User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
For smokers, the ends may not justify the ENDS
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
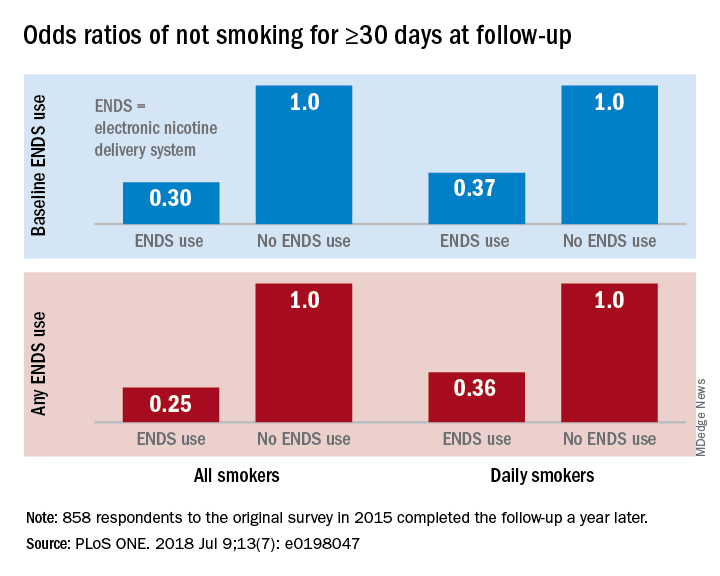
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
Smokers who used e-cigarettes and other electronic nicotine delivery systems (ENDS) were less likely to quit than were those who did not use such products, according to a 2015 survey and a follow-up conducted a year later.
“Under ‘real world’ use and conditions [ENDS] may have suppressed or delayed quitting among some adult smokers,” Scott R. Weaver, PhD, and his associates at Georgia State University, Atlanta, wrote in PLoS One. The original survey, conducted in August and September of 2015, involved 1,284 U.S. adult smokers from the GfK KnowledgePanel, of whom 858 completed the follow-up survey in September 2016.
Smokers who used ENDS at baseline were slightly more likely to attempt to quit (53.7%) than were those who did not (48.6%) but were much less likely to have quit (defined as no smoking for at least 30 days at the time of follow-up): 9.4% vs. 18.9%, for an adjusted odds ratio of 0.30. Those who used ENDS at any time during the study were much more likely than were non-ENDS users to make an attempt (58.5% vs. 44.4%), but they were, again, much less likely to succeed (7.7% vs. 22.2%; AOR, 0.25), the investigators reported.
The results were similar for the subset of respondents who smoked every day: ENDS users were more likely to attempt to quit but less likely to succeed. Odds ratios for quitting were 0.37 for those using ENDS at baseline and 0.36 for those who used ENDS at any time since the first survey, Dr. Weaver and his associates said.
“Use of current ENDS products in real world conditions [does] not seem to improve the chances of quitting for smokers, and, under the current landscape, may not be the disruptive technology that increases the population quit rate and reduces the harm of combustibles,” they wrote.
The study was supported by the National Institute of Drug Abuse and the Food and Drug Administration’s Center for Tobacco Products. One of the investigators has received funding in the form of grant funding from Pfizer and the National Institutes of Health and another has served as a paid consultant to the Centers for Disease Control and Prevention.
SOURCE: Weaver SR et al. PLoS ONE. 2018 Jul 9;13(7): e0198047. doi: 10.1371/journal.pone.0198047.
FROM PLOS ONE
Primary efficacy not met by new M. tuberculosis vaccine strategies
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint; however, both vaccines were immunogenic.
Major finding: For the secondary endpoint of sustained QuantiFERON-TB Gold In-Tube Assay (QFT) conversion, efficacy was 45.4% (P = .03) for BCG revaccination and 34.2% (P = .11) for H4:IC31, a candidate subunit vaccine.
Study details: A phase 2, randomized, placebo-controlled trial including 990 adolescents in South Africa who had received BCG vaccine in infancy.
Disclosures: The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
Source: Nemes E et al. N Engl J Med. 2018;379:138-49.
Fluoroquinolones can cause fatal hypoglycemia, FDA warns
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.

The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Study profiles sleep disruption in depression
Waking theta reduced in depression
BALTIMORE – Disruption of slow-wave activity may potentially explain the positive influence that sleep deprivation may have on major depressive disorder, according to results of a study presented at the annual meeting of the Associated Professional Sleep Societies.
Jennifer Goldschmied, PhD, of the University of Pennsylvania, Philadelphia, reported preliminary results of a study of slow-wave activity (SWA) disruption in 26 subjects – 12 healthy controls and 14 people diagnosed with major depressive disorder – that found a significant decrease of about 20% in waking theta activity, as measured with EEG, in the MDD group. In the 3-night sleep study, conducted at the University of Michigan, Ann Arbor, an adaptation night was followed by baseline and SWA disruption nights with EEGs performed each night. After the baseline night, patients also had a morning and afternoon EEG.
Across the baseline day, patients with depression showed “no modulation of theta activity whatsoever,” Dr. Goldschmied said. “And then we see, following slow-wave disruption, a significant decrease in theta activity,” whereas, healthy controls showed no change in waking theta following slow-wave disruption. “So what this means is that the presence of SWA may actually be facilitating the reduction of theta or sleep propensity during typical sleep in healthy individuals,” she added. In MDD patients, the decline in theta power following slow-wave disruption was from around 5.4 to 4.3.
Dr. Goldschmied noted that this finding somewhat supports what is known as the synaptic homeostasis hypothesis that University of Wisconsin researchers Giulio Tononi, MD, PhD, and Chiara Cirelli, MD, PhD, reported (Brain Res Bull. 2003;62:143-50). This hypothesis holds that SWA is a marker of synaptic strength and promotes the downscaling of synaptic strength during sleep. No method for measuring synaptic strength in humans exists, Dr. Goldschmied added, but waking theta can be considered a proxy for net synaptic strength across the cortex.
Dr. Goldschmied noted other research that has found SWA disruption improves mood (Psychiatry Res. 2015;228:715-8; J Psychiatr Res. 2011;45:1019-26), but the study she reported on found no role of decreased theta activity in that change. “To go even further,” she said, “we looked at the entire data set and found no relationship between the decrease in theta and any of the measures of sleep architecture – so there’s really no way to predict this decrease in our sample of people with depression.”
SWA plays a significant role in depression and merits more study, Dr. Goldschmied said. She noted that future research should examine the effects of SWA disruption in a larger sample, investigate theta findings with other proxy measures of synaptic strength such as brain-derived neurotrophic factor and transcranial magnetic stimulation, explore differences in SWA between sexes, and explore how SWA enhancement influences mood and theta activity.
Dr. Goldschmied reported having no financial relationships.
SOURCE: Goldschmied J et al. Sleep 2018, Abstract 0245.
Waking theta reduced in depression
Waking theta reduced in depression
BALTIMORE – Disruption of slow-wave activity may potentially explain the positive influence that sleep deprivation may have on major depressive disorder, according to results of a study presented at the annual meeting of the Associated Professional Sleep Societies.
Jennifer Goldschmied, PhD, of the University of Pennsylvania, Philadelphia, reported preliminary results of a study of slow-wave activity (SWA) disruption in 26 subjects – 12 healthy controls and 14 people diagnosed with major depressive disorder – that found a significant decrease of about 20% in waking theta activity, as measured with EEG, in the MDD group. In the 3-night sleep study, conducted at the University of Michigan, Ann Arbor, an adaptation night was followed by baseline and SWA disruption nights with EEGs performed each night. After the baseline night, patients also had a morning and afternoon EEG.
Across the baseline day, patients with depression showed “no modulation of theta activity whatsoever,” Dr. Goldschmied said. “And then we see, following slow-wave disruption, a significant decrease in theta activity,” whereas, healthy controls showed no change in waking theta following slow-wave disruption. “So what this means is that the presence of SWA may actually be facilitating the reduction of theta or sleep propensity during typical sleep in healthy individuals,” she added. In MDD patients, the decline in theta power following slow-wave disruption was from around 5.4 to 4.3.
Dr. Goldschmied noted that this finding somewhat supports what is known as the synaptic homeostasis hypothesis that University of Wisconsin researchers Giulio Tononi, MD, PhD, and Chiara Cirelli, MD, PhD, reported (Brain Res Bull. 2003;62:143-50). This hypothesis holds that SWA is a marker of synaptic strength and promotes the downscaling of synaptic strength during sleep. No method for measuring synaptic strength in humans exists, Dr. Goldschmied added, but waking theta can be considered a proxy for net synaptic strength across the cortex.
Dr. Goldschmied noted other research that has found SWA disruption improves mood (Psychiatry Res. 2015;228:715-8; J Psychiatr Res. 2011;45:1019-26), but the study she reported on found no role of decreased theta activity in that change. “To go even further,” she said, “we looked at the entire data set and found no relationship between the decrease in theta and any of the measures of sleep architecture – so there’s really no way to predict this decrease in our sample of people with depression.”
SWA plays a significant role in depression and merits more study, Dr. Goldschmied said. She noted that future research should examine the effects of SWA disruption in a larger sample, investigate theta findings with other proxy measures of synaptic strength such as brain-derived neurotrophic factor and transcranial magnetic stimulation, explore differences in SWA between sexes, and explore how SWA enhancement influences mood and theta activity.
Dr. Goldschmied reported having no financial relationships.
SOURCE: Goldschmied J et al. Sleep 2018, Abstract 0245.
BALTIMORE – Disruption of slow-wave activity may potentially explain the positive influence that sleep deprivation may have on major depressive disorder, according to results of a study presented at the annual meeting of the Associated Professional Sleep Societies.
Jennifer Goldschmied, PhD, of the University of Pennsylvania, Philadelphia, reported preliminary results of a study of slow-wave activity (SWA) disruption in 26 subjects – 12 healthy controls and 14 people diagnosed with major depressive disorder – that found a significant decrease of about 20% in waking theta activity, as measured with EEG, in the MDD group. In the 3-night sleep study, conducted at the University of Michigan, Ann Arbor, an adaptation night was followed by baseline and SWA disruption nights with EEGs performed each night. After the baseline night, patients also had a morning and afternoon EEG.
Across the baseline day, patients with depression showed “no modulation of theta activity whatsoever,” Dr. Goldschmied said. “And then we see, following slow-wave disruption, a significant decrease in theta activity,” whereas, healthy controls showed no change in waking theta following slow-wave disruption. “So what this means is that the presence of SWA may actually be facilitating the reduction of theta or sleep propensity during typical sleep in healthy individuals,” she added. In MDD patients, the decline in theta power following slow-wave disruption was from around 5.4 to 4.3.
Dr. Goldschmied noted that this finding somewhat supports what is known as the synaptic homeostasis hypothesis that University of Wisconsin researchers Giulio Tononi, MD, PhD, and Chiara Cirelli, MD, PhD, reported (Brain Res Bull. 2003;62:143-50). This hypothesis holds that SWA is a marker of synaptic strength and promotes the downscaling of synaptic strength during sleep. No method for measuring synaptic strength in humans exists, Dr. Goldschmied added, but waking theta can be considered a proxy for net synaptic strength across the cortex.
Dr. Goldschmied noted other research that has found SWA disruption improves mood (Psychiatry Res. 2015;228:715-8; J Psychiatr Res. 2011;45:1019-26), but the study she reported on found no role of decreased theta activity in that change. “To go even further,” she said, “we looked at the entire data set and found no relationship between the decrease in theta and any of the measures of sleep architecture – so there’s really no way to predict this decrease in our sample of people with depression.”
SWA plays a significant role in depression and merits more study, Dr. Goldschmied said. She noted that future research should examine the effects of SWA disruption in a larger sample, investigate theta findings with other proxy measures of synaptic strength such as brain-derived neurotrophic factor and transcranial magnetic stimulation, explore differences in SWA between sexes, and explore how SWA enhancement influences mood and theta activity.
Dr. Goldschmied reported having no financial relationships.
SOURCE: Goldschmied J et al. Sleep 2018, Abstract 0245.
REPORTING FROM SLEEP 2018
Key clinical point: Patients with major depression have reduced brain activity after sleep disruption.
Major finding: Morning theta activity was 20% lower following slow-wave disruption in patients with major depressive disorder than in healthy controls.
Data source: EEG measures of 14 individuals with major depressive disorder and 12 healthy controls in the evening before sleep and the morning following sleep after 1 night of baseline and 1 night of selective slow-wave disrupted sleep.
Disclosure: Dr. Goldschmied had no financial relationships to disclose.
Source: Goldschmied J et al. Sleep 2018, Abstract 0245.
FDA reverses warning on LABA-containing asthma medications
The combination a by 17%, without increasing the risk of asthma-related intubation or death.
An independent analysis of four large, drug company–sponsored trials supports the Food and Drug Administration’s recent decision to remove the black box warning on LABA/inhaled glucocorticoid products, wrote William W. Busse, MD, and his colleagues. The report was published in the New England Journal of Medicine.
“Our analysis confirmed a lower relative risk of asthma exacerbations of 17% with combination therapy than with an inhaled glucocorticoid alone. This finding corresponds to the lower relative rates of asthma exacerbations that were reported in the sponsored individual trials: by 21% in the GlaxoSmithKline trial [hazard ratio 0.79], by 16% in the AstraZeneca trial [HR. 0.84], and by 11% in the Merck trial [HR 0.89],” wrote Dr. Busse of the University of Wisconsin, Madison, and his coauthors.
The FDA based its December 2017 reversal on an initial review of the studies, which were reviewed by an independent committee and are now public. Dr. Busse led the expert analysis of the studies, which the FDA required after it put the black box warning on the combination products.
In 2010, the FDA advised that LABAs shouldn’t be used as first-line therapy for asthma and required a black box warning on all LABA-containing products. Despite an FDA-conducted meta-analysis that found no increase in serious asthma-related incidents, the agency said there wasn’t enough subgroup evidence to support the safety of LABAs when combined with an inhaled glucocorticoid.
“FDA stated that the small numbers of patients who were enrolled in these studies prevented a definitive conclusion regarding mitigation of serious asthma-related events with the addition of inhaled glucocorticoids,” the investigators stated.
The agency required the four companies marketing a LABA for asthma to conduct prospective randomized trials comparing the safety of LABA/inhaled glucocorticoid to inhaled glucocorticoid alone. The trials by AstraZeneca, GlaxoSmithKline, Merck, and Novartis were identical. Three had complete, 26-week data; Novartis submitted partial data, as it withdrew its product from the American market in 2015. The committee reviewed all of the studies, which comprised a total of 36,010 teens and adults (aged 12-91 years). The primary endpoint was a composite of asthma-related intubation or death; secondary endpoints were a composite of hospitalization, intubation, or death, and individual assessments of each of those events.
Among the four studies, there were three asthma-related intubations: two in the inhaled-glucocorticoid group and one in the combination-therapy group. There were also two asthma-related deaths, both in the combination group.
Serious asthma-related events occurred in 108 of the inhaled glucocorticoid group (0.60%) and in 119 of the combination-therapy group (0.66%), a nonsignificant difference.
However, the combination therapy did confer a significant 17% reduction in asthma exacerbations. Exacerbations occurred in 11.7% of the inhaled glucocorticoid group and in 9.8% of the combination therapy group (relative risk 0.83; P less than 0.001). All four trials showed a similarly decreased risk of exacerbation.
The committee looked at several subgroups, dividing the cohort by age, race/ethnicity/ obesity, and smoking history. The advantage associated with combination therapy remained significant in all these analyses.
“… Our data provide support for the treatment guidelines of both the Global Initiative for Asthma and the Expert Panel Report 3 of the National Asthma Education and Prevention Program, which recommend the use of a low-dose glucocorticoid (step 3) and a medium-dose glucocorticoid (step 4), plus a LABA, with the caution that LABAs should not be used as monotherapy in asthma; the convenience and safety of a combination inhaler is a likely plus,” the committee wrote. “Finally, our combined analysis provides strong evidence to support the recent FDA decision to remove the boxed safety warning for combination therapy with a LABA plus an inhaled glucocorticoid for asthma treatment.”
Dr. Busse disclosed financial relationships with a number of pharmaceutical companies, including Novartis, but noted that none of them were relevant to this work.
SOURCE: Busse WW et al. N Engl J Med. 2018;78:2497-505.
It takes a lot for the FDA to remove a boxed warning on a product, but the data generated by these four manufacturer-led trials of long-acting beta agonists and inhaled corticosteroid combination therapy warrant the regulatory reversal, Sally Seymour, MD, and her colleagues wrote in an accompanying editorial.
“Each completed trial met the prespecified objective and demonstrated noninferiority of combination products to inhaled corticosteroids alone with respect to the composite endpoint of asthma-related death, intubation, or hospitalization.
The majority of events were asthma-related hospitalizations; there were five intubations and deaths overall,” Dr. Seymour and her colleagues wrote.
All of the studies met their primary safety objective, and faced with this – and the consistency of the results among the studies – the path was clear.
“In addition, the observed reduction in asthma exacerbations that required systemic corticosteroids demonstrates a benefit associated with combination products. On the basis of this strong and consistent evidence, we opted to remove the boxed warning right away, without convening an FDA advisory committee meeting.”
There will always be areas of uncertainty, however.
“Admittedly, the results from these trials cannot answer all questions regarding the safety of LABAs. Some uncertainties remain, and we cannot conclude that there is no increase in risk associated with combination products containing an inhaled corticosteroid and a LABA as compared with inhaled corticosteroids alone. Although the trials found that combination therapy reduces the rate of exacerbations that require the administration of systemic corticosteroids, none of them showed a decrease in asthma-related hospitalizations. People with life-threatening asthma were excluded because of safety and ethical concerns, so we don’t know whether the results can be generalized to these patients.”
Nevertheless, the evidence in favor of combination therapy was clear and compelling enough to convince a national regulatory agency to change a stance on safety.
Dr. Seymour is the acting director of the FDA’s Division of Pulmonary, Allergy, & Rheumatology Products.
It takes a lot for the FDA to remove a boxed warning on a product, but the data generated by these four manufacturer-led trials of long-acting beta agonists and inhaled corticosteroid combination therapy warrant the regulatory reversal, Sally Seymour, MD, and her colleagues wrote in an accompanying editorial.
“Each completed trial met the prespecified objective and demonstrated noninferiority of combination products to inhaled corticosteroids alone with respect to the composite endpoint of asthma-related death, intubation, or hospitalization.
The majority of events were asthma-related hospitalizations; there were five intubations and deaths overall,” Dr. Seymour and her colleagues wrote.
All of the studies met their primary safety objective, and faced with this – and the consistency of the results among the studies – the path was clear.
“In addition, the observed reduction in asthma exacerbations that required systemic corticosteroids demonstrates a benefit associated with combination products. On the basis of this strong and consistent evidence, we opted to remove the boxed warning right away, without convening an FDA advisory committee meeting.”
There will always be areas of uncertainty, however.
“Admittedly, the results from these trials cannot answer all questions regarding the safety of LABAs. Some uncertainties remain, and we cannot conclude that there is no increase in risk associated with combination products containing an inhaled corticosteroid and a LABA as compared with inhaled corticosteroids alone. Although the trials found that combination therapy reduces the rate of exacerbations that require the administration of systemic corticosteroids, none of them showed a decrease in asthma-related hospitalizations. People with life-threatening asthma were excluded because of safety and ethical concerns, so we don’t know whether the results can be generalized to these patients.”
Nevertheless, the evidence in favor of combination therapy was clear and compelling enough to convince a national regulatory agency to change a stance on safety.
Dr. Seymour is the acting director of the FDA’s Division of Pulmonary, Allergy, & Rheumatology Products.
It takes a lot for the FDA to remove a boxed warning on a product, but the data generated by these four manufacturer-led trials of long-acting beta agonists and inhaled corticosteroid combination therapy warrant the regulatory reversal, Sally Seymour, MD, and her colleagues wrote in an accompanying editorial.
“Each completed trial met the prespecified objective and demonstrated noninferiority of combination products to inhaled corticosteroids alone with respect to the composite endpoint of asthma-related death, intubation, or hospitalization.
The majority of events were asthma-related hospitalizations; there were five intubations and deaths overall,” Dr. Seymour and her colleagues wrote.
All of the studies met their primary safety objective, and faced with this – and the consistency of the results among the studies – the path was clear.
“In addition, the observed reduction in asthma exacerbations that required systemic corticosteroids demonstrates a benefit associated with combination products. On the basis of this strong and consistent evidence, we opted to remove the boxed warning right away, without convening an FDA advisory committee meeting.”
There will always be areas of uncertainty, however.
“Admittedly, the results from these trials cannot answer all questions regarding the safety of LABAs. Some uncertainties remain, and we cannot conclude that there is no increase in risk associated with combination products containing an inhaled corticosteroid and a LABA as compared with inhaled corticosteroids alone. Although the trials found that combination therapy reduces the rate of exacerbations that require the administration of systemic corticosteroids, none of them showed a decrease in asthma-related hospitalizations. People with life-threatening asthma were excluded because of safety and ethical concerns, so we don’t know whether the results can be generalized to these patients.”
Nevertheless, the evidence in favor of combination therapy was clear and compelling enough to convince a national regulatory agency to change a stance on safety.
Dr. Seymour is the acting director of the FDA’s Division of Pulmonary, Allergy, & Rheumatology Products.
The combination a by 17%, without increasing the risk of asthma-related intubation or death.
An independent analysis of four large, drug company–sponsored trials supports the Food and Drug Administration’s recent decision to remove the black box warning on LABA/inhaled glucocorticoid products, wrote William W. Busse, MD, and his colleagues. The report was published in the New England Journal of Medicine.
“Our analysis confirmed a lower relative risk of asthma exacerbations of 17% with combination therapy than with an inhaled glucocorticoid alone. This finding corresponds to the lower relative rates of asthma exacerbations that were reported in the sponsored individual trials: by 21% in the GlaxoSmithKline trial [hazard ratio 0.79], by 16% in the AstraZeneca trial [HR. 0.84], and by 11% in the Merck trial [HR 0.89],” wrote Dr. Busse of the University of Wisconsin, Madison, and his coauthors.
The FDA based its December 2017 reversal on an initial review of the studies, which were reviewed by an independent committee and are now public. Dr. Busse led the expert analysis of the studies, which the FDA required after it put the black box warning on the combination products.
In 2010, the FDA advised that LABAs shouldn’t be used as first-line therapy for asthma and required a black box warning on all LABA-containing products. Despite an FDA-conducted meta-analysis that found no increase in serious asthma-related incidents, the agency said there wasn’t enough subgroup evidence to support the safety of LABAs when combined with an inhaled glucocorticoid.
“FDA stated that the small numbers of patients who were enrolled in these studies prevented a definitive conclusion regarding mitigation of serious asthma-related events with the addition of inhaled glucocorticoids,” the investigators stated.
The agency required the four companies marketing a LABA for asthma to conduct prospective randomized trials comparing the safety of LABA/inhaled glucocorticoid to inhaled glucocorticoid alone. The trials by AstraZeneca, GlaxoSmithKline, Merck, and Novartis were identical. Three had complete, 26-week data; Novartis submitted partial data, as it withdrew its product from the American market in 2015. The committee reviewed all of the studies, which comprised a total of 36,010 teens and adults (aged 12-91 years). The primary endpoint was a composite of asthma-related intubation or death; secondary endpoints were a composite of hospitalization, intubation, or death, and individual assessments of each of those events.
Among the four studies, there were three asthma-related intubations: two in the inhaled-glucocorticoid group and one in the combination-therapy group. There were also two asthma-related deaths, both in the combination group.
Serious asthma-related events occurred in 108 of the inhaled glucocorticoid group (0.60%) and in 119 of the combination-therapy group (0.66%), a nonsignificant difference.
However, the combination therapy did confer a significant 17% reduction in asthma exacerbations. Exacerbations occurred in 11.7% of the inhaled glucocorticoid group and in 9.8% of the combination therapy group (relative risk 0.83; P less than 0.001). All four trials showed a similarly decreased risk of exacerbation.
The committee looked at several subgroups, dividing the cohort by age, race/ethnicity/ obesity, and smoking history. The advantage associated with combination therapy remained significant in all these analyses.
“… Our data provide support for the treatment guidelines of both the Global Initiative for Asthma and the Expert Panel Report 3 of the National Asthma Education and Prevention Program, which recommend the use of a low-dose glucocorticoid (step 3) and a medium-dose glucocorticoid (step 4), plus a LABA, with the caution that LABAs should not be used as monotherapy in asthma; the convenience and safety of a combination inhaler is a likely plus,” the committee wrote. “Finally, our combined analysis provides strong evidence to support the recent FDA decision to remove the boxed safety warning for combination therapy with a LABA plus an inhaled glucocorticoid for asthma treatment.”
Dr. Busse disclosed financial relationships with a number of pharmaceutical companies, including Novartis, but noted that none of them were relevant to this work.
SOURCE: Busse WW et al. N Engl J Med. 2018;78:2497-505.
The combination a by 17%, without increasing the risk of asthma-related intubation or death.
An independent analysis of four large, drug company–sponsored trials supports the Food and Drug Administration’s recent decision to remove the black box warning on LABA/inhaled glucocorticoid products, wrote William W. Busse, MD, and his colleagues. The report was published in the New England Journal of Medicine.
“Our analysis confirmed a lower relative risk of asthma exacerbations of 17% with combination therapy than with an inhaled glucocorticoid alone. This finding corresponds to the lower relative rates of asthma exacerbations that were reported in the sponsored individual trials: by 21% in the GlaxoSmithKline trial [hazard ratio 0.79], by 16% in the AstraZeneca trial [HR. 0.84], and by 11% in the Merck trial [HR 0.89],” wrote Dr. Busse of the University of Wisconsin, Madison, and his coauthors.
The FDA based its December 2017 reversal on an initial review of the studies, which were reviewed by an independent committee and are now public. Dr. Busse led the expert analysis of the studies, which the FDA required after it put the black box warning on the combination products.
In 2010, the FDA advised that LABAs shouldn’t be used as first-line therapy for asthma and required a black box warning on all LABA-containing products. Despite an FDA-conducted meta-analysis that found no increase in serious asthma-related incidents, the agency said there wasn’t enough subgroup evidence to support the safety of LABAs when combined with an inhaled glucocorticoid.
“FDA stated that the small numbers of patients who were enrolled in these studies prevented a definitive conclusion regarding mitigation of serious asthma-related events with the addition of inhaled glucocorticoids,” the investigators stated.
The agency required the four companies marketing a LABA for asthma to conduct prospective randomized trials comparing the safety of LABA/inhaled glucocorticoid to inhaled glucocorticoid alone. The trials by AstraZeneca, GlaxoSmithKline, Merck, and Novartis were identical. Three had complete, 26-week data; Novartis submitted partial data, as it withdrew its product from the American market in 2015. The committee reviewed all of the studies, which comprised a total of 36,010 teens and adults (aged 12-91 years). The primary endpoint was a composite of asthma-related intubation or death; secondary endpoints were a composite of hospitalization, intubation, or death, and individual assessments of each of those events.
Among the four studies, there were three asthma-related intubations: two in the inhaled-glucocorticoid group and one in the combination-therapy group. There were also two asthma-related deaths, both in the combination group.
Serious asthma-related events occurred in 108 of the inhaled glucocorticoid group (0.60%) and in 119 of the combination-therapy group (0.66%), a nonsignificant difference.
However, the combination therapy did confer a significant 17% reduction in asthma exacerbations. Exacerbations occurred in 11.7% of the inhaled glucocorticoid group and in 9.8% of the combination therapy group (relative risk 0.83; P less than 0.001). All four trials showed a similarly decreased risk of exacerbation.
The committee looked at several subgroups, dividing the cohort by age, race/ethnicity/ obesity, and smoking history. The advantage associated with combination therapy remained significant in all these analyses.
“… Our data provide support for the treatment guidelines of both the Global Initiative for Asthma and the Expert Panel Report 3 of the National Asthma Education and Prevention Program, which recommend the use of a low-dose glucocorticoid (step 3) and a medium-dose glucocorticoid (step 4), plus a LABA, with the caution that LABAs should not be used as monotherapy in asthma; the convenience and safety of a combination inhaler is a likely plus,” the committee wrote. “Finally, our combined analysis provides strong evidence to support the recent FDA decision to remove the boxed safety warning for combination therapy with a LABA plus an inhaled glucocorticoid for asthma treatment.”
Dr. Busse disclosed financial relationships with a number of pharmaceutical companies, including Novartis, but noted that none of them were relevant to this work.
SOURCE: Busse WW et al. N Engl J Med. 2018;78:2497-505.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Combination LABA/inhaled glucocorticoid products appear safe for patients with asthma.
Major finding: The products reduced the risk of an asthma exacerbation by 17%, without increasing the risk of a serious adverse outcome.
Study details: The four randomized studies comprised more than 13,000 patients.
Disclosures: The studies were sponsored by AstraZeneca, GlaxoSmithKline, Merck, and Novartis. Dr. Busse disclosed a financial relationship with Novartis, but said it was not relevant to this work.
Source: Busse WW. N Engl J Med. 2018;78:2497-505.
What are the benefits/risks of giving betamethasone to women at risk of late preterm labor?
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
EVIDENCE SUMMARY
A 2016 systematic review and meta-analysis of 3 RCTs that included 3200 women with late preterm labor (between 34 weeks 0 days and 36 weeks 6 days) found that women who were given betamethasone had a significantly lower incidence of transient tachypnea of the newborn (number needed to treat [NNT]=37; relative risk [RR]=0.72; 95% confidence interval [CI], 0.56-0.92), severe respiratory distress syndrome (NNT=114; RR=0.60; 95% CI, 0.33-0.94), and use of surfactant (NNT=92; RR=0.61; 95% CI, 0.38-0.99).1
A composite outcome measure also favors betamethasone
In addition to these 3 outcomes, the largest RCT in the meta-analysis evaluated a composite outcome and found that betamethasone improved it by 20%. The RCT, comparing 1427 women in the experimental arm with 1400 controls, found benefit to administering 12 mg betamethasone intramuscularly every 24 hours for 2 days for women at high risk of late preterm delivery.2 Enrollment criteria included women with 3 cm dilation or 75% effacement, preterm premature rupture of membranes, or a planned delivery scheduled in the late preterm period.
The primary outcome was a composite score based on one or more of the following within 72 hours after birth: continuous positive airway pressure or high-flow nasal cannula for at least 2 continuous hours, supplemental oxygen with a fraction of inspired oxygen of 0.30 or more for at least 4 continuous hours, mechanical ventilation, stillbirth or neonatal death, or the need for extracorporeal circulation membrane oxygenation. The betamethasone group had 165 women (11.6%) with the primary outcome compared with 202 (14.4%) in the control arm (NNT=34; RR=0.80; 95% CI, 0.66–0.97; P<.02).
Neonatal hypoglycemia may increase, but not dangerously
The same RCT explored the risks of late preterm betamethasone. There was no increase in chorioamnionitis nor neonatal sepsis in the betamethasone group.2 Although neonatal hypoglycemia increased (24% vs 15%; number needed to harm=11.1; RR=1.60; 95% CI, 1.37-1.87; P<.001), no increase was seen in intermediate care nursery or ICU stays (41.8% vs 44.9%; RR=0.93; 95% CI, 0.85-1.01; P=.09) nor length of hospital stay (7 vs 8 days; P=.20).
Three letters to the editor questioned whether hypoglycemia from late-term corticosteroids may lead to long-term neurocognitive delays.3 The authors responded that meta-analyses of RCTs haven’t found an association between antenatal steroid use and neurocognitive delay and that studies that have found an association between hypoglycemia and neurocognitive delay looked at profound and prolonged hypoglycemia, which wasn’t seen in the late preterm betamethasone study.
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
Both the American College of Obstetricians and Gynecologists and the Society for Maternal-Fetal Medicine have published recommendations supporting corticosteroids for threatened late preterm delivery with certain caveats.4-6 Because of a lack of evidence, maternity care providers shouldn’t give corticosteroids for threatened late preterm delivery to women with multiple gestation, diabetes, previous exposure to steroids during pregnancy, or pregnancies with major nonlethal fetal malformations.4-6 Evidence doesn’t support tocolysis when steroids are given in the late preterm period.4,5
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
1. Saccone G, Berghella V. Antenatal corticosteroids for maturity of term or near term fetuses: systematic review and meta-analysis of randomized controlled trials. BMJ. 2016;355:5044.
2. Gyamfi-Bannerman C, Thom E, Blackwell S, et al. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;374:1311-1320.
3. Gyamfi-Bannerman C, Thom E. Antenatal betamethasone for women at risk for late preterm delivery. N Engl J Med. 2016;375:486-487.
4. American College of Obstetricians and Gynecologists. Committee Opinion No. 677: Antenatal Corticosteroid Therapy for Fetal Maturation. Obstet Gynecol. 2016;128:e187-e194.
5. Society for Maternal-Fetal Medicine (SMFM) Publications Committee. Implementation of the use of antenatal corticosteroids in the late preterm birth period in women at risk for preterm delivery. Am J Obstet Gynecol. 2016;215:B13-B15.
6. American College of Obstetricians and Gynecologists. Practice Bulletin No. 159: Management of Preterm Labor. Obstet Gynecol. 2016;127:e29-e38.
EVIDENCE-BASED ANSWER:
Giving betamethasone to women at risk for delivery between 34 weeks 0 days and 36 weeks 6 days can lower by almost 40% the incidence of transient tachypnea of the newborn, severe respiratory distress syndrome, and the use of surfactant (strength of recommendation [SOR]: A, systematic review of randomized controlled trials [RCTs]).
Betamethasone may increase neonatal hypoglycemia, but the hypoglycemia isn’t associated with a prolonged hospital stay or other negative outcomes.

FDA approves Zephyr endobronchial valve to treat severe emphysema
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
The valve is the first minimally invasive device approved in the United States for treating such patients, according to Pulmonx, the device manufacturer.
The FDA previously granted the novel device expedited review, as patients who did not respond to drug treatment had only limited alternative options, including lung volume reduction and lung transplant, Tina Kiang, PhD, of the FDA’s Center for Devices and Radiological Health, said in a press release. “This novel device is a less invasive treatment that expands the options available to patients,” said Dr. Kiang, acting director of the center’s Division of Anesthesiology, General Hospital, Respiratory, Infection Control, and Dental Devices.![]()
The approval is based on a multicenter study of 190 patients with severe emphysema. A total of 128 received Zephyr valves and medical management, while 62 received medical management only. The primary measure was the number of patients who achieved at least a 15% improvement in their pulmonary function score: At 1 year, 47.7% of the Zephyr valve patients had achieved such improvement versus 16.8% of the control group, according to the FDA.
Adverse events included death, pneumothorax, pneumonia, worsening of emphysema, coughing up blood, shortness of breath, and chest pain. The valve is contraindicated in patients with active lung infections; those allergic to nitinol, nickel, titanium, or silicone; and active smokers.
Read more about this approval in the full FDA press announcement.
Noninvasive ventilation during exercise benefited a subgroup of COPD patients
SAN DIEGO – Use of noninvasive ventilation during an exercise session in hypercapnic patients with very severe chronic obstructive pulmonary disease (COPD) led to a clinically relevant increase in endurance time, a randomized trial showed.
At an international conference of the American Thoracic Society, lead study author Tessa Schneeberger noted that nocturnal noninvasive ventilation (NIV) in hypercapnic COPD patients has been shown to improve quality of life and survival (Lancet Resp Med. 2014;2[9]:698-705). Another study found that NIV with unchanged nocturnal settings during a 6-minute walk test in hypercapnic COPD patients can increase oxygenation, decrease dyspnea, and increase walking distance (Eur Respir J. 2007;29:930-6).

For the current study, Ms. Schneeberger, a physiotherapist at the Institute for Pulmonary Rehabilitation Research, Schoenau am Koenigssee, Germany, and her associates set out to investigate short-term effects of using NIV during exercise in hypercapnic patients with very severe COPD, as part of a 3-week inpatient physical rehabilitation program. The researchers limited their analysis to 20 Global Initiative for Chronic Obstructive Lung Disease stage IV patients aged 40-80 years with a carbon dioxide partial pressure (PCO2) of greater than 50 mm Hg at rest and/or during exercise and who were non-naive to NIV, and excluded patients with concomitant conditions that make cycling impossible, those with acute exacerbations, and those with exercise-limiting cardiovascular diseases.
The day after an initial incremental cycle ergometer test, patients performed two constant work rate tests (CWRT) at 60% of the peak work rate, with and without NIV, in randomized order and with a resting time of 1 hour between tests. The inspiratory positive airway pressure (IPAP) was individually adjusted from each patient’s nocturnal settings to provide sufficient pressure to relieve the work on breathing muscles and to decrease transcutaneous PCO2 (TcPCO2) levels during NIV. The primary outcome was cycle endurance time. Other outcomes of interest were TcPCO2, oxygen saturation (SpO2) and perceived dyspnea/leg fatigue via the 10-point Borg scale during CWRTs.
The mean age of the study participants was 60 years, their mean body mass index was 23 kg/m2, their mean forced expiratory volume in1 second was 19% predicted, their mean PaCO2 was 51 mm Hg, their mean PaO2 was 54.5 mm Hg, their mean distance on the 6-minute walk test was 243 meters, and their mean peak work rate was 42 watts.
NIV via full face mask and assisted pressure control ventilation mode was performed with mean IPAP/expiratory PAP levels of 27/6 cm H2O.
During CWRTs patients cycled with NIV for 663 seconds and without NIV for 476 seconds, a significant difference (P = .013) and one that was clinically relevant. At isotime (the time of CWRT with shortest duration), TcPCO2 was significantly lower with NIV (a mean of –6.1 mm Hg), while SpO2 was significantly higher with NIV (a mean of 3.6%). In addition, after CWRT, NIV patients perceived less dyspnea (P = .008) with comparable leg fatigue (P = .79).
“We found that NIV during cycling exercise in hypercapnic patients with very severe COPD can lead to an acutely significant increase in exercise duration, with lower TcPCO2 and a reduced sensation of dyspnea,” Ms. Schneeberger concluded. “It can be performed with high-pressure assisted-controlled ventilation comparable as that used nocturnally to effectively reduce TcPCO2 in people with COPD.”
She emphasized that this approach requires appropriate equipment and special staff expertise for setup titration. “We will continue this research to look into the underlying physiological mechanisms to define nonresponders and responders, and also to look how at this might improve outcomes of an exercise training program.”
Ms. Schneeberger reported having no financial disclosures.
SOURCE: Schneeberger T et al. ATS 2018, Abstract A2453.
SAN DIEGO – Use of noninvasive ventilation during an exercise session in hypercapnic patients with very severe chronic obstructive pulmonary disease (COPD) led to a clinically relevant increase in endurance time, a randomized trial showed.
At an international conference of the American Thoracic Society, lead study author Tessa Schneeberger noted that nocturnal noninvasive ventilation (NIV) in hypercapnic COPD patients has been shown to improve quality of life and survival (Lancet Resp Med. 2014;2[9]:698-705). Another study found that NIV with unchanged nocturnal settings during a 6-minute walk test in hypercapnic COPD patients can increase oxygenation, decrease dyspnea, and increase walking distance (Eur Respir J. 2007;29:930-6).
For the current study, Ms. Schneeberger, a physiotherapist at the Institute for Pulmonary Rehabilitation Research, Schoenau am Koenigssee, Germany, and her associates set out to investigate short-term effects of using NIV during exercise in hypercapnic patients with very severe COPD, as part of a 3-week inpatient physical rehabilitation program. The researchers limited their analysis to 20 Global Initiative for Chronic Obstructive Lung Disease stage IV patients aged 40-80 years with a carbon dioxide partial pressure (PCO2) of greater than 50 mm Hg at rest and/or during exercise and who were non-naive to NIV, and excluded patients with concomitant conditions that make cycling impossible, those with acute exacerbations, and those with exercise-limiting cardiovascular diseases.
The day after an initial incremental cycle ergometer test, patients performed two constant work rate tests (CWRT) at 60% of the peak work rate, with and without NIV, in randomized order and with a resting time of 1 hour between tests. The inspiratory positive airway pressure (IPAP) was individually adjusted from each patient’s nocturnal settings to provide sufficient pressure to relieve the work on breathing muscles and to decrease transcutaneous PCO2 (TcPCO2) levels during NIV. The primary outcome was cycle endurance time. Other outcomes of interest were TcPCO2, oxygen saturation (SpO2) and perceived dyspnea/leg fatigue via the 10-point Borg scale during CWRTs.
The mean age of the study participants was 60 years, their mean body mass index was 23 kg/m2, their mean forced expiratory volume in1 second was 19% predicted, their mean PaCO2 was 51 mm Hg, their mean PaO2 was 54.5 mm Hg, their mean distance on the 6-minute walk test was 243 meters, and their mean peak work rate was 42 watts.
NIV via full face mask and assisted pressure control ventilation mode was performed with mean IPAP/expiratory PAP levels of 27/6 cm H2O.
During CWRTs patients cycled with NIV for 663 seconds and without NIV for 476 seconds, a significant difference (P = .013) and one that was clinically relevant. At isotime (the time of CWRT with shortest duration), TcPCO2 was significantly lower with NIV (a mean of –6.1 mm Hg), while SpO2 was significantly higher with NIV (a mean of 3.6%). In addition, after CWRT, NIV patients perceived less dyspnea (P = .008) with comparable leg fatigue (P = .79).
“We found that NIV during cycling exercise in hypercapnic patients with very severe COPD can lead to an acutely significant increase in exercise duration, with lower TcPCO2 and a reduced sensation of dyspnea,” Ms. Schneeberger concluded. “It can be performed with high-pressure assisted-controlled ventilation comparable as that used nocturnally to effectively reduce TcPCO2 in people with COPD.”
She emphasized that this approach requires appropriate equipment and special staff expertise for setup titration. “We will continue this research to look into the underlying physiological mechanisms to define nonresponders and responders, and also to look how at this might improve outcomes of an exercise training program.”
Ms. Schneeberger reported having no financial disclosures.
SOURCE: Schneeberger T et al. ATS 2018, Abstract A2453.
SAN DIEGO – Use of noninvasive ventilation during an exercise session in hypercapnic patients with very severe chronic obstructive pulmonary disease (COPD) led to a clinically relevant increase in endurance time, a randomized trial showed.
At an international conference of the American Thoracic Society, lead study author Tessa Schneeberger noted that nocturnal noninvasive ventilation (NIV) in hypercapnic COPD patients has been shown to improve quality of life and survival (Lancet Resp Med. 2014;2[9]:698-705). Another study found that NIV with unchanged nocturnal settings during a 6-minute walk test in hypercapnic COPD patients can increase oxygenation, decrease dyspnea, and increase walking distance (Eur Respir J. 2007;29:930-6).
For the current study, Ms. Schneeberger, a physiotherapist at the Institute for Pulmonary Rehabilitation Research, Schoenau am Koenigssee, Germany, and her associates set out to investigate short-term effects of using NIV during exercise in hypercapnic patients with very severe COPD, as part of a 3-week inpatient physical rehabilitation program. The researchers limited their analysis to 20 Global Initiative for Chronic Obstructive Lung Disease stage IV patients aged 40-80 years with a carbon dioxide partial pressure (PCO2) of greater than 50 mm Hg at rest and/or during exercise and who were non-naive to NIV, and excluded patients with concomitant conditions that make cycling impossible, those with acute exacerbations, and those with exercise-limiting cardiovascular diseases.
The day after an initial incremental cycle ergometer test, patients performed two constant work rate tests (CWRT) at 60% of the peak work rate, with and without NIV, in randomized order and with a resting time of 1 hour between tests. The inspiratory positive airway pressure (IPAP) was individually adjusted from each patient’s nocturnal settings to provide sufficient pressure to relieve the work on breathing muscles and to decrease transcutaneous PCO2 (TcPCO2) levels during NIV. The primary outcome was cycle endurance time. Other outcomes of interest were TcPCO2, oxygen saturation (SpO2) and perceived dyspnea/leg fatigue via the 10-point Borg scale during CWRTs.
The mean age of the study participants was 60 years, their mean body mass index was 23 kg/m2, their mean forced expiratory volume in1 second was 19% predicted, their mean PaCO2 was 51 mm Hg, their mean PaO2 was 54.5 mm Hg, their mean distance on the 6-minute walk test was 243 meters, and their mean peak work rate was 42 watts.
NIV via full face mask and assisted pressure control ventilation mode was performed with mean IPAP/expiratory PAP levels of 27/6 cm H2O.
During CWRTs patients cycled with NIV for 663 seconds and without NIV for 476 seconds, a significant difference (P = .013) and one that was clinically relevant. At isotime (the time of CWRT with shortest duration), TcPCO2 was significantly lower with NIV (a mean of –6.1 mm Hg), while SpO2 was significantly higher with NIV (a mean of 3.6%). In addition, after CWRT, NIV patients perceived less dyspnea (P = .008) with comparable leg fatigue (P = .79).
“We found that NIV during cycling exercise in hypercapnic patients with very severe COPD can lead to an acutely significant increase in exercise duration, with lower TcPCO2 and a reduced sensation of dyspnea,” Ms. Schneeberger concluded. “It can be performed with high-pressure assisted-controlled ventilation comparable as that used nocturnally to effectively reduce TcPCO2 in people with COPD.”
She emphasized that this approach requires appropriate equipment and special staff expertise for setup titration. “We will continue this research to look into the underlying physiological mechanisms to define nonresponders and responders, and also to look how at this might improve outcomes of an exercise training program.”
Ms. Schneeberger reported having no financial disclosures.
SOURCE: Schneeberger T et al. ATS 2018, Abstract A2453.
REPORTING FROM ATS 2018
Key clinical point: Noninvasive ventilation (NIV) during exercise seems feasible and could provide an opportunity to improve endurance training outcomes in selected chronic obstructive pulmonary disease patients.
Major finding: During constant work rate tests patients cycled with NIV for 663 seconds and without NIV for 476 seconds, a significant difference (P = .013).
Study details: A randomized trial of short-term effects of NIV during exercise in 20 hypercapnic patients with very severe COPD.
Disclosures: Ms. Schneeberger reported having no financial disclosures.
Source: Schneeberger T et al. ATS 2018, Abstract A2453.
Effort to phenotype pulmonary hypertension patients under way
SAN DIEGO – A massive effort to better understand and treat patients with pulmonary hypertension and right heart dysfunction is underway.
The endeavor, funded by the National Heart, Lung, and Blood Institute and the Pulmonary Hypertension Association and known as Redefining Pulmonary Hypertension Through Pulmonary Vascular Disease Phenomics (PVDOMICS), began recruiting participants in 2017, with a goal of 1,500 by 2019. The aim is to perform comprehensive phenotyping and endophenotyping across the World Health Organization–classified pulmonary hypertension (PH) clinical groups 1 through 5 in order to deconstruct the traditional classification and define new meaningful subclassifications of patients with pulmonary vascular disease.
At an international conference of the American Thoracic Society, one of the study’s investigators, Robert P. Frantz, MD, discussed the role of echocardiography and MRI in the overall PVDOMICS program, which he characterized as a work in progress. “Imaging is critically important as we try to integrate severity of pulmonary vascular disease along with how well the ventricle functions as way to try and understand why some patients have a failing RV at a given pulmonary resistance and others don’t,” said Dr. Frantz, who directs the Mayo Pulmonary Hypertension Clinic in Rochester, Minn. The goals are to be able to integrate cardiac morphology and function with contemporaneous hemodynamics, he said. This will allow for validation of noninvasive hemodynamics versus right heart catheterization across all the phenotypes.
“In addition, we’ll have imaging parameters as predictors of hemodynamics at rest and with exercise, particularly in conditions like heart failure with preserved ejection fraction or concerns about left atrial stiffness,” he said. “In these cases, our ability on the basis of echocardiography or MRI to guess what the wedge pressure is at rest or exercise, or to think about other more recently described phenotypes like left atrial stiffness in patients who have left atrial ablation procedures, will be enabled by looking at parameters such as left atrial strain.”
Ultimately, he continued, a key goal of PVDOMICS is to be able to correlate the “-omics” with markers of RV compensation in an effort to understand what the determinants of RV compensation are across the varying types of pulmonary vascular disease.
“If we could do that, we might be able to develop new targets for therapy,” said Dr. Frantz. To illustrate how this might work, he cited findings from researchers who set out to identify and characterize homogeneous phenotypes by a cluster analysis in scleroderma patients with pulmonary hypertension, who were identified from two prospective cohorts in the United States and France (PLoS One 2018 May 15;13[5]:e0197112).
The researchers identified four different clusters of scleroderma patients: those with mild to moderate PAH with no or minimal interstitial lung disease and low-diffusing capacity for carbon monoxide; those with precapillary PH with severe ILD and worse survival; those with severe PAH, who trended toward worse survival, and those similar to the first cluster but with higher DLCO.
Dr. Frantz then shared preliminary findings of echocardiographic parameters by primary WHO group in PVDOMICS, on behalf of his PVDOMICS collaborators. They found, for example, that the mean right ventricular systolic pressure in group 3 was 45 mm Hg, as opposed to group 1, which was 64 mm Hg. “In general we had some patients in group 3 with less severe elevation of PA pressures,” he said.
Other parameters that can be compared across WHO groups include ventricular fractional area change, tricuspid annular plane systolic excursion, and RV free wall strain. “That strain of the right ventricle is one of the most important ways of looking at how the right ventricle works,” Dr. Frantz explained. “With this, we can integrate the concept of severity of RV dysfunction with severity of pulmonary vascular disease. This is where the rubber hits the road. It’s going to be very complicated and time consuming, but I think critically important. Ultimately, we can make proteomic heat maps that track these correlates, and ultimately identify pathways that may be driving RV compensation in pulmonary vascular disease.”
Dr. Frantz reported having no relevant financial disclosures.
SAN DIEGO – A massive effort to better understand and treat patients with pulmonary hypertension and right heart dysfunction is underway.
The endeavor, funded by the National Heart, Lung, and Blood Institute and the Pulmonary Hypertension Association and known as Redefining Pulmonary Hypertension Through Pulmonary Vascular Disease Phenomics (PVDOMICS), began recruiting participants in 2017, with a goal of 1,500 by 2019. The aim is to perform comprehensive phenotyping and endophenotyping across the World Health Organization–classified pulmonary hypertension (PH) clinical groups 1 through 5 in order to deconstruct the traditional classification and define new meaningful subclassifications of patients with pulmonary vascular disease.
At an international conference of the American Thoracic Society, one of the study’s investigators, Robert P. Frantz, MD, discussed the role of echocardiography and MRI in the overall PVDOMICS program, which he characterized as a work in progress. “Imaging is critically important as we try to integrate severity of pulmonary vascular disease along with how well the ventricle functions as way to try and understand why some patients have a failing RV at a given pulmonary resistance and others don’t,” said Dr. Frantz, who directs the Mayo Pulmonary Hypertension Clinic in Rochester, Minn. The goals are to be able to integrate cardiac morphology and function with contemporaneous hemodynamics, he said. This will allow for validation of noninvasive hemodynamics versus right heart catheterization across all the phenotypes.
“In addition, we’ll have imaging parameters as predictors of hemodynamics at rest and with exercise, particularly in conditions like heart failure with preserved ejection fraction or concerns about left atrial stiffness,” he said. “In these cases, our ability on the basis of echocardiography or MRI to guess what the wedge pressure is at rest or exercise, or to think about other more recently described phenotypes like left atrial stiffness in patients who have left atrial ablation procedures, will be enabled by looking at parameters such as left atrial strain.”
Ultimately, he continued, a key goal of PVDOMICS is to be able to correlate the “-omics” with markers of RV compensation in an effort to understand what the determinants of RV compensation are across the varying types of pulmonary vascular disease.
“If we could do that, we might be able to develop new targets for therapy,” said Dr. Frantz. To illustrate how this might work, he cited findings from researchers who set out to identify and characterize homogeneous phenotypes by a cluster analysis in scleroderma patients with pulmonary hypertension, who were identified from two prospective cohorts in the United States and France (PLoS One 2018 May 15;13[5]:e0197112).
The researchers identified four different clusters of scleroderma patients: those with mild to moderate PAH with no or minimal interstitial lung disease and low-diffusing capacity for carbon monoxide; those with precapillary PH with severe ILD and worse survival; those with severe PAH, who trended toward worse survival, and those similar to the first cluster but with higher DLCO.
Dr. Frantz then shared preliminary findings of echocardiographic parameters by primary WHO group in PVDOMICS, on behalf of his PVDOMICS collaborators. They found, for example, that the mean right ventricular systolic pressure in group 3 was 45 mm Hg, as opposed to group 1, which was 64 mm Hg. “In general we had some patients in group 3 with less severe elevation of PA pressures,” he said.
Other parameters that can be compared across WHO groups include ventricular fractional area change, tricuspid annular plane systolic excursion, and RV free wall strain. “That strain of the right ventricle is one of the most important ways of looking at how the right ventricle works,” Dr. Frantz explained. “With this, we can integrate the concept of severity of RV dysfunction with severity of pulmonary vascular disease. This is where the rubber hits the road. It’s going to be very complicated and time consuming, but I think critically important. Ultimately, we can make proteomic heat maps that track these correlates, and ultimately identify pathways that may be driving RV compensation in pulmonary vascular disease.”
Dr. Frantz reported having no relevant financial disclosures.
SAN DIEGO – A massive effort to better understand and treat patients with pulmonary hypertension and right heart dysfunction is underway.
The endeavor, funded by the National Heart, Lung, and Blood Institute and the Pulmonary Hypertension Association and known as Redefining Pulmonary Hypertension Through Pulmonary Vascular Disease Phenomics (PVDOMICS), began recruiting participants in 2017, with a goal of 1,500 by 2019. The aim is to perform comprehensive phenotyping and endophenotyping across the World Health Organization–classified pulmonary hypertension (PH) clinical groups 1 through 5 in order to deconstruct the traditional classification and define new meaningful subclassifications of patients with pulmonary vascular disease.
At an international conference of the American Thoracic Society, one of the study’s investigators, Robert P. Frantz, MD, discussed the role of echocardiography and MRI in the overall PVDOMICS program, which he characterized as a work in progress. “Imaging is critically important as we try to integrate severity of pulmonary vascular disease along with how well the ventricle functions as way to try and understand why some patients have a failing RV at a given pulmonary resistance and others don’t,” said Dr. Frantz, who directs the Mayo Pulmonary Hypertension Clinic in Rochester, Minn. The goals are to be able to integrate cardiac morphology and function with contemporaneous hemodynamics, he said. This will allow for validation of noninvasive hemodynamics versus right heart catheterization across all the phenotypes.
“In addition, we’ll have imaging parameters as predictors of hemodynamics at rest and with exercise, particularly in conditions like heart failure with preserved ejection fraction or concerns about left atrial stiffness,” he said. “In these cases, our ability on the basis of echocardiography or MRI to guess what the wedge pressure is at rest or exercise, or to think about other more recently described phenotypes like left atrial stiffness in patients who have left atrial ablation procedures, will be enabled by looking at parameters such as left atrial strain.”
Ultimately, he continued, a key goal of PVDOMICS is to be able to correlate the “-omics” with markers of RV compensation in an effort to understand what the determinants of RV compensation are across the varying types of pulmonary vascular disease.
“If we could do that, we might be able to develop new targets for therapy,” said Dr. Frantz. To illustrate how this might work, he cited findings from researchers who set out to identify and characterize homogeneous phenotypes by a cluster analysis in scleroderma patients with pulmonary hypertension, who were identified from two prospective cohorts in the United States and France (PLoS One 2018 May 15;13[5]:e0197112).
The researchers identified four different clusters of scleroderma patients: those with mild to moderate PAH with no or minimal interstitial lung disease and low-diffusing capacity for carbon monoxide; those with precapillary PH with severe ILD and worse survival; those with severe PAH, who trended toward worse survival, and those similar to the first cluster but with higher DLCO.
Dr. Frantz then shared preliminary findings of echocardiographic parameters by primary WHO group in PVDOMICS, on behalf of his PVDOMICS collaborators. They found, for example, that the mean right ventricular systolic pressure in group 3 was 45 mm Hg, as opposed to group 1, which was 64 mm Hg. “In general we had some patients in group 3 with less severe elevation of PA pressures,” he said.
Other parameters that can be compared across WHO groups include ventricular fractional area change, tricuspid annular plane systolic excursion, and RV free wall strain. “That strain of the right ventricle is one of the most important ways of looking at how the right ventricle works,” Dr. Frantz explained. “With this, we can integrate the concept of severity of RV dysfunction with severity of pulmonary vascular disease. This is where the rubber hits the road. It’s going to be very complicated and time consuming, but I think critically important. Ultimately, we can make proteomic heat maps that track these correlates, and ultimately identify pathways that may be driving RV compensation in pulmonary vascular disease.”
Dr. Frantz reported having no relevant financial disclosures.
AT ATS 2018
Anthrax vaccine recommendations updated in the event of a wide-area release
at their meeting.
The recommendations to the committee sought to optimize the use of Anthrax Vaccine Adsorbed (AVA) in post-exposure prophylaxis (PEP) in the event of a wide-area release of Bacillus anthracis spores. In this event, a mass vaccination effort would be undertaken, requiring expedited administration of AVA. ACIP now recommends that the intramuscular administration may be used over the traditional subcutaneous approach if there are any operational or logistical challenges that delay effective vaccination. Another recommendation from ACIP would allow two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. The committee also recommended that AbxPEP, an antimicrobial, be stopped 42 days after the first dose of AVA or 2 weeks after the last dose.
William A. Bower, MD, of the division of high-consequence pathogens and pathology at the Centers for Disease Control and Prevention, and the anthrax work group looked at three nonhuman primate studies and eight human immunogenicity and adverse event studies during the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE). All of the animal studies were used to predict human survival by vaccinating the nonhuman primates with AVA, then challenging them with B. anthracis. Using animal studies to predict human survival is common practice under the “animal rule.”
When Dr. Bower and the work group assessed the studies comparing intramuscular administration with subcutaneous administration of AVA, they rated the overall evidence as GRADE 2. However, they rated the adverse events data as GRADE 1.
Dr. Bower and his colleagues also reviewed dose-sparing studies to identify the feasibility of allowing two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. For these studies, the anthrax work group gave a GRADE score of 2.
The overall evidence score for microbial duration for PEP was judged to be GRADE 2.
“These forthcoming recommendations will be used by the CDC to inform state and local health departments to better prepare for an emergency response to a wide-area release of Bacillus anthracis spores,” said Dr. Bower.
The committee’s recommendations must be approved by the CDC’s director before they are considered official recommendations.
Dr. Bower did not report any relevant financial conflicts of interest.
at their meeting.
The recommendations to the committee sought to optimize the use of Anthrax Vaccine Adsorbed (AVA) in post-exposure prophylaxis (PEP) in the event of a wide-area release of Bacillus anthracis spores. In this event, a mass vaccination effort would be undertaken, requiring expedited administration of AVA. ACIP now recommends that the intramuscular administration may be used over the traditional subcutaneous approach if there are any operational or logistical challenges that delay effective vaccination. Another recommendation from ACIP would allow two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. The committee also recommended that AbxPEP, an antimicrobial, be stopped 42 days after the first dose of AVA or 2 weeks after the last dose.
William A. Bower, MD, of the division of high-consequence pathogens and pathology at the Centers for Disease Control and Prevention, and the anthrax work group looked at three nonhuman primate studies and eight human immunogenicity and adverse event studies during the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE). All of the animal studies were used to predict human survival by vaccinating the nonhuman primates with AVA, then challenging them with B. anthracis. Using animal studies to predict human survival is common practice under the “animal rule.”
When Dr. Bower and the work group assessed the studies comparing intramuscular administration with subcutaneous administration of AVA, they rated the overall evidence as GRADE 2. However, they rated the adverse events data as GRADE 1.
Dr. Bower and his colleagues also reviewed dose-sparing studies to identify the feasibility of allowing two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. For these studies, the anthrax work group gave a GRADE score of 2.
The overall evidence score for microbial duration for PEP was judged to be GRADE 2.
“These forthcoming recommendations will be used by the CDC to inform state and local health departments to better prepare for an emergency response to a wide-area release of Bacillus anthracis spores,” said Dr. Bower.
The committee’s recommendations must be approved by the CDC’s director before they are considered official recommendations.
Dr. Bower did not report any relevant financial conflicts of interest.
at their meeting.
The recommendations to the committee sought to optimize the use of Anthrax Vaccine Adsorbed (AVA) in post-exposure prophylaxis (PEP) in the event of a wide-area release of Bacillus anthracis spores. In this event, a mass vaccination effort would be undertaken, requiring expedited administration of AVA. ACIP now recommends that the intramuscular administration may be used over the traditional subcutaneous approach if there are any operational or logistical challenges that delay effective vaccination. Another recommendation from ACIP would allow two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. The committee also recommended that AbxPEP, an antimicrobial, be stopped 42 days after the first dose of AVA or 2 weeks after the last dose.
William A. Bower, MD, of the division of high-consequence pathogens and pathology at the Centers for Disease Control and Prevention, and the anthrax work group looked at three nonhuman primate studies and eight human immunogenicity and adverse event studies during the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE). All of the animal studies were used to predict human survival by vaccinating the nonhuman primates with AVA, then challenging them with B. anthracis. Using animal studies to predict human survival is common practice under the “animal rule.”
When Dr. Bower and the work group assessed the studies comparing intramuscular administration with subcutaneous administration of AVA, they rated the overall evidence as GRADE 2. However, they rated the adverse events data as GRADE 1.
Dr. Bower and his colleagues also reviewed dose-sparing studies to identify the feasibility of allowing two full doses or three half doses of AVA to be used to expand vaccine coverage for PEP in the event there is an inadequate vaccine supply. For these studies, the anthrax work group gave a GRADE score of 2.
The overall evidence score for microbial duration for PEP was judged to be GRADE 2.
“These forthcoming recommendations will be used by the CDC to inform state and local health departments to better prepare for an emergency response to a wide-area release of Bacillus anthracis spores,” said Dr. Bower.
The committee’s recommendations must be approved by the CDC’s director before they are considered official recommendations.
Dr. Bower did not report any relevant financial conflicts of interest.
REPORTING FROM AN ACIP MEETING