User login
A 37-year-old white man presented to his primary care provider’s office for follow-up after a visit to the emergency department (ED). He had been evaluated at a local ED a week earlier for atypical chest pain and left arm pain. At the ED, blood work was done, along with an ECG, chest x-ray, and chest CT scan, but the results of these evaluations were not available during his initial primary care visit. On discharge from the ED, he was told that his heart was not the cause of his pain and that he should follow up with his primary care provider.
In the office, the patient reported that for the past several months he had been experiencing pain in his left arm when doing heavy or continuous physical labor; he noted that his job as a laborer required vigorous activity. Rest seemed to make his pain go away. He denied pain in the right arm or being awakened by the pain at night. Review of systems was unremarkable, and medical and surgical history was negative.
On physical exam, inspection of his torso and upper and lower extremities did not reveal any apparent abnormalities. Left shoulder and neck exams were normal. Cardiac auscultation was unremarkable, but palpation of the left upper extremity revealed neither a brachial, radial, nor ulnar pulse. Pulses in his right upper extremity were within normal limits. No bruits were appreciated over the carotids or either subclavian artery. Basic Doppler ultrasound over the left upper extremity at the brachial, radial, and ulnar sites showed symmetrical Doppler sounds. The remainder of his exam was unremarkable.
The patient’s ED documents and imaging results were received later in the day, after his office visit. The ECG, blood work results, and chest x-ray were normal. The chest CT results showed no evidence of pulmonary embolism. The radiologist did note mild narrowing at the left subclavian artery secondary to nonspecific surrounding soft tissue, which was noted to possibly represent intramural hemorrhage or atherosclerotic changes. No intimal flap was identified.
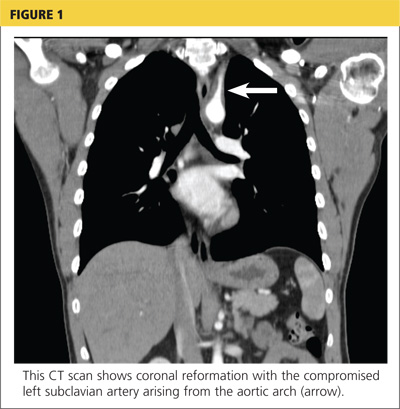
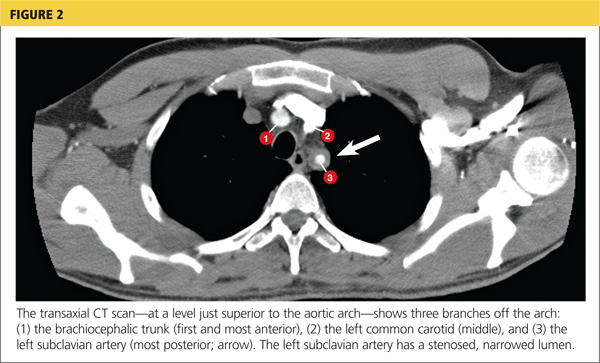
Because the diagnosis remained unclear, the patient was asked to bring the disc containing his chest CT images to the office. The radiologist, who was informed about the patient’s history and exam findings by phone, reviewed the CT images and felt there were changes surrounding the three branches off the aortic arch suggestive of inflammation, in addition to the stenosis at the left subclavian artery (see Figure 1 and Figure 2).
Based on the radiologist’s interpretation, additional lab tests were ordered. A complete blood count, comprehensive metabolic panel, prothrombin time/partial thromboplastin time, and lipid panel all yielded results within normal limits. Erythrocyte sedimentation rate (ESR) was 12 mm/h (reference range, 0 to 15 mm/h) and C-reactive protein (CRP) level was 4.9 mg/dL (reference range, 0.1 to 4.9 mg/dL). These laboratory results were essentially unremarkable, and therefore made his diagnosis more elusive.
The patient was referred to a vascular surgeon because of his immediate symptoms. The surgeon performed a thoracic outlet study in which Doppler waveform analysis of the left brachial, radial, and ulnar arteries of the thoracic outlet were analyzed during range-of-motion testing. Results suggested the possibility of thoracic outlet syndrome involving the left upper extremity, with significant baseline arterial insufficiency. A CT angiogram showed critical stenosis of the left subclavian artery and arterial wall thickening. Inflammatory changes were noted as well, and concern for “an inflammatory vasculitis” was described on the CT angiogram. The patient underwent left carotid-to-axillary bypass grafting, after which his left arm pain improved.
Following surgery, the patient returned to the primary care office for evaluation. Although the surgery was successful, the diagnosis was still not clear, requiring additional medical evaluation. The physical exam showed normal pulses in his left upper extremity. Lab tests revealed an elevated ESR of 54 mm/h and a CRP level of 4 mg/dL (reference range, 0.1 to 0.8 mg/dL; a different lab testing site was used, which accounts for the different reference range). In light of the patient’s lab test results, premature arterial vascular disease, and imaging studies suggesting inflammation, Takayasu arteritis (TA) was arrived at as a working diagnosis.
The patient was referred to a rheumatologist, who ordered a repeat ESR and CRP, antineutrophil cytoplasmic antibodies, and a magnetic resonance angiography study of the right brachial artery and major aortic branches to rule out other types of arteritis. Based on the test results, the patient was diagnosed with TA. He was placed on high-dose corticosteroid therapy (prednisone 60 mg/d). Methotrexate 10 mg/wk po was added three months after initiation of the prednisone.
Since being diagnosed with TA, the patient has presented with complaints related to the adverse effects of high-dose corticosteroids (ie, insomnia, weight gain, elevated blood pressure).
Continued on the next page >>
DISCUSSION
The first description of TA is credited to Japanese ophthalmologist Mikito Takayasu, who in 1908 described a wreathlike arteriovenous anastomosis around the optic disc of a 21-year-old woman who had experienced acute vision loss.1-3 Much earlier, in 1761, Italian anatomist Giovanni Battista Morgagni described large-vessel aneurysms and stenosis on a postmortem exam of a 40-year-old woman.2,4 However, TA was not formally labeled a disease until 1975.
TA is a chronic large vessel vasculitis of unknown origin, mainly involving the aorta and its primary branches: the left common carotid, brachiocephalic, and left subclavian arteries. Ongoing inflammation of affected vessels causes fibrotic changes, stenosis, and eventual occlusion and may lead to aneurysm formation.5,6 TA is rare, with an annual incidence in North America of 2.6 cases per million population.6 It occurs most frequently in Asian countries but has been reported in a wide range of ethnic groups.5,7 TA has been characterized as a disease of young women: Between 80% and 97% of patients are women,6,8 and the average age at diagnosis is 25 to 30.8-10
The process of vascular injury in TA begins with inflammation in the vasa vasorum of the aortic vessels. This inflammation, thought to be triggered by an as-yet-unknown antigen, leads to an initial inflammatory cellular infiltration of the aortic media and adventitia; the infiltrate is comprised predominantly of macrophages and T cells.5,9 Inflammatory infiltration causes myointimal proliferation, thickening of the blood vessel wall, and eventual luminal stenosis.5 Cytokines, interleukin 6, interferon , and other chemokines released by infiltrating inflammatory cells within the injured tissue also contribute to the inflammatory response and tissue damage.5,11
Histologically, granulomatous inflammation and giant cells are found in the media.12,13 Destruction of the elastic lamina and the muscular media results in the aneurysmal dilation seen in TA, while dense scarring and continued inflammation of the arterial vasculature results in arterial stenosis.12
Continued on the next page >>
CLINICAL PRESENTATION
Presentation of TA varies widely and can range from asymptomatic disease identified by pulse deficits or impalpable pulses to severe neurologic impairment. The early or prepulseless phase of TA is characterized by inflammatory changes.14 Signs and symptoms are frequently vague and nonspecific, particularly in this early phase, when fatigue, weight loss, and low-grade fever may be seen.12 Headache is another common symptom at the time of disease onset.5
In the later or chronic phase of the disease, individuals will begin to demonstrate signs and symptoms of vascular insufficiency.14 More common physical signs reflect the underlying arterial occlusive disease and include diminished or absent arterial pulses, asymmetrical arm blood pressures, bruits, extremity claudication, and hypertension.5,9,10 Hypertension, generally reflecting renal artery stenosis,10 is present in approximately 40% of cases in the United States and Europe.5,7,15 Neurologic features secondary to hypertension or ischemia affect more than half of patients; in addition to headache, these may include dizziness, syncope, vertigo, transient ischemic attack, and stroke.5
TA can also present with eye, lung, and skin manifestations; however, these features are less common. Although ocular involvement, including amaurosis fugax, has been reported in up to 26% of patients in TA series,5,7,16 permanent loss of vision in North American patients is uncommon.5,7 Pulmonary involvement affecting the large- or medium-sized pulmonary arteries has been reported to occur in approximately 55% of cases5; however, there is uncertainty regarding the prevalence of angiographically demonstrated pulmonary artery involvement, as studies have reported rates ranging from 14.3% to 70%.9,17-20 Pulmonary involvement is often asymptomatic, but features can include dyspnea, cough, and chest pain.5 Skin lesions are seen in up to 28% of cases, most commonly erythema nodosum, erythema induratum, tuberculoidlike eruptions, pyoderma gangrenosum, and cutaneous signs of necrotizing or granulomatous vasculitis.5,21
Continued on the next page >>
DIAGNOSIS
The American College of Rheumatology (ACR) has developed classification criteria for the diagnosis of TA.22 The presence of three or more of the six criteria (age of onset ≤ 40, claudication of the extremities, decreased brachial artery pulse, > 10 mm Hg difference in systolic blood pressure between the arms, bruit over subclavian arteries or aorta, and arteriographic abnormalities) yields a sensitivity of 90.5% and a specificity of 97.8%. Although the ACR classification remains the most widely applied for TA, a limitation of its diagnostic criteria is its failure to distinguish patients with early nonocclusive disease.23
In 1988, the Ishikawa classification criteria were developed, with a modified version subsequently published in 1996.23 Considered superior to the original Ishikawa and ACR criteria based on its application in 106 patients with angiographically proven TA, the modified version has a reported sensitivity and specificity of 92.5% and 95%, respectively.23
With the modified Ishikawa diagnostic criteria, the presence of two major or one major and two or four minor criteria suggests a high probability of TA. The three major criteria consist of lesions of the left mid-subclavian artery and the right mid-subclavian artery and characteristic signs and symptoms of at least 1 mo duration. The 10 minor criteria are high ESR (> 20 mm/h); carotid artery tenderness; hypertension; aortic regurgitation or annuloaortic ectasia; and lesions of pulmonary artery, left mid-common carotid, distal brachiocephalic trunk, descending thoracic aorta, abdominal aorta, and coronary artery.
The diagnosis of TA is based on recognition of clinical findings suggestive of large-vessel vasculitis. Imaging of the arterial tree with CT, MRI, or angiography also demonstrates findings consistent with TA, typically including early-onset vascular wall thickening/enhancement.24 Late imaging studies may reveal arterial stenoses, occlusions, and aneurysms.
Several types of imaging modalities have been used in the diagnosis and management of TA, each with strengths and limitations. Traditional angiography is invasive and requires an arterial puncture. Large doses of radiation are used, exposing the patient to iodinated contrast material, which may be dangerous in patients with poor renal function. However, the primary advantage of traditional angiography is that it allows for interventions such as stent placement and/or angioplasty to be performed.24 Findings on angiography often include long, smooth, tapered stenoses ranging from mild to severe or frank occlusions, as well as collateral vessels or the subclavian steal phenomenon.24
CT imaging is very useful for assessing thickening of the arterial wall. In early TA disease, evaluation of vessel wall thickness may be identified prior to frank stenosis of the artery(s).24 The spectrum of findings on CT angiography includes stenoses; occlusions; aneurysms; and concentric arterial wall thickening affecting the aorta and its branches, the pulmonary arteries, and occasionally the coronary arteries.24
MRI does not require the use of iodinated contrast, nor is there radiation exposure. MRI also has the advantage of evaluating arterial wall thickening, which is often present prior to stenosis (similar to CT imaging).24 Findings of TA on MRI include mural thrombi, signal alterations within and surrounding inflamed vessels, fusiform vascular dilation, thickened aortic valvular cusps, multifocal stenoses, and concentric thickening of the aortic wall.24
Laboratory testing is neither specific nor sensitive. Hoffman and Ahmed studied multiple serologic tests and found that no test reliably distinguishes between patients with active TA and healthy volunteers.25 Increases in the acute phase reactants (ESR and CRP) support the presence of an underlying inflammatory process, and these laboratory tests may be useful in disease monitoring. The ESR and CRP often do not correlate with systemic symptoms or disease progression but are used in conjunction with the clinical exam and serial imaging to gauge treatment success and to monitor disease progression.5,25 Biopsy material typically is not available in the initial diagnosis of TA, but histologic examination at the time of a surgery or procedure is often undertaken to confirm the diagnosis.5
The differential diagnosis of TA includes connective tissue diseases associated with the formation of multiple aneurysms, such as Marfan syndrome and Ehlers-Danlos syndrome.5 However, these diseases do not manifest with large vessel stenosis, the hallmark of TA. Infections known to cause aneurysms of the aorta should also be considered; these include bacterial, fungal, syphilitic, mycotic, and mycobacterial pathogens.5 Blood cultures are used to rule out bacterial agents. Rapid plasma reagin (RPR) and venereal disease research laboratory tests (VDRL) will identify a syphilitic etiology. Fungal cultures or fungal serology will help to rule out a mycotic pathogen.
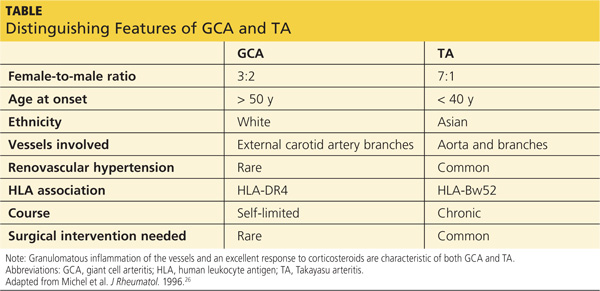
Autoimmune diseases that can mimic TA include Behçet’s disease, Cogan syndrome, the spondyloarthropathies, and systemic lupus erythematosus. These diseases are not associated with stenosis of large vessels, which differentiates them from TA.5 Giant cell (temporal) arteritis (GCA) may present very similarly to TA, as both diseases affect large arteries.12 The table provides distinguishing features of TA and GCA.26
Continued on the next page >>
TREATMENT
Active phase TA is initially treated with high-dose glucocorticoid therapy (prednisone or methylprednisolone). Typical prednisone doses are 0.5 to 1 mg/kg/d.5 Clinical improvement is seen in almost all patients with glucocorticoid therapy,6,10,23 but relapse is common when prednisone is tapered to less than 20 mg/d.5 The corticosteroid dose is gradually tapered depending on patient response. Common side effects of corticosteroids may include weight gain, elevations in blood glucose, insomnia, increased infection risk, osteoporosis, and slowing of wound healing.
Because nearly half of all patients treated with glucocorticoids alone demonstrate chronic active disease, immunosuppressive therapies are almost always used concomitantly.27 Immune-suppressing drugs that may be used include methotrexate (15 to 25 mg/wk), azathioprine (2 mg/kg/d), and cyclophosphamide (1 to 2 mg/kg/d orally).5,28 Tumor necrosis factor (TNF)–blocking agents used to treat TA include etanercept, infliximab, or adalimumab.28,29 Adverse effects associated with immunosuppressive therapies and TNF-blocking agents include an increased risk for infection(s) and malignancy, bone marrow suppression, and hepatitis B reactivation. Although data are limited on anti-TNF agents, this class of drug has shown promise when used in conjunction with corticosteroids.28
In one open-label study by Hoffman and colleagues, remission rates with methotrexate plus steroids were 81%. Relapse occurred in 44% of study participants when the steroid dose was tapered or decreased to near discontinuation.27 More recently, in an uncontrolled study series involving 15 TA patients from India who were treated with azathioprine plus steroids, remission was achieved following 12 weeks of therapy. Angiographically, there was no progression of arterial disease after one year.30
Surgical and endovascular procedures used to return blood flow in stenotic or occluded vessels include synthetic or autologous vessel bypass, endarterectomy, and percutaneous transluminal angioplasty.5 When aortic insufficiency is present, aortic root replacement or repair is undertaken.5 These procedures are performed by vascular or cardiovascular surgeons and interventional radiologists. Rheumatologists are the medical specialists most involved in the direct care and management of TA patients. Cardiologists are sometimes consulted as well.
Continued on the next page >>
PROGNOSIS
Disability is common in TA. In a National Institutes of Health cohort study, 74% of TA patients reported experiencing functional effects from their disease, and 47% were fully disabled.2,8 In their retrospective review of 107 cases of TA, Lupi-Herrera and colleagues reported a 14% mortality rate.31 Half the deaths in this study were attributed to congestive heart failure (CHF). A cohort study in India that included 88 patients with TA reported cumulative 5- and 10-year survival rates of 91% and 84%, respectively. Of the 10 deaths in this cohort, four were due to CHF.2,32
CONCLUSION
Signs and symptoms of rheumatologic diseases such as TA are often vague, and diagnosis may prove difficult and elusive. Repeat office visits at short intervals may prove to be helpful in making the diagnosis. Referral for radiology and/or rheumatology consultation (face-to-face, if possible) is often necessary.
In cases such as this, completing a personal review of documents and test results done elsewhere, particularly ED/inpatient hospital data, is necessary; relying on the patient’s word that “they told me everything was fine” is insufficient. Clinicians should implement a system that works best for obtaining test results and other documents, follow their instincts, and if the correct diagnosis is not arrived at immediately, keep looking.
References >>
REFERENCES
1. Takayasu M. A case with peculiar changes of the retinal central vessels. Acta Soc Ophthalmol Jpn. 1908;12:554-555.
2. Maksimowicz-McKinnon K, Hoffman GS. Takayasu arteritis: what is the long-term prognosis? Rheum Dis Clin North Am. 2007;33:777-786.
3. Numano F. The story of Takayasu arteritis. Rheumatology. 2002;41:103-106.
4. Morgagni GB. De sedibus et causis morborum per anatomen indagatis.
(Letter 30).1761. Article 12.
5. Hernandez-Rodriguez J, Maksimowicz-McKinnon K, Hoffman GS. Takayasu’s arteritis. In: Carey WD, ed. Current Clinical Medicine. 2nd ed. Philadelphia: Saunders Elsevier; 2010:1195-1199.
6. Hall S, Barr W, Lie JT, et al. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore). 1985;64:89-99.
7. Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56:1000-1009.
8. Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994:120:919-929.
9. Gornik HL, Creager MA. Aortic diseases: aortitis. Circulation. 2008;117:
3039-3051.
10. Mwipatayi BP, Jeffery PC, Beningfield SJ, et al. Takayasu arteritis: clinical features and management: report of 272 cases. ANZ J Surg. 2005;75:110-117.
11. Noris M. Pathogenesis of Takayasu’s arteritis. J Nephrol. 2001;14:506-513.
12. Hunder GG, Stone JH, Ramirez MP. Clinical features and diagnosis of Takayasu arteritis (2013). www.uptodate.com/contents/clinical-features-and-
diagnosis-of-takayasu-arteritis. Accessed March 24, 2014.
13. Nasu T. Takayasu’s truncoarteritis. Pulseless disease or aortitis syndrome. Acta Pathol Jpn. 1982;32 (suppl 1):117.
14. Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol. 2002;55:481-486.
15. Vanoli M, Daina E, Salvarani C, et al. Takayasu’s arteritis: a study of 104 Italian patients. Arthritis Rheum. 2005;53:100-107.
16. Chun YS, Park SJ, Chung H, Lee J. The clinical and ocular manifestations of Takayasu’s arteritis. Retina. 2001;21:132-140.
17. Liu YQ, Jin BL, Ling J. Pulmonary artery involvement in aortoarteritis: an angiographic study. Cardiovasc Intervent Radiol. 1994;17:2-6.
18. Yamada I, Shibuya H, Matsubara O, et al. Pulmonary artery disease in Takayasu’s arteritis: angiographic findings. AJR Am J Roentgenol. 1992;159:
263-269.
19. Sharma S, Kamalakar T, Rajani M, et al. The incidence and patterns of pulmonary artery involvement in Takayasu’s arteritis. Clin Radiol. 1990;42:177-181.
20. He NS, Liu F, Wu EH, et al. Pulmonary artery involvement in aorto-arteritis: an analysis of DSA. Chin Med J (Engl). 1990;103:666-672.
21. Werfel T, Kuipers JG, Zeidler H, et al. Cutaneous manifestations of Takayasu arteritis. Acta Derm Venereol. 1996;76:496-497.
22. Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129-1134.
23. Andrews J, Mason JC. Takayasu’s arteritis—recent advances in imaging offer promise. Rheumatology (Oxford). 2007;46:6-15.
24. Gotway MB, Araoz PA, Macedo TA, et al. Imaging findings in Takayasu’s arteritis. AJR Am J Roentgenol. 2005;184:1945-1950.
25. Hoffman GS, Ahmed AE. Surrogate markers of disease activity in patients with Takayasu arteritis. A preliminary report from The International Network for the Study of the Systemic Vasculitides (INSSYS). Int J Cardiol. 1998;66 (suppl 1):S191-S194.
26. Michel BA, Arend WP, Hunder GG. Clinical differentiation between giant cell (temporal) arteritis and Takayasu’s arteritis. J Rheumatol. 1996;23:106-111.
27. Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578-582.
28. Hunder GG, Stone JH, Ramirez MP. Treatment of Takayasu arteritis (2013). www.uptodate.com/contents/treatment-of-takayasu-arteritis. Accessed March 24, 2014.
29. Schmidt J, Kerman TA, Bacani AK, et al. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: experience from a referral center with long-term followup. Arthritis Care Res (Hoboken). 2012;64:1079-1083.
30. Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol. 2003;30:1793-1798.
31. Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, et al. Takayasu’s arteritis: clinical study of 107 cases. Am Heart J. 1977;93:94-103.
32. Subramanyan R, Joy J, Balakrishnan KG. Natural history of aortoarteritis (Takayasu’s disease). Circulation. 1989;80:429-437.
A 37-year-old white man presented to his primary care provider’s office for follow-up after a visit to the emergency department (ED). He had been evaluated at a local ED a week earlier for atypical chest pain and left arm pain. At the ED, blood work was done, along with an ECG, chest x-ray, and chest CT scan, but the results of these evaluations were not available during his initial primary care visit. On discharge from the ED, he was told that his heart was not the cause of his pain and that he should follow up with his primary care provider.
In the office, the patient reported that for the past several months he had been experiencing pain in his left arm when doing heavy or continuous physical labor; he noted that his job as a laborer required vigorous activity. Rest seemed to make his pain go away. He denied pain in the right arm or being awakened by the pain at night. Review of systems was unremarkable, and medical and surgical history was negative.
On physical exam, inspection of his torso and upper and lower extremities did not reveal any apparent abnormalities. Left shoulder and neck exams were normal. Cardiac auscultation was unremarkable, but palpation of the left upper extremity revealed neither a brachial, radial, nor ulnar pulse. Pulses in his right upper extremity were within normal limits. No bruits were appreciated over the carotids or either subclavian artery. Basic Doppler ultrasound over the left upper extremity at the brachial, radial, and ulnar sites showed symmetrical Doppler sounds. The remainder of his exam was unremarkable.
The patient’s ED documents and imaging results were received later in the day, after his office visit. The ECG, blood work results, and chest x-ray were normal. The chest CT results showed no evidence of pulmonary embolism. The radiologist did note mild narrowing at the left subclavian artery secondary to nonspecific surrounding soft tissue, which was noted to possibly represent intramural hemorrhage or atherosclerotic changes. No intimal flap was identified.
Because the diagnosis remained unclear, the patient was asked to bring the disc containing his chest CT images to the office. The radiologist, who was informed about the patient’s history and exam findings by phone, reviewed the CT images and felt there were changes surrounding the three branches off the aortic arch suggestive of inflammation, in addition to the stenosis at the left subclavian artery (see Figure 1 and Figure 2).
Based on the radiologist’s interpretation, additional lab tests were ordered. A complete blood count, comprehensive metabolic panel, prothrombin time/partial thromboplastin time, and lipid panel all yielded results within normal limits. Erythrocyte sedimentation rate (ESR) was 12 mm/h (reference range, 0 to 15 mm/h) and C-reactive protein (CRP) level was 4.9 mg/dL (reference range, 0.1 to 4.9 mg/dL). These laboratory results were essentially unremarkable, and therefore made his diagnosis more elusive.
The patient was referred to a vascular surgeon because of his immediate symptoms. The surgeon performed a thoracic outlet study in which Doppler waveform analysis of the left brachial, radial, and ulnar arteries of the thoracic outlet were analyzed during range-of-motion testing. Results suggested the possibility of thoracic outlet syndrome involving the left upper extremity, with significant baseline arterial insufficiency. A CT angiogram showed critical stenosis of the left subclavian artery and arterial wall thickening. Inflammatory changes were noted as well, and concern for “an inflammatory vasculitis” was described on the CT angiogram. The patient underwent left carotid-to-axillary bypass grafting, after which his left arm pain improved.
Following surgery, the patient returned to the primary care office for evaluation. Although the surgery was successful, the diagnosis was still not clear, requiring additional medical evaluation. The physical exam showed normal pulses in his left upper extremity. Lab tests revealed an elevated ESR of 54 mm/h and a CRP level of 4 mg/dL (reference range, 0.1 to 0.8 mg/dL; a different lab testing site was used, which accounts for the different reference range). In light of the patient’s lab test results, premature arterial vascular disease, and imaging studies suggesting inflammation, Takayasu arteritis (TA) was arrived at as a working diagnosis.
The patient was referred to a rheumatologist, who ordered a repeat ESR and CRP, antineutrophil cytoplasmic antibodies, and a magnetic resonance angiography study of the right brachial artery and major aortic branches to rule out other types of arteritis. Based on the test results, the patient was diagnosed with TA. He was placed on high-dose corticosteroid therapy (prednisone 60 mg/d). Methotrexate 10 mg/wk po was added three months after initiation of the prednisone.
Since being diagnosed with TA, the patient has presented with complaints related to the adverse effects of high-dose corticosteroids (ie, insomnia, weight gain, elevated blood pressure).
Continued on the next page >>
DISCUSSION
The first description of TA is credited to Japanese ophthalmologist Mikito Takayasu, who in 1908 described a wreathlike arteriovenous anastomosis around the optic disc of a 21-year-old woman who had experienced acute vision loss.1-3 Much earlier, in 1761, Italian anatomist Giovanni Battista Morgagni described large-vessel aneurysms and stenosis on a postmortem exam of a 40-year-old woman.2,4 However, TA was not formally labeled a disease until 1975.
TA is a chronic large vessel vasculitis of unknown origin, mainly involving the aorta and its primary branches: the left common carotid, brachiocephalic, and left subclavian arteries. Ongoing inflammation of affected vessels causes fibrotic changes, stenosis, and eventual occlusion and may lead to aneurysm formation.5,6 TA is rare, with an annual incidence in North America of 2.6 cases per million population.6 It occurs most frequently in Asian countries but has been reported in a wide range of ethnic groups.5,7 TA has been characterized as a disease of young women: Between 80% and 97% of patients are women,6,8 and the average age at diagnosis is 25 to 30.8-10
The process of vascular injury in TA begins with inflammation in the vasa vasorum of the aortic vessels. This inflammation, thought to be triggered by an as-yet-unknown antigen, leads to an initial inflammatory cellular infiltration of the aortic media and adventitia; the infiltrate is comprised predominantly of macrophages and T cells.5,9 Inflammatory infiltration causes myointimal proliferation, thickening of the blood vessel wall, and eventual luminal stenosis.5 Cytokines, interleukin 6, interferon , and other chemokines released by infiltrating inflammatory cells within the injured tissue also contribute to the inflammatory response and tissue damage.5,11
Histologically, granulomatous inflammation and giant cells are found in the media.12,13 Destruction of the elastic lamina and the muscular media results in the aneurysmal dilation seen in TA, while dense scarring and continued inflammation of the arterial vasculature results in arterial stenosis.12
Continued on the next page >>
CLINICAL PRESENTATION
Presentation of TA varies widely and can range from asymptomatic disease identified by pulse deficits or impalpable pulses to severe neurologic impairment. The early or prepulseless phase of TA is characterized by inflammatory changes.14 Signs and symptoms are frequently vague and nonspecific, particularly in this early phase, when fatigue, weight loss, and low-grade fever may be seen.12 Headache is another common symptom at the time of disease onset.5
In the later or chronic phase of the disease, individuals will begin to demonstrate signs and symptoms of vascular insufficiency.14 More common physical signs reflect the underlying arterial occlusive disease and include diminished or absent arterial pulses, asymmetrical arm blood pressures, bruits, extremity claudication, and hypertension.5,9,10 Hypertension, generally reflecting renal artery stenosis,10 is present in approximately 40% of cases in the United States and Europe.5,7,15 Neurologic features secondary to hypertension or ischemia affect more than half of patients; in addition to headache, these may include dizziness, syncope, vertigo, transient ischemic attack, and stroke.5
TA can also present with eye, lung, and skin manifestations; however, these features are less common. Although ocular involvement, including amaurosis fugax, has been reported in up to 26% of patients in TA series,5,7,16 permanent loss of vision in North American patients is uncommon.5,7 Pulmonary involvement affecting the large- or medium-sized pulmonary arteries has been reported to occur in approximately 55% of cases5; however, there is uncertainty regarding the prevalence of angiographically demonstrated pulmonary artery involvement, as studies have reported rates ranging from 14.3% to 70%.9,17-20 Pulmonary involvement is often asymptomatic, but features can include dyspnea, cough, and chest pain.5 Skin lesions are seen in up to 28% of cases, most commonly erythema nodosum, erythema induratum, tuberculoidlike eruptions, pyoderma gangrenosum, and cutaneous signs of necrotizing or granulomatous vasculitis.5,21
Continued on the next page >>
DIAGNOSIS
The American College of Rheumatology (ACR) has developed classification criteria for the diagnosis of TA.22 The presence of three or more of the six criteria (age of onset ≤ 40, claudication of the extremities, decreased brachial artery pulse, > 10 mm Hg difference in systolic blood pressure between the arms, bruit over subclavian arteries or aorta, and arteriographic abnormalities) yields a sensitivity of 90.5% and a specificity of 97.8%. Although the ACR classification remains the most widely applied for TA, a limitation of its diagnostic criteria is its failure to distinguish patients with early nonocclusive disease.23
In 1988, the Ishikawa classification criteria were developed, with a modified version subsequently published in 1996.23 Considered superior to the original Ishikawa and ACR criteria based on its application in 106 patients with angiographically proven TA, the modified version has a reported sensitivity and specificity of 92.5% and 95%, respectively.23
With the modified Ishikawa diagnostic criteria, the presence of two major or one major and two or four minor criteria suggests a high probability of TA. The three major criteria consist of lesions of the left mid-subclavian artery and the right mid-subclavian artery and characteristic signs and symptoms of at least 1 mo duration. The 10 minor criteria are high ESR (> 20 mm/h); carotid artery tenderness; hypertension; aortic regurgitation or annuloaortic ectasia; and lesions of pulmonary artery, left mid-common carotid, distal brachiocephalic trunk, descending thoracic aorta, abdominal aorta, and coronary artery.
The diagnosis of TA is based on recognition of clinical findings suggestive of large-vessel vasculitis. Imaging of the arterial tree with CT, MRI, or angiography also demonstrates findings consistent with TA, typically including early-onset vascular wall thickening/enhancement.24 Late imaging studies may reveal arterial stenoses, occlusions, and aneurysms.
Several types of imaging modalities have been used in the diagnosis and management of TA, each with strengths and limitations. Traditional angiography is invasive and requires an arterial puncture. Large doses of radiation are used, exposing the patient to iodinated contrast material, which may be dangerous in patients with poor renal function. However, the primary advantage of traditional angiography is that it allows for interventions such as stent placement and/or angioplasty to be performed.24 Findings on angiography often include long, smooth, tapered stenoses ranging from mild to severe or frank occlusions, as well as collateral vessels or the subclavian steal phenomenon.24
CT imaging is very useful for assessing thickening of the arterial wall. In early TA disease, evaluation of vessel wall thickness may be identified prior to frank stenosis of the artery(s).24 The spectrum of findings on CT angiography includes stenoses; occlusions; aneurysms; and concentric arterial wall thickening affecting the aorta and its branches, the pulmonary arteries, and occasionally the coronary arteries.24
MRI does not require the use of iodinated contrast, nor is there radiation exposure. MRI also has the advantage of evaluating arterial wall thickening, which is often present prior to stenosis (similar to CT imaging).24 Findings of TA on MRI include mural thrombi, signal alterations within and surrounding inflamed vessels, fusiform vascular dilation, thickened aortic valvular cusps, multifocal stenoses, and concentric thickening of the aortic wall.24
Laboratory testing is neither specific nor sensitive. Hoffman and Ahmed studied multiple serologic tests and found that no test reliably distinguishes between patients with active TA and healthy volunteers.25 Increases in the acute phase reactants (ESR and CRP) support the presence of an underlying inflammatory process, and these laboratory tests may be useful in disease monitoring. The ESR and CRP often do not correlate with systemic symptoms or disease progression but are used in conjunction with the clinical exam and serial imaging to gauge treatment success and to monitor disease progression.5,25 Biopsy material typically is not available in the initial diagnosis of TA, but histologic examination at the time of a surgery or procedure is often undertaken to confirm the diagnosis.5
The differential diagnosis of TA includes connective tissue diseases associated with the formation of multiple aneurysms, such as Marfan syndrome and Ehlers-Danlos syndrome.5 However, these diseases do not manifest with large vessel stenosis, the hallmark of TA. Infections known to cause aneurysms of the aorta should also be considered; these include bacterial, fungal, syphilitic, mycotic, and mycobacterial pathogens.5 Blood cultures are used to rule out bacterial agents. Rapid plasma reagin (RPR) and venereal disease research laboratory tests (VDRL) will identify a syphilitic etiology. Fungal cultures or fungal serology will help to rule out a mycotic pathogen.
Autoimmune diseases that can mimic TA include Behçet’s disease, Cogan syndrome, the spondyloarthropathies, and systemic lupus erythematosus. These diseases are not associated with stenosis of large vessels, which differentiates them from TA.5 Giant cell (temporal) arteritis (GCA) may present very similarly to TA, as both diseases affect large arteries.12 The table provides distinguishing features of TA and GCA.26
Continued on the next page >>
TREATMENT
Active phase TA is initially treated with high-dose glucocorticoid therapy (prednisone or methylprednisolone). Typical prednisone doses are 0.5 to 1 mg/kg/d.5 Clinical improvement is seen in almost all patients with glucocorticoid therapy,6,10,23 but relapse is common when prednisone is tapered to less than 20 mg/d.5 The corticosteroid dose is gradually tapered depending on patient response. Common side effects of corticosteroids may include weight gain, elevations in blood glucose, insomnia, increased infection risk, osteoporosis, and slowing of wound healing.
Because nearly half of all patients treated with glucocorticoids alone demonstrate chronic active disease, immunosuppressive therapies are almost always used concomitantly.27 Immune-suppressing drugs that may be used include methotrexate (15 to 25 mg/wk), azathioprine (2 mg/kg/d), and cyclophosphamide (1 to 2 mg/kg/d orally).5,28 Tumor necrosis factor (TNF)–blocking agents used to treat TA include etanercept, infliximab, or adalimumab.28,29 Adverse effects associated with immunosuppressive therapies and TNF-blocking agents include an increased risk for infection(s) and malignancy, bone marrow suppression, and hepatitis B reactivation. Although data are limited on anti-TNF agents, this class of drug has shown promise when used in conjunction with corticosteroids.28
In one open-label study by Hoffman and colleagues, remission rates with methotrexate plus steroids were 81%. Relapse occurred in 44% of study participants when the steroid dose was tapered or decreased to near discontinuation.27 More recently, in an uncontrolled study series involving 15 TA patients from India who were treated with azathioprine plus steroids, remission was achieved following 12 weeks of therapy. Angiographically, there was no progression of arterial disease after one year.30
Surgical and endovascular procedures used to return blood flow in stenotic or occluded vessels include synthetic or autologous vessel bypass, endarterectomy, and percutaneous transluminal angioplasty.5 When aortic insufficiency is present, aortic root replacement or repair is undertaken.5 These procedures are performed by vascular or cardiovascular surgeons and interventional radiologists. Rheumatologists are the medical specialists most involved in the direct care and management of TA patients. Cardiologists are sometimes consulted as well.
Continued on the next page >>
PROGNOSIS
Disability is common in TA. In a National Institutes of Health cohort study, 74% of TA patients reported experiencing functional effects from their disease, and 47% were fully disabled.2,8 In their retrospective review of 107 cases of TA, Lupi-Herrera and colleagues reported a 14% mortality rate.31 Half the deaths in this study were attributed to congestive heart failure (CHF). A cohort study in India that included 88 patients with TA reported cumulative 5- and 10-year survival rates of 91% and 84%, respectively. Of the 10 deaths in this cohort, four were due to CHF.2,32
CONCLUSION
Signs and symptoms of rheumatologic diseases such as TA are often vague, and diagnosis may prove difficult and elusive. Repeat office visits at short intervals may prove to be helpful in making the diagnosis. Referral for radiology and/or rheumatology consultation (face-to-face, if possible) is often necessary.
In cases such as this, completing a personal review of documents and test results done elsewhere, particularly ED/inpatient hospital data, is necessary; relying on the patient’s word that “they told me everything was fine” is insufficient. Clinicians should implement a system that works best for obtaining test results and other documents, follow their instincts, and if the correct diagnosis is not arrived at immediately, keep looking.
References >>
REFERENCES
1. Takayasu M. A case with peculiar changes of the retinal central vessels. Acta Soc Ophthalmol Jpn. 1908;12:554-555.
2. Maksimowicz-McKinnon K, Hoffman GS. Takayasu arteritis: what is the long-term prognosis? Rheum Dis Clin North Am. 2007;33:777-786.
3. Numano F. The story of Takayasu arteritis. Rheumatology. 2002;41:103-106.
4. Morgagni GB. De sedibus et causis morborum per anatomen indagatis.
(Letter 30).1761. Article 12.
5. Hernandez-Rodriguez J, Maksimowicz-McKinnon K, Hoffman GS. Takayasu’s arteritis. In: Carey WD, ed. Current Clinical Medicine. 2nd ed. Philadelphia: Saunders Elsevier; 2010:1195-1199.
6. Hall S, Barr W, Lie JT, et al. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore). 1985;64:89-99.
7. Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56:1000-1009.
8. Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994:120:919-929.
9. Gornik HL, Creager MA. Aortic diseases: aortitis. Circulation. 2008;117:
3039-3051.
10. Mwipatayi BP, Jeffery PC, Beningfield SJ, et al. Takayasu arteritis: clinical features and management: report of 272 cases. ANZ J Surg. 2005;75:110-117.
11. Noris M. Pathogenesis of Takayasu’s arteritis. J Nephrol. 2001;14:506-513.
12. Hunder GG, Stone JH, Ramirez MP. Clinical features and diagnosis of Takayasu arteritis (2013). www.uptodate.com/contents/clinical-features-and-
diagnosis-of-takayasu-arteritis. Accessed March 24, 2014.
13. Nasu T. Takayasu’s truncoarteritis. Pulseless disease or aortitis syndrome. Acta Pathol Jpn. 1982;32 (suppl 1):117.
14. Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol. 2002;55:481-486.
15. Vanoli M, Daina E, Salvarani C, et al. Takayasu’s arteritis: a study of 104 Italian patients. Arthritis Rheum. 2005;53:100-107.
16. Chun YS, Park SJ, Chung H, Lee J. The clinical and ocular manifestations of Takayasu’s arteritis. Retina. 2001;21:132-140.
17. Liu YQ, Jin BL, Ling J. Pulmonary artery involvement in aortoarteritis: an angiographic study. Cardiovasc Intervent Radiol. 1994;17:2-6.
18. Yamada I, Shibuya H, Matsubara O, et al. Pulmonary artery disease in Takayasu’s arteritis: angiographic findings. AJR Am J Roentgenol. 1992;159:
263-269.
19. Sharma S, Kamalakar T, Rajani M, et al. The incidence and patterns of pulmonary artery involvement in Takayasu’s arteritis. Clin Radiol. 1990;42:177-181.
20. He NS, Liu F, Wu EH, et al. Pulmonary artery involvement in aorto-arteritis: an analysis of DSA. Chin Med J (Engl). 1990;103:666-672.
21. Werfel T, Kuipers JG, Zeidler H, et al. Cutaneous manifestations of Takayasu arteritis. Acta Derm Venereol. 1996;76:496-497.
22. Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129-1134.
23. Andrews J, Mason JC. Takayasu’s arteritis—recent advances in imaging offer promise. Rheumatology (Oxford). 2007;46:6-15.
24. Gotway MB, Araoz PA, Macedo TA, et al. Imaging findings in Takayasu’s arteritis. AJR Am J Roentgenol. 2005;184:1945-1950.
25. Hoffman GS, Ahmed AE. Surrogate markers of disease activity in patients with Takayasu arteritis. A preliminary report from The International Network for the Study of the Systemic Vasculitides (INSSYS). Int J Cardiol. 1998;66 (suppl 1):S191-S194.
26. Michel BA, Arend WP, Hunder GG. Clinical differentiation between giant cell (temporal) arteritis and Takayasu’s arteritis. J Rheumatol. 1996;23:106-111.
27. Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578-582.
28. Hunder GG, Stone JH, Ramirez MP. Treatment of Takayasu arteritis (2013). www.uptodate.com/contents/treatment-of-takayasu-arteritis. Accessed March 24, 2014.
29. Schmidt J, Kerman TA, Bacani AK, et al. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: experience from a referral center with long-term followup. Arthritis Care Res (Hoboken). 2012;64:1079-1083.
30. Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol. 2003;30:1793-1798.
31. Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, et al. Takayasu’s arteritis: clinical study of 107 cases. Am Heart J. 1977;93:94-103.
32. Subramanyan R, Joy J, Balakrishnan KG. Natural history of aortoarteritis (Takayasu’s disease). Circulation. 1989;80:429-437.
A 37-year-old white man presented to his primary care provider’s office for follow-up after a visit to the emergency department (ED). He had been evaluated at a local ED a week earlier for atypical chest pain and left arm pain. At the ED, blood work was done, along with an ECG, chest x-ray, and chest CT scan, but the results of these evaluations were not available during his initial primary care visit. On discharge from the ED, he was told that his heart was not the cause of his pain and that he should follow up with his primary care provider.
In the office, the patient reported that for the past several months he had been experiencing pain in his left arm when doing heavy or continuous physical labor; he noted that his job as a laborer required vigorous activity. Rest seemed to make his pain go away. He denied pain in the right arm or being awakened by the pain at night. Review of systems was unremarkable, and medical and surgical history was negative.
On physical exam, inspection of his torso and upper and lower extremities did not reveal any apparent abnormalities. Left shoulder and neck exams were normal. Cardiac auscultation was unremarkable, but palpation of the left upper extremity revealed neither a brachial, radial, nor ulnar pulse. Pulses in his right upper extremity were within normal limits. No bruits were appreciated over the carotids or either subclavian artery. Basic Doppler ultrasound over the left upper extremity at the brachial, radial, and ulnar sites showed symmetrical Doppler sounds. The remainder of his exam was unremarkable.
The patient’s ED documents and imaging results were received later in the day, after his office visit. The ECG, blood work results, and chest x-ray were normal. The chest CT results showed no evidence of pulmonary embolism. The radiologist did note mild narrowing at the left subclavian artery secondary to nonspecific surrounding soft tissue, which was noted to possibly represent intramural hemorrhage or atherosclerotic changes. No intimal flap was identified.
Because the diagnosis remained unclear, the patient was asked to bring the disc containing his chest CT images to the office. The radiologist, who was informed about the patient’s history and exam findings by phone, reviewed the CT images and felt there were changes surrounding the three branches off the aortic arch suggestive of inflammation, in addition to the stenosis at the left subclavian artery (see Figure 1 and Figure 2).
Based on the radiologist’s interpretation, additional lab tests were ordered. A complete blood count, comprehensive metabolic panel, prothrombin time/partial thromboplastin time, and lipid panel all yielded results within normal limits. Erythrocyte sedimentation rate (ESR) was 12 mm/h (reference range, 0 to 15 mm/h) and C-reactive protein (CRP) level was 4.9 mg/dL (reference range, 0.1 to 4.9 mg/dL). These laboratory results were essentially unremarkable, and therefore made his diagnosis more elusive.
The patient was referred to a vascular surgeon because of his immediate symptoms. The surgeon performed a thoracic outlet study in which Doppler waveform analysis of the left brachial, radial, and ulnar arteries of the thoracic outlet were analyzed during range-of-motion testing. Results suggested the possibility of thoracic outlet syndrome involving the left upper extremity, with significant baseline arterial insufficiency. A CT angiogram showed critical stenosis of the left subclavian artery and arterial wall thickening. Inflammatory changes were noted as well, and concern for “an inflammatory vasculitis” was described on the CT angiogram. The patient underwent left carotid-to-axillary bypass grafting, after which his left arm pain improved.
Following surgery, the patient returned to the primary care office for evaluation. Although the surgery was successful, the diagnosis was still not clear, requiring additional medical evaluation. The physical exam showed normal pulses in his left upper extremity. Lab tests revealed an elevated ESR of 54 mm/h and a CRP level of 4 mg/dL (reference range, 0.1 to 0.8 mg/dL; a different lab testing site was used, which accounts for the different reference range). In light of the patient’s lab test results, premature arterial vascular disease, and imaging studies suggesting inflammation, Takayasu arteritis (TA) was arrived at as a working diagnosis.
The patient was referred to a rheumatologist, who ordered a repeat ESR and CRP, antineutrophil cytoplasmic antibodies, and a magnetic resonance angiography study of the right brachial artery and major aortic branches to rule out other types of arteritis. Based on the test results, the patient was diagnosed with TA. He was placed on high-dose corticosteroid therapy (prednisone 60 mg/d). Methotrexate 10 mg/wk po was added three months after initiation of the prednisone.
Since being diagnosed with TA, the patient has presented with complaints related to the adverse effects of high-dose corticosteroids (ie, insomnia, weight gain, elevated blood pressure).
Continued on the next page >>
DISCUSSION
The first description of TA is credited to Japanese ophthalmologist Mikito Takayasu, who in 1908 described a wreathlike arteriovenous anastomosis around the optic disc of a 21-year-old woman who had experienced acute vision loss.1-3 Much earlier, in 1761, Italian anatomist Giovanni Battista Morgagni described large-vessel aneurysms and stenosis on a postmortem exam of a 40-year-old woman.2,4 However, TA was not formally labeled a disease until 1975.
TA is a chronic large vessel vasculitis of unknown origin, mainly involving the aorta and its primary branches: the left common carotid, brachiocephalic, and left subclavian arteries. Ongoing inflammation of affected vessels causes fibrotic changes, stenosis, and eventual occlusion and may lead to aneurysm formation.5,6 TA is rare, with an annual incidence in North America of 2.6 cases per million population.6 It occurs most frequently in Asian countries but has been reported in a wide range of ethnic groups.5,7 TA has been characterized as a disease of young women: Between 80% and 97% of patients are women,6,8 and the average age at diagnosis is 25 to 30.8-10
The process of vascular injury in TA begins with inflammation in the vasa vasorum of the aortic vessels. This inflammation, thought to be triggered by an as-yet-unknown antigen, leads to an initial inflammatory cellular infiltration of the aortic media and adventitia; the infiltrate is comprised predominantly of macrophages and T cells.5,9 Inflammatory infiltration causes myointimal proliferation, thickening of the blood vessel wall, and eventual luminal stenosis.5 Cytokines, interleukin 6, interferon , and other chemokines released by infiltrating inflammatory cells within the injured tissue also contribute to the inflammatory response and tissue damage.5,11
Histologically, granulomatous inflammation and giant cells are found in the media.12,13 Destruction of the elastic lamina and the muscular media results in the aneurysmal dilation seen in TA, while dense scarring and continued inflammation of the arterial vasculature results in arterial stenosis.12
Continued on the next page >>
CLINICAL PRESENTATION
Presentation of TA varies widely and can range from asymptomatic disease identified by pulse deficits or impalpable pulses to severe neurologic impairment. The early or prepulseless phase of TA is characterized by inflammatory changes.14 Signs and symptoms are frequently vague and nonspecific, particularly in this early phase, when fatigue, weight loss, and low-grade fever may be seen.12 Headache is another common symptom at the time of disease onset.5
In the later or chronic phase of the disease, individuals will begin to demonstrate signs and symptoms of vascular insufficiency.14 More common physical signs reflect the underlying arterial occlusive disease and include diminished or absent arterial pulses, asymmetrical arm blood pressures, bruits, extremity claudication, and hypertension.5,9,10 Hypertension, generally reflecting renal artery stenosis,10 is present in approximately 40% of cases in the United States and Europe.5,7,15 Neurologic features secondary to hypertension or ischemia affect more than half of patients; in addition to headache, these may include dizziness, syncope, vertigo, transient ischemic attack, and stroke.5
TA can also present with eye, lung, and skin manifestations; however, these features are less common. Although ocular involvement, including amaurosis fugax, has been reported in up to 26% of patients in TA series,5,7,16 permanent loss of vision in North American patients is uncommon.5,7 Pulmonary involvement affecting the large- or medium-sized pulmonary arteries has been reported to occur in approximately 55% of cases5; however, there is uncertainty regarding the prevalence of angiographically demonstrated pulmonary artery involvement, as studies have reported rates ranging from 14.3% to 70%.9,17-20 Pulmonary involvement is often asymptomatic, but features can include dyspnea, cough, and chest pain.5 Skin lesions are seen in up to 28% of cases, most commonly erythema nodosum, erythema induratum, tuberculoidlike eruptions, pyoderma gangrenosum, and cutaneous signs of necrotizing or granulomatous vasculitis.5,21
Continued on the next page >>
DIAGNOSIS
The American College of Rheumatology (ACR) has developed classification criteria for the diagnosis of TA.22 The presence of three or more of the six criteria (age of onset ≤ 40, claudication of the extremities, decreased brachial artery pulse, > 10 mm Hg difference in systolic blood pressure between the arms, bruit over subclavian arteries or aorta, and arteriographic abnormalities) yields a sensitivity of 90.5% and a specificity of 97.8%. Although the ACR classification remains the most widely applied for TA, a limitation of its diagnostic criteria is its failure to distinguish patients with early nonocclusive disease.23
In 1988, the Ishikawa classification criteria were developed, with a modified version subsequently published in 1996.23 Considered superior to the original Ishikawa and ACR criteria based on its application in 106 patients with angiographically proven TA, the modified version has a reported sensitivity and specificity of 92.5% and 95%, respectively.23
With the modified Ishikawa diagnostic criteria, the presence of two major or one major and two or four minor criteria suggests a high probability of TA. The three major criteria consist of lesions of the left mid-subclavian artery and the right mid-subclavian artery and characteristic signs and symptoms of at least 1 mo duration. The 10 minor criteria are high ESR (> 20 mm/h); carotid artery tenderness; hypertension; aortic regurgitation or annuloaortic ectasia; and lesions of pulmonary artery, left mid-common carotid, distal brachiocephalic trunk, descending thoracic aorta, abdominal aorta, and coronary artery.
The diagnosis of TA is based on recognition of clinical findings suggestive of large-vessel vasculitis. Imaging of the arterial tree with CT, MRI, or angiography also demonstrates findings consistent with TA, typically including early-onset vascular wall thickening/enhancement.24 Late imaging studies may reveal arterial stenoses, occlusions, and aneurysms.
Several types of imaging modalities have been used in the diagnosis and management of TA, each with strengths and limitations. Traditional angiography is invasive and requires an arterial puncture. Large doses of radiation are used, exposing the patient to iodinated contrast material, which may be dangerous in patients with poor renal function. However, the primary advantage of traditional angiography is that it allows for interventions such as stent placement and/or angioplasty to be performed.24 Findings on angiography often include long, smooth, tapered stenoses ranging from mild to severe or frank occlusions, as well as collateral vessels or the subclavian steal phenomenon.24
CT imaging is very useful for assessing thickening of the arterial wall. In early TA disease, evaluation of vessel wall thickness may be identified prior to frank stenosis of the artery(s).24 The spectrum of findings on CT angiography includes stenoses; occlusions; aneurysms; and concentric arterial wall thickening affecting the aorta and its branches, the pulmonary arteries, and occasionally the coronary arteries.24
MRI does not require the use of iodinated contrast, nor is there radiation exposure. MRI also has the advantage of evaluating arterial wall thickening, which is often present prior to stenosis (similar to CT imaging).24 Findings of TA on MRI include mural thrombi, signal alterations within and surrounding inflamed vessels, fusiform vascular dilation, thickened aortic valvular cusps, multifocal stenoses, and concentric thickening of the aortic wall.24
Laboratory testing is neither specific nor sensitive. Hoffman and Ahmed studied multiple serologic tests and found that no test reliably distinguishes between patients with active TA and healthy volunteers.25 Increases in the acute phase reactants (ESR and CRP) support the presence of an underlying inflammatory process, and these laboratory tests may be useful in disease monitoring. The ESR and CRP often do not correlate with systemic symptoms or disease progression but are used in conjunction with the clinical exam and serial imaging to gauge treatment success and to monitor disease progression.5,25 Biopsy material typically is not available in the initial diagnosis of TA, but histologic examination at the time of a surgery or procedure is often undertaken to confirm the diagnosis.5
The differential diagnosis of TA includes connective tissue diseases associated with the formation of multiple aneurysms, such as Marfan syndrome and Ehlers-Danlos syndrome.5 However, these diseases do not manifest with large vessel stenosis, the hallmark of TA. Infections known to cause aneurysms of the aorta should also be considered; these include bacterial, fungal, syphilitic, mycotic, and mycobacterial pathogens.5 Blood cultures are used to rule out bacterial agents. Rapid plasma reagin (RPR) and venereal disease research laboratory tests (VDRL) will identify a syphilitic etiology. Fungal cultures or fungal serology will help to rule out a mycotic pathogen.
Autoimmune diseases that can mimic TA include Behçet’s disease, Cogan syndrome, the spondyloarthropathies, and systemic lupus erythematosus. These diseases are not associated with stenosis of large vessels, which differentiates them from TA.5 Giant cell (temporal) arteritis (GCA) may present very similarly to TA, as both diseases affect large arteries.12 The table provides distinguishing features of TA and GCA.26
Continued on the next page >>
TREATMENT
Active phase TA is initially treated with high-dose glucocorticoid therapy (prednisone or methylprednisolone). Typical prednisone doses are 0.5 to 1 mg/kg/d.5 Clinical improvement is seen in almost all patients with glucocorticoid therapy,6,10,23 but relapse is common when prednisone is tapered to less than 20 mg/d.5 The corticosteroid dose is gradually tapered depending on patient response. Common side effects of corticosteroids may include weight gain, elevations in blood glucose, insomnia, increased infection risk, osteoporosis, and slowing of wound healing.
Because nearly half of all patients treated with glucocorticoids alone demonstrate chronic active disease, immunosuppressive therapies are almost always used concomitantly.27 Immune-suppressing drugs that may be used include methotrexate (15 to 25 mg/wk), azathioprine (2 mg/kg/d), and cyclophosphamide (1 to 2 mg/kg/d orally).5,28 Tumor necrosis factor (TNF)–blocking agents used to treat TA include etanercept, infliximab, or adalimumab.28,29 Adverse effects associated with immunosuppressive therapies and TNF-blocking agents include an increased risk for infection(s) and malignancy, bone marrow suppression, and hepatitis B reactivation. Although data are limited on anti-TNF agents, this class of drug has shown promise when used in conjunction with corticosteroids.28
In one open-label study by Hoffman and colleagues, remission rates with methotrexate plus steroids were 81%. Relapse occurred in 44% of study participants when the steroid dose was tapered or decreased to near discontinuation.27 More recently, in an uncontrolled study series involving 15 TA patients from India who were treated with azathioprine plus steroids, remission was achieved following 12 weeks of therapy. Angiographically, there was no progression of arterial disease after one year.30
Surgical and endovascular procedures used to return blood flow in stenotic or occluded vessels include synthetic or autologous vessel bypass, endarterectomy, and percutaneous transluminal angioplasty.5 When aortic insufficiency is present, aortic root replacement or repair is undertaken.5 These procedures are performed by vascular or cardiovascular surgeons and interventional radiologists. Rheumatologists are the medical specialists most involved in the direct care and management of TA patients. Cardiologists are sometimes consulted as well.
Continued on the next page >>
PROGNOSIS
Disability is common in TA. In a National Institutes of Health cohort study, 74% of TA patients reported experiencing functional effects from their disease, and 47% were fully disabled.2,8 In their retrospective review of 107 cases of TA, Lupi-Herrera and colleagues reported a 14% mortality rate.31 Half the deaths in this study were attributed to congestive heart failure (CHF). A cohort study in India that included 88 patients with TA reported cumulative 5- and 10-year survival rates of 91% and 84%, respectively. Of the 10 deaths in this cohort, four were due to CHF.2,32
CONCLUSION
Signs and symptoms of rheumatologic diseases such as TA are often vague, and diagnosis may prove difficult and elusive. Repeat office visits at short intervals may prove to be helpful in making the diagnosis. Referral for radiology and/or rheumatology consultation (face-to-face, if possible) is often necessary.
In cases such as this, completing a personal review of documents and test results done elsewhere, particularly ED/inpatient hospital data, is necessary; relying on the patient’s word that “they told me everything was fine” is insufficient. Clinicians should implement a system that works best for obtaining test results and other documents, follow their instincts, and if the correct diagnosis is not arrived at immediately, keep looking.
References >>
REFERENCES
1. Takayasu M. A case with peculiar changes of the retinal central vessels. Acta Soc Ophthalmol Jpn. 1908;12:554-555.
2. Maksimowicz-McKinnon K, Hoffman GS. Takayasu arteritis: what is the long-term prognosis? Rheum Dis Clin North Am. 2007;33:777-786.
3. Numano F. The story of Takayasu arteritis. Rheumatology. 2002;41:103-106.
4. Morgagni GB. De sedibus et causis morborum per anatomen indagatis.
(Letter 30).1761. Article 12.
5. Hernandez-Rodriguez J, Maksimowicz-McKinnon K, Hoffman GS. Takayasu’s arteritis. In: Carey WD, ed. Current Clinical Medicine. 2nd ed. Philadelphia: Saunders Elsevier; 2010:1195-1199.
6. Hall S, Barr W, Lie JT, et al. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore). 1985;64:89-99.
7. Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56:1000-1009.
8. Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994:120:919-929.
9. Gornik HL, Creager MA. Aortic diseases: aortitis. Circulation. 2008;117:
3039-3051.
10. Mwipatayi BP, Jeffery PC, Beningfield SJ, et al. Takayasu arteritis: clinical features and management: report of 272 cases. ANZ J Surg. 2005;75:110-117.
11. Noris M. Pathogenesis of Takayasu’s arteritis. J Nephrol. 2001;14:506-513.
12. Hunder GG, Stone JH, Ramirez MP. Clinical features and diagnosis of Takayasu arteritis (2013). www.uptodate.com/contents/clinical-features-and-
diagnosis-of-takayasu-arteritis. Accessed March 24, 2014.
13. Nasu T. Takayasu’s truncoarteritis. Pulseless disease or aortitis syndrome. Acta Pathol Jpn. 1982;32 (suppl 1):117.
14. Johnston SL, Lock RJ, Gompels MM. Takayasu arteritis: a review. J Clin Pathol. 2002;55:481-486.
15. Vanoli M, Daina E, Salvarani C, et al. Takayasu’s arteritis: a study of 104 Italian patients. Arthritis Rheum. 2005;53:100-107.
16. Chun YS, Park SJ, Chung H, Lee J. The clinical and ocular manifestations of Takayasu’s arteritis. Retina. 2001;21:132-140.
17. Liu YQ, Jin BL, Ling J. Pulmonary artery involvement in aortoarteritis: an angiographic study. Cardiovasc Intervent Radiol. 1994;17:2-6.
18. Yamada I, Shibuya H, Matsubara O, et al. Pulmonary artery disease in Takayasu’s arteritis: angiographic findings. AJR Am J Roentgenol. 1992;159:
263-269.
19. Sharma S, Kamalakar T, Rajani M, et al. The incidence and patterns of pulmonary artery involvement in Takayasu’s arteritis. Clin Radiol. 1990;42:177-181.
20. He NS, Liu F, Wu EH, et al. Pulmonary artery involvement in aorto-arteritis: an analysis of DSA. Chin Med J (Engl). 1990;103:666-672.
21. Werfel T, Kuipers JG, Zeidler H, et al. Cutaneous manifestations of Takayasu arteritis. Acta Derm Venereol. 1996;76:496-497.
22. Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129-1134.
23. Andrews J, Mason JC. Takayasu’s arteritis—recent advances in imaging offer promise. Rheumatology (Oxford). 2007;46:6-15.
24. Gotway MB, Araoz PA, Macedo TA, et al. Imaging findings in Takayasu’s arteritis. AJR Am J Roentgenol. 2005;184:1945-1950.
25. Hoffman GS, Ahmed AE. Surrogate markers of disease activity in patients with Takayasu arteritis. A preliminary report from The International Network for the Study of the Systemic Vasculitides (INSSYS). Int J Cardiol. 1998;66 (suppl 1):S191-S194.
26. Michel BA, Arend WP, Hunder GG. Clinical differentiation between giant cell (temporal) arteritis and Takayasu’s arteritis. J Rheumatol. 1996;23:106-111.
27. Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578-582.
28. Hunder GG, Stone JH, Ramirez MP. Treatment of Takayasu arteritis (2013). www.uptodate.com/contents/treatment-of-takayasu-arteritis. Accessed March 24, 2014.
29. Schmidt J, Kerman TA, Bacani AK, et al. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: experience from a referral center with long-term followup. Arthritis Care Res (Hoboken). 2012;64:1079-1083.
30. Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol. 2003;30:1793-1798.
31. Lupi-Herrera E, Sanchez-Torres G, Marcushamer J, et al. Takayasu’s arteritis: clinical study of 107 cases. Am Heart J. 1977;93:94-103.
32. Subramanyan R, Joy J, Balakrishnan KG. Natural history of aortoarteritis (Takayasu’s disease). Circulation. 1989;80:429-437.