that provide the least benefit while being more likely to contribute to psychotic symptoms. Anticholinergic medications, dopamine agonists, selegiline should all be uppermost in that consideration until one is left with L-dopa and COMT inhibitors (the latter function to increase levodopa availability). Then COMT inhibitors and levodopa can be reduced; however, at any point motor control can suffer with the loss of symptomatic therapies [74].
Clozapine is effective against psychotic symptoms in PD, at doses much lower than used in schizophrenia (300-600mg/day). The average dose in the US randomized controlled clinical trial was 25 mg/day, with no associated motor worsening. Patients in the United States are required by the FDA to be placed in a computer-based registry and monitored for agranulocytosis for the duration of clozapine therapy. This rare adverse event is not dose related. Orthostasis can occur at these low doses however. Fortunately the metabolic syndrome is not associated with this range of administration [75,76].
Quetiapine was not found to be effective in 3 blinded randomized controlled trials despite its rather common use for this purpose. It was not associated with motor worsening, however.
Other neuroleptic medications have not resulted in widespread use, because trials have been open label, or outcomes demonstrated motor worsening. Cholinesterase inhibitors have been the subject of a few positive case series, however results appear to be sporadic, the effect size is relatively small, and side effects of this medication class are common [77–79]. It is clear that there is an unmet need for a medication for psychotic symptoms. Clozapine is effective but onerous in its monitoring requirements. Practically speaking, there are relatively few PD patients who take advantage of it because of its feasibility challenges. Yet the problem of psychotic symptoms is a significant one that imposes important challenges to the patient and caregiver, and may limit the number of medications that the patient needs in order to optimize quality of life.
Pimavanserin, a novel medication which acts as a selective serotonin inverse agonist, is in the early application stages for FDA approval for treatment of psychotic symptoms in PD [80]. In its pivotal phase III controlled trial, the drug reduced not only positive symptoms (hallucinations/delusions) without causing motoric worsening, but also reduced caregiver burden. Pimavanserin improved certain sleep features without causing daytime sedation. If this drug meets final approval, it may present an exciting option for many patients for whom treatment was previously limited.
Depression
A study of early PD suggested that depression is often unrecognized and frequently untreated [1]. Indeed it is not unusual for depression to predate the diagnosis of PD by an average of 4 to 6 years [81]. Expanding to the larger PD population, it is generally accepted that about 30% to 50% of PD patients experience clinically significant depression, and once diagnosed may have a long term course, or may recur [82,83]. This is important as untreated depression is an important cause of poor quality of life in early PD. In addition depression can exacerbate motor disability, lead to earlier motor treatment with medication, and increase caregiver stress [83–85].
Diagnosis
A number of clinimetric rating scales for depression have been used and their advantages have been largely related to their objective nature (quantifiable); thus, they tend to be most useful in epidemiologic research studies or for larger scale screening purposes. Examples include the the Beck Depression Inventory, the Geriatric Depression Scale, and the Hamilton Depression Scale, all of which have been shown to be valid tools in PD (with the exception of the UPDRS Depression). It is important to note that they do not substitute for a diagnostic clinical interview [89].
Suicide is not common in PD, however suicidal ideation is estimated at about 11% in PD patients [90], and while there was concern initially after deep brain stimulation procedures began that suicide incidence was increased, evidence does not support this [91].
Pathophysiology
The pathophysiology of depression in PD is largely unknown however is thought to be less causally due to psychosocial factors and more etiologically driven by brainstem monoamine and serotonergic dysfunction [92]. Nonetheless, similar to other chronic conditions, PD patients can certainly develop fear of disability, guilt about impact on others, or other reactive mood changes. Overall, rates of depression are higher in PD compared with patients with similar conditions matched for disability [93].
Treatment
First, the clinician must determine if depression is a result of short-term fluctuations, chronic undertreatment of motor disease, or longer-term mood phenomenon. One important pattern to recognize are mood fluctuations, which can parallel motor OFF-ON cycling. It can be valuable to distinguish this as “subsyndromic” depression or anxiety (sometimes referred to as “OFF dysphoria”), as it can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations[94–96]. Similarly, elevating chronic motor undertreatment to goal therapy can result in mood normalization.
If symptoms persist despite optimization of motor/nonmotor fluctuations or chronic undertreatment and are severe enough to warrant treatment, then therapies used can range from nonpharmacologic education, support, and mental health referrals, as well as pharmacologic support in the form of medications.
A frequent but uncontrolled observation was that when undertreatment of motor disease was finally redressed, mood often improved. A multicenter randomized controlled trial of pramipexole in PD patients without motor fluctuations but with mild to moderate depressive symptoms showed the drug improved scores on the Beck Depression Inventory over a 12-week period. The improvement in mood was 6 points overall, but by 2 points over placebo, illustrating the importance of the size of the placebo effect [97]. Given the potential side effect profile of dopamine agonists, it may be useful to weigh the antidepressant effects only when their motor benefits are already being employed.
Controlled trials have demonstrated efficacy of both selective serotonin reuptake inhibitors (SSRI) and selective norepinephrine reuptake inhibitors (SNRI) antidepressants in PD. Clinical trials have demonstrated efficacy against placebo or with other antidepressant comparators. Examples of drugs with demonstrated efficacy include citalopram, paroxetine, venlafaxine, and nortriptyline. Results have attempted to illuminate the small unique differences between classes of antidepressants or dynamic properties between drugs within a class. For example, desipramine may nudge scores on a mood scale a few weeks sooner than a purer SSRI. Paroxetine (SSRI) versus venlafaxine (SNRI) improved mood scores comparably in a multicenter trial with a placebo comparator. In general, all have all been demonstrated to be effective and with a relatively low side-effect profile, comparable to the general population[98–102]. While case reports exist in the literature, the interaction of monoamine oxidase B inhibitors and SSRIs has not caused significant hypertensive crises or risk of serotonin syndrome [103,104]. Electroconvulsive therapy (ECT) can be used for severe refractory depression in PD as for non-PD patients, with case reports of very effective results. Due to the rarity of use, systematic evidence for its use is lacking [105,106].
Other novel agents and techniques such as omega-3 fatty acids [107] and repetitive transcranial magnetic stimulation [108] have been reported with promising early results. Cognitive behavioral therapy (CBT), which may involve stress management techniques, sleep hygiene, and caregiver support, additionally almost always provided improvement in measured outcomes, whether the trial was controlled or open label in design. In one RCT of CBT in PD of 14 weeks’ duration, there were significantly more treatment responders in the CBT group, with a number needed to treat of only 2 [109].
Anxiety
Anxiety is also common in PD, at least as common as depression considering that prevalence estimates suggest up to 50% of patients experience it [110–112]. Manifestations of anxiety may include panic attacks, generalized anxiety disorder, social anxiety, or other phobias [113]. Anxiety has an important negative impact on health-related quality of life and is often underrecognized by clinicians [114]. While reliable and valid scales to measure anxiety have been lacking in PD, a new effort has yielded the “Parkinson Anxiety Scale” though full clinimetric properties of the scale remain to be demonstrated (sensitivity to change) [115].
Anxiety that parallels the timing of motor OFF-ON cycling is important to recognize. This “subsyndromic” anxiety or anxiety disorder not otherwise specified (ie, the anxiety does not meet DSM-IV criteria) can respond to improvement in antiparkinsonian medication dosing patterns that reduce fluctuations [116,117]. Indeed, the presence of motor fluctuations is the principle marker of anxiety in many studies [118–120]. In an analogous manner, anxiety can predate PD by years and be part of the nonmotor amalgam of features heralding the disease [6,121].
Treatment
Systematic controlled trials of anxiolytic treatment for PD are lacking; therefore, SSRIs are prescribed for this purpose as in non-PD patients. Until SSRIs are demonstrated to be of benefit in anxiety, they are likely safer than use of benzodiazepines, which are associated with risk for falling, cognitive dysfunction, or autonomic dysregulation in PD patients when used during waking hours. Psychotherapy and other nonpharmacologic approaches are likely to be of benefit. A small study of neuromuscular (massage) therapy demonstrated improvement on the Beck Anxiety Inventory in PD [122]. A case report of ECT for severe anxiety has been published [123].
Conclusion
Neuropsychiatric symptoms are common in PD and new knowledge about clinical features, epidemiology, and treatment options has been gained in the last decade, though much remains to be discovered. The development of valid instruments to measure neuropsychiatric symptoms has been vital in these research efforts to bridge the gaps in our understanding. Further elucidation of the pathophysiologies of neuropsychiatric symptoms will help to define treatment targets and likely fuel drug development and the discovery of drugs with more potent benefit and fewer side effects.
Corresponding author: Kathryn A. Chung, MD, Department of Neurology, Oregon Health & Science University, Portland, OR, [email protected].
Financial disclosures: None.
1. American Psychiatric Association. DSM-IV. 2000.
2. Voon V, Hassan K, Zurowski M, et al. Prevalence of behaviors in Parkinson disease Neurology 2006;67;1254–7.
3. D. Weintraub D, J. Koester J, M. N. Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67589–95.
4. Hälbig TD, Tse W, Frisina PG, et al. Subthalamic deep brain stimulation and impulse control in Parkinson’s disease. Eur J Neuro 2009;16:493–7.
5. Callesen MB, Scheel-Krüger J, Kringelbach ML, Møller A. A systematic review of impulse control disorders in Parkinson’s disease. J Parkinsons Dis 2013;3:105–38.
6. De Riva P, Smith K, Xie SX, Weintraub D. Course of psychiatric symptoms and global cognition in early course of psychiatric symptoms and global cognition in early Parkinson disease Neurology 2014;83:1–8.
7. Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev 2006;30:1–23.
8. Rowe JB, Hughes L, Ghosh BCP, et al. Parkinson’s disease and dopaminergic therapy – Differential effects on movement, reward and cognition. Brain 2008;131:2094–105.
9. Cools R, Frank MJ, Gibbs SE, et al. Striatal dopamine predicts outcome-specific reversal learning and its sensitivity to dopaminergic drug administration. J Neurosci 2009;29:1538–43.
10. Di Matteo V, Pierucci M, Esposito E, et al. Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson’s disease and other motor disorders. Prog Brain Res 2008;172:423–463.
11. Campbell-Meiklejohn D, Wakeley J, Herbert V, et al. Serotonin and dopamine play complementary roles in gambling to recover losses. Neuropsychopharmacology 2011;36:402–10.
12. Macoveanu J, Rowe JB, Hornboll B, et al. Playing it safe but losing anyway-Serotonergic signaling of negative outcomes in dorsomedial prefrontal cortex in the context of risk-aversion. Eur Neuropsychopharmacol 2013;23:919–30.
13. Mamikonyan E, Siderowf AD, Duda JE, et al. NIH Public Access 2009;23:75–80.
14. Ivanco LS, Bohnen NI. Effects of donepezil on compulsive hypersexual behavior in Parkinson disease: a single case study. Am J Ther 2005;12:467–8.
15. Hicks CW, Pandya MM, Itin I, Fernandez HH. Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease. Parkinsonism Relat Disord 2011;5:379–81.
16. Bermejo PE, Ruiz-Huete C, Anciones B. Zonisamide in managing impulse control disorders in Parkinson’s disease. J Neurol 2010;257:1682–5.
17. Pawlikowska T, Chalder T, Hirsch SR, et al. Population based study of fatigue and psychological distress. BMJ 1994;308:763–6.
18. Lou JS, Kearns G, Oken B, et al. Exacerbated physical fatigue and mental fatigue in Parkinson’s disease. Mov Disord 2001;16:190–6.
19. Herlofson K, Larsen JP. The influence of fatigue on health-related quality of life in patients with Parkinson’s disease. Acta Neurol Scand 2003;107:1–6.
20. Hagell P, Brundin L. Towards an understanding of fatigue in Parkinson disease. J Neurol Neurosurg Psychiatry 2009;80:489–92.
21. Alves G, Wentzel-Larsen T, Larsen JP. Is fatigue an independent and persistent symptom in patients with Parkinson disease? Neurology 2004;63:1908–11.
22. Martinez-Martin P, Catalan MJ, Benito-Leon J, et al. Impact of fatigue in Parkinson’s disease: The fatigue impact scale for daily use (D-FIS). Qual Life Res 2006:15:597–606.
23. Havlikova E, Rosenberger J, Nagyova I, et al. Clinical and psychosocial factors associated with fatigue in patients with Parkinson’s disease. Parkinsonism Relat Disord 2008;14:187–92.
24. Lou JS, Kearns G, Benice T, et al. Levodopa improves physical fatigue in Parkinson’s disease: A double-blind, placebo controlled, crossover study. Mov Disord 2003;18:1108–14.
25. Khedr EM, Galal O, Said A, et al. Lack of post-exercise depression of corticospinal excitability in patients with Parkinson’s disease. Eur J Neurol 2007;14:793–6.
26. Lou JS, Benice T, Kearns G, et al. Levodopa normalizes exercise related cortico-motoneuron excitability abnormalities in Parkinson’s disease. Clin. Neurophysiol 2003;114:930–7.
27. Schifitto G, Friedman JH, Oakes D, et al. Fatigue in levodopa-naive subjects with Parkinson disease. Neurology 2008;71:481–5.
28. Pavese N, Metta V, Bose SK, et al. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010;133:3434–43.
29. Brown RG, Dittner A, Findley L, Wessely SC. The Parkinson fatigue scale. Park Relat Disord 2005;11:49–55.
30. Grace J, Mendelsohn A, Friedman JH. A comparison of fatigue measures in Parkinson’s disease. Parkinsonism Relat Disord 2007;13:443–5.
31. Mendonça DA, Menezes K, Jog MS. Methylphenidate improves fatigue scores in Parkinson disease: a randomized controlled trial. Mov Disord 2007;22:2070–6.
32. Lou JS, Dimitrova DM, Park BS, et al. Using modafinil to treat fatigue in Parkinson disease: a double-blind, placebo-controlled pilot study. Clin Neuropharmacol 2009;32:305–10.
33. Ziv I, Avraham M, Michaelov Y, et al. Enhanced fatigue during motor performance in patients with Parkinson’s disease. Neurology 1998;51:1583–6.
34. Stocchi F. Benefits of treatment with rasagiline for fatigue symptoms in patients with early Parkinson’s disease. Eur J Neurol 2014;21:357–60.
35. Marin RS. Apathy: a neuropsychiatric syndrome. J. Neuropsychiatry Clin Neurosci 1991;3:243–4.
36. Marin RS. Apathy: concept, syndrome, neural mechanisms, and treatment. Semin Clin Neuropsychiatry 1996:1:304–14.
37. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cerebral Cortex 2006;16:916–28.
38. Pedersen KF, Larsen JP, Alves G, Aarsland D. Prevalence and clinical correlates of apathy in Parkinson’s disease: a community-based study. Parkinsonism Relat Disord 2009;15:295–9.
39. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
40. Pedersen KF, Alves G, Brønnick K, et al. Apathy in drug-naïve patients with incident Parkinson’s disease: The Norwegian ParkWest study. J Neurol 2010;257:217–23.
41. Ziropadja L, Stefanova E, Petrovic M, et al. Apathy and depression in Parkinson’s disease: The Belgrade PD study report. Parkinsonism Relat Disord 2012;18:339–42.
42. Dujardin K, Sockeel P, Devos D, et al. Characteristics of apathy in Parkinson’s disease. Mov Disord 2007;22:778–84.
43. Carriere N, Besson P, Dujardin K, et al. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov Disord 2014;29;897–903.
44. Reijnders JSAM, Scholtissen B, Weber WEJ, et al. Neuroanatomical correlates of apathy in Parkinson’s disease: a magnetic resonance imaging study using voxel-based morphometry. Mov Disord 2010;25:2318–25.
45. Leroi I, Harbishettar V, Andrews M, et al. Carer burden in apathy and impulse control disorders in Parkinson’s disease. Int J Geriatr Psychiatry 2012;27:160–6.
46. Schiehser DM, Liu L, Lessig SL, et al. Predictors of discrepancies in Parkinson’s disease patient and caregiver ratings of apathy, disinhibition, and executive dysfunction before and after diagnosis. J Int Neuropsychol Soc 2013;19:295–304.
47. Benito-León J, Cubo E, Coronell C, et al. Impact of apathy on health-related quality of life in recently diagnosed Parkinson’s disease: The ANIMO study. Mov Disord 2012;27:211–8.
48. Sockeel P, Dujardin K, Devos D, et al. The Lille apathy rating scale (LARS), a new instrument for detecting and quantifying apathy: validation in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2006;77:579–84.
49. Zahodne LB, Young S, Kirsch-Darrow L, et al. Examination of the Lille Apathy Rating Scale in Parkinson disease. Mov Disord 2009;24:677–83.
50. Drijgers RL, Dujardin K, Reijnders JSAM, et al. Validation of diagnostic criteria for apathy in Parkinson’s disease. Parkinsonism Relat Disord 2010;16:656–60.
51. Leentjens AFG, Koester J, Fruh B, et al. The effect of pramipexole on mood and motivational symptoms in parkinson’s disease: A meta-analysis of placebo-controlled studies. Clin Ther 2009;31:89–98.
52. Thobois S, Ardouin C, Lhommée E, et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: Predictors and underlying mesolimbic denervation. Brain 2010;133:1111–27.
53. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry 2014;85:668–74.
54. Smith KM, Eyal E, Weintraub D; ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 2015;72:88–95.
55. Forsaa EB, Larsen JP, Wentzel-Larsen T, et al. A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol 2010;67:996–1001.
56. Holroyd S, Currie L, Wooten GF. Prospective study of hallucinations and delusions in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2001;70:734–8.
57. Fénelon G, Alves G. Epidemiology of psychosis in Parkinson’s disease. J Neurol Sci 2010;289:12–7.
58. Mack J, Rabins P, Anderson K, et al. Prevalence of psychotic symptoms in a community-based Parkinson disease sample. Am J Geriatr Psychiatry 2012;20:123–32.
59. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: Report of an NINDS, NIMH Work Group. Mov Disord 2007;22:1061–8.
60. Diederich NJ, Fénelon G, Stebbins G, Goetz CG. Hallucinations in Parkinson disease. Nat Rev Neurol 2009;5:331–42.
61. Gallagher DA, Parkkinen L, O’Sullivan SS, et al. Testing an aetiological model of visual hallucinations in Parkinson’s disease. Brain 2011;134:3299–309.
62. Barnes J, Boubert L, Harris J, et al. Reality monitoring and visual hallucinations in Parkinson’s disease. Neuropsychologia 2003:41:565–74.
63. Kurita A, Murakami M, Takagi S, et al. Visual hallucinations and altered visual information processing in Parkinson disease and dementia with Lewy bodies. Mov Disord 2010;25:167–71.
64. Stebbins GT, Goetz CG, Carrillo MC, et al. Altered cortical visual processing in PD with hallucinations: an fMRI study. Neurology 2004;63:1409–16.
65. Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: A retrospective autopsy study. Lancet Neurol 2005:4:605–10.
66. Biglan KM, Holloway RG, McDermott MP, Richard IH. Risk factors for somnolence, edema, and hallucinations in early Parkinson disease. Neurology 2007;69:187–95.
67. Kiziltan G, Ozekmekci S, Ertan S, et al. Relationship between age and subtypes of psychotic symptoms in Parkinson’s disease. J Neurol 2007;254:448–52.
68. Ecker D, Unrath A, Kassubek J, Sabolek M. Dopamine agonists and their risk to induce psychotic episodes in Parkinson’s disease: a case-control study. BMC Neurol 2009;9:23.
69. Huot P, Johnston TH, Darr T, et al. Increased 5-HT2A receptors in the temporal cortex of Parkinsonian patients with visual hallucinations. Mov Disord 2010;25:1399–408.
70. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch. Neurol 2010;67:416–21.
71. Boecker H, Ceballos-Baumann O, Volk D, et al. Metabolic alterations in patients with Parkinson disease and visual hallucinations. Arch Neurol 2007;64:984–8.
72. Sanchez-Castaneda C, Rene R, Ramirez-Ruiz B, et al. Frontal and associative visual areas related to visual hallucinations in dementia with Lewy bodies and Parkinson’s disease with dementia. Mov Disord 2010;25:615–22.
73. Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993;43:2227–9.
74. Friedman JH. Parkinson disease psychosis: update. Behav.Neurol 2013;27:469–77.
75. Pollak P, Tison F, Rascol O, et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004;75:689–95.
76. Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis. N Engl J Med 1999;340:757–63.
77. Sobow T. Parkinson’s disease-related visual hallucinations unresponsive to atypical antipsychotics treated with cholinesterase inhibitors: a case series. Neurol Neurochir Pol 2007;41:276–9.
78. Fabbrini G, Barbanti P, Aurilia C, et al. Donepezil in the treatment of hallucinations and delusions in Parkinson’s disease. Neurol Sci 2002;23:41–3.
79. Bullock R, Cameron A, Hospital WH. Rivastigmine for the treatment of dementia and visual hallucinations associated with Parkinson’s disease: a case series. Curr Med Res Opin 2002;18,:258–64.
80. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383:533–40.
81. L. Ishihara L, and C. Brayne C. A systematic review of depression and mental illness preceding Parkinson’s disease. Acta Neurol Scand 2006;113:211–20.
82. Reijnders JSAM, Ehrt U, Weber WEJ, et al. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord 2008;23:183–9.
83. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992:55:377–82.
84. Müller B, Assmus J, Herlofson K, et al. Importance of motor vs. non-motor symptoms for health-related quality of life in early Parkinson’s disease. Parkinsonism Relat Disord 2013;19:1027–32.
85. Ravina B, Camicioli R, Como PG, et al. The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
86. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148–58.
87. Marsh L. Depression and Parkinson’s disease: current knowledge. Curr. Neurol. Neurosci. Rep 2013; 13:409.
88. Starkstein SE, Merello M, Jorge R, et al. A validation study of depressive syndromes in Parkinson’s disease. Mov Disord 2008;23,:538–46.
89. Williams JR, Hirsch ES, Anderson K, et al. A comparison of nine scales to detect depression in Parkinson disease: which scale to use? Neurology 2012;78:998–1006.
90. Nazem S, Siderowf AD, Duda JE, et al. Suicidal and death ideation in Parkinson’s disease. Mov Disord 2008;23:1573–9.
91. Weintraub D, Duda JE, Carlson K, et al. Suicide ideation and behaviours after STN and GPi DBS surgery for Parkinson’s disease: results from a randomised, controlled trial. J Neurol Neurosurg Psychiatry 2013;84:1113–8.
92. Aarsland D, Påhlhagen S, Ballard CG, et al. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol 2011;8:35–47.
93. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson’s disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
94. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology 2002:59:408–13.
95. Nissenbaum H, Quinn NP, Brown RG, et al. Mood swings associated with the ‘on-off’ phenomenon in Parkinson’s disease. Psychol Med 1987;17:899–904.
96. Maricle RA, Nutt JG, Carter JH. Mood and anxiety fluctuation in Parkinson’s disease associated with levodopa infusion: preliminary findings. Mov Disord 1995;10:329–32.
97. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
98. Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008;23:850–7.
99. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
100. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
101. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord 2011;26 Suppl 3:S42–80.
102. Skapinakis P, Bakola E, Salanti G, et al. Efficacy and acceptability of selective serotonin reuptake inhibitors for the treatment of depression in Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. BMC Neurol 2010;10:49.
103. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Parkinson Study Group. Neurology 1997;48:1070–7.
104. Montgomery EB, Panisset M. Retrospective statistical analysis of the incidence of serotonin toxicity in patients taking rasagiline and anti-depressants in clinical trials. Mov Disord 2009;24:359.
105. Bailine S, Kremen N, Kohen I, et al. Bitemporal electroconvulsive therapy for depression in a Parkinson disease patient with a deep-brain stimulator. J ECT 2008;24:171–2.
106. Faber R, Trimble MR. Electroconvulsive therapy in Parkinson’s disease and other movement disorders. Mov Disord 1991;6:293–303.
107. da Silva TM, Munhoz RP, Alvarez C, et al. Depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled pilot study of omega-3 fatty-acid supplementation. J Affect Disord 2008;111:351–9.
108. Pal E, Nagy F, Aschermann Z, et al. The impact of left prefrontal repetitive transcranial magnetic stimulation on depression in Parkinson’s disease: a randomized, double-blind, placebo-controlled study. Mov Disord 2010;25:2311–7.
109. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry 2011;168:1066–74.
110. Stein MB, Heuser IJ, Juncos JL, Uhde TW. Anxiety disorders in patients with parkinson’s disease. Am J Psychiatry 1990;147:217–20.
111. Nuti A, Ceravolo R, Piccinni A, et al. Psychiatric comorbidity in a population of Parkinson’s disease patients. Eur J Neurol 2004;11:315–20.
112. Marinus J, Leentjens AFG, Visser M, et al. Evaluation of the Hospital Anxiety and Depression Scale in patients With Parkinson’s disease. Clin Neuropharmacol 2002:25:318–24.
113. Dissanayaka NNW, White E, O’Sullivan JD, et al. The clinical spectrum of anxiety in Parkinson’s disease. Mov Disord 2014;29:967–75.
114. Dissanayaka NNW, Sellbach A, Matheson S, et al. Anxiety disorders in Parkinson’s disease: prevalence and risk factors. Mov Disord 2010;25:838–45.
115. Leentjens AFG, Dujardin K, Pontone GM, et al. The Parkinson anxiety scale (PAS): Development and validation of a new anxiety scale. Mov Disord 2014;29:1035–43.
116. Maricle RA, Nutt JG, Valentine RJ, Carter JH. Dose-response relationship of levodopa with mood and anxiety in fluctuating Parkinson’s disease: a double-blind, placebo-controlled study. Neurology 1995;45:1757–60.
117. Erdal KJ. Depression and anxiety in persons with Parkinson’s disease with and without ‘on–off’ phenomena. J Clin Psychol Med Settings 2001:8:293–9.
118. Leentjens AFG, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson’s disease: a cross-sectional study. Mov. Disord 2011;26:484–92.
119. Solla P, Cannas A, Floris GL, et al. Behavioral, neuropsychiatric and cognitive disorders in Parkinson’s disease patients with and without motor complications. Prog Neuropsychopharmacol Biol Psychiatry 2011;35:1009–13.
120. E. Stefanova E, L. Ziropadja L, M. Petrović M, et al. Screening for anxiety symptoms in Parkinson disease: a cross-sectional study. J Geriatr Psychiatry Neurol 2013;26:34–40.
121. Jacob EL, Gatto NM, Thompson A, et al. Occurrence of depression and anxiety prior to Parkinson’s disease. Parkinsonism Relat Disord 2010;16:576–81.
122. Craig LH, Svircev A, Haber M, Juncos JL. Controlled pilot study of the effects of neuromuscular therapy in patients with Parkinson’s disease. Mov Disord 2006;21:2127–33.
123. Marino L, Friedman JH. Letter to the editor: Successful use of electroconvulsive therapy for refractory anxiety in Parkinson’s disease. Int J Neurosci 2013;123:70–1.
1. American Psychiatric Association. DSM-IV. 2000.
2. Voon V, Hassan K, Zurowski M, et al. Prevalence of behaviors in Parkinson disease Neurology 2006;67;1254–7.
3. D. Weintraub D, J. Koester J, M. N. Potenza MN, et al. Impulse control disorders in Parkinson disease: a cross-sectional study of 3090 patients. Arch Neurol 2010;67589–95.
4. Hälbig TD, Tse W, Frisina PG, et al. Subthalamic deep brain stimulation and impulse control in Parkinson’s disease. Eur J Neuro 2009;16:493–7.
5. Callesen MB, Scheel-Krüger J, Kringelbach ML, Møller A. A systematic review of impulse control disorders in Parkinson’s disease. J Parkinsons Dis 2013;3:105–38.
6. De Riva P, Smith K, Xie SX, Weintraub D. Course of psychiatric symptoms and global cognition in early course of psychiatric symptoms and global cognition in early Parkinson disease Neurology 2014;83:1–8.
7. Cools R. Dopaminergic modulation of cognitive function-implications for L-DOPA treatment in Parkinson’s disease. Neurosci Biobehav Rev 2006;30:1–23.
8. Rowe JB, Hughes L, Ghosh BCP, et al. Parkinson’s disease and dopaminergic therapy – Differential effects on movement, reward and cognition. Brain 2008;131:2094–105.
9. Cools R, Frank MJ, Gibbs SE, et al. Striatal dopamine predicts outcome-specific reversal learning and its sensitivity to dopaminergic drug administration. J Neurosci 2009;29:1538–43.
10. Di Matteo V, Pierucci M, Esposito E, et al. Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson’s disease and other motor disorders. Prog Brain Res 2008;172:423–463.
11. Campbell-Meiklejohn D, Wakeley J, Herbert V, et al. Serotonin and dopamine play complementary roles in gambling to recover losses. Neuropsychopharmacology 2011;36:402–10.
12. Macoveanu J, Rowe JB, Hornboll B, et al. Playing it safe but losing anyway-Serotonergic signaling of negative outcomes in dorsomedial prefrontal cortex in the context of risk-aversion. Eur Neuropsychopharmacol 2013;23:919–30.
13. Mamikonyan E, Siderowf AD, Duda JE, et al. NIH Public Access 2009;23:75–80.
14. Ivanco LS, Bohnen NI. Effects of donepezil on compulsive hypersexual behavior in Parkinson disease: a single case study. Am J Ther 2005;12:467–8.
15. Hicks CW, Pandya MM, Itin I, Fernandez HH. Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease. Parkinsonism Relat Disord 2011;5:379–81.
16. Bermejo PE, Ruiz-Huete C, Anciones B. Zonisamide in managing impulse control disorders in Parkinson’s disease. J Neurol 2010;257:1682–5.
17. Pawlikowska T, Chalder T, Hirsch SR, et al. Population based study of fatigue and psychological distress. BMJ 1994;308:763–6.
18. Lou JS, Kearns G, Oken B, et al. Exacerbated physical fatigue and mental fatigue in Parkinson’s disease. Mov Disord 2001;16:190–6.
19. Herlofson K, Larsen JP. The influence of fatigue on health-related quality of life in patients with Parkinson’s disease. Acta Neurol Scand 2003;107:1–6.
20. Hagell P, Brundin L. Towards an understanding of fatigue in Parkinson disease. J Neurol Neurosurg Psychiatry 2009;80:489–92.
21. Alves G, Wentzel-Larsen T, Larsen JP. Is fatigue an independent and persistent symptom in patients with Parkinson disease? Neurology 2004;63:1908–11.
22. Martinez-Martin P, Catalan MJ, Benito-Leon J, et al. Impact of fatigue in Parkinson’s disease: The fatigue impact scale for daily use (D-FIS). Qual Life Res 2006:15:597–606.
23. Havlikova E, Rosenberger J, Nagyova I, et al. Clinical and psychosocial factors associated with fatigue in patients with Parkinson’s disease. Parkinsonism Relat Disord 2008;14:187–92.
24. Lou JS, Kearns G, Benice T, et al. Levodopa improves physical fatigue in Parkinson’s disease: A double-blind, placebo controlled, crossover study. Mov Disord 2003;18:1108–14.
25. Khedr EM, Galal O, Said A, et al. Lack of post-exercise depression of corticospinal excitability in patients with Parkinson’s disease. Eur J Neurol 2007;14:793–6.
26. Lou JS, Benice T, Kearns G, et al. Levodopa normalizes exercise related cortico-motoneuron excitability abnormalities in Parkinson’s disease. Clin. Neurophysiol 2003;114:930–7.
27. Schifitto G, Friedman JH, Oakes D, et al. Fatigue in levodopa-naive subjects with Parkinson disease. Neurology 2008;71:481–5.
28. Pavese N, Metta V, Bose SK, et al. Fatigue in Parkinson’s disease is linked to striatal and limbic serotonergic dysfunction. Brain 2010;133:3434–43.
29. Brown RG, Dittner A, Findley L, Wessely SC. The Parkinson fatigue scale. Park Relat Disord 2005;11:49–55.
30. Grace J, Mendelsohn A, Friedman JH. A comparison of fatigue measures in Parkinson’s disease. Parkinsonism Relat Disord 2007;13:443–5.
31. Mendonça DA, Menezes K, Jog MS. Methylphenidate improves fatigue scores in Parkinson disease: a randomized controlled trial. Mov Disord 2007;22:2070–6.
32. Lou JS, Dimitrova DM, Park BS, et al. Using modafinil to treat fatigue in Parkinson disease: a double-blind, placebo-controlled pilot study. Clin Neuropharmacol 2009;32:305–10.
33. Ziv I, Avraham M, Michaelov Y, et al. Enhanced fatigue during motor performance in patients with Parkinson’s disease. Neurology 1998;51:1583–6.
34. Stocchi F. Benefits of treatment with rasagiline for fatigue symptoms in patients with early Parkinson’s disease. Eur J Neurol 2014;21:357–60.
35. Marin RS. Apathy: a neuropsychiatric syndrome. J. Neuropsychiatry Clin Neurosci 1991;3:243–4.
36. Marin RS. Apathy: concept, syndrome, neural mechanisms, and treatment. Semin Clin Neuropsychiatry 1996:1:304–14.
37. Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cerebral Cortex 2006;16:916–28.
38. Pedersen KF, Larsen JP, Alves G, Aarsland D. Prevalence and clinical correlates of apathy in Parkinson’s disease: a community-based study. Parkinsonism Relat Disord 2009;15:295–9.
39. Pedersen KF, Alves G, Aarsland D, Larsen JP. Occurrence and risk factors for apathy in Parkinson disease: a 4-year prospective longitudinal study. J Neurol Neurosurg Psychiatry 2009;80:1279–82.
40. Pedersen KF, Alves G, Brønnick K, et al. Apathy in drug-naïve patients with incident Parkinson’s disease: The Norwegian ParkWest study. J Neurol 2010;257:217–23.
41. Ziropadja L, Stefanova E, Petrovic M, et al. Apathy and depression in Parkinson’s disease: The Belgrade PD study report. Parkinsonism Relat Disord 2012;18:339–42.
42. Dujardin K, Sockeel P, Devos D, et al. Characteristics of apathy in Parkinson’s disease. Mov Disord 2007;22:778–84.
43. Carriere N, Besson P, Dujardin K, et al. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov Disord 2014;29;897–903.
44. Reijnders JSAM, Scholtissen B, Weber WEJ, et al. Neuroanatomical correlates of apathy in Parkinson’s disease: a magnetic resonance imaging study using voxel-based morphometry. Mov Disord 2010;25:2318–25.
45. Leroi I, Harbishettar V, Andrews M, et al. Carer burden in apathy and impulse control disorders in Parkinson’s disease. Int J Geriatr Psychiatry 2012;27:160–6.
46. Schiehser DM, Liu L, Lessig SL, et al. Predictors of discrepancies in Parkinson’s disease patient and caregiver ratings of apathy, disinhibition, and executive dysfunction before and after diagnosis. J Int Neuropsychol Soc 2013;19:295–304.
47. Benito-León J, Cubo E, Coronell C, et al. Impact of apathy on health-related quality of life in recently diagnosed Parkinson’s disease: The ANIMO study. Mov Disord 2012;27:211–8.
48. Sockeel P, Dujardin K, Devos D, et al. The Lille apathy rating scale (LARS), a new instrument for detecting and quantifying apathy: validation in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2006;77:579–84.
49. Zahodne LB, Young S, Kirsch-Darrow L, et al. Examination of the Lille Apathy Rating Scale in Parkinson disease. Mov Disord 2009;24:677–83.
50. Drijgers RL, Dujardin K, Reijnders JSAM, et al. Validation of diagnostic criteria for apathy in Parkinson’s disease. Parkinsonism Relat Disord 2010;16:656–60.
51. Leentjens AFG, Koester J, Fruh B, et al. The effect of pramipexole on mood and motivational symptoms in parkinson’s disease: A meta-analysis of placebo-controlled studies. Clin Ther 2009;31:89–98.
52. Thobois S, Ardouin C, Lhommée E, et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: Predictors and underlying mesolimbic denervation. Brain 2010;133:1111–27.
53. Devos D, Moreau C, Maltête D, et al. Rivastigmine in apathetic but dementia and depression-free patients with Parkinson’s disease: a double-blind, placebo-controlled, randomised clinical trial. J Neurol Neurosurg Psychiatry 2014;85:668–74.
54. Smith KM, Eyal E, Weintraub D; ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO study: effects on nonmotor symptoms and tolerability. JAMA Neurol 2015;72:88–95.
55. Forsaa EB, Larsen JP, Wentzel-Larsen T, et al. A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol 2010;67:996–1001.
56. Holroyd S, Currie L, Wooten GF. Prospective study of hallucinations and delusions in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2001;70:734–8.
57. Fénelon G, Alves G. Epidemiology of psychosis in Parkinson’s disease. J Neurol Sci 2010;289:12–7.
58. Mack J, Rabins P, Anderson K, et al. Prevalence of psychotic symptoms in a community-based Parkinson disease sample. Am J Geriatr Psychiatry 2012;20:123–32.
59. Ravina B, Marder K, Fernandez HH, et al. Diagnostic criteria for psychosis in Parkinson’s disease: Report of an NINDS, NIMH Work Group. Mov Disord 2007;22:1061–8.
60. Diederich NJ, Fénelon G, Stebbins G, Goetz CG. Hallucinations in Parkinson disease. Nat Rev Neurol 2009;5:331–42.
61. Gallagher DA, Parkkinen L, O’Sullivan SS, et al. Testing an aetiological model of visual hallucinations in Parkinson’s disease. Brain 2011;134:3299–309.
62. Barnes J, Boubert L, Harris J, et al. Reality monitoring and visual hallucinations in Parkinson’s disease. Neuropsychologia 2003:41:565–74.
63. Kurita A, Murakami M, Takagi S, et al. Visual hallucinations and altered visual information processing in Parkinson disease and dementia with Lewy bodies. Mov Disord 2010;25:167–71.
64. Stebbins GT, Goetz CG, Carrillo MC, et al. Altered cortical visual processing in PD with hallucinations: an fMRI study. Neurology 2004;63:1409–16.
65. Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: A retrospective autopsy study. Lancet Neurol 2005:4:605–10.
66. Biglan KM, Holloway RG, McDermott MP, Richard IH. Risk factors for somnolence, edema, and hallucinations in early Parkinson disease. Neurology 2007;69:187–95.
67. Kiziltan G, Ozekmekci S, Ertan S, et al. Relationship between age and subtypes of psychotic symptoms in Parkinson’s disease. J Neurol 2007;254:448–52.
68. Ecker D, Unrath A, Kassubek J, Sabolek M. Dopamine agonists and their risk to induce psychotic episodes in Parkinson’s disease: a case-control study. BMC Neurol 2009;9:23.
69. Huot P, Johnston TH, Darr T, et al. Increased 5-HT2A receptors in the temporal cortex of Parkinsonian patients with visual hallucinations. Mov Disord 2010;25:1399–408.
70. Ballanger B, Strafella AP, van Eimeren T, et al. Serotonin 2A receptors and visual hallucinations in Parkinson disease. Arch. Neurol 2010;67:416–21.
71. Boecker H, Ceballos-Baumann O, Volk D, et al. Metabolic alterations in patients with Parkinson disease and visual hallucinations. Arch Neurol 2007;64:984–8.
72. Sanchez-Castaneda C, Rene R, Ramirez-Ruiz B, et al. Frontal and associative visual areas related to visual hallucinations in dementia with Lewy bodies and Parkinson’s disease with dementia. Mov Disord 2010;25:615–22.
73. Goetz CG, Stebbins GT. Risk factors for nursing home placement in advanced Parkinson’s disease. Neurology 1993;43:2227–9.
74. Friedman JH. Parkinson disease psychosis: update. Behav.Neurol 2013;27:469–77.
75. Pollak P, Tison F, Rascol O, et al. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 2004;75:689–95.
76. Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis. N Engl J Med 1999;340:757–63.
77. Sobow T. Parkinson’s disease-related visual hallucinations unresponsive to atypical antipsychotics treated with cholinesterase inhibitors: a case series. Neurol Neurochir Pol 2007;41:276–9.
78. Fabbrini G, Barbanti P, Aurilia C, et al. Donepezil in the treatment of hallucinations and delusions in Parkinson’s disease. Neurol Sci 2002;23:41–3.
79. Bullock R, Cameron A, Hospital WH. Rivastigmine for the treatment of dementia and visual hallucinations associated with Parkinson’s disease: a case series. Curr Med Res Opin 2002;18,:258–64.
80. Cummings J, Isaacson S, Mills R, et al. Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial. Lancet 2014;383:533–40.
81. L. Ishihara L, and C. Brayne C. A systematic review of depression and mental illness preceding Parkinson’s disease. Acta Neurol Scand 2006;113:211–20.
82. Reijnders JSAM, Ehrt U, Weber WEJ, et al. A systematic review of prevalence studies of depression in Parkinson’s disease. Mov Disord 2008;23:183–9.
83. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992:55:377–82.
84. Müller B, Assmus J, Herlofson K, et al. Importance of motor vs. non-motor symptoms for health-related quality of life in early Parkinson’s disease. Parkinsonism Relat Disord 2013;19:1027–32.
85. Ravina B, Camicioli R, Como PG, et al. The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–7.
86. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148–58.
87. Marsh L. Depression and Parkinson’s disease: current knowledge. Curr. Neurol. Neurosci. Rep 2013; 13:409.
88. Starkstein SE, Merello M, Jorge R, et al. A validation study of depressive syndromes in Parkinson’s disease. Mov Disord 2008;23,:538–46.
89. Williams JR, Hirsch ES, Anderson K, et al. A comparison of nine scales to detect depression in Parkinson disease: which scale to use? Neurology 2012;78:998–1006.
90. Nazem S, Siderowf AD, Duda JE, et al. Suicidal and death ideation in Parkinson’s disease. Mov Disord 2008;23:1573–9.
91. Weintraub D, Duda JE, Carlson K, et al. Suicide ideation and behaviours after STN and GPi DBS surgery for Parkinson’s disease: results from a randomised, controlled trial. J Neurol Neurosurg Psychiatry 2013;84:1113–8.
92. Aarsland D, Påhlhagen S, Ballard CG, et al. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol 2011;8:35–47.
93. Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson’s disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol 1990;3:3–9.
94. Witjas T, Kaphan E, Azulay JP, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology 2002:59:408–13.
95. Nissenbaum H, Quinn NP, Brown RG, et al. Mood swings associated with the ‘on-off’ phenomenon in Parkinson’s disease. Psychol Med 1987;17:899–904.
96. Maricle RA, Nutt JG, Carter JH. Mood and anxiety fluctuation in Parkinson’s disease associated with levodopa infusion: preliminary findings. Mov Disord 1995;10:329–32.
97. Barone P, Poewe W, Albrecht S, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010;9:573–80.
98. Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008;23:850–7.
99. Richard IH, McDermott MP, Kurlan R, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology 2012;78:1229–36.
100. Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009;72:886–92.
101. Seppi K, Weintraub D, Coelho M, et al. The Movement Disorder Society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord 2011;26 Suppl 3:S42–80.
102. Skapinakis P, Bakola E, Salanti G, et al. Efficacy and acceptability of selective serotonin reuptake inhibitors for the treatment of depression in Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. BMC Neurol 2010;10:49.
103. Richard IH, Kurlan R, Tanner C, et al. Serotonin syndrome and the combined use of deprenyl and an antidepressant in Parkinson’s disease. Parkinson Study Group. Neurology 1997;48:1070–7.
104. Montgomery EB, Panisset M. Retrospective statistical analysis of the incidence of serotonin toxicity in patients taking rasagiline and anti-depressants in clinical trials. Mov Disord 2009;24:359.
105. Bailine S, Kremen N, Kohen I, et al. Bitemporal electroconvulsive therapy for depression in a Parkinson disease patient with a deep-brain stimulator. J ECT 2008;24:171–2.
106. Faber R, Trimble MR. Electroconvulsive therapy in Parkinson’s disease and other movement disorders. Mov Disord 1991;6:293–303.
107. da Silva TM, Munhoz RP, Alvarez C, et al. Depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled pilot study of omega-3 fatty-acid supplementation. J Affect Disord 2008;111:351–9.
108. Pal E, Nagy F, Aschermann Z, et al. The impact of left prefrontal repetitive transcranial magnetic stimulation on depression in Parkinson’s disease: a randomized, double-blind, placebo-controlled study. Mov Disord 2010;25:2311–7.
109. Dobkin RD, Menza M, Allen LA, et al. Cognitive-behavioral therapy for depression in Parkinson’s disease: a randomized, controlled trial. Am J Psychiatry 2011;168:1066–74.
110. Stein MB, Heuser IJ, Juncos JL, Uhde TW. Anxiety disorders in patients with parkinson’s disease. Am J Psychiatry 1990;147:217–20.
111. Nuti A, Ceravolo R, Piccinni A, et al. Psychiatric comorbidity in a population of Parkinson’s disease patients. Eur J Neurol 2004;11:315–20.
112. Marinus J, Leentjens AFG, Visser M, et al. Evaluation of the Hospital Anxiety and Depression Scale in patients With Parkinson’s disease. Clin Neuropharmacol 2002:25:318–24.
113. Dissanayaka NNW, White E, O’Sullivan JD, et al. The clinical spectrum of anxiety in Parkinson’s disease. Mov Disord 2014;29:967–75.
114. Dissanayaka NNW, Sellbach A, Matheson S, et al. Anxiety disorders in Parkinson’s disease: prevalence and risk factors. Mov Disord 2010;25:838–45.
115. Leentjens AFG, Dujardin K, Pontone GM, et al. The Parkinson anxiety scale (PAS): Development and validation of a new anxiety scale. Mov Disord 2014;29:1035–43.
116. Maricle RA, Nutt JG, Valentine RJ, Carter JH. Dose-response relationship of levodopa with mood and anxiety in fluctuating Parkinson’s disease: a double-blind, placebo-controlled study. Neurology 1995;45:1757–60.
117. Erdal KJ. Depression and anxiety in persons with Parkinson’s disease with and without ‘on–off’ phenomena. J Clin Psychol Med Settings 2001:8:293–9.
118. Leentjens AFG, Dujardin K, Marsh L, et al. Symptomatology and markers of anxiety disorders in Parkinson’s disease: a cross-sectional study. Mov. Disord 2011;26:484–92.
119. Solla P, Cannas A, Floris GL, et al. Behavioral, neuropsychiatric and cognitive disorders in Parkinson’s disease patients with and without motor complications. Prog Neuropsychopharmacol Biol Psychiatry 2011;35:1009–13.
120. E. Stefanova E, L. Ziropadja L, M. Petrović M, et al. Screening for anxiety symptoms in Parkinson disease: a cross-sectional study. J Geriatr Psychiatry Neurol 2013;26:34–40.
121. Jacob EL, Gatto NM, Thompson A, et al. Occurrence of depression and anxiety prior to Parkinson’s disease. Parkinsonism Relat Disord 2010;16:576–81.
122. Craig LH, Svircev A, Haber M, Juncos JL. Controlled pilot study of the effects of neuromuscular therapy in patients with Parkinson’s disease. Mov Disord 2006;21:2127–33.
123. Marino L, Friedman JH. Letter to the editor: Successful use of electroconvulsive therapy for refractory anxiety in Parkinson’s disease. Int J Neurosci 2013;123:70–1.