User login
Enhancing Cosmetic and Functional Improvement of Recalcitrant Nail Lichen Planus With Resin Nail
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
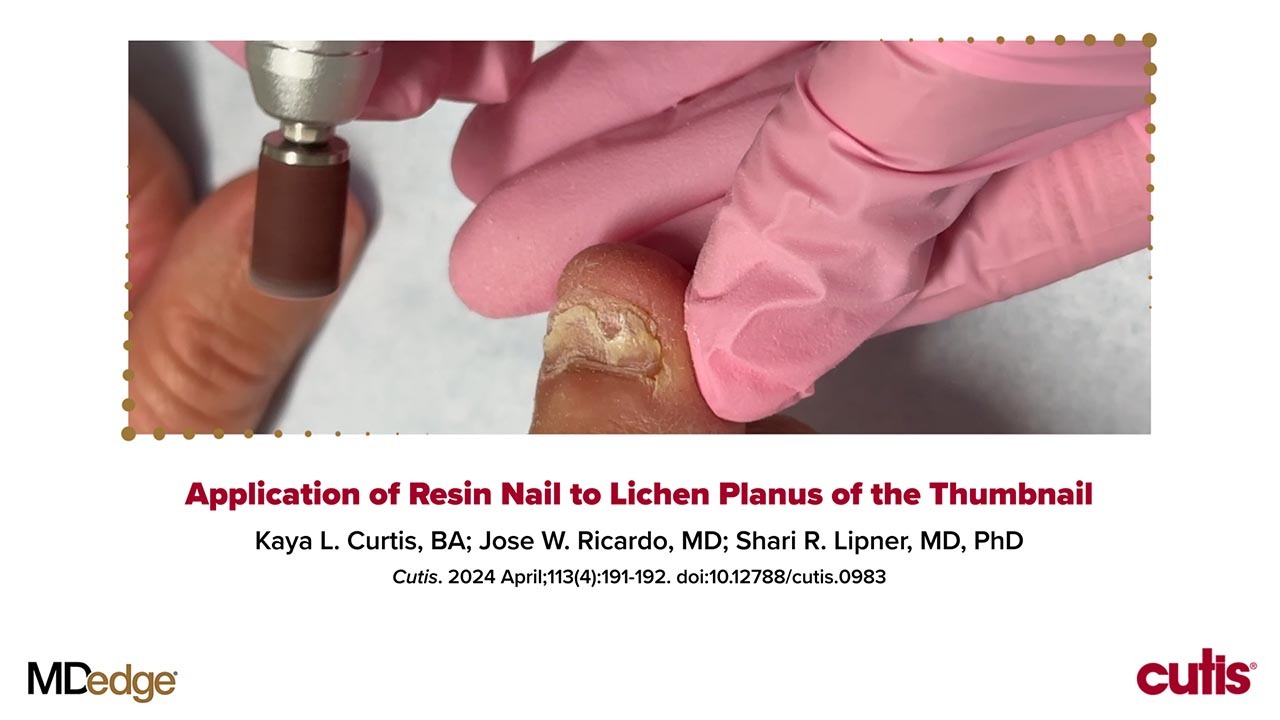
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.

Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.
Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
Practice Gap
Lichen planus (LP)—a chronic inflammatory disorder affecting the nails—is prevalent in 10% to 15% of patients and is more common in the fingernails than toenails. Clinical manifestation includes longitudinal ridges, nail plate atrophy, and splitting, which all contribute to cosmetic disfigurement and difficulty with functionality. Quality of life and daily activities may be impacted profoundly.1 First-line therapies include intralesional and systemic corticosteroids; however, efficacy is limited and recurrence is common.1,2 Lichen planus is one of the few conditions that may cause permanent and debilitating nail loss.
Tools
A resin nail can be used to improve cosmetic appearance and functionality in patients with recalcitrant nail LP. The composite resin creates a flexible nonporous nail and allows the underlying natural nail to grow. Application of resin nails has been used for toenail onychodystrophies to improve cosmesis and functionality but has not been reported for fingernails. The resin typically lasts 6 to 8 weeks on toenails.
The Technique
Application of a resin nail involves several steps (see video online). First, the affected nail should be debrided and a bonding agent applied. Next, multiple layers of resin are applied until the patient’s desired thickness is achieved (typically 2 layers), followed by a sealing agent. Finally, the nail is cured with UV light. We recommend applying sunscreen to the hand(s) prior to curing with UV light. The liquid resin allows the nail to be customized to the patient’s desired length and shape. The overall procedure takes approximately 20 minutes for a single nail.
We applied resin nail to the thumbnail of a 46-year-old woman with recalcitrant isolated nail LP of 7 years’ duration (Figure). She previously had difficulties performing everyday activities, and the resin improved her functionality. She also was pleased with the cosmetic appearance. After 2 weeks, the resin started falling off with corresponding natural nail growth. The patient denied any adverse events.
Practice Implications
Resin nail application may serve as a temporary solution to improve cosmesis and functionality in patients with recalcitrant nail LP. As shown in our patient, the resin may fall off faster on the fingernails than the toenails, likely because of the faster growth rate of fingernails and more frequent exposure from daily activities. Further studies of resin nail application for the fingernails are needed to establish duration in patients with varying levels of activity (eg, washing dishes, woodworking).
Because the resin nail may be removed easily at any time, resin nail application does not interfere with treatments such as intralesional steroid injections. For patients using a topical medication regimen, the resin nail may be applied slightly distal to the cuticle so that the medication can still be applied by the proximal nail fold of the underlying natural nail.
The resin nail should be kept short and removed after 2 to 4 weeks for the fingernails and 6 to 8 weeks for the toenails to examine the underlying natural nail. Patients may go about their daily activities with the resin nail, including applying nail polish to the resin nail, bathing, and swimming. Resin nail application may complement medical treatments and improve quality of life for patients with nail LP.
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056

Evaluating the Cost Burden of Alopecia Areata Treatment: A Comprehensive Review for Dermatologists
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
 treatment.")
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29


Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29
Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
Alopecia areata (AA) affects 4.5 million individuals in the United States, with 66% younger than 30 years.1,2 Inflammation causes hair loss in well-circumscribed, nonscarring patches on the body with a predilection for the scalp.3-6 The disease can devastate a patient’s self-esteem, in turn reducing quality of life.1,7 Alopecia areata is an autoimmune T-cell–mediated disease in which hair follicles lose their immune privilege.8-10 Several specific mechanisms in the cytokine interactions between T cells and the hair follicle have been discovered, revealing the Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway as pivotal in the pathogenesis of the disease and leading to the use of JAK inhibitors for treatment.11
There is no cure for AA, and the condition is managed with prolonged medical treatments and cosmetic therapies.2 Although some patients may be able to manage the annual cost, the cumulative cost of AA treatment can be burdensome.12 This cumulative cost may increase if newer, potentially expensive treatments become the standard of care. Patients with AA report dipping into their savings (41.3%) and cutting back on food or clothing expenses (33.9%) to account for the cost of alopecia treatment. Although prior estimates of the annual out-of-pocket cost of AA treatments range from $1354 to $2685, the cost burden of individual therapies is poorly understood.12-14
Patients who must juggle expensive medical bills with basic living expenses may be lost to follow-up or fall into treatment nonadherence.15 Other patients’ out-of-pocket costs may be manageable, but the costs to the health care system may compromise care in other ways. We conducted a literature review of the recommended therapies for AA based on American Academy of Dermatology (AAD) guidelines to identify the costs of alopecia treatment and consolidate the available data for the practicing dermatologist.
Methods
We conducted a PubMed search of articles indexed for MEDLINE through September 15, 2022, using the terms alopecia and cost plus one of the treatments (n=21) identified by the AAD2 for the treatment of AA (Figure). The reference lists of included articles were reviewed to identify other potentially relevant studies. Forty-five articles were identified.
Given the dearth of cost research in alopecia and the paucity of large prospective studies, we excluded articles that were not available in their full-text form or were not in English (n=3), articles whose primary study topic was not AA or an expert-approved alopecia treatment (n=15), and articles with no concrete cost data (n=17), which yielded 10 relevant articles that we studied using qualitative analysis.
Due to substantial differences in study methods and outcome measures, we did not compare the costs of alopecia among studies and did not perform statistical analysis. The quality of each study was investigated and assigned a level of evidence per the 2009 criteria from the Centre for Evidence-Based Medicine.16
All cost data were converted into US dollars ($) using the conversion rate from the time of the original article’s publication.
Results
Total and Out-of-pocket Costs of AA—Li et al13 studied out-of-pocket health care costs for AA patients (N=675). Of these participants, 56.9% said their AA was moderately to seriously financially burdensome, and 41.3% reported using their savings to manage these expenses. Participants reported median out-of-pocket spending of $1354 (interquartile range, $537–$3300) annually. The most common categories of expenses were hair appointments (81.8%) and vitamins/supplements (67.7%).13
Mesinkovska et al14 studied the qualitative and quantitative financial burdens of moderate to severe AA (N=216). Fifty-seven percent of patients reported the financial impact of AA as moderately to severely burdensome with a willingness to borrow money or use savings to cover out-of-pocket costs. Patients without insurance cited cost as a major barrier to obtaining reatment. In addition to direct treatment-related expenses, AA patients spent a mean of $1961 per year on therapy to cope with the disease’s psychological burden. Lost work hours represented another source of financial burden; 61% of patients were employed, and 45% of them reported missing time from their job because of AA.14
Mostaghimi et al12 studied health care resource utilization and all-cause direct health care costs in privately insured AA patients with or without alopecia totalis (AT) or alopecia universalis (AU)(n=14,972) matched with non-AA controls (n=44,916)(1:3 ratio). Mean total all-cause medical and pharmacy costs were higher in both AA groups compared with controls (AT/AU, $18,988 vs $11,030; non-AT/AU, $13,686 vs $9336; P<.001 for both). Out-of-pocket costs were higher for AA vs controls (AT/AU, $2685 vs $1457; non-AT/AU, $2223 vs $1341; P<.001 for both). Medical costs in the AT/AU and non-AT/AU groups largely were driven by outpatient costs (AT/AU, $10,277 vs $5713; non-AT/AU, $8078 vs $4672; P<.001 for both).12
Costs of Concealment—When studying the out-of-pocket costs of AA (N=675), Li et al13 discovered that the median yearly spending was highest on headwear or cosmetic items such as hats, wigs, and makeup ($450; interquartile range, $50–$1500). Mesinkovska et al14 reported that 49% of patients had insurance that covered AA treatment. However, 75% of patients reported that their insurance would not cover costs of concealment (eg, weave, wig, hair piece). Patients (N=112) spent a mean of $2211 per year and 10.3 hours per week on concealment.14
Minoxidil—Minoxidil solution is available over-the-counter, and its ease of access makes it a popular treatment for AA.17 Because manufacturers can sell directly to the public, minoxidil is marketed with bold claims and convincing packaging. Shrank18 noted that the product can take 4 months to work, meaning customers must incur a substantial cost burden before realizing the treatment’s benefit, which is not always obvious when purchasing minoxidil products, leaving customers—who were marketed a miracle drug—disappointed. Per Shrank,18 patients who did not experience hair regrowth after 4 months were advised to continue treatment for a year, leading them to spend hundreds of dollars for uncertain results. Those who did experience hair regrowth were advised to continue using the product twice daily 7 days per week indefinitely.18
Wehner et al19 studied the association between gender and drug cost for over-the-counter minoxidil. The price that women paid for 2% regular-strength minoxidil solutions was similar to the price that men paid for 5% extra-strength minoxidil solutions (women’s 2%, $7.63/30 mL; men’s 5%, $7.61/30 mL; P=.67). Minoxidil 5% foams with identical ingredients were priced significantly more per volume of the same product when sold as a product directed at women vs a product directed at men (men’s 5%, $8.05/30 mL; women’s 5%, $11.27/30 mL; P<.001).19
Beach20 compared the cost of oral minoxidil to topical minoxidil. At $28.60 for a 3-month supply, oral minoxidil demonstrated cost savings compared to topical minoxidil ($48.30).20
Diphencyprone—Bhat et al21 studied the cost-efficiency of diphencyprone (DPC) in patients with AA resistant to at least 2 conventional treatments (N=29). After initial sensitization with 2% DPC, patients received weekly or fortnightly treatments. Most of the annual cost burden of DPC treatment was due to staff time and overhead rather than the cost of the DPC itself: $258 for the DPC, $978 in staff time and overhead for the department, and $1233 directly charged to the patient.21
Lekhavat et al22 studied the economic impact of home-use vs office-use DPC in extensive AA (N=82). Both groups received weekly treatments in the hospital until DPC concentrations had been adjusted. Afterward, the home group was given training on self-applying DPC at home. The home group had monthly office visits for DPC concentration evaluation and refills, while the office group had weekly appointments for DPC treatment at the hospital. Calculated costs included those to the health care provider (ie, material, labor, capital costs) and the patient’s final out-of-pocket expense. The total cost to the health care provider was higher for the office group than the home group at 48 weeks (office, $683.52; home, $303.67; P<.001). Median out-of-pocket costs did not vary significantly between groups, which may have been due to small sample size affecting the range (office, $418.07; home, $189.69; P=.101). There was no significant difference between groups in the proportion of patients who responded favorably to the DPC.22
JAK Inhibitors—Chen et al23 studied the efficacy of low-dose (5 mg) tofacitinib to treat severe AA (N=6). Compared to prior studies,24-27 this analysis reported the efficacy of low-dose tofacitinib was not inferior to higher doses (10–20 mg), and low-dose tofacitinib reduced treatment costs by more than 50%.23
Per the GlobalData Healthcare database, the estimated annual cost of therapy for JAK inhibitors following US Food and Drug Administration approval was $50,000. At the time of their reporting, the next most expensive immunomodulatory drug for AA was cyclosporine, with an annual cost of therapy of $1400.28 Dillon29 reviewed the use of JAK inhibitors for the treatment of AA. The cost estimates by Dillon29 prior to FDA approval aligned with the pricing of Eli Lilly and Company for the now-approved JAK inhibitor baricitinib.30 The list price of baricitinib is $2739.99 for a 30-day supply of 2-mg tablets or $5479.98 for a 30-day supply of 4-mg tablets. This amounts to $32,879.88 for an annual supply of 2-mg tablets and $65,759.76 for an annual supply for 4-mg tablets, though the out-of-pocket costs will vary.30
Comment
We reviewed the global and treatment-specific costs of AA, consolidating the available data for the practicing dermatologist. Ten studies of approximately 16,000 patients with AA across a range of levels of evidence (1a to 4) were included (Table). Three of 10 articles studied global costs of AA, 1 studied costs of concealment, 3 studied costs of minoxidil, 2 studied costs of DPC, and 2 studied costs of JAK inhibitors. Only 2 studies achieved level of evidence 1a: the first assessed the economic impact of home-use vs office-use DPC,22 and the second researched the efficacy and outcomes of JAK inhibitors.29
Hair-loss treatments and concealment techniques cost the average patient thousands of dollars. Spending was highest on headwear or cosmetic items, which were rarely covered by insurance.13 Psychosocial sequelae further increased cost via therapy charges and lost time at work.14 Patients with AA had greater all-cause medical costs than those without AA, with most of the cost driven by outpatient visits. Patients with AA also paid nearly twice as much as non-AA patients on out-of-pocket health care expenses.14 Despite the high costs and limited efficacy of many AA therapies, patients reported willingness to incur debt or use savings to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress.13
Minoxidil solution does not require physician office visits and is available over-the-counter.17 Despite identical ingredients, minoxidil is priced more per volume when marketed to women compared with men, which reflects the larger issue of gender-based pricing that does not exist for other AAD-approved alopecia therapies but may exist for cosmetic treatments and nonapproved therapies (eg, vitamins/supplements) that are popular in the treatment of AA.19 Oral minoxidil was more cost-effective than the topical form, and gender-based pricing was a nonissue.20 However, oral minoxidil requires a prescription, mandating patients incur the cost of an office visit. Patients should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective choice.
Diphencyprone is a relatively affordable drug for AA, but the regular office visits traditionally required for its administration increase associated cost.21 Self-administration of DPC at home was more cost- and time-effective than in-office DPC administration and did not decrease efficacy. A regimen combining office visits for initial DPC titration, at-home DPC administration, and periodic office follow-up could minimize costs while preserving outcomes and safety.22
Janus kinase inhibitors are cutting-edge and expensive therapies for AA. The annual cost of these medications poses a tremendous burden on the payer (list price of annual supply ritlecitinib is $49,000),31 be that the patient or the insurance company. Low-dose tofacitinib may be similarly efficacious and could substantially reduce treatment costs.23 The true utility of these medications, specifically considering their steep costs, remains to be determined.
Conclusion
Alopecia areata poses a substantial and recurring cost burden on patients that is multifactorial including treatment, office visits, concealment, alternative therapies, psychosocial costs, and missed time at work. Although several treatment options exist, none of them are definitive. Oral minoxidil and at-home DPC administration can be cost-effective, though the cumulative cost is still high. The cost utility of JAK inhibitors remains unclear. When JAK inhibitors are prescribed, low-dose therapy may be used as maintenance to curb treatment costs. Concealment and therapy costs pose an additional, largely out-of-pocket financial burden. Despite the limited efficacy of many AA therapies, patients incur substantial expenses to manage their AA. This willingness to pay reflects AA’s impact on quality of life and puts these patients at high risk for financial distress. There are no head-to-head studies comparing the cost-effectiveness of the different AA therapies; thus, it is unclear if one treatment is most efficacious. This topic remains an avenue for future investigation. Much of the cost burden of AA treatment falls directly on patients. Increasing coverage of AA-associated expenses, such as minoxidil therapy or wigs, could decrease the cost burden on patients. Providers also can inform patients about cost-saving tactics, such as purchasing minoxidil based on concentration and vehicle rather than marketing directed at men vs women. Finally, some patients may have insurance plans that at least partially cover the costs of wigs but may not be aware of this benefit. Querying a patient’s insurance provider can further minimize costs.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003;49:96-98. doi:10.1067/mjd.2003.423
- Strazzulla LC, Wang EHC, Avila L, et al. Alopecia areata: an appraisal of new treatment approaches and overview of current therapies. J Am Acad Dermatol. 2018;78:15-24. doi:10.1016/j.jaad.2017.04.1142
- Olsen EA, Carson SC, Turney EA. Systemic steroids with or without 2% topical minoxidil in the treatment of alopecia areata. Arch Dermatol. 1992;128:1467-1473.
- Levy LL, Urban J, King BA. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J Am Acad Dermatol. 2015;73:395-399. doi:10.1016/j.jaad.2015.06.045
- Ports WC, Khan S, Lan S, et al. A randomized phase 2a efficacy and safety trial of the topical Janus kinase inhibitor tofacitinib in the treatment of chronic plaque psoriasis. Br J Dermatol. 2013;169:137-145. doi:10.1111/bjd.12266
- Strober B, Buonanno M, Clark JD, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. Br J Dermatol. 2013;169:992-999. doi:10.1111/bjd.12517
- van der Steen PH, van Baar HM, Happle R, et al. Prognostic factors in the treatment of alopecia areata with diphenylcyclopropenone. J Am Acad Dermatol. 1991;24(2, pt 1):227-230. doi:10.1016/0190-9622(91)70032-w
- Strazzulla LC, Avila L, Lo Sicco K, et al. Image gallery: treatment of refractory alopecia universalis with oral tofacitinib citrate and adjunct intralesional triamcinolone injections. Br J Dermatol. 2017;176:E125. doi:10.1111/bjd.15483
- Madani S, Shapiro J. Alopecia areata update. J Am Acad Dermatol. 2000;42:549-566; quiz 567-570.
- Carnahan MC, Goldstein DA. Ocular complications of topical, peri-ocular, and systemic corticosteroids. Curr Opin Ophthalmol. 2000;11:478-483. doi:10.1097/00055735-200012000-00016
- Harel S, Higgins CA, Cerise JE, et al. Pharmacologic inhibition of JAK-STAT signaling promotes hair growth. Sci Adv. 2015;1:E1500973. doi:10.1126/sciadv.1500973
- Mostaghimi A, Gandhi K, Done N, et al. All-cause health care resource utilization and costs among adults with alopecia areata: a retrospective claims database study in the United States. J Manag Care Spec Pharm. 2022;28:426-434. doi:10.18553/jmcp.2022.28.4.426
- Li SJ, Mostaghimi A, Tkachenko E, et al. Association of out-of-pocket health care costs and financial burden for patients with alopecia areata. JAMA Dermatol. 2019;155:493-494. doi:10.1001/jamadermatol.2018.5218
- Mesinkovska N, King B, Mirmirani P, et al. Burden of illness in alopecia areata: a cross-sectional online survey study. J Investig Dermatol Symp Proc. 2020;20:S62-S68. doi:10.1016/j.jisp.2020.05.007
- Iuga AO, McGuire MJ. Adherence and health care costs. Risk Manag Healthc Policy. 2014;7:35-44. doi:10.2147/rmhp.S19801
- Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009). University of Oxford website. Accessed March 25, 2024. https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009
- Klifto KM, Othman S, Kovach SJ. Minoxidil, platelet-rich plasma (PRP), or combined minoxidil and PRP for androgenetic alopecia in men: a cost-effectiveness Markov decision analysis of prospective studies. Cureus. 2021;13:E20839. doi:10.7759/cureus.20839
- Shrank AB. Minoxidil over the counter. BMJ. 1995;311:526. doi:10.1136/bmj.311.7004.526
- Wehner MR, Nead KT, Lipoff JB. Association between gender and drug cost for over-the-counter minoxidil. JAMA Dermatol. 2017;153:825-826.
- Beach RA. Case series of oral minoxidil for androgenetic and traction alopecia: tolerability & the five C’s of oral therapy. Dermatol Ther. 2018;31:E12707. doi:10.1111/dth.12707
- Bhat A, Sripathy K, Wahie S, et al. Efficacy and cost-efficiency of diphencyprone for alopecia areata. Br J Dermatol. 2011;165:43-44.
- Lekhavat C, Rattanaumpawan P, Juengsamranphong I. Economic impact of home-use versus office-use diphenylcyclopropenone in extensive alopecia areata. Skin Appendage Disord. 2022;8:108-117.
- Chen YY, Lin SY, Chen YC, et al. Low-dose tofacitinib for treating patients with severe alopecia areata: an efficient and cost-saving regimen. Eur J Dermatol. 2019;29:667-669. doi:10.1684/ejd.2019.3668
- Liu LY, Craiglow BG, Dai F, et al. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22-28. doi:10.1016/j.jaad.2016.09.007
- Kennedy Crispin M, Ko JM, Craiglow BG, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight. 2016;1:e89776. doi:10.1172/jci.insight.89776
- Jabbari A, Sansaricq F, Cerise J, et al. An open-label pilot study to evaluate the efficacy of tofacitinib in moderate to severe patch-type alopecia areata, totalis, and universalis. J Invest Dermatol. 2018;138:1539-1545. doi:10.1016/j.jid.2018.01.032
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol. 2017;76:29-32. doi:10.1016/j.jaad.2016.09.006
- GlobalData Healthcare. Can JAK inhibitors penetrate the alopecia areata market effectively? Pharmaceutical Technology. July 15, 2019. Accessed February 8, 2024. https://www.pharmaceutical-technology.com/analyst-comment/alopecia-areata-treatment-2019/
- Dillon KL. A comprehensive literature review of JAK inhibitors in treatment of alopecia areata. Clin Cosmet Investig Dermatol. 2021;14:691-714. doi:10.2147/ccid.S309215
- How much should I expect to pay for Olumiant? Accessed March 20, 2024. https://www.lillypricinginfo.com/olumiant
- McNamee A. FDA approves first-ever adolescent alopecia treatment from Pfizer. Pharmaceutical Technology. June 26, 2023. Accessed March 20, 2024. https://www.pharmaceutical-technology.com/news/fda-approves-first-ever-adolescent-alopecia-treatment-from-pfizer/?cf-view
Practice Points
- Hair loss treatments and concealment techniques cost the average patient thousands of dollars. Much of this cost burden comes from items not covered by insurance.
- Providers should be wary of gender- or marketing-related surcharges for minoxidil solutions, and oral minoxidil may be a cost-effective option.
- Self-administering diphencyprone at home is more cost- and time-effective than in-office diphencyprone administration and does not decrease efficacy.

The Impact of Primary Tumor Site on Survival in Mycosis Fungoides
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.


Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
 for overall survival and disease-specific survival.")

For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).


Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).


Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.
Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).
Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).
Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
Mycosis fungoides (MF), the most common cutaneous T-cell lymphoma (CTCL), is characterized by clonal proliferation of predominantly CD4+ T cells with localization to the skin.1 Mycosis fungoides typically affects older adults with a male to female ratio of 2:1 but also can occur in children and younger adults.2,3 Known as the great imitator, the manifestations of MF can be variable with considerable clinical and pathologic overlap with benign inflammatory skin diseases, rendering definitive diagnosis challenging.4-7 The early stages of classic MF manifest as pruritic erythematous patches and plaques with variable scaling that can progress in later stages to ulceration and tumors.8 Histopathologically, classic MF is characterized by epidermotropic proliferation of small- to intermediate-sized pleomorphic lymphocytes with cerebriform nuclei and a haloed appearance; intraepidermal nests of atypical lymphocytes known as Pautrier microabscesses occasionally are observed.5 Mycosis fungoides typically follows an indolent clinical course, with advanced-stage MF portending a poor prognosis.9,10 Current treatment is focused on halting disease progression, with topical therapies, phototherapy, and radiation therapy as the standard therapies for early-stage MF.11-13 For advanced-stage MF, treatment may include systemic therapies such as interferon alfa and oral retinoids along with chemotherapies for more refractive cases.14 Allogenic hematopoietic cell transplantation is the only curative treatment.11
Current staging guidelines for MF do not address anatomic location as there is little known about its impact on patient outcomes.11,15 Due to the indolent nature of MF leading to diagnostic challenges, the exact frequency of each primary disease site for MF also remains unclear, though the suggested incidence of MF of the head and neck ranges from 30% to 70%.16,17 Involvement of the head and neck16,18 or external ear and external auditory canal19 is associated with worse prognosis. The purpose of this study was to examine the impact of anatomic location of primary disease site on survival in MF.
Methods
The National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database includes patient records from 18 registries and encompasses approximately 48% of the US population.20 Using SEER*STAT software (version 8.4.0.1), we conducted a search of patients diagnosed with MF (International Classification of Diseases for Oncology, Third Edition [ICD-O-3] histologic code 9700/3 [mycosis fungoides]) between 2000 and 2019. For inclusion in the study, patients were required to have a known age, specified primary site, and a known cause of death (if applicable). Patients with known Sézary syndrome (SS)—an aggressive form of CTCL that is characterized by the presence of clonally related neoplastic T cells in the skin, lymph nodes, and peripheral blood—were not included because the World Health Organization/European Organisation for Research and Treatment of Cancer considers SS and MF to be separate entities1,15; SS does not necessarily arise from preexisting MF and is associated with markedly poorer survival. This study was exempt from institutional review board approval because the data were publicly available and anonymized.
Data Collection—For age at diagnosis, patients were divided into the following categories: younger than 40 years, 40 to 59 years, 60 to 79 years, and 80 years and older. Demographics, tumor characteristics, and surgical management (if applicable) were obtained for each patient. The designations of chemotherapy and radiation treatment in the SEER database are not reliable and prone to false negatives. As such, these were excluded from analysis.
The primary outcomes of interest were overall survival (OS) and disease-specific survival (DSS), which were calculated as time from MF diagnosis to death. Although OS included all patients who died of any cause, DSS only included patients who died of MF.
Statistical Analysis—Demographics (age, sex, race, ethnicity), tumor characteristics (tumor size, primary site, T stage, lymph node involvement, metastasis), and surgical management (if applicable) were summarized. Overall survival and DSS were calculated using Kaplan-Meier analysis. Univariate and multivariable Cox proportional hazards regression models were generated to determine which prognostic factors for MF were associated with poorer OS and DSS. Only statistically significant variables in the univariate analysis were used to construct the multivariable analysis. Hazard ratios (HRs) and their associated 95% CIs were reported. Incidence rates were calculated and age adjusted to the 2000 US standard population. The SEER JoinPoint Regression program was used to determine the annual percent change (APC)—change in incidence rate over time. P<.05 was considered statistically significant. All statistical analyses were conducted with R version 4.0.2.
Results
Patient Demographics and Tumor Characteristics—There were 4265 patients diagnosed with MF from 2000 to 2019. The overall incidence of MF was 2.55 per million (95% CI, 2.48-2.63) when age adjusted to the 2000 US standard population, which increased with time (mean APC, 0.97% per year; P=.01). The mean age at diagnosis was 56.4 years with a male to female ratio of 1.2:1. Males (3.07 per million; 95% CI, 2.94-3.20) had a higher incidence of MF than females (2.16 per million; 95% CI, 2.06-2.26), with incidence in females increasing over time (mean APC, 1.52% per year; P=.02) while incidence in males remained stable (mean APC, 1.09%; P=.37). Patients predominantly self-identified as White (73.08%). Patients with MF of the head and neck were more likely to have smaller tumors (P=.02), a more advanced T stage (P<.001), and lymph node involvement (P=.01) at the time of diagnosis. Additional demographics and tumor characteristics are summarized in eTable 1.
Survival Outcomes—The mean follow-up time was 86.9 months. The 5- and 10-year OS rates were 85.4% (95% CI, 84.2%-86.6%) and 75.0% (95% CI, 73.4%-76.7%), respectively (Figure 1)(Table). The 5- and 10-year DSS rates were 93.3% (95% CI, 92.4%-94.1%) and 89.5% (95% CI, 88.3%-90.6%), respectively. For OS, univariate analysis indicated that significant prognostic factors included increasing age (P<.001), female sex (P<.001), self-identifying as Asian or Pacific Islander (P<.001), self-identifying as Hispanic Latino (P<.001), primary tumor sites of either the head and neck or upper limb (P<.001), T3 or T4 staging (P=.001), lymph node involvement at the time of diagnosis (P<.001), and metastasis (P<.001).
For DSS, univariate analysis had similar risk factors with self-identifying as Black being an additional risk factor (P=.02), though self-identifying as Asian/Pacific Islander or Hispanic Latino were not significant nor was location on the lower limb. For recorded tumor size, the HR increased by 1.001 per each 1-mm increase in size (eTable 2).
Multivariate analysis showed age at diagnosis (60–79 years: HR, 23.11 [95% CI, 3.03-176.32]; P=.002; ≥80 years: HR, 92.41 [95% CI, 11.78-724.75]; P<.001), T3 staging (HR, 2.37 [95% CI, 1.32-4.27]; P=.004), and metastasis (HR, 40.14 [95% CI, 4.14-389.50]; P=.001) significantly influenced OS. For DSS, multivariate analysis indicated the significant prognostic factors were age at diagnosis (60–79 years: HR, 8.94 [95% CI, 1.16-69.23]; P=.04];≥80 years: HR, 26.71; [95% CI, 3.26-218.99]; P=.002), tumor size (HR, 1.001 [95% CI, 1.000-1.002]; P=.04), T3 staging (HR, 3.71 [95% CI, 1.58-8.67]; P=.003), lymph node involvement (HR, 3.87 [95% CI, 1.11-13.50]; P=.03) and metastasis (HR, 49.76 [95% CI, 4.03-615.00]; P=.002)(Figure 2). When controlling for the aforementioned factors, the primary disease site was not significant (eTable 3).
Comment
Although the prognostic significance of primary disease sites on various types of CTCLs has been examined, limited research exists on MF due to its rarity. For the 4265 patients with MF included in our study, statistically significant prognostic factors on multivariate analysis for DSS included age at diagnosis, tumor size, T staging, lymph node involvement, and presence of metastasis. For OS, only age at diagnosis, T staging, and presence of metastasis were statistically significant predictors. Although initially statistically significant on univariate analysis for both OS and DSS, tumor location was not significant when controlling for confounders.
Our population-based analysis found that 5- and 10-year OS for patients with head and neck MF were 85.4% and 75.0%, respectively, and 5- and 10-year DSS were 93.3% and 89.5%, respectively. Our 10-year OS survival rate of 75.0% was slightly worse than the 81.6% reported by Jung et al16 in a study of 39 cases of MF of the head and neck from the Asan Medical Center database. The difference in survival rate may not only be due to differences in sample size but also because the Asan Medical Center database had a higher proportion of Asian patients as a Korean registry. In our univariate analysis, Asian/Pacific Islander race was shown to be a statistically significant predictor of worse prognosis for OS (P<.001). When comparing survival in patients with head and neck MF vs all primary tumor sites, our OS rate for head and neck MF was more favorable than the 5-year OS of 75% reported by Agar et al21 in their analysis of 1502 patients with MF of all locations, though their cohort also included patients with SS, which is known to have a poorer prognosis. Additionally, our 10-year OS rate of 75.0% for patients with MF with a primary tumor site of the head and neck was slightly less favorable than the 81.0% reported by a prior analysis of the SEER database for MF of all locations,22 which initially may be suggestive of worse outcomes associated with MF originating from the head and neck.
Although MF originating in the head and neck region was found to be a statistically significant prognostic factor under univariate analysis (P<.001), tumor location was not significant upon adjusting for confounders in the multivariate analysis. These results are consistent with those reported in a multivariable analysis conducted by Jung et al,16 which compared 39 cases of head and neck MF to 85 cases without head and neck involvement. The investigators found that the head and neck as the primary site was a significant prognostic factor associated with worsened rates of OS when patients had stages IA to IIA (P=.009) and T2 stage tumors (P=.012) but not in either T1 stage or advanced stage IIB to IVB tumors.16 In contrast, a study by Su et al18 evaluating patients with MF from the National Cancer Database found that patients with MF originating in the head and neck region had similar survival compared with MF originating in the lower limbs after pairwise propensity matching. It previously has been postulated that primary MF lesions originating in the head and neck region have relatively higher frequencies of biological markers believed to be associated with more aggressive tumor behavior and poorer prognosis, such as histopathologic folliculotropism, T-cell receptor gene rearrangements, and large-cell transformations.16 However, MF typically is an indolent disease with advanced-stage MF following an aggressive disease course that often is refractory to treatment. A review from a single academic center noted that 5-year DSS was 97.3% for T1a but only 37.5% for T4.23 Similarly, a meta-analysis evaluating survival in patients with MF noted the 5-year OS for stage IB was 85.8% while for stage IVB it was only 23.3%.24 As such, having advanced-stage MF influences survival to a far greater extent than the presence of head and neck involvement alone. Accordingly, the significantly higher prevalence of advanced T stage disease and increased likelihood of lymph node involvement in MF lesions originating in the head and neck region (both P<.001) may explain why previous studies noted a poorer survival rate with head and neck involvement, as they did not have the sample size to adjust for these factors. Controlling for the above factors likely explains the nonsignificance of this region as a prognostic indicator in our multivariate analysis of OS and DSS.
Similar to MF originating in the head and neck region, the upper limb as a primary tumor site initially was found to be a significant predictor of both OS and DSS on univariate analysis but not on multivariate analysis. By contrast, Su et al18 found survival outcomes were worse for patients diagnosed with MF with the upper limb as the primary tumor site compared with the lower limb on multivariate Cox proportional hazards analysis but not on pairwise propensity score matching. The difference in our results compared with Su et al18 may be because the National Cancer Database only reports OS, while DSS may be more useful in determining prognostic factors associated with poorer survival, especially in an older patient population with greater comorbidities. Furthermore, the nonsignificance of the upper limb as a primary tumor site on further multivariate analysis may be due to similar reasonings as for the head and neck, including more advanced T staging and an anatomic location close to lymph nodes.
Study Limitations—The SEER database is a national registry, which lends itself to potential data heterogeneity in recording and miscoding. Additionally, there may be higher rates of unconfirmed or missing information given the retrospective nature of the SEER database; the database also does not delineate facility type, insurance status, or Charlson/Deyo comorbidity index as demographic factors, which could influence the multivariable analysis. Finally, the SEER database does not further demarcate subtypes of MF, such as the aggressive folliculotropic variant commonly seen in head and neck MF lesions, which precludes independent analysis of disease course by subtype.
Conclusion
Our study evaluated primary disease site as a prognostic factor for OS and DSS in patients with MF. Although head and neck and upper limb as primary disease sites were found to be significant on univariate analysis, they were found to be an insignificant prognostic variable for OS or DSS in our multivariable analysis, potentially due to the aggressive nature of advanced-stage MF and localization close to lymph nodes. Further research including a deeper dive into MF of all stages and subtypes is needed to fully investigate primary disease site as a prognostic indicator. Older age, larger tumor size, a higher T stage, lymph node involvement, and presence of metastasis were associated with worse DSS, and patients with these attributes should be counseled regarding expected disease course and prognosis.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785. doi:10.1182/blood-2004-09-3502
- Hwang ST, Janik JE, Jaffe ES, et al. Mycosis fungoides and Sézary syndrome. Lancet. 2008;371:945-957. doi:10.1016/S0140-6736(08)60420-1
- Jung JM, Lim DJ, Won CH, et al. Mycosis fungoides in children and adolescents: a systematic review. JAMA Dermatol. 2021;157:431-438. doi:10.1001/jamadermatol.2021.0083
- Hodak E, Amitay-Laish I. Mycosis fungoides: a great imitator. Clin Dermatol. 2019;37:255-267. doi:10.1016/j.clindermatol.2019.01.004
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063. doi:10.1016/j.jaad.2005.08.057
- Spieth K, Grundmann-Kollmann M, Runne U, et al. Mycosis-fungoides-type Cutaneous T cell lymphoma of the hands and soles: a variant causing delay in diagnosis and adequate treatment of patients with palmoplantar eczema. Dermatology. 2002;205:239-244. doi:10.1159/000065862
- Scarisbrick JJ, Quaglino P, Prince HM, et al. The PROCLIPI international registry of early-stage mycosis fungoides identifies substantial diagnostic delay in most patients. Br J Dermatol. 2019;181:350-357. doi:10.1111/bjd.17258
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome): part i. diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol. 2014;70:205.e1-205.e16. doi:10.1016/j.jaad.2013.07.049
- Suh KS, Jang MS, Jung JH, et al. Clinical characteristics and long-term outcome of 223 patients with mycosis fungoides at a single tertiary center in Korea: a 29-year review. J Am Acad Dermatol. 2022;86:1275-1284. doi:10.1016/j.jaad.2021.06.860
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866. doi:10.1001/archderm.139.7.857
- Trautinger F, Eder J, Assaf C, et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—update 2017. Eur J Cancer. 2017;77:57-74. doi:10.1016/j.ejca.2017.02.027
- Quaglino P, Prince HM, Cowan R, et al. Treatment of early-stage mycosis fungoides: results from the PROspective Cutaneous Lymphoma International Prognostic Index (PROCLIPI) study. Br J Dermatol. 2021;184:722-730. doi:10.1111/bjd.19252
- Specht L, Dabaja B, Illidge T, et al. Modern radiation therapy for primary cutaneous lymphomas: field and dose guidelines from the International Lymphoma Radiation Oncology Group. Int J Radiat Oncol Biol Phys. 2015;92:32-39. doi:10.1016/j.ijrobp.2015.01.008
- Alberti-Violetti S, Talpur R, Schlichte M, et al. Advanced-stagemycosis fungoides and Sézary syndrome: survival and response to treatment. Clin Lymphoma Myeloma Leuk. 2015;15:E105-E112. doi:10.1016/j.clml.2015.02.027
- Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110:1713-1722. doi:10.1182/blood-2007-03-055749
- Jung JM, Yoo H, Lim DJ, et al. Clinicoprognostic implications of head and neck involvement by mycosis fungoides: a retrospective cohort study. J Am Acad Dermatol. 2022;86:1258-1265. doi:10.1016/j.jaad.2021.03.056
- Brennan JA. The head and neck manifestations of mycosis fungoides. Laryngoscope. 1995;105(5, pt 1):478-480. doi:10.1288/00005537-199505000-00005
- Su C, Tang R, Bai HX, et al. Disease site as a prognostic factor for mycosis fungoides: an analysis of 2428 cases from the US National Cancer Database. Br J Haematol. 2019;185:592-595. doi:10.1111/bjh.15570
- Wilkinson AJ, Nader ME, Roberts D, et al. Survival outcomes of patients with mycosis fungoides involving the external ear and ear canal. Laryngoscope. 2023;133:1486-1491. doi:10.1002/lary.30377
- National Cancer Institute. Surveillance, Epidemiology, and End Results (SEER) surveillance research program. Published July 2021. Accessed March 14, 2024. https://seer.cancer.gov/about/factsheets/SEER_Overview.pdf
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging proposal. J Clin Oncol. 2010;28:4730-4739. doi:10.1200/JCO.2009.27.7665
- Vollmer RT. A review of survival in mycosis fungoides. Am J Clin Pathol. 2014;141:706-711. doi:10.1309/AJCPH2PHXFCX3BOX
- Desai M, Liu S, Parker S. Clinical characteristics, prognostic factors, and survival of 393 patients with mycosis fungoides and Sézary syndrome in the southeastern United States: a single-institution cohort. J Am Acad Dermatol. 2015;72:276-285. doi:10.1016/j.jaad.2014.10.019
- Mourad A, Gniadecki R. Overall survival in mycosis fungoides: a systematic review and meta-analysis. J Invest Dermatol. 2020;140:495-497.e5. doi:10.1016/j.jid.2019.07.712
Practice Points
- Mycosis fungoides (MF) is the most common cutaneous T-cell lymphoma.
- Because MF is associated with diagnostic challenges due to its indolent course, data regarding primary tumor site as a prognostic factor are limited.
- Although MF originating from the head and neck region did not appear to influence survival, it was found that patients who were older or who had a larger tumor size at diagnosis, a higher T stage, lymph node involvement, or presence of metastasis had poorer survival overall and may benefit from additional counseling regarding their prognosis.
Virtual Reality Brings Relief to Hospitalized Patients With Cancer
suggests a new randomized controlled trial.
While both interventions brought some pain relief, VR therapy yielded greater, longer-lasting comfort, reported lead author Hunter Groninger, MD, of MedStar Health Research Institute, Hyattsville, Maryland, and colleagues.

“Investigators have explored immersive VR interventions in cancer populations for a variety of indications including anxiety, depression, fatigue, and procedure‐associated pain, particularly among patients with pediatric cancer and adult breast cancer,” the investigators wrote in Cancer. “Nevertheless, despite growing evidence supporting the efficacy of VR‐delivered interventions for analgesia, few data address its role to mitigate cancer‐related pain specifically.”
To address this knowledge gap, Dr. Groninger and colleagues enrolled 128 adult hospitalized patients with cancer of any kind, all of whom had moderate to severe pain (self-reported score at least 4 out of 10) within the past 24 hours.
Study Methods and Results
Patients were randomized to receive either 10 minutes of immersive VR distraction therapy or 10 minutes of two-dimensional guided imagery distraction therapy.
“[The VR therapy] provides noncompetitive experiences in which the user can move around and explore natural environments (e.g., beachscape, forest) from standing, seated, or fixed positions, including within a hospital bed or chair,” the investigators wrote. “We provided over‐the‐ear headphones to assure high sound quality for the experience in the virtual natural environment.”
The two-dimensional intervention, delivered via electronic tablet, featured a meditation with images of natural landscapes and instrumental background music.
“We chose this active control because it is readily available and reflects content similar to relaxation‐focused television channels that are increasingly common in hospital settings,” the investigators noted.
Compared with this more common approach, patients who received VR therapy had significantly greater immediate reduction in pain (mean change in pain score, –1.4 vs –0.7; P = .03). Twenty-four hours later, improvements in the VR group generally persisted, while pain level in the two-dimensional group returned almost to baseline (P = .004). In addition, patients in the VR group reported significantly greater improvements in general distress and pain bothersomeness.
“VR therapies may modulate the pain experience by reducing the level of attention paid to noxious stimuli, thereby suppressing transmission of painful sensations via pain processing pathways to the cerebral cortex, particularly with more active VR experiences compared to passive experiences,” the investigators wrote.
Downsides to Using VR
Although VR brought more benefit, participants in the VR group more often reported difficulty using the intervention compared with those who interacted with an electronic tablet.
Plus, one VR user described mild dizziness that resolved with pharmacologic intervention. Still, approximately 9 out of 10 participants in each group reported willingness to try the intervention again.
Future VR Research
“Virtual reality is a rapidly evolving technology with a wealth of potential patient‐facing applications,” the investigators wrote. “Future studies should explore repeated use, optimal dosing, and impact on VR therapy on opioid analgesic requirements as well as usability testing, VR content preferences and facilitators of analgesia, and barriers and facilitators to use in acute care settings.”
This study was supported by the American Cancer Society. The investigators disclosed no conflicts of interest.
suggests a new randomized controlled trial.
While both interventions brought some pain relief, VR therapy yielded greater, longer-lasting comfort, reported lead author Hunter Groninger, MD, of MedStar Health Research Institute, Hyattsville, Maryland, and colleagues.
“Investigators have explored immersive VR interventions in cancer populations for a variety of indications including anxiety, depression, fatigue, and procedure‐associated pain, particularly among patients with pediatric cancer and adult breast cancer,” the investigators wrote in Cancer. “Nevertheless, despite growing evidence supporting the efficacy of VR‐delivered interventions for analgesia, few data address its role to mitigate cancer‐related pain specifically.”
To address this knowledge gap, Dr. Groninger and colleagues enrolled 128 adult hospitalized patients with cancer of any kind, all of whom had moderate to severe pain (self-reported score at least 4 out of 10) within the past 24 hours.
Study Methods and Results
Patients were randomized to receive either 10 minutes of immersive VR distraction therapy or 10 minutes of two-dimensional guided imagery distraction therapy.
“[The VR therapy] provides noncompetitive experiences in which the user can move around and explore natural environments (e.g., beachscape, forest) from standing, seated, or fixed positions, including within a hospital bed or chair,” the investigators wrote. “We provided over‐the‐ear headphones to assure high sound quality for the experience in the virtual natural environment.”
The two-dimensional intervention, delivered via electronic tablet, featured a meditation with images of natural landscapes and instrumental background music.
“We chose this active control because it is readily available and reflects content similar to relaxation‐focused television channels that are increasingly common in hospital settings,” the investigators noted.
Compared with this more common approach, patients who received VR therapy had significantly greater immediate reduction in pain (mean change in pain score, –1.4 vs –0.7; P = .03). Twenty-four hours later, improvements in the VR group generally persisted, while pain level in the two-dimensional group returned almost to baseline (P = .004). In addition, patients in the VR group reported significantly greater improvements in general distress and pain bothersomeness.
“VR therapies may modulate the pain experience by reducing the level of attention paid to noxious stimuli, thereby suppressing transmission of painful sensations via pain processing pathways to the cerebral cortex, particularly with more active VR experiences compared to passive experiences,” the investigators wrote.
Downsides to Using VR
Although VR brought more benefit, participants in the VR group more often reported difficulty using the intervention compared with those who interacted with an electronic tablet.
Plus, one VR user described mild dizziness that resolved with pharmacologic intervention. Still, approximately 9 out of 10 participants in each group reported willingness to try the intervention again.
Future VR Research
“Virtual reality is a rapidly evolving technology with a wealth of potential patient‐facing applications,” the investigators wrote. “Future studies should explore repeated use, optimal dosing, and impact on VR therapy on opioid analgesic requirements as well as usability testing, VR content preferences and facilitators of analgesia, and barriers and facilitators to use in acute care settings.”
This study was supported by the American Cancer Society. The investigators disclosed no conflicts of interest.
suggests a new randomized controlled trial.
While both interventions brought some pain relief, VR therapy yielded greater, longer-lasting comfort, reported lead author Hunter Groninger, MD, of MedStar Health Research Institute, Hyattsville, Maryland, and colleagues.
“Investigators have explored immersive VR interventions in cancer populations for a variety of indications including anxiety, depression, fatigue, and procedure‐associated pain, particularly among patients with pediatric cancer and adult breast cancer,” the investigators wrote in Cancer. “Nevertheless, despite growing evidence supporting the efficacy of VR‐delivered interventions for analgesia, few data address its role to mitigate cancer‐related pain specifically.”
To address this knowledge gap, Dr. Groninger and colleagues enrolled 128 adult hospitalized patients with cancer of any kind, all of whom had moderate to severe pain (self-reported score at least 4 out of 10) within the past 24 hours.
Study Methods and Results
Patients were randomized to receive either 10 minutes of immersive VR distraction therapy or 10 minutes of two-dimensional guided imagery distraction therapy.
“[The VR therapy] provides noncompetitive experiences in which the user can move around and explore natural environments (e.g., beachscape, forest) from standing, seated, or fixed positions, including within a hospital bed or chair,” the investigators wrote. “We provided over‐the‐ear headphones to assure high sound quality for the experience in the virtual natural environment.”
The two-dimensional intervention, delivered via electronic tablet, featured a meditation with images of natural landscapes and instrumental background music.
“We chose this active control because it is readily available and reflects content similar to relaxation‐focused television channels that are increasingly common in hospital settings,” the investigators noted.
Compared with this more common approach, patients who received VR therapy had significantly greater immediate reduction in pain (mean change in pain score, –1.4 vs –0.7; P = .03). Twenty-four hours later, improvements in the VR group generally persisted, while pain level in the two-dimensional group returned almost to baseline (P = .004). In addition, patients in the VR group reported significantly greater improvements in general distress and pain bothersomeness.
“VR therapies may modulate the pain experience by reducing the level of attention paid to noxious stimuli, thereby suppressing transmission of painful sensations via pain processing pathways to the cerebral cortex, particularly with more active VR experiences compared to passive experiences,” the investigators wrote.
Downsides to Using VR
Although VR brought more benefit, participants in the VR group more often reported difficulty using the intervention compared with those who interacted with an electronic tablet.
Plus, one VR user described mild dizziness that resolved with pharmacologic intervention. Still, approximately 9 out of 10 participants in each group reported willingness to try the intervention again.
Future VR Research
“Virtual reality is a rapidly evolving technology with a wealth of potential patient‐facing applications,” the investigators wrote. “Future studies should explore repeated use, optimal dosing, and impact on VR therapy on opioid analgesic requirements as well as usability testing, VR content preferences and facilitators of analgesia, and barriers and facilitators to use in acute care settings.”
This study was supported by the American Cancer Society. The investigators disclosed no conflicts of interest.
FROM CANCER
Prostate Cancer Tsunami Coming, Experts Caution
An “inevitable” global surge in prostate cancer is coming, with a worldwide doubling of cases to 2.9 million and an 85% increase in deaths to nearly 700,000 by the year 2040, the Lancet Commission on Prostate Cancer warned this week.
At a meeting of urologists in Paris, the commission said that the acceleration is already underway in high-income countries such as the United States and the United Kingdom but will gain momentum in low- and medium-income countries.
Nick James, MD, lead author of The Lancet report and professor of prostate and bladder cancer research at The Institute of Cancer Research in London, said that the surge, in part, is a medical success story.
Dr. James told this news organization.
“There is a big rise in the high-income countries. But we’re going to see a big rise in the number of 50-, 60-, 70-year-olds in the coming decades in the poorer countries, and with that comes more prostate cancer. High-income countries such as the UK and USA will also see smaller increases for the same reason.”
According to the report, to be presented April 6 at the 2024 European Association of Urology Congress in Paris, “The case for prostate cancer screening for all men aged 50-70 years (and all men of African origin aged 45–70 years) in high-income countries is strengthening with improved use of technologies such as MRI and growing evidence for the safety of active surveillance.”
Andrew Vickers, PhD, a biostatistician at Memorial Sloan Kettering Cancer Center in New York City, said that the Lancet Commission came to similar conclusions as he and an international group of researchers did in a 2023 policy paper in The BMJ. A major gap, Dr. Vickers said, is misuse of prostate-specific antigen (PSA) screening.
“We found that the ubiquitous policy compromise of letting patients decide for themselves about PSA has led to the worst possible outcomes of overuse in men unlikely to benefit, high rates of overdiagnosis and overtreatment, and economic and racial inequity,” Dr. Vickers said. “Our view is that PSA screening should be done well — by implementing straightforward harm-reduction strategies like restricting screening in older men and use of secondary tests before biopsy — or not at all.”
Dr. James said that undertreatment of advanced disease is widespread; only about 30%-40% of men in the United States receive combination hormone therapy for metastatic disease, for example. “Simply doing what we know works would improve outcomes,” he said.
Dr. James said that men of African ancestry are twice as likely to develop prostate cancer, but whether treatment should follow a different approach in these men is unclear. The new report stressed the need to include more men of African ancestry in research.
Brandon Mahal, MD, vice chair of research in radiation oncology the University of Miami Sylvester Comprehensive Cancer Center and a coauthor of the report, said that new approaches are needed to enable earlier diagnosis of prostate cancer in men in low- to middle-income countries, where most patients present with metastatic disease and are less likely to survive for long periods.
Dr. James recommended pop-up clinics and mobile testing to encourage men who are at high risk for prostate cancer but feel well to detect lethal cancers early.
In England, for example, Dr. James helped introduce an outreach program called The Man Van which provided free health checks, including PSA tests, to high-risk men in London.
“By bringing a van with quick and easy testing straight to men at work and in the community, and targeting those who have a higher risk of prostate cancer, we provided thousands of health checks which resulted in almost 100 cancer diagnoses in men who might otherwise have only seen a doctor once their cancer has progressed to a more advanced stage,” he said.
He noted that the medical community worldwide is ill-prepared for the onslaught of prostate cancer cases.
“The solution cannot be training more urologists, radiation oncologists, pathologists, and radiologists because it simply takes too long,” Dr. James said. However, increased use of nurses and artificial intelligence may help. “In my own hospital, biopsies are a nurse-led and -delivered service. AI is extraordinarily good at diagnosis already and will only get better,” he said.
In poorer countries, smartphones could fill gaps too. “The same technology that does face recognition already can say that’s a Gleason 7 prostate cancer,” Dr. James said. “It’s not being rolled out in countries like America of course because pathologists’ income is at risk.”
Dr. James, Dr. Vickers, and Dr. Mahal reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
An “inevitable” global surge in prostate cancer is coming, with a worldwide doubling of cases to 2.9 million and an 85% increase in deaths to nearly 700,000 by the year 2040, the Lancet Commission on Prostate Cancer warned this week.
At a meeting of urologists in Paris, the commission said that the acceleration is already underway in high-income countries such as the United States and the United Kingdom but will gain momentum in low- and medium-income countries.
Nick James, MD, lead author of The Lancet report and professor of prostate and bladder cancer research at The Institute of Cancer Research in London, said that the surge, in part, is a medical success story.
Dr. James told this news organization.
“There is a big rise in the high-income countries. But we’re going to see a big rise in the number of 50-, 60-, 70-year-olds in the coming decades in the poorer countries, and with that comes more prostate cancer. High-income countries such as the UK and USA will also see smaller increases for the same reason.”
According to the report, to be presented April 6 at the 2024 European Association of Urology Congress in Paris, “The case for prostate cancer screening for all men aged 50-70 years (and all men of African origin aged 45–70 years) in high-income countries is strengthening with improved use of technologies such as MRI and growing evidence for the safety of active surveillance.”
Andrew Vickers, PhD, a biostatistician at Memorial Sloan Kettering Cancer Center in New York City, said that the Lancet Commission came to similar conclusions as he and an international group of researchers did in a 2023 policy paper in The BMJ. A major gap, Dr. Vickers said, is misuse of prostate-specific antigen (PSA) screening.
“We found that the ubiquitous policy compromise of letting patients decide for themselves about PSA has led to the worst possible outcomes of overuse in men unlikely to benefit, high rates of overdiagnosis and overtreatment, and economic and racial inequity,” Dr. Vickers said. “Our view is that PSA screening should be done well — by implementing straightforward harm-reduction strategies like restricting screening in older men and use of secondary tests before biopsy — or not at all.”
Dr. James said that undertreatment of advanced disease is widespread; only about 30%-40% of men in the United States receive combination hormone therapy for metastatic disease, for example. “Simply doing what we know works would improve outcomes,” he said.
Dr. James said that men of African ancestry are twice as likely to develop prostate cancer, but whether treatment should follow a different approach in these men is unclear. The new report stressed the need to include more men of African ancestry in research.
Brandon Mahal, MD, vice chair of research in radiation oncology the University of Miami Sylvester Comprehensive Cancer Center and a coauthor of the report, said that new approaches are needed to enable earlier diagnosis of prostate cancer in men in low- to middle-income countries, where most patients present with metastatic disease and are less likely to survive for long periods.
Dr. James recommended pop-up clinics and mobile testing to encourage men who are at high risk for prostate cancer but feel well to detect lethal cancers early.
In England, for example, Dr. James helped introduce an outreach program called The Man Van which provided free health checks, including PSA tests, to high-risk men in London.
“By bringing a van with quick and easy testing straight to men at work and in the community, and targeting those who have a higher risk of prostate cancer, we provided thousands of health checks which resulted in almost 100 cancer diagnoses in men who might otherwise have only seen a doctor once their cancer has progressed to a more advanced stage,” he said.
He noted that the medical community worldwide is ill-prepared for the onslaught of prostate cancer cases.
“The solution cannot be training more urologists, radiation oncologists, pathologists, and radiologists because it simply takes too long,” Dr. James said. However, increased use of nurses and artificial intelligence may help. “In my own hospital, biopsies are a nurse-led and -delivered service. AI is extraordinarily good at diagnosis already and will only get better,” he said.
In poorer countries, smartphones could fill gaps too. “The same technology that does face recognition already can say that’s a Gleason 7 prostate cancer,” Dr. James said. “It’s not being rolled out in countries like America of course because pathologists’ income is at risk.”
Dr. James, Dr. Vickers, and Dr. Mahal reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
An “inevitable” global surge in prostate cancer is coming, with a worldwide doubling of cases to 2.9 million and an 85% increase in deaths to nearly 700,000 by the year 2040, the Lancet Commission on Prostate Cancer warned this week.
At a meeting of urologists in Paris, the commission said that the acceleration is already underway in high-income countries such as the United States and the United Kingdom but will gain momentum in low- and medium-income countries.
Nick James, MD, lead author of The Lancet report and professor of prostate and bladder cancer research at The Institute of Cancer Research in London, said that the surge, in part, is a medical success story.
Dr. James told this news organization.
“There is a big rise in the high-income countries. But we’re going to see a big rise in the number of 50-, 60-, 70-year-olds in the coming decades in the poorer countries, and with that comes more prostate cancer. High-income countries such as the UK and USA will also see smaller increases for the same reason.”
According to the report, to be presented April 6 at the 2024 European Association of Urology Congress in Paris, “The case for prostate cancer screening for all men aged 50-70 years (and all men of African origin aged 45–70 years) in high-income countries is strengthening with improved use of technologies such as MRI and growing evidence for the safety of active surveillance.”
Andrew Vickers, PhD, a biostatistician at Memorial Sloan Kettering Cancer Center in New York City, said that the Lancet Commission came to similar conclusions as he and an international group of researchers did in a 2023 policy paper in The BMJ. A major gap, Dr. Vickers said, is misuse of prostate-specific antigen (PSA) screening.
“We found that the ubiquitous policy compromise of letting patients decide for themselves about PSA has led to the worst possible outcomes of overuse in men unlikely to benefit, high rates of overdiagnosis and overtreatment, and economic and racial inequity,” Dr. Vickers said. “Our view is that PSA screening should be done well — by implementing straightforward harm-reduction strategies like restricting screening in older men and use of secondary tests before biopsy — or not at all.”
Dr. James said that undertreatment of advanced disease is widespread; only about 30%-40% of men in the United States receive combination hormone therapy for metastatic disease, for example. “Simply doing what we know works would improve outcomes,” he said.
Dr. James said that men of African ancestry are twice as likely to develop prostate cancer, but whether treatment should follow a different approach in these men is unclear. The new report stressed the need to include more men of African ancestry in research.
Brandon Mahal, MD, vice chair of research in radiation oncology the University of Miami Sylvester Comprehensive Cancer Center and a coauthor of the report, said that new approaches are needed to enable earlier diagnosis of prostate cancer in men in low- to middle-income countries, where most patients present with metastatic disease and are less likely to survive for long periods.
Dr. James recommended pop-up clinics and mobile testing to encourage men who are at high risk for prostate cancer but feel well to detect lethal cancers early.
In England, for example, Dr. James helped introduce an outreach program called The Man Van which provided free health checks, including PSA tests, to high-risk men in London.
“By bringing a van with quick and easy testing straight to men at work and in the community, and targeting those who have a higher risk of prostate cancer, we provided thousands of health checks which resulted in almost 100 cancer diagnoses in men who might otherwise have only seen a doctor once their cancer has progressed to a more advanced stage,” he said.
He noted that the medical community worldwide is ill-prepared for the onslaught of prostate cancer cases.
“The solution cannot be training more urologists, radiation oncologists, pathologists, and radiologists because it simply takes too long,” Dr. James said. However, increased use of nurses and artificial intelligence may help. “In my own hospital, biopsies are a nurse-led and -delivered service. AI is extraordinarily good at diagnosis already and will only get better,” he said.
In poorer countries, smartphones could fill gaps too. “The same technology that does face recognition already can say that’s a Gleason 7 prostate cancer,” Dr. James said. “It’s not being rolled out in countries like America of course because pathologists’ income is at risk.”
Dr. James, Dr. Vickers, and Dr. Mahal reported no relevant financial conflicts of interest.
A version of this article appeared on Medscape.com.
How to Cure Hedonic Eating?
Logan is a 62-year-old woman who has reached the pinnacle of professional success. She started a $50 million consumer products company and, after selling it, managed to develop another successful brand. She is healthy and happily married, with four adult children. And yet, despite all her achievements and stable family life, Logan was always bothered by her inability to lose weight.
Despite peddling in beauty, she felt perpetually overweight and, frankly, unattractive. She has no family history of obesity, drinks minimal alcohol, and follows an (allegedly) healthy diet. Logan had tried “everything” to lose weight — human growth hormone injections (not prescribed by me), Ozempic-like medications, Belviq, etc. — all to no avail.
Here’s the catch: After she finished with her busy days of meetings and spreadsheets, Logan sat down to read through countless emails and rewarded herself with all her favorite foods. Without realizing it, she often doubled her daily caloric intake in one sitting. She wasn’t hungry in these moments, rather just a little worn out and perhaps a little careless. She then proceeded to email her doctor (me) to report on this endless cycle of unwanted behavior.
In January 2024, a novel study from Turkey examined the relationship between hedonic eating, self-condemnation, and self-esteem. Surprising to no one, the study determined that higher hedonic hunger scores were associated with lower self-esteem and an increased propensity to self-stigmatize.
Oprah could have handily predicted this conclusion. Many years ago, she described food as a fake friend: Perhaps you’ve had a long and difficult day. While you’re busy eating your feelings, the heaping plate of pasta feels like your best buddy in the world. However, the moment the plate is empty, you realize that you feel worse than before. Not only do you have to unbutton your new jeans, but you also realize that you have just lost your ability to self-regulate.
While the positive association between hedonic eating and low self-esteem may seem self-evident, the solution is less obvious. Mindfulness is one possible approach to this issue. Mindfulness has been described as “paying attention in a particular way: on purpose, in the present moment, and nonjudgmentally” and has existed for thousands of years. Mindful eating, in particular, involves paying close attention to our food choices and how they affect our emotions, and typically includes some combination of:
- Slowing down eating/chewing thoroughly
- Eliminating distractions such as TV, computers, and phones — perhaps even eating in silence
- Eating only until physically satiated
- Distinguishing between true hunger and cravings
- Noticing the texture, flavors, and smell of food
- Paying attention to the effect of food on your mood
- Appreciating food
In our society, where processed food is so readily available and stress is so ubiquitous, eating can become a hedonic and fast-paced activity. Our brains don’t have time to process our bodies’ signals of fullness and, as a result, we often ingest many more calories than we need for a healthy lifestyle.
If mindless eating is part of the problem, mindful eating is part of the solution. Indeed, a meta-review of 10 scientific studies showed that mindful eating is as effective as conventional weight loss programs in regard to body mass index and waist circumference. On the basis of these studies — as well as some good old-fashioned common sense — intuitive eating is an important component of sustainable weight reduction.
Eventually, I convinced Logan to meet up with the psychologist in our group who specializes in emotional eating. Through weekly cognitive-behavioral therapy sessions, Logan was able to understand the impetus behind her self-defeating behavior and has finally been able to reverse some of her lifelong habits. Once she started practicing mindful eating, I was able to introduce Ozempic, and now Logan is happily shedding several pounds a week.
Dr. Messer has disclosed no relevant financial relationships.
Dr. Messer is clinical assistant professor, Mount Sinai School of Medicine and associate professor, Hofstra School of Medicine, both in New York City.
A version of this article first appeared on Medscape.com.
Logan is a 62-year-old woman who has reached the pinnacle of professional success. She started a $50 million consumer products company and, after selling it, managed to develop another successful brand. She is healthy and happily married, with four adult children. And yet, despite all her achievements and stable family life, Logan was always bothered by her inability to lose weight.
Despite peddling in beauty, she felt perpetually overweight and, frankly, unattractive. She has no family history of obesity, drinks minimal alcohol, and follows an (allegedly) healthy diet. Logan had tried “everything” to lose weight — human growth hormone injections (not prescribed by me), Ozempic-like medications, Belviq, etc. — all to no avail.
Here’s the catch: After she finished with her busy days of meetings and spreadsheets, Logan sat down to read through countless emails and rewarded herself with all her favorite foods. Without realizing it, she often doubled her daily caloric intake in one sitting. She wasn’t hungry in these moments, rather just a little worn out and perhaps a little careless. She then proceeded to email her doctor (me) to report on this endless cycle of unwanted behavior.
In January 2024, a novel study from Turkey examined the relationship between hedonic eating, self-condemnation, and self-esteem. Surprising to no one, the study determined that higher hedonic hunger scores were associated with lower self-esteem and an increased propensity to self-stigmatize.
Oprah could have handily predicted this conclusion. Many years ago, she described food as a fake friend: Perhaps you’ve had a long and difficult day. While you’re busy eating your feelings, the heaping plate of pasta feels like your best buddy in the world. However, the moment the plate is empty, you realize that you feel worse than before. Not only do you have to unbutton your new jeans, but you also realize that you have just lost your ability to self-regulate.
While the positive association between hedonic eating and low self-esteem may seem self-evident, the solution is less obvious. Mindfulness is one possible approach to this issue. Mindfulness has been described as “paying attention in a particular way: on purpose, in the present moment, and nonjudgmentally” and has existed for thousands of years. Mindful eating, in particular, involves paying close attention to our food choices and how they affect our emotions, and typically includes some combination of:
- Slowing down eating/chewing thoroughly
- Eliminating distractions such as TV, computers, and phones — perhaps even eating in silence
- Eating only until physically satiated
- Distinguishing between true hunger and cravings
- Noticing the texture, flavors, and smell of food
- Paying attention to the effect of food on your mood
- Appreciating food
In our society, where processed food is so readily available and stress is so ubiquitous, eating can become a hedonic and fast-paced activity. Our brains don’t have time to process our bodies’ signals of fullness and, as a result, we often ingest many more calories than we need for a healthy lifestyle.
If mindless eating is part of the problem, mindful eating is part of the solution. Indeed, a meta-review of 10 scientific studies showed that mindful eating is as effective as conventional weight loss programs in regard to body mass index and waist circumference. On the basis of these studies — as well as some good old-fashioned common sense — intuitive eating is an important component of sustainable weight reduction.
Eventually, I convinced Logan to meet up with the psychologist in our group who specializes in emotional eating. Through weekly cognitive-behavioral therapy sessions, Logan was able to understand the impetus behind her self-defeating behavior and has finally been able to reverse some of her lifelong habits. Once she started practicing mindful eating, I was able to introduce Ozempic, and now Logan is happily shedding several pounds a week.
Dr. Messer has disclosed no relevant financial relationships.
Dr. Messer is clinical assistant professor, Mount Sinai School of Medicine and associate professor, Hofstra School of Medicine, both in New York City.
A version of this article first appeared on Medscape.com.
Logan is a 62-year-old woman who has reached the pinnacle of professional success. She started a $50 million consumer products company and, after selling it, managed to develop another successful brand. She is healthy and happily married, with four adult children. And yet, despite all her achievements and stable family life, Logan was always bothered by her inability to lose weight.
Despite peddling in beauty, she felt perpetually overweight and, frankly, unattractive. She has no family history of obesity, drinks minimal alcohol, and follows an (allegedly) healthy diet. Logan had tried “everything” to lose weight — human growth hormone injections (not prescribed by me), Ozempic-like medications, Belviq, etc. — all to no avail.
Here’s the catch: After she finished with her busy days of meetings and spreadsheets, Logan sat down to read through countless emails and rewarded herself with all her favorite foods. Without realizing it, she often doubled her daily caloric intake in one sitting. She wasn’t hungry in these moments, rather just a little worn out and perhaps a little careless. She then proceeded to email her doctor (me) to report on this endless cycle of unwanted behavior.
In January 2024, a novel study from Turkey examined the relationship between hedonic eating, self-condemnation, and self-esteem. Surprising to no one, the study determined that higher hedonic hunger scores were associated with lower self-esteem and an increased propensity to self-stigmatize.
Oprah could have handily predicted this conclusion. Many years ago, she described food as a fake friend: Perhaps you’ve had a long and difficult day. While you’re busy eating your feelings, the heaping plate of pasta feels like your best buddy in the world. However, the moment the plate is empty, you realize that you feel worse than before. Not only do you have to unbutton your new jeans, but you also realize that you have just lost your ability to self-regulate.
While the positive association between hedonic eating and low self-esteem may seem self-evident, the solution is less obvious. Mindfulness is one possible approach to this issue. Mindfulness has been described as “paying attention in a particular way: on purpose, in the present moment, and nonjudgmentally” and has existed for thousands of years. Mindful eating, in particular, involves paying close attention to our food choices and how they affect our emotions, and typically includes some combination of:
- Slowing down eating/chewing thoroughly
- Eliminating distractions such as TV, computers, and phones — perhaps even eating in silence
- Eating only until physically satiated
- Distinguishing between true hunger and cravings
- Noticing the texture, flavors, and smell of food
- Paying attention to the effect of food on your mood
- Appreciating food
In our society, where processed food is so readily available and stress is so ubiquitous, eating can become a hedonic and fast-paced activity. Our brains don’t have time to process our bodies’ signals of fullness and, as a result, we often ingest many more calories than we need for a healthy lifestyle.
If mindless eating is part of the problem, mindful eating is part of the solution. Indeed, a meta-review of 10 scientific studies showed that mindful eating is as effective as conventional weight loss programs in regard to body mass index and waist circumference. On the basis of these studies — as well as some good old-fashioned common sense — intuitive eating is an important component of sustainable weight reduction.
Eventually, I convinced Logan to meet up with the psychologist in our group who specializes in emotional eating. Through weekly cognitive-behavioral therapy sessions, Logan was able to understand the impetus behind her self-defeating behavior and has finally been able to reverse some of her lifelong habits. Once she started practicing mindful eating, I was able to introduce Ozempic, and now Logan is happily shedding several pounds a week.
Dr. Messer has disclosed no relevant financial relationships.
Dr. Messer is clinical assistant professor, Mount Sinai School of Medicine and associate professor, Hofstra School of Medicine, both in New York City.
A version of this article first appeared on Medscape.com.
Is Measuring How Many Times Patients Get Screened for Depression Really a Reflection of Good Clinical Care?
Every time a patient visits Jason Connelly, MD, they must fill out a depression screening, thanks to a 2017 rule which mandates such assessments.
Providing a screening and, if needed, a follow-up plan means a patient may gain access to medication or cognitive-behavioral therapy that will improve their lives. But Dr. Connelly, a family medicine physician at Novant Health West Rowan Family Medicine in Cleveland, North Carolina, said the screening measure — and others like it that insurers and quality groups use to assess clinician performance — does not allow for enough flexibility.
For instance, he must follow-up with patients every 4 months, regardless of the severity of their depression.
“A lot of times when these are written for the purpose of measures, they don’t take into consideration the reality of clinical medicine,” Dr. Connelly, who is also a clinical physician executive with Novant, said. “There certainly needs to be room for the ability to specify the level of depression such that if it is mild, well, maybe that follow-up is at 6 months or 12 months or at patient discretion.”
A recent report from the American College of Physicians (ACP) supported Dr. Connelly’s view. The body looked at eight quality measures in primary care for patients with major depressive disorder (MDD) and found only one — a risk assessment for suicide — to be clinically meaningful and based on evidence.
The ACP panel said nearly all of the performance measures “lacked current clinical evidence, did not consider patient preferences, were not tested appropriately, or were outside a physician’s control.”
The group called for improvements in such assessments “to accurately assess the quality of clinical care” for patients with major depression.
Necessary Evil or Burdensome Time Suck?
The Centers for Medicare & Medicaid Services scores clinicians and health systems on the percentage of their patients who receive a screening during a visit; if the screening is positive, clinicians must document a follow-up plan using special manual entry codes.
Physicians say the process of meeting government standards for invalid measures can create unnecessary visits and physician paperwork, shrink monetary bonuses, and may not portray an accurate portrait of what best practice looks like in primary care for mental health. But many also said the program overall brings value to patients and provides a picture of how well they are practicing but only when measures are clinically relevant.
Standards ACP Used for Validating Depression Measurement
A committee with ACP used a modified appropriateness method from RAND and UCLA.
They weighed if a metric was evidence-based, methodologically sound, and clinically meaningful.
They rated each measure using a 9-point scale, including appropriate care, feasibility or applicability, and measure specifications.
A total of 11 committee members voted anonymously if each metric was a valid way of measuring individual clinicians, at the practice/system level, and health plan.
“There’s been such a flood of performance measurements that we can get sidetracked, diverted, and spend resources and effort on measurements that don’t improve care,” said Nick Fitterman, MD, chair of the ACP’s Performance Measurement Committee.
Primary care clinicians can choose from more than 60 metrics for 2024. Many involve caring for patients with mental illness or screening for those who could be underdiagnosed. Programs that certify health systems as providing quality care use the measures, in addition to the Merit-Based Incentive Payment System. Health systems choose six measures of quality to tie to their reimbursement — along with assessments of costs and use of technology.
In turn, Medicare adjusts its reimbursement based on how well a clinician’s numbers turn out and if they improved over time.
“You don’t get the benefit of the upside if you don’t meet the measure, so your payment is neutral and that can be significant from a broader system lens,” Dr. Connelly said. “Then you start to have to make decisions on what services do we then have to limit because we no longer have the financial capability.”
The implications for health systems and patient care are the reason ACP and clinicians are calling for some measures to be amended. Dr. Fitterman said his organization plans to work with CMS.
Implementing Measurement
At Bassett Health in New York, the health system uses the depression and follow-up plan measure to qualify for certification from the Health Resources and Services Administration as a patient-centered medical home, which the company uses in part to market itself to patients.
Amy Grace, MD, an attending physician in internal and family medicine at Bassett Health in Little Falls, New York, said if a patient refuses to take a depression screening, she will not meet the measure for that visit. But providing a screening is not always clinically appropriate, and some patients do not need a follow-up plan.
“If someone has just had a death in the family, they might answer the questions in a way that would be consistent with depression, but they’re experiencing grief as opposed to clinical depression,” Dr. Grace said.
Suggestions From ACP for Improvement in MDD Metrics
- Create and implement criteria for patients who do not need a follow-up plan based on clinician judgment.
- Add methods for clinicians to measure and indicate severity of MDD.
- Enable use of a wider array of evidence-based tools and screenings to screen for MDD.
- Allow clinicians to document changes in treatment plan.
Bassett is building into the electronic health record a button that documents the screening was not conducted and that it was not appropriate to administer that day. Of course, building these in-house options entails utilizing resources that smaller systems or independent groups of clinicians may lack.
Eric Wei, MD, senior vice president and chief quality officer at NYC Health + Hospitals in New York City, said the ACP report underscores that many measures, even beyond depression, must be improved.
“With burnout and cognitive overload of our providers, on top of the medicine and just trying to come to the right diagnosis and providing the right treatment and the best care experience, you have to remember all these quality metrics and make sure you put all these things in certain places in the electronic health record,” Dr. Wei said.
Still, Dr. Wei said that the annual rate of depression screening across 400,000 patients in his system is 91%. He and his team spent 6 years working to improve uptake among clinicians, and now, they have moved on to increasing rates of administration of the suicide assessment.
Each clinician uses a dashboard to track their individual metric performance, according to Ted Long, MD, senior vice president for ambulatory care and population health at NYC Health + Hospitals. Dr. Long said he is proud of the improvements he and his colleagues have made in catching undiagnosed depression and in other disease states.
At his primary care practice in the Bronx, nearly 9 out of 10 patients with hypertension have their condition under control, he said. How does he know? Measurement tracking.
“Knowing that when a new patient is in front of me with high blood pressure, that there’s a 9 out of 10 chance that after seeing me because of my clinic, not just because of me, I’m going to be able to keep them healthy by controlling their blood pressure, that’s very meaningful to me,” Dr. Long said. “I think that’s the other side: It enables me as a doctor to know that I’m delivering the highest quality of care to my patients.”
Takeaways for Depression Screening and Follow-Up in Clinical Settings
Just because a patient scores positive for the depression screener, a clinician should dig deeper before making a diagnosis.
Patients have the right to refuse a screener and their wishes should be respected.
Providing a screener may not be appropriate at every visit, such as for a patient with a sprained ankle or a potential respiratory infection where time is limited.
Clinicians can clarify within the measure that the patient did not have mental capacity on that visit to fill out the screener.
A version of this article appeared on Medscape.com.
Every time a patient visits Jason Connelly, MD, they must fill out a depression screening, thanks to a 2017 rule which mandates such assessments.
Providing a screening and, if needed, a follow-up plan means a patient may gain access to medication or cognitive-behavioral therapy that will improve their lives. But Dr. Connelly, a family medicine physician at Novant Health West Rowan Family Medicine in Cleveland, North Carolina, said the screening measure — and others like it that insurers and quality groups use to assess clinician performance — does not allow for enough flexibility.
For instance, he must follow-up with patients every 4 months, regardless of the severity of their depression.
“A lot of times when these are written for the purpose of measures, they don’t take into consideration the reality of clinical medicine,” Dr. Connelly, who is also a clinical physician executive with Novant, said. “There certainly needs to be room for the ability to specify the level of depression such that if it is mild, well, maybe that follow-up is at 6 months or 12 months or at patient discretion.”
A recent report from the American College of Physicians (ACP) supported Dr. Connelly’s view. The body looked at eight quality measures in primary care for patients with major depressive disorder (MDD) and found only one — a risk assessment for suicide — to be clinically meaningful and based on evidence.
The ACP panel said nearly all of the performance measures “lacked current clinical evidence, did not consider patient preferences, were not tested appropriately, or were outside a physician’s control.”
The group called for improvements in such assessments “to accurately assess the quality of clinical care” for patients with major depression.
Necessary Evil or Burdensome Time Suck?
The Centers for Medicare & Medicaid Services scores clinicians and health systems on the percentage of their patients who receive a screening during a visit; if the screening is positive, clinicians must document a follow-up plan using special manual entry codes.
Physicians say the process of meeting government standards for invalid measures can create unnecessary visits and physician paperwork, shrink monetary bonuses, and may not portray an accurate portrait of what best practice looks like in primary care for mental health. But many also said the program overall brings value to patients and provides a picture of how well they are practicing but only when measures are clinically relevant.
Standards ACP Used for Validating Depression Measurement
A committee with ACP used a modified appropriateness method from RAND and UCLA.
They weighed if a metric was evidence-based, methodologically sound, and clinically meaningful.
They rated each measure using a 9-point scale, including appropriate care, feasibility or applicability, and measure specifications.
A total of 11 committee members voted anonymously if each metric was a valid way of measuring individual clinicians, at the practice/system level, and health plan.
“There’s been such a flood of performance measurements that we can get sidetracked, diverted, and spend resources and effort on measurements that don’t improve care,” said Nick Fitterman, MD, chair of the ACP’s Performance Measurement Committee.
Primary care clinicians can choose from more than 60 metrics for 2024. Many involve caring for patients with mental illness or screening for those who could be underdiagnosed. Programs that certify health systems as providing quality care use the measures, in addition to the Merit-Based Incentive Payment System. Health systems choose six measures of quality to tie to their reimbursement — along with assessments of costs and use of technology.
In turn, Medicare adjusts its reimbursement based on how well a clinician’s numbers turn out and if they improved over time.
“You don’t get the benefit of the upside if you don’t meet the measure, so your payment is neutral and that can be significant from a broader system lens,” Dr. Connelly said. “Then you start to have to make decisions on what services do we then have to limit because we no longer have the financial capability.”
The implications for health systems and patient care are the reason ACP and clinicians are calling for some measures to be amended. Dr. Fitterman said his organization plans to work with CMS.
Implementing Measurement
At Bassett Health in New York, the health system uses the depression and follow-up plan measure to qualify for certification from the Health Resources and Services Administration as a patient-centered medical home, which the company uses in part to market itself to patients.
Amy Grace, MD, an attending physician in internal and family medicine at Bassett Health in Little Falls, New York, said if a patient refuses to take a depression screening, she will not meet the measure for that visit. But providing a screening is not always clinically appropriate, and some patients do not need a follow-up plan.
“If someone has just had a death in the family, they might answer the questions in a way that would be consistent with depression, but they’re experiencing grief as opposed to clinical depression,” Dr. Grace said.
Suggestions From ACP for Improvement in MDD Metrics
- Create and implement criteria for patients who do not need a follow-up plan based on clinician judgment.
- Add methods for clinicians to measure and indicate severity of MDD.
- Enable use of a wider array of evidence-based tools and screenings to screen for MDD.
- Allow clinicians to document changes in treatment plan.
Bassett is building into the electronic health record a button that documents the screening was not conducted and that it was not appropriate to administer that day. Of course, building these in-house options entails utilizing resources that smaller systems or independent groups of clinicians may lack.
Eric Wei, MD, senior vice president and chief quality officer at NYC Health + Hospitals in New York City, said the ACP report underscores that many measures, even beyond depression, must be improved.
“With burnout and cognitive overload of our providers, on top of the medicine and just trying to come to the right diagnosis and providing the right treatment and the best care experience, you have to remember all these quality metrics and make sure you put all these things in certain places in the electronic health record,” Dr. Wei said.
Still, Dr. Wei said that the annual rate of depression screening across 400,000 patients in his system is 91%. He and his team spent 6 years working to improve uptake among clinicians, and now, they have moved on to increasing rates of administration of the suicide assessment.
Each clinician uses a dashboard to track their individual metric performance, according to Ted Long, MD, senior vice president for ambulatory care and population health at NYC Health + Hospitals. Dr. Long said he is proud of the improvements he and his colleagues have made in catching undiagnosed depression and in other disease states.
At his primary care practice in the Bronx, nearly 9 out of 10 patients with hypertension have their condition under control, he said. How does he know? Measurement tracking.
“Knowing that when a new patient is in front of me with high blood pressure, that there’s a 9 out of 10 chance that after seeing me because of my clinic, not just because of me, I’m going to be able to keep them healthy by controlling their blood pressure, that’s very meaningful to me,” Dr. Long said. “I think that’s the other side: It enables me as a doctor to know that I’m delivering the highest quality of care to my patients.”
Takeaways for Depression Screening and Follow-Up in Clinical Settings
Just because a patient scores positive for the depression screener, a clinician should dig deeper before making a diagnosis.
Patients have the right to refuse a screener and their wishes should be respected.
Providing a screener may not be appropriate at every visit, such as for a patient with a sprained ankle or a potential respiratory infection where time is limited.
Clinicians can clarify within the measure that the patient did not have mental capacity on that visit to fill out the screener.
A version of this article appeared on Medscape.com.
Every time a patient visits Jason Connelly, MD, they must fill out a depression screening, thanks to a 2017 rule which mandates such assessments.
Providing a screening and, if needed, a follow-up plan means a patient may gain access to medication or cognitive-behavioral therapy that will improve their lives. But Dr. Connelly, a family medicine physician at Novant Health West Rowan Family Medicine in Cleveland, North Carolina, said the screening measure — and others like it that insurers and quality groups use to assess clinician performance — does not allow for enough flexibility.
For instance, he must follow-up with patients every 4 months, regardless of the severity of their depression.
“A lot of times when these are written for the purpose of measures, they don’t take into consideration the reality of clinical medicine,” Dr. Connelly, who is also a clinical physician executive with Novant, said. “There certainly needs to be room for the ability to specify the level of depression such that if it is mild, well, maybe that follow-up is at 6 months or 12 months or at patient discretion.”
A recent report from the American College of Physicians (ACP) supported Dr. Connelly’s view. The body looked at eight quality measures in primary care for patients with major depressive disorder (MDD) and found only one — a risk assessment for suicide — to be clinically meaningful and based on evidence.
The ACP panel said nearly all of the performance measures “lacked current clinical evidence, did not consider patient preferences, were not tested appropriately, or were outside a physician’s control.”
The group called for improvements in such assessments “to accurately assess the quality of clinical care” for patients with major depression.
Necessary Evil or Burdensome Time Suck?
The Centers for Medicare & Medicaid Services scores clinicians and health systems on the percentage of their patients who receive a screening during a visit; if the screening is positive, clinicians must document a follow-up plan using special manual entry codes.
Physicians say the process of meeting government standards for invalid measures can create unnecessary visits and physician paperwork, shrink monetary bonuses, and may not portray an accurate portrait of what best practice looks like in primary care for mental health. But many also said the program overall brings value to patients and provides a picture of how well they are practicing but only when measures are clinically relevant.
Standards ACP Used for Validating Depression Measurement
A committee with ACP used a modified appropriateness method from RAND and UCLA.
They weighed if a metric was evidence-based, methodologically sound, and clinically meaningful.
They rated each measure using a 9-point scale, including appropriate care, feasibility or applicability, and measure specifications.
A total of 11 committee members voted anonymously if each metric was a valid way of measuring individual clinicians, at the practice/system level, and health plan.
“There’s been such a flood of performance measurements that we can get sidetracked, diverted, and spend resources and effort on measurements that don’t improve care,” said Nick Fitterman, MD, chair of the ACP’s Performance Measurement Committee.
Primary care clinicians can choose from more than 60 metrics for 2024. Many involve caring for patients with mental illness or screening for those who could be underdiagnosed. Programs that certify health systems as providing quality care use the measures, in addition to the Merit-Based Incentive Payment System. Health systems choose six measures of quality to tie to their reimbursement — along with assessments of costs and use of technology.
In turn, Medicare adjusts its reimbursement based on how well a clinician’s numbers turn out and if they improved over time.
“You don’t get the benefit of the upside if you don’t meet the measure, so your payment is neutral and that can be significant from a broader system lens,” Dr. Connelly said. “Then you start to have to make decisions on what services do we then have to limit because we no longer have the financial capability.”
The implications for health systems and patient care are the reason ACP and clinicians are calling for some measures to be amended. Dr. Fitterman said his organization plans to work with CMS.
Implementing Measurement
At Bassett Health in New York, the health system uses the depression and follow-up plan measure to qualify for certification from the Health Resources and Services Administration as a patient-centered medical home, which the company uses in part to market itself to patients.
Amy Grace, MD, an attending physician in internal and family medicine at Bassett Health in Little Falls, New York, said if a patient refuses to take a depression screening, she will not meet the measure for that visit. But providing a screening is not always clinically appropriate, and some patients do not need a follow-up plan.
“If someone has just had a death in the family, they might answer the questions in a way that would be consistent with depression, but they’re experiencing grief as opposed to clinical depression,” Dr. Grace said.
Suggestions From ACP for Improvement in MDD Metrics
- Create and implement criteria for patients who do not need a follow-up plan based on clinician judgment.
- Add methods for clinicians to measure and indicate severity of MDD.
- Enable use of a wider array of evidence-based tools and screenings to screen for MDD.
- Allow clinicians to document changes in treatment plan.
Bassett is building into the electronic health record a button that documents the screening was not conducted and that it was not appropriate to administer that day. Of course, building these in-house options entails utilizing resources that smaller systems or independent groups of clinicians may lack.
Eric Wei, MD, senior vice president and chief quality officer at NYC Health + Hospitals in New York City, said the ACP report underscores that many measures, even beyond depression, must be improved.
“With burnout and cognitive overload of our providers, on top of the medicine and just trying to come to the right diagnosis and providing the right treatment and the best care experience, you have to remember all these quality metrics and make sure you put all these things in certain places in the electronic health record,” Dr. Wei said.
Still, Dr. Wei said that the annual rate of depression screening across 400,000 patients in his system is 91%. He and his team spent 6 years working to improve uptake among clinicians, and now, they have moved on to increasing rates of administration of the suicide assessment.
Each clinician uses a dashboard to track their individual metric performance, according to Ted Long, MD, senior vice president for ambulatory care and population health at NYC Health + Hospitals. Dr. Long said he is proud of the improvements he and his colleagues have made in catching undiagnosed depression and in other disease states.
At his primary care practice in the Bronx, nearly 9 out of 10 patients with hypertension have their condition under control, he said. How does he know? Measurement tracking.
“Knowing that when a new patient is in front of me with high blood pressure, that there’s a 9 out of 10 chance that after seeing me because of my clinic, not just because of me, I’m going to be able to keep them healthy by controlling their blood pressure, that’s very meaningful to me,” Dr. Long said. “I think that’s the other side: It enables me as a doctor to know that I’m delivering the highest quality of care to my patients.”
Takeaways for Depression Screening and Follow-Up in Clinical Settings
Just because a patient scores positive for the depression screener, a clinician should dig deeper before making a diagnosis.
Patients have the right to refuse a screener and their wishes should be respected.
Providing a screener may not be appropriate at every visit, such as for a patient with a sprained ankle or a potential respiratory infection where time is limited.
Clinicians can clarify within the measure that the patient did not have mental capacity on that visit to fill out the screener.
A version of this article appeared on Medscape.com.
Telestroke Outcomes Rival Traditional Care
These studies set the stage for larger studies comparing outcomes and efficiency of various telemedicine and transport models and gauging stakeholder satisfaction, authors said.
Surprising Results
In a single-site retrospective comparison of 252 patients with acute stroke assessed under an in-house telestroke protocol and 2437 assessed in person, telestroke provided statistically significant advantages in the following areas:
- Door-to-imaging times (median: 38 minutes vs 44)
- Rates of intravenous (18.2% vs 8.6%) and mechanical (10.4% vs 5.1%) treatment
- Length of stay (median: 6 days vs 8)
- Symptomatic hemorrhagic transformation rate (1.1% vs 5.1%)
- Mortality (6.7% vs 11.1%)

The better metrics observed in the telestroke group were especially surprising, said lead author Rodrigo Meirelles Massaud, MD, because the same team of neurologists conducted both types of evaluations. “This consistency ensures that the quality and expertise of medical care were maintained across both groups,” said Dr. Massaud, a neurologist at the Hospital Israelita Albert Einstein in São Paulo, Brazil. The study appeared online in Frontiers in Neurology.
The findings also counter the preconceived notion that distance medicine could be inferior because of the inability to conduct direct physical examinations and the potential for communication failures, he said. The telestroke group’s younger average age (63.5 years vs 69.5 years) and lower initial National Institutes of Health Stroke Scale (NIHSS) scores — 2 versus 3 — might explain the disparity, Dr. Massaud added, because both factors augur improved outcomes.
Conversely, the authors wrote that the in-person group’s lower median door-to-groin puncture time in ischemic stroke (103.5 minutes vs 151.5 for telemedicine) likely resulted from the need to transport patients from satellite facilities to a hub hospital with neurologists on continuous standby. After adjustment for initial NIHSS score and age, both groups achieved similar percentages of patients with modified Rankin Scale (mRS) scores of 0-2 at discharge: 58.5% for in-person evaluation versus 61.9% for telemedicine (P = .028).
Acute Ischemic Stroke
In another study, a systematic review that included 7396 thrombolysed patients with acute ischemic stroke, odds ratios (ORs) revealed no significant differences between telestroke and in-person care for the percentage of mRS scores 0-2 at discharge (1.06; P = .5), 90-day mortality (OR, 1.16; P = .17), and symptomatic intracranial hemorrhage (OR, 0.99; P = .93). The study appeared in the March International Journal of Stroke.

The lack of significant differences between telestroke and in-person care regarding mortality and mRS scores of 0-2 (which defines a good outcome) surprised researchers, said lead author Ahmed Mohamed, who is completing a master of health sciences degree in medical physiology at the University of Toronto Temerty Faculty of Medicine, Toronto, Ontario, Canada.
“When we were starting this project,” he said, “we thought that telemedicine would probably take longer than conventional treatment.” And waiting longer for treatment — especially for patients with acute ischemic stroke — leads to worse outcomes. “However,” Mr. Mohamed said, “that wasn’t the case.” Additional measures that showed no significant differences included rates of intravenous tissue plasminogen activator (ivtPA) use and endovascular mechanical thrombectomy.
Telestroke Expansion
Authors of a study that analyzed the impact of expanding telestroke coverage beyond community ERs credited many postexpansion improvements to the addition of advanced practice providers (APPs). ProMedica Stroke Network, Toledo, Ohio, added seven APPs in June 2020 to provide two-way audiovisual inpatient stroke and TIA consultations and follow-ups at 19 spoke facilities supported by vascular neurologists at the hub comprehensive stroke center (CSC).
Revamping the TS workflow resulted in a threefold increase in TS cart utilization, a 31% decrease in transfers to the CSC, and a higher home discharge rate from spoke hospitals than from the CSC (57.38% versus 52.8%, respectively). Diagnostic sensitivity also improved, with overall decreases in stroke and TIA diagnosis of 11.5% and 39.8%, respectively, and a 12.9% increase in identification of stroke mimics. The study was published in the March Annals of Neurology.
Future Directions
All three author groups called for larger, more granular follow-up studies. Mr. Mohamed said that the 7396-patient review of 33 studies does not show whether video consultations with neurologists produce better outcomes than phone calls, for example, or whether utilizing different telestroke modalities such as a third-party telemedicine service provides better outcomes than other methods. Additionally, authors wrote, future research should compare telestroke versus non-telestroke patient transport models to optimize treatment plans and outcomes and validate potential advantages and disadvantages of telemedicine for patients with acute ischemic stroke.
“There is also a need to understand the long-term outcomes of patients treated via telestroke versus in-person care,” said Dr. Massaud. Future studies could include randomized, controlled trials comparing telestroke to traditional care in various settings with larger sample sizes, he said. “Additionally, research into the cost-effectiveness of telestroke services, patient satisfaction, and the impact of telestroke on different subtypes of stroke could provide a more comprehensive understanding of its benefits and limitations.”
Dr. Massaud and Mr. Mohamed reported no relevant financial interests. Authors of all three studies reported no funding sources or potential conflicts of interest.
These studies set the stage for larger studies comparing outcomes and efficiency of various telemedicine and transport models and gauging stakeholder satisfaction, authors said.
Surprising Results
In a single-site retrospective comparison of 252 patients with acute stroke assessed under an in-house telestroke protocol and 2437 assessed in person, telestroke provided statistically significant advantages in the following areas:
- Door-to-imaging times (median: 38 minutes vs 44)
- Rates of intravenous (18.2% vs 8.6%) and mechanical (10.4% vs 5.1%) treatment
- Length of stay (median: 6 days vs 8)
- Symptomatic hemorrhagic transformation rate (1.1% vs 5.1%)
- Mortality (6.7% vs 11.1%)
The better metrics observed in the telestroke group were especially surprising, said lead author Rodrigo Meirelles Massaud, MD, because the same team of neurologists conducted both types of evaluations. “This consistency ensures that the quality and expertise of medical care were maintained across both groups,” said Dr. Massaud, a neurologist at the Hospital Israelita Albert Einstein in São Paulo, Brazil. The study appeared online in Frontiers in Neurology.
The findings also counter the preconceived notion that distance medicine could be inferior because of the inability to conduct direct physical examinations and the potential for communication failures, he said. The telestroke group’s younger average age (63.5 years vs 69.5 years) and lower initial National Institutes of Health Stroke Scale (NIHSS) scores — 2 versus 3 — might explain the disparity, Dr. Massaud added, because both factors augur improved outcomes.
Conversely, the authors wrote that the in-person group’s lower median door-to-groin puncture time in ischemic stroke (103.5 minutes vs 151.5 for telemedicine) likely resulted from the need to transport patients from satellite facilities to a hub hospital with neurologists on continuous standby. After adjustment for initial NIHSS score and age, both groups achieved similar percentages of patients with modified Rankin Scale (mRS) scores of 0-2 at discharge: 58.5% for in-person evaluation versus 61.9% for telemedicine (P = .028).
Acute Ischemic Stroke
In another study, a systematic review that included 7396 thrombolysed patients with acute ischemic stroke, odds ratios (ORs) revealed no significant differences between telestroke and in-person care for the percentage of mRS scores 0-2 at discharge (1.06; P = .5), 90-day mortality (OR, 1.16; P = .17), and symptomatic intracranial hemorrhage (OR, 0.99; P = .93). The study appeared in the March International Journal of Stroke.
The lack of significant differences between telestroke and in-person care regarding mortality and mRS scores of 0-2 (which defines a good outcome) surprised researchers, said lead author Ahmed Mohamed, who is completing a master of health sciences degree in medical physiology at the University of Toronto Temerty Faculty of Medicine, Toronto, Ontario, Canada.
“When we were starting this project,” he said, “we thought that telemedicine would probably take longer than conventional treatment.” And waiting longer for treatment — especially for patients with acute ischemic stroke — leads to worse outcomes. “However,” Mr. Mohamed said, “that wasn’t the case.” Additional measures that showed no significant differences included rates of intravenous tissue plasminogen activator (ivtPA) use and endovascular mechanical thrombectomy.
Telestroke Expansion
Authors of a study that analyzed the impact of expanding telestroke coverage beyond community ERs credited many postexpansion improvements to the addition of advanced practice providers (APPs). ProMedica Stroke Network, Toledo, Ohio, added seven APPs in June 2020 to provide two-way audiovisual inpatient stroke and TIA consultations and follow-ups at 19 spoke facilities supported by vascular neurologists at the hub comprehensive stroke center (CSC).
Revamping the TS workflow resulted in a threefold increase in TS cart utilization, a 31% decrease in transfers to the CSC, and a higher home discharge rate from spoke hospitals than from the CSC (57.38% versus 52.8%, respectively). Diagnostic sensitivity also improved, with overall decreases in stroke and TIA diagnosis of 11.5% and 39.8%, respectively, and a 12.9% increase in identification of stroke mimics. The study was published in the March Annals of Neurology.
Future Directions
All three author groups called for larger, more granular follow-up studies. Mr. Mohamed said that the 7396-patient review of 33 studies does not show whether video consultations with neurologists produce better outcomes than phone calls, for example, or whether utilizing different telestroke modalities such as a third-party telemedicine service provides better outcomes than other methods. Additionally, authors wrote, future research should compare telestroke versus non-telestroke patient transport models to optimize treatment plans and outcomes and validate potential advantages and disadvantages of telemedicine for patients with acute ischemic stroke.
“There is also a need to understand the long-term outcomes of patients treated via telestroke versus in-person care,” said Dr. Massaud. Future studies could include randomized, controlled trials comparing telestroke to traditional care in various settings with larger sample sizes, he said. “Additionally, research into the cost-effectiveness of telestroke services, patient satisfaction, and the impact of telestroke on different subtypes of stroke could provide a more comprehensive understanding of its benefits and limitations.”
Dr. Massaud and Mr. Mohamed reported no relevant financial interests. Authors of all three studies reported no funding sources or potential conflicts of interest.
These studies set the stage for larger studies comparing outcomes and efficiency of various telemedicine and transport models and gauging stakeholder satisfaction, authors said.
Surprising Results
In a single-site retrospective comparison of 252 patients with acute stroke assessed under an in-house telestroke protocol and 2437 assessed in person, telestroke provided statistically significant advantages in the following areas:
- Door-to-imaging times (median: 38 minutes vs 44)
- Rates of intravenous (18.2% vs 8.6%) and mechanical (10.4% vs 5.1%) treatment
- Length of stay (median: 6 days vs 8)
- Symptomatic hemorrhagic transformation rate (1.1% vs 5.1%)
- Mortality (6.7% vs 11.1%)
The better metrics observed in the telestroke group were especially surprising, said lead author Rodrigo Meirelles Massaud, MD, because the same team of neurologists conducted both types of evaluations. “This consistency ensures that the quality and expertise of medical care were maintained across both groups,” said Dr. Massaud, a neurologist at the Hospital Israelita Albert Einstein in São Paulo, Brazil. The study appeared online in Frontiers in Neurology.
The findings also counter the preconceived notion that distance medicine could be inferior because of the inability to conduct direct physical examinations and the potential for communication failures, he said. The telestroke group’s younger average age (63.5 years vs 69.5 years) and lower initial National Institutes of Health Stroke Scale (NIHSS) scores — 2 versus 3 — might explain the disparity, Dr. Massaud added, because both factors augur improved outcomes.
Conversely, the authors wrote that the in-person group’s lower median door-to-groin puncture time in ischemic stroke (103.5 minutes vs 151.5 for telemedicine) likely resulted from the need to transport patients from satellite facilities to a hub hospital with neurologists on continuous standby. After adjustment for initial NIHSS score and age, both groups achieved similar percentages of patients with modified Rankin Scale (mRS) scores of 0-2 at discharge: 58.5% for in-person evaluation versus 61.9% for telemedicine (P = .028).
Acute Ischemic Stroke
In another study, a systematic review that included 7396 thrombolysed patients with acute ischemic stroke, odds ratios (ORs) revealed no significant differences between telestroke and in-person care for the percentage of mRS scores 0-2 at discharge (1.06; P = .5), 90-day mortality (OR, 1.16; P = .17), and symptomatic intracranial hemorrhage (OR, 0.99; P = .93). The study appeared in the March International Journal of Stroke.
The lack of significant differences between telestroke and in-person care regarding mortality and mRS scores of 0-2 (which defines a good outcome) surprised researchers, said lead author Ahmed Mohamed, who is completing a master of health sciences degree in medical physiology at the University of Toronto Temerty Faculty of Medicine, Toronto, Ontario, Canada.
“When we were starting this project,” he said, “we thought that telemedicine would probably take longer than conventional treatment.” And waiting longer for treatment — especially for patients with acute ischemic stroke — leads to worse outcomes. “However,” Mr. Mohamed said, “that wasn’t the case.” Additional measures that showed no significant differences included rates of intravenous tissue plasminogen activator (ivtPA) use and endovascular mechanical thrombectomy.
Telestroke Expansion
Authors of a study that analyzed the impact of expanding telestroke coverage beyond community ERs credited many postexpansion improvements to the addition of advanced practice providers (APPs). ProMedica Stroke Network, Toledo, Ohio, added seven APPs in June 2020 to provide two-way audiovisual inpatient stroke and TIA consultations and follow-ups at 19 spoke facilities supported by vascular neurologists at the hub comprehensive stroke center (CSC).
Revamping the TS workflow resulted in a threefold increase in TS cart utilization, a 31% decrease in transfers to the CSC, and a higher home discharge rate from spoke hospitals than from the CSC (57.38% versus 52.8%, respectively). Diagnostic sensitivity also improved, with overall decreases in stroke and TIA diagnosis of 11.5% and 39.8%, respectively, and a 12.9% increase in identification of stroke mimics. The study was published in the March Annals of Neurology.
Future Directions
All three author groups called for larger, more granular follow-up studies. Mr. Mohamed said that the 7396-patient review of 33 studies does not show whether video consultations with neurologists produce better outcomes than phone calls, for example, or whether utilizing different telestroke modalities such as a third-party telemedicine service provides better outcomes than other methods. Additionally, authors wrote, future research should compare telestroke versus non-telestroke patient transport models to optimize treatment plans and outcomes and validate potential advantages and disadvantages of telemedicine for patients with acute ischemic stroke.
“There is also a need to understand the long-term outcomes of patients treated via telestroke versus in-person care,” said Dr. Massaud. Future studies could include randomized, controlled trials comparing telestroke to traditional care in various settings with larger sample sizes, he said. “Additionally, research into the cost-effectiveness of telestroke services, patient satisfaction, and the impact of telestroke on different subtypes of stroke could provide a more comprehensive understanding of its benefits and limitations.”
Dr. Massaud and Mr. Mohamed reported no relevant financial interests. Authors of all three studies reported no funding sources or potential conflicts of interest.
FROM FRONTIERS IN NEUROLOGY, INTERNATIONAL JOURNAL OF STROKE, AND ANNALS OF NEUROLOGY
For Some MDs, Long COVID Burnout Is a New Reality
Dhaval Desai, MD, was teaching his 4-year-old to ride a bike after another exhausting shift at the hospital during the summer after the first COVID-19 surge. He was putting on a happy face and forcing out a “Yay!” he did not feel. The pandemic had taken its toll, and he just wanted to lie down and be alone. Realizing that he was “scraping to find joy” was when he knew something was wrong.
“I was giving, giving, giving at work a lot, and I had little left to give at home,” said Dr. Desai, director of hospital medicine at Emory Saint Joseph’s Hospital and an assistant professor of medicine at Emory University in Atlanta, Georgia.
At work, he worried about his wife managing two kids — including a newborn — during the pandemic. At home, he stressed about work and the crush of patients with COVID the hospital was grappling to handle. He was exhausted, resentful, and angry, and it was jeopardizing what mattered most to him: His home life.
“It was all colliding…I realized, OK, I’m struggling,” he said.
Dr. Desai is one of thousands of physicians across the United States who have experienced burnout and depression, exacerbated by the pandemic. After 4 years, the impact is still being felt. Medscape’s 2024 annual report on this issue found that burnout and depression among doctors — while encouragingly better than the prior year — remain higher than before COVID. For doctors caring for patients with long COVID, those suffering from the debilitating aftereffects of an infection, the sense of helplessness when recovery is elusive can also weigh heavily.
Overall, more female physicians reported feeling burned out and depressed. Experts attributed this gap to issues including fewer women in supportive leadership and mentoring roles, compensation disparities, fewer career advancement opportunities, and more responsibilities caring for children and elders.
Multiple international studies and reports have highlighted the surge in burnout experienced by physicians and healthcare workers globally during the pandemic. Even before COVID, studies found the suicide rate among male and female US physicians was higher than the general population and higher than any other profession, including the military. The risk among female physicians, in particular, was 250%-400% higher.
“That’s really, on average, one a day, and that’s really unacceptable. No one should die by suicide, but a physician who knows the risks and knows that, should never do that,” said Dr. Desai about suicides overall among doctors.
The story of Lorna Breen had rattled Dr. Desai. Dr. Breen was a Manhattan physician who died by suicide in April 2020 after grappling with the city’s devastating first wave and then contracting COVID-19 herself. While Dr. Desai did not have thoughts of suicide, he was facing his own battles. Those experiences and the stigma around mental health prompted him to write his book, Burning Out on the Covid Front Lines: A Doctor’s Memoir of Fatherhood, Race and Perseverance in the Pandemic, with the hope that it can help others like him.
Mental Health Stigma
But despite the body of research and growing awareness toward addressing mental health among physicians, almost four in 10 doctors are wary of revealing their personal struggles.
More than half of those surveyed in the Medscape Medical News report said they had not consulted a mental health professional before and would not do so going forward either. The fear of tarnishing their reputation or even losing their license keeps doctors silent. Advocates and groups like the Dr. Lorna Breen Heroes’ Foundation are pushing for hospitals and healthcare systems to remove and rephrase invasive and stigmatizing language around mental health in licensure, credentialing, or insurance applications.
Burnout Triggers: Systemic Problems, Social Tensions
Burnout can make a person feel “depleted and used up” and is characterized by extreme tiredness, low energy, frustration about work, emotional distance or numbness, and difficulty with concentration, responsibilities, or creativity. It can make an individual feel helpless, alone, defeated, cynical, and without purpose and can also cause physical symptoms such as headaches, loss of appetite, insomnia, and body aches. Unaddressed, it can lead to depression, anxiety, and a variety of physical health issues.
“We can still be highly functional and not okay,” said Dr. Desai.
For doctors, burnout often builds over time from large and small systemic problems and inefficiencies, multiplied by a dozen or more patients each day: Not enough time for documentation, complicated paperwork, navigating byzantine health and insurance systems, and hitting roadblocks. The administrative work, combined with an enormous patient load, and staffing and resource shortages create barriers to care and cuts into the amount of time they can spend providing actual care.
These existing problems worsened as patients with COVID overwhelmed hospitals and clinics. At the same time, healthcare workers worried about caring for the sick, getting infected themselves, or having multiple staff falling ill at once. As each surge came and went, backlash, hostility, abuse, and even violence toward healthcare workers also increased. The discrimination some medical staff were subjected to compounded the burnout.
“When we’re not getting the support we need as physicians and healthcare workers, that adds to burnout, and I saw that in my colleagues,” said Dr. Desai.
Impact of Burnout
At the Mount Sinai Center for Post-COVID Care in New York City, doctors grapple with feelings of helplessness in caring for patients with long COVID who show little sign of recovery. That emotional toll can also be difficult, said director Zijian Chen, MD, who helped launch the clinic in May 2020.
“Sometimes you’re faced with patients who you’re trying to do everything for, but they’re not just not getting better,” said Dr. Chen. “It’s really frustrating because we want everybody to get better. So, there’s that lack of fulfillment there that can cause a sense of burnout.”
While the worst outcomes and death rates initially brought on by acute infections have lessened, long COVID clinics exemplify some of the ongoing challenges within healthcare. Many operate with insufficient financial and staffing resources despite wait-lists and a steady flow of new and returning patients. Even with the demand, a number of these clinics have shuttered, leaving patients without access to much-needed medical help.
“There are clinicians who are burning out. That is definitely something that I’ve seen,” said Monica Verduzco-Gutierrez, MD, a professor and chair of the Department of Rehabilitation Medicine at the University of Texas Health Science Center in San Antonio, Texas.
“[It] takes a lot of resources for a successful long COVID clinic. A lot of special funding may be drying up and couple that with clinicians burning out, then they’re going to shut their doors.”
And it’s not just long COVID clinics. Data have shown an overall exodus in healthcare, especially during the pandemic. One study found burnout was one of the “most impactful” predictors of a physician’s intention to leave the profession during the pandemic. The loss of talent and skills during a major health crisis can put the entire system under stress, with patients ultimately suffering from poorer care.
“Healthcare system fragility and the chaos is far worse than it was before. We are continuing to be forced to do more with less,” said Dr. Desai.
Alleviating Burnout
While it is difficult to assess whether burnout from the pandemic is transient, experts say this is an opportunity for health institutions to learn from these experiences and implement policies and actions that can help reduce the mental health strain on staff. One study found that changes made by organizations had a bigger positive impact on reducing burnout than individual changes.
Advocates say more support staff, more work flexibility, and higher compensation would significantly ease the burden that drives burnout and depression.
In addition, half the physicians surveyed in the Medscape Medical News report felt their employers were not acknowledging how pervasive burnout is at their workplace. Having a trusted peer or leader set an example by sharing his or her own challenging experiences and saying it›s time to address these struggles can be an enormously validating step forward, said Dr. Desai. Acknowledging his own difficulties was not only a huge weight off his shoulders but also helped surpris colleagues who sought him out for counsel.
“I’m not suggesting everybody get on medication,” he said. “But talking to a therapist, acknowledging there’s issues, restructuring your life to realize something’s off, and just knowing that you’re not alone? That’s huge.”
Dr. Desai said he still faces personal challenges but is in a much better place, doing well at work and at home. He talks to a therapist, is taking medication, and has developed better coping mechanisms. He is spending more time with his family, detaching for a few hours from work-related emails, learning to draw boundaries and say no, and trying to be more present and “intentional” in connecting with colleagues and patients.
“It’s okay to not be okay,” said Dr. Desai. “It’s okay to be vulnerable and acknowledge when we can’t do more.”
Are you in a crisis? Call or text 988 or text TALK to 741741. For immediate support for healthcare professionals, as well as resources for institutions and organizations, visit: afsp.org/suicide-prevention-for-healthcare-professionals/#facts-about-mental-health-and-suicide.
A version of this article appeared on Medscape.com.
Dhaval Desai, MD, was teaching his 4-year-old to ride a bike after another exhausting shift at the hospital during the summer after the first COVID-19 surge. He was putting on a happy face and forcing out a “Yay!” he did not feel. The pandemic had taken its toll, and he just wanted to lie down and be alone. Realizing that he was “scraping to find joy” was when he knew something was wrong.
“I was giving, giving, giving at work a lot, and I had little left to give at home,” said Dr. Desai, director of hospital medicine at Emory Saint Joseph’s Hospital and an assistant professor of medicine at Emory University in Atlanta, Georgia.
At work, he worried about his wife managing two kids — including a newborn — during the pandemic. At home, he stressed about work and the crush of patients with COVID the hospital was grappling to handle. He was exhausted, resentful, and angry, and it was jeopardizing what mattered most to him: His home life.
“It was all colliding…I realized, OK, I’m struggling,” he said.
Dr. Desai is one of thousands of physicians across the United States who have experienced burnout and depression, exacerbated by the pandemic. After 4 years, the impact is still being felt. Medscape’s 2024 annual report on this issue found that burnout and depression among doctors — while encouragingly better than the prior year — remain higher than before COVID. For doctors caring for patients with long COVID, those suffering from the debilitating aftereffects of an infection, the sense of helplessness when recovery is elusive can also weigh heavily.
Overall, more female physicians reported feeling burned out and depressed. Experts attributed this gap to issues including fewer women in supportive leadership and mentoring roles, compensation disparities, fewer career advancement opportunities, and more responsibilities caring for children and elders.
Multiple international studies and reports have highlighted the surge in burnout experienced by physicians and healthcare workers globally during the pandemic. Even before COVID, studies found the suicide rate among male and female US physicians was higher than the general population and higher than any other profession, including the military. The risk among female physicians, in particular, was 250%-400% higher.
“That’s really, on average, one a day, and that’s really unacceptable. No one should die by suicide, but a physician who knows the risks and knows that, should never do that,” said Dr. Desai about suicides overall among doctors.
The story of Lorna Breen had rattled Dr. Desai. Dr. Breen was a Manhattan physician who died by suicide in April 2020 after grappling with the city’s devastating first wave and then contracting COVID-19 herself. While Dr. Desai did not have thoughts of suicide, he was facing his own battles. Those experiences and the stigma around mental health prompted him to write his book, Burning Out on the Covid Front Lines: A Doctor’s Memoir of Fatherhood, Race and Perseverance in the Pandemic, with the hope that it can help others like him.
Mental Health Stigma
But despite the body of research and growing awareness toward addressing mental health among physicians, almost four in 10 doctors are wary of revealing their personal struggles.
More than half of those surveyed in the Medscape Medical News report said they had not consulted a mental health professional before and would not do so going forward either. The fear of tarnishing their reputation or even losing their license keeps doctors silent. Advocates and groups like the Dr. Lorna Breen Heroes’ Foundation are pushing for hospitals and healthcare systems to remove and rephrase invasive and stigmatizing language around mental health in licensure, credentialing, or insurance applications.
Burnout Triggers: Systemic Problems, Social Tensions
Burnout can make a person feel “depleted and used up” and is characterized by extreme tiredness, low energy, frustration about work, emotional distance or numbness, and difficulty with concentration, responsibilities, or creativity. It can make an individual feel helpless, alone, defeated, cynical, and without purpose and can also cause physical symptoms such as headaches, loss of appetite, insomnia, and body aches. Unaddressed, it can lead to depression, anxiety, and a variety of physical health issues.
“We can still be highly functional and not okay,” said Dr. Desai.
For doctors, burnout often builds over time from large and small systemic problems and inefficiencies, multiplied by a dozen or more patients each day: Not enough time for documentation, complicated paperwork, navigating byzantine health and insurance systems, and hitting roadblocks. The administrative work, combined with an enormous patient load, and staffing and resource shortages create barriers to care and cuts into the amount of time they can spend providing actual care.
These existing problems worsened as patients with COVID overwhelmed hospitals and clinics. At the same time, healthcare workers worried about caring for the sick, getting infected themselves, or having multiple staff falling ill at once. As each surge came and went, backlash, hostility, abuse, and even violence toward healthcare workers also increased. The discrimination some medical staff were subjected to compounded the burnout.
“When we’re not getting the support we need as physicians and healthcare workers, that adds to burnout, and I saw that in my colleagues,” said Dr. Desai.
Impact of Burnout
At the Mount Sinai Center for Post-COVID Care in New York City, doctors grapple with feelings of helplessness in caring for patients with long COVID who show little sign of recovery. That emotional toll can also be difficult, said director Zijian Chen, MD, who helped launch the clinic in May 2020.
“Sometimes you’re faced with patients who you’re trying to do everything for, but they’re not just not getting better,” said Dr. Chen. “It’s really frustrating because we want everybody to get better. So, there’s that lack of fulfillment there that can cause a sense of burnout.”
While the worst outcomes and death rates initially brought on by acute infections have lessened, long COVID clinics exemplify some of the ongoing challenges within healthcare. Many operate with insufficient financial and staffing resources despite wait-lists and a steady flow of new and returning patients. Even with the demand, a number of these clinics have shuttered, leaving patients without access to much-needed medical help.
“There are clinicians who are burning out. That is definitely something that I’ve seen,” said Monica Verduzco-Gutierrez, MD, a professor and chair of the Department of Rehabilitation Medicine at the University of Texas Health Science Center in San Antonio, Texas.
“[It] takes a lot of resources for a successful long COVID clinic. A lot of special funding may be drying up and couple that with clinicians burning out, then they’re going to shut their doors.”
And it’s not just long COVID clinics. Data have shown an overall exodus in healthcare, especially during the pandemic. One study found burnout was one of the “most impactful” predictors of a physician’s intention to leave the profession during the pandemic. The loss of talent and skills during a major health crisis can put the entire system under stress, with patients ultimately suffering from poorer care.
“Healthcare system fragility and the chaos is far worse than it was before. We are continuing to be forced to do more with less,” said Dr. Desai.
Alleviating Burnout
While it is difficult to assess whether burnout from the pandemic is transient, experts say this is an opportunity for health institutions to learn from these experiences and implement policies and actions that can help reduce the mental health strain on staff. One study found that changes made by organizations had a bigger positive impact on reducing burnout than individual changes.
Advocates say more support staff, more work flexibility, and higher compensation would significantly ease the burden that drives burnout and depression.
In addition, half the physicians surveyed in the Medscape Medical News report felt their employers were not acknowledging how pervasive burnout is at their workplace. Having a trusted peer or leader set an example by sharing his or her own challenging experiences and saying it›s time to address these struggles can be an enormously validating step forward, said Dr. Desai. Acknowledging his own difficulties was not only a huge weight off his shoulders but also helped surpris colleagues who sought him out for counsel.
“I’m not suggesting everybody get on medication,” he said. “But talking to a therapist, acknowledging there’s issues, restructuring your life to realize something’s off, and just knowing that you’re not alone? That’s huge.”
Dr. Desai said he still faces personal challenges but is in a much better place, doing well at work and at home. He talks to a therapist, is taking medication, and has developed better coping mechanisms. He is spending more time with his family, detaching for a few hours from work-related emails, learning to draw boundaries and say no, and trying to be more present and “intentional” in connecting with colleagues and patients.
“It’s okay to not be okay,” said Dr. Desai. “It’s okay to be vulnerable and acknowledge when we can’t do more.”
Are you in a crisis? Call or text 988 or text TALK to 741741. For immediate support for healthcare professionals, as well as resources for institutions and organizations, visit: afsp.org/suicide-prevention-for-healthcare-professionals/#facts-about-mental-health-and-suicide.
A version of this article appeared on Medscape.com.
Dhaval Desai, MD, was teaching his 4-year-old to ride a bike after another exhausting shift at the hospital during the summer after the first COVID-19 surge. He was putting on a happy face and forcing out a “Yay!” he did not feel. The pandemic had taken its toll, and he just wanted to lie down and be alone. Realizing that he was “scraping to find joy” was when he knew something was wrong.
“I was giving, giving, giving at work a lot, and I had little left to give at home,” said Dr. Desai, director of hospital medicine at Emory Saint Joseph’s Hospital and an assistant professor of medicine at Emory University in Atlanta, Georgia.
At work, he worried about his wife managing two kids — including a newborn — during the pandemic. At home, he stressed about work and the crush of patients with COVID the hospital was grappling to handle. He was exhausted, resentful, and angry, and it was jeopardizing what mattered most to him: His home life.
“It was all colliding…I realized, OK, I’m struggling,” he said.
Dr. Desai is one of thousands of physicians across the United States who have experienced burnout and depression, exacerbated by the pandemic. After 4 years, the impact is still being felt. Medscape’s 2024 annual report on this issue found that burnout and depression among doctors — while encouragingly better than the prior year — remain higher than before COVID. For doctors caring for patients with long COVID, those suffering from the debilitating aftereffects of an infection, the sense of helplessness when recovery is elusive can also weigh heavily.
Overall, more female physicians reported feeling burned out and depressed. Experts attributed this gap to issues including fewer women in supportive leadership and mentoring roles, compensation disparities, fewer career advancement opportunities, and more responsibilities caring for children and elders.
Multiple international studies and reports have highlighted the surge in burnout experienced by physicians and healthcare workers globally during the pandemic. Even before COVID, studies found the suicide rate among male and female US physicians was higher than the general population and higher than any other profession, including the military. The risk among female physicians, in particular, was 250%-400% higher.
“That’s really, on average, one a day, and that’s really unacceptable. No one should die by suicide, but a physician who knows the risks and knows that, should never do that,” said Dr. Desai about suicides overall among doctors.
The story of Lorna Breen had rattled Dr. Desai. Dr. Breen was a Manhattan physician who died by suicide in April 2020 after grappling with the city’s devastating first wave and then contracting COVID-19 herself. While Dr. Desai did not have thoughts of suicide, he was facing his own battles. Those experiences and the stigma around mental health prompted him to write his book, Burning Out on the Covid Front Lines: A Doctor’s Memoir of Fatherhood, Race and Perseverance in the Pandemic, with the hope that it can help others like him.
Mental Health Stigma
But despite the body of research and growing awareness toward addressing mental health among physicians, almost four in 10 doctors are wary of revealing their personal struggles.
More than half of those surveyed in the Medscape Medical News report said they had not consulted a mental health professional before and would not do so going forward either. The fear of tarnishing their reputation or even losing their license keeps doctors silent. Advocates and groups like the Dr. Lorna Breen Heroes’ Foundation are pushing for hospitals and healthcare systems to remove and rephrase invasive and stigmatizing language around mental health in licensure, credentialing, or insurance applications.
Burnout Triggers: Systemic Problems, Social Tensions
Burnout can make a person feel “depleted and used up” and is characterized by extreme tiredness, low energy, frustration about work, emotional distance or numbness, and difficulty with concentration, responsibilities, or creativity. It can make an individual feel helpless, alone, defeated, cynical, and without purpose and can also cause physical symptoms such as headaches, loss of appetite, insomnia, and body aches. Unaddressed, it can lead to depression, anxiety, and a variety of physical health issues.
“We can still be highly functional and not okay,” said Dr. Desai.
For doctors, burnout often builds over time from large and small systemic problems and inefficiencies, multiplied by a dozen or more patients each day: Not enough time for documentation, complicated paperwork, navigating byzantine health and insurance systems, and hitting roadblocks. The administrative work, combined with an enormous patient load, and staffing and resource shortages create barriers to care and cuts into the amount of time they can spend providing actual care.
These existing problems worsened as patients with COVID overwhelmed hospitals and clinics. At the same time, healthcare workers worried about caring for the sick, getting infected themselves, or having multiple staff falling ill at once. As each surge came and went, backlash, hostility, abuse, and even violence toward healthcare workers also increased. The discrimination some medical staff were subjected to compounded the burnout.
“When we’re not getting the support we need as physicians and healthcare workers, that adds to burnout, and I saw that in my colleagues,” said Dr. Desai.
Impact of Burnout
At the Mount Sinai Center for Post-COVID Care in New York City, doctors grapple with feelings of helplessness in caring for patients with long COVID who show little sign of recovery. That emotional toll can also be difficult, said director Zijian Chen, MD, who helped launch the clinic in May 2020.
“Sometimes you’re faced with patients who you’re trying to do everything for, but they’re not just not getting better,” said Dr. Chen. “It’s really frustrating because we want everybody to get better. So, there’s that lack of fulfillment there that can cause a sense of burnout.”
While the worst outcomes and death rates initially brought on by acute infections have lessened, long COVID clinics exemplify some of the ongoing challenges within healthcare. Many operate with insufficient financial and staffing resources despite wait-lists and a steady flow of new and returning patients. Even with the demand, a number of these clinics have shuttered, leaving patients without access to much-needed medical help.
“There are clinicians who are burning out. That is definitely something that I’ve seen,” said Monica Verduzco-Gutierrez, MD, a professor and chair of the Department of Rehabilitation Medicine at the University of Texas Health Science Center in San Antonio, Texas.
“[It] takes a lot of resources for a successful long COVID clinic. A lot of special funding may be drying up and couple that with clinicians burning out, then they’re going to shut their doors.”
And it’s not just long COVID clinics. Data have shown an overall exodus in healthcare, especially during the pandemic. One study found burnout was one of the “most impactful” predictors of a physician’s intention to leave the profession during the pandemic. The loss of talent and skills during a major health crisis can put the entire system under stress, with patients ultimately suffering from poorer care.
“Healthcare system fragility and the chaos is far worse than it was before. We are continuing to be forced to do more with less,” said Dr. Desai.
Alleviating Burnout
While it is difficult to assess whether burnout from the pandemic is transient, experts say this is an opportunity for health institutions to learn from these experiences and implement policies and actions that can help reduce the mental health strain on staff. One study found that changes made by organizations had a bigger positive impact on reducing burnout than individual changes.
Advocates say more support staff, more work flexibility, and higher compensation would significantly ease the burden that drives burnout and depression.
In addition, half the physicians surveyed in the Medscape Medical News report felt their employers were not acknowledging how pervasive burnout is at their workplace. Having a trusted peer or leader set an example by sharing his or her own challenging experiences and saying it›s time to address these struggles can be an enormously validating step forward, said Dr. Desai. Acknowledging his own difficulties was not only a huge weight off his shoulders but also helped surpris colleagues who sought him out for counsel.
“I’m not suggesting everybody get on medication,” he said. “But talking to a therapist, acknowledging there’s issues, restructuring your life to realize something’s off, and just knowing that you’re not alone? That’s huge.”
Dr. Desai said he still faces personal challenges but is in a much better place, doing well at work and at home. He talks to a therapist, is taking medication, and has developed better coping mechanisms. He is spending more time with his family, detaching for a few hours from work-related emails, learning to draw boundaries and say no, and trying to be more present and “intentional” in connecting with colleagues and patients.
“It’s okay to not be okay,” said Dr. Desai. “It’s okay to be vulnerable and acknowledge when we can’t do more.”
Are you in a crisis? Call or text 988 or text TALK to 741741. For immediate support for healthcare professionals, as well as resources for institutions and organizations, visit: afsp.org/suicide-prevention-for-healthcare-professionals/#facts-about-mental-health-and-suicide.
A version of this article appeared on Medscape.com.
Tuberculosis Prevention Brings Economic Gains, Says WHO
A new study conducted by the World Health Organization (WHO) suggests that in addition to providing significant improvements in public health, investment in the diagnosis and prevention of tuberculosis also generates economic benefits.
According to a survey conducted by governments and researchers from Brazil, Georgia, Kenya, and South Africa, even modest increases in funding for measures against tuberculosis can bring gains. Every $1 invested produces returns of as much as $39, it found.
The findings may remind governments and policymakers about the importance of investing in public health policies. According to the WHO, the study “provides strong economic arguments” about the true costs of tuberculosis and proves the benefits of increasing funding to accelerate the diagnosis and preventive treatment of the disease.
UN Targets Tuberculosis
In September 2023, during the last meeting of the United Nations General Assembly, following a widespread worsening of disease indicators because of the COVID-19 pandemic, world leaders signed a declaration committing to the expansion of efforts to combat tuberculosis during the next 5 years. The current WHO study was developed to provide a road map for the implementation of key measures against the disease.
The survey highlights two fundamental actions: The expansion of screening, especially in populations considered more vulnerable, and the provision of tuberculosis preventive treatment (TPT), which entails administering drugs to people who have been exposed to the disease but have not yet developed it.
“TPT is a proven and effective intervention to prevent the development of tuberculosis among exposed people, reducing the risk of developing the disease by about 60%-90% compared with individuals who did not receive it,” the document emphasized.
Investments Yield Returns
To achieve the necessary coverage levels, the study estimated that Brazil would need to increase annual per capita investment by $0.28 (about R$1.41) between 2024 and 2050. Brazilian society, in turn, would receive a return of $11 (R$55.27) for every dollar invested.
For South Africa, whose per capita investment increase is estimated at $1.10 per year, the return would be even more significant: $39 for every dollar allocated.
The WHO emphasized that funding for combating the disease is much lower than the value of the damage it causes to nations. “Tuberculosis has high costs for society. Only a small proportion of these costs go directly to the health system (ranging from 1.7% in South Africa to 7.8% in Kenya). Most are costs for patients and society.”
The study projected that between 2024 and 2050, the total cost of tuberculosis to Brazilian society would be $81.2 billion, with an average annual value of $3.01 billion. This figure represents, in 2024, 0.16% of the country’s gross domestic product.
Achieving screening and preventive treatment goals in Brazil would lead to a reduction of as much as 18% in the national incidence of the disease, as well as 1.9 million fewer deaths, between 2024 and 2050.
Although treatable and preventable, tuberculosis remains the leading cause of death from infectious agents worldwide. It is estimated that over 1.3 million people died from the disease in 2022.
The document provides the “health and economic justification for investing in evidence-based interventions recommended by WHO in tuberculosis screening and prevention,” according to WHO Director-General Tedros Adhanom Ghebreyesus, PhD.
“Today we have the knowledge, tools, and political commitment that can end this age-old disease that continues to be one of the leading causes of death from infectious diseases worldwide,” he added.
Emerging Concerns
Although the WHO highlighted the global increase in access to tuberculosis diagnosis and treatment in 2022, which coincided with the recovery of healthcare systems in several countries after the beginning of the pandemic, it emphasized that the implementation of preventive treatment for exposed individuals and high-vulnerability populations remains slow.
Another concern is the increase in drug resistance. Multidrug-resistant tuberculosis is considered a public health crisis. It is estimated that about 410,000 people had multidrug-resistant tuberculosis or rifampicin-resistant tuberculosis in 2022, but only two of every five patients had access to treatment.This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com .
A new study conducted by the World Health Organization (WHO) suggests that in addition to providing significant improvements in public health, investment in the diagnosis and prevention of tuberculosis also generates economic benefits.
According to a survey conducted by governments and researchers from Brazil, Georgia, Kenya, and South Africa, even modest increases in funding for measures against tuberculosis can bring gains. Every $1 invested produces returns of as much as $39, it found.
The findings may remind governments and policymakers about the importance of investing in public health policies. According to the WHO, the study “provides strong economic arguments” about the true costs of tuberculosis and proves the benefits of increasing funding to accelerate the diagnosis and preventive treatment of the disease.
UN Targets Tuberculosis
In September 2023, during the last meeting of the United Nations General Assembly, following a widespread worsening of disease indicators because of the COVID-19 pandemic, world leaders signed a declaration committing to the expansion of efforts to combat tuberculosis during the next 5 years. The current WHO study was developed to provide a road map for the implementation of key measures against the disease.
The survey highlights two fundamental actions: The expansion of screening, especially in populations considered more vulnerable, and the provision of tuberculosis preventive treatment (TPT), which entails administering drugs to people who have been exposed to the disease but have not yet developed it.
“TPT is a proven and effective intervention to prevent the development of tuberculosis among exposed people, reducing the risk of developing the disease by about 60%-90% compared with individuals who did not receive it,” the document emphasized.
Investments Yield Returns
To achieve the necessary coverage levels, the study estimated that Brazil would need to increase annual per capita investment by $0.28 (about R$1.41) between 2024 and 2050. Brazilian society, in turn, would receive a return of $11 (R$55.27) for every dollar invested.
For South Africa, whose per capita investment increase is estimated at $1.10 per year, the return would be even more significant: $39 for every dollar allocated.
The WHO emphasized that funding for combating the disease is much lower than the value of the damage it causes to nations. “Tuberculosis has high costs for society. Only a small proportion of these costs go directly to the health system (ranging from 1.7% in South Africa to 7.8% in Kenya). Most are costs for patients and society.”
The study projected that between 2024 and 2050, the total cost of tuberculosis to Brazilian society would be $81.2 billion, with an average annual value of $3.01 billion. This figure represents, in 2024, 0.16% of the country’s gross domestic product.
Achieving screening and preventive treatment goals in Brazil would lead to a reduction of as much as 18% in the national incidence of the disease, as well as 1.9 million fewer deaths, between 2024 and 2050.
Although treatable and preventable, tuberculosis remains the leading cause of death from infectious agents worldwide. It is estimated that over 1.3 million people died from the disease in 2022.
The document provides the “health and economic justification for investing in evidence-based interventions recommended by WHO in tuberculosis screening and prevention,” according to WHO Director-General Tedros Adhanom Ghebreyesus, PhD.
“Today we have the knowledge, tools, and political commitment that can end this age-old disease that continues to be one of the leading causes of death from infectious diseases worldwide,” he added.
Emerging Concerns
Although the WHO highlighted the global increase in access to tuberculosis diagnosis and treatment in 2022, which coincided with the recovery of healthcare systems in several countries after the beginning of the pandemic, it emphasized that the implementation of preventive treatment for exposed individuals and high-vulnerability populations remains slow.
Another concern is the increase in drug resistance. Multidrug-resistant tuberculosis is considered a public health crisis. It is estimated that about 410,000 people had multidrug-resistant tuberculosis or rifampicin-resistant tuberculosis in 2022, but only two of every five patients had access to treatment.This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com .
A new study conducted by the World Health Organization (WHO) suggests that in addition to providing significant improvements in public health, investment in the diagnosis and prevention of tuberculosis also generates economic benefits.
According to a survey conducted by governments and researchers from Brazil, Georgia, Kenya, and South Africa, even modest increases in funding for measures against tuberculosis can bring gains. Every $1 invested produces returns of as much as $39, it found.
The findings may remind governments and policymakers about the importance of investing in public health policies. According to the WHO, the study “provides strong economic arguments” about the true costs of tuberculosis and proves the benefits of increasing funding to accelerate the diagnosis and preventive treatment of the disease.
UN Targets Tuberculosis
In September 2023, during the last meeting of the United Nations General Assembly, following a widespread worsening of disease indicators because of the COVID-19 pandemic, world leaders signed a declaration committing to the expansion of efforts to combat tuberculosis during the next 5 years. The current WHO study was developed to provide a road map for the implementation of key measures against the disease.
The survey highlights two fundamental actions: The expansion of screening, especially in populations considered more vulnerable, and the provision of tuberculosis preventive treatment (TPT), which entails administering drugs to people who have been exposed to the disease but have not yet developed it.
“TPT is a proven and effective intervention to prevent the development of tuberculosis among exposed people, reducing the risk of developing the disease by about 60%-90% compared with individuals who did not receive it,” the document emphasized.
Investments Yield Returns
To achieve the necessary coverage levels, the study estimated that Brazil would need to increase annual per capita investment by $0.28 (about R$1.41) between 2024 and 2050. Brazilian society, in turn, would receive a return of $11 (R$55.27) for every dollar invested.
For South Africa, whose per capita investment increase is estimated at $1.10 per year, the return would be even more significant: $39 for every dollar allocated.
The WHO emphasized that funding for combating the disease is much lower than the value of the damage it causes to nations. “Tuberculosis has high costs for society. Only a small proportion of these costs go directly to the health system (ranging from 1.7% in South Africa to 7.8% in Kenya). Most are costs for patients and society.”
The study projected that between 2024 and 2050, the total cost of tuberculosis to Brazilian society would be $81.2 billion, with an average annual value of $3.01 billion. This figure represents, in 2024, 0.16% of the country’s gross domestic product.
Achieving screening and preventive treatment goals in Brazil would lead to a reduction of as much as 18% in the national incidence of the disease, as well as 1.9 million fewer deaths, between 2024 and 2050.
Although treatable and preventable, tuberculosis remains the leading cause of death from infectious agents worldwide. It is estimated that over 1.3 million people died from the disease in 2022.
The document provides the “health and economic justification for investing in evidence-based interventions recommended by WHO in tuberculosis screening and prevention,” according to WHO Director-General Tedros Adhanom Ghebreyesus, PhD.
“Today we have the knowledge, tools, and political commitment that can end this age-old disease that continues to be one of the leading causes of death from infectious diseases worldwide,” he added.
Emerging Concerns
Although the WHO highlighted the global increase in access to tuberculosis diagnosis and treatment in 2022, which coincided with the recovery of healthcare systems in several countries after the beginning of the pandemic, it emphasized that the implementation of preventive treatment for exposed individuals and high-vulnerability populations remains slow.
Another concern is the increase in drug resistance. Multidrug-resistant tuberculosis is considered a public health crisis. It is estimated that about 410,000 people had multidrug-resistant tuberculosis or rifampicin-resistant tuberculosis in 2022, but only two of every five patients had access to treatment.This story was translated from the Medscape Portuguese edition using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication.
A version of this article appeared on Medscape.com .