User login
For MD-IQ use only
AI Scribes or VHA Docs: Which Created Better Clinical Notes?
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Artificial intelligence (AI) scribes produced lower-quality documentation of clinical notes than human clinicians, and especially struggled in settings with background noise or clinicians wearing masks, a new Veterans Health Administration (VHA) study finds.
In 5 simulated clinical cases, notes written by various AI programs scored lower than reports produced by humans on the modified Physician Documentation Quality Instrument (PDQI-9), a measurement of note quality scale, reported Ashok Reddy, MD, MSc, of the University of Washington and Veterans Affairs Puget Sound Health Care System, Seattle, et al in the April issue of Annals of Internal Medicine.
AI scribes scored lower compared with humans across all domains, including accuracy, thoroughness, and usefulness. There was an especially large gap in scores on the 50-point PDQI-9 in an acute low back pain case (human, 43.8 points; AI, 20.3 points; difference, 23.5 points).
“For clinicians, AI scribes should be regarded as tools for generating draft documentation that requires review and editing, rather than as a substitute for clinician-authored notes,” the authors wrote. “Although ambient AI scribes hold promise for reducing clinician burden, rigorous and ongoing evaluation of their quality is essential to ensure that these tools enhance rather than compromise the quality of clinical care.”
AI Scribe Use is Widespread
Taylor N. Anderson, MD, a clinical informatics fellow at Oregon Health & Science University, Portland, is familiar with the study findings and noted that the use of AI scribes in medicine has grown rapidly. All major health organizations are either using it or facing “enormous pressure” from clinicians to do so, she told Federal Practitioner.
Previous research has linked the use of AI scribes for clinical notes to less electronic health record usage and documentation time for clinicians, leading to more time for patient visits. Still, the quality of clinical notes written by AI is “quite variable across vendors,” Anderson said.
Anderson led a 2025 study that examined 5 AI scribe platforms and found an average of 3.0 errors per case with “potential for moderate-to-severe harm.”
For the new study on the simulated cases, part of a VHA-sponsored “technology sprint” via Challenge.gov, researchers developed audio descriptions of 5 clinical cases reflecting common patient encounters in primary care: acute low back pain, chest pain, a new diagnosis of diabetes, a pharmacy consultation, and a follow-up with a nurse case manager for heart failure.
Two cases included non-English accents, 1 included background noise, and 1 featured speech through a medical mask. All the “patients” were played by what the authors described as “trained standardized patient actors.”
For each case, 3 humans and 11 AI scribe programs produced clinical notes. The clinical notes were then evaluated by 6 raters.
Researchers found that AI scribe-generated notes scored worse than human-generated notes across all 10 domains of the modified PDQI-9 (accuracy, thoroughness, usefulness, organization, comprehensiveness, succinctness, synthesization, internal consistency, and freedom from hallucination and bias).
There were especially large gaps between the AI and human notes in the domains of thoroughness, organization, and usefulness. Even wider gaps were observed for the encounters with noise and mask usage.
“These findings highlight that although ambient AI scribes can generate complete notes, the overall quality remains broadly below that of human-authored documentation,” the authors wrote.
No Comparison Between AI Scribes
The researchers noted that “given contractual limitations, we cannot interpret the results for specific vendors.” They also noted that the study did not use professional scribes, who may produce even higher-quality results, and the humans were not producing notes in a real-world clinical environment.
Anderson, the clinical informatics fellow, pointed out that the study does not examine the common scenario in which a clinician edits notes produced by an AI scribe. In fact, she said, there is no current research on this, failing to examine “the postediting note that would actually go into the chart.”
In an accompanying commentary, collaborative scientist Aaron Tierney, PhD, and Kristine Lee, MD, an associate executive director, both with the Permanente Medical Group, California, called for future research to focus on “real-world performance, promote the development of documentation policies that prioritize patient care over billing requirements, and systematically incorporate patient perspectives into assessments of quality.”
Why AI Misses the Mark
In an interview with Federal Practitioner, AI researcher Maxim Topaz, PhD, RN, MA, an associate professor of Nursing and Data Science at Columbia University School of Nursing, New York City, who is familiar with the study but did not participate in it, praised the research.
He pointed out that AI has trouble accurately representing clinical encounters because they “tend to fill gaps with plausible-sounding language, which can mask omissions and make errors harder to catch.” Also, “ambient scribes can only document what is verbalized aloud. Physical exam findings the clinician notices but does not narrate, nonverbal cues, and patient-initiated concerns that drift past in conversation are systematically underrepresented.”
Moving forward, Topaz advised clinicians to “treat AI-generated notes as a first draft, not a finished product. Read them carefully, especially for omissions, which the current evidence suggests are by far the most common error type and which are harder to spot than fabrications because the surrounding note still reads coherently.”
Two study authors disclosed employment by the US Department of Veterans Affairs. Other authors had no disclosures. The commentary authors have no disclosures. Anderson has no disclosures. Topaz discloses relationships with the National Institutes of Health and other federal sources.
Underground Hospitals: Is Combat Medicine Entering a New Era?
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Drone warfare and repeated attacks on medical infrastructure are reshaping battlefield medicine in Ukraine, driving the development of underground military hospitals designed to stabilize and treat wounded soldiers close to active combat zones, rather than relying on rapid evacuation.
Since the start of Russia’s full-scale invasion of Ukraine, the World Health Organization has documented nearly 3000 attacks on healthcare facilities and violations of the Geneva Conventions that protect medical personnel and healthcare infrastructure during armed conflict.
In response, Ukraine has developed underground military hospitals designed to withstand bombardment and maintain the continuity of medical care. By combining infrastructure inherited from the Cold War with rapidly constructed new facilities, the country has managed to preserve healthcare capacity and support military operations close to the frontlines.
Underground Hospital
In September 2024, the Ukrainian Ministry of Defense, in partnership with the Metinvest Group, opened Ukraine’s first underground military stabilization hospital near the front lines. The project was developed under Metinvest’s military support initiative, known as the Steel Front, which supplies protective infrastructure and equipment for frontline operations.
In addition to producing steel bunkers for these facilities, the company manufactures military support equipment, including mine clearing plows, drone protection screens, systems designed to intercept loitering munitions, armor plates, and vehicle reinforcements for frontline operations.
The underground hospital consists of six steel bunkers, each measuring 7.6 m in length and 2.5 m in diameter, with a total area of 500 m2. The structures function as multifunctional units designed to maintain operational capability in high-threat environments. The facility includes ventilation, water supply, drainage, and electrical systems. During construction and installation, security measures aimed to reduce detectability and lower the risk for attack. The hospital also incorporates electronic warfare systems intended to strengthen operational protection.
The total investment reached 20 million Ukrainian hryvnias, approximately 385,000 euros. Of these, 7 million hryvnias funded medical equipment, while 13 million supported metal structures, construction materials, and infrastructure.
The hospital is equipped with oxygen concentrators, ventilators, cardiac monitors, defibrillators, surgical equipment, lighting systems, sterilizers, patient warming systems, and medical furniture. The complex includes two operating rooms, two resuscitation stations, a work area, and a staff rest area. Depending on the staffing and operational configuration, the hospital can stabilize wounded individuals and perform up to four simultaneous procedures. The design follows North Atlantic Treaty Organization standards for second-level field hospitals, designated Role/Echelon 2.
In a statement released by the Metinvest Group after the facility opened in 2024, Roman Kuzev, acting commander of the “East” medical task force, said: “This underground hospital is the best stabilization center available. This will allow us to provide medical care to over 100 patients a day, saving hundreds of lives for our heroes. I hope the number of such facilities will grow.”
Kuzev’s expectations materialized in 2025, when the Metinvest Group completed the construction of a second underground military hospital in one of the most active frontline sectors. The new facility provides greater protection and camouflage, and incorporates structural modifications based on lessons learned from the first hospital. It is buried more than 6 m underground and reinforced with additional protective layers.
The hospital includes four functional units housing surgical and stabilization areas, a delivery room, and a break area for healthcare personnel. The facility covers 350 m2 and required an investment exceeding 21 million Ukrainian hryvnias.
The center can simultaneously support up to three surgical procedures of varying complexities. Military authorities supplied equipment, including high-flow infusion pumps, x-ray systems, oxygen concentrators, defibrillators, and additional devices. Medical services are provided by teams of up to 20 professionals, including orthopedic surgeons, general surgeons, anesthesiologists, surgical nurses, and nursing assistants.
Historic Origin
World War I marked a turning point in modern warfare by introducing technologies that increased battlefield violence to unprecedented levels. The widespread use of machine guns, poisonous gas, tanks, and trench warfare has turned the battlefield into an extremely deadly environment.
At the same time, the conflict drove advances in military medicine that continue to influence practice today, including blood transfusions, psychological support for soldiers experiencing so called “shell shock,” and the development of field hospitals and mobile medical units.
One of the earliest documented underground hospitals was established in Arras, France, where a network of preexisting tunnels known as boves was expanded by New Zealand engineers to provide Allied forces with a tactical advantage. The tunnels were designed to shelter troops in preparation for the 1917 Arras Offensive, allowing them to assemble safely without being detected by German forces.
The underground hospital in Arras, which opened in 1916, includes waiting rooms, operating rooms, rest areas, spaces accommodating up to 700 stretchers, and a morgue. It also features internal electrical and plumbing systems, making it one of the most advanced medical facilities of its time.
Shift in Care
The expanding use of drones on the battlefield has increased the risks linked to casualty evacuation, particularly aeromedical evacuation, reducing the effectiveness of traditional military care models. In response, Ukraine has adopted an approach centered on extended field care and the development of a decentralized medical system, supported by close collaboration with the private sector to rapidly secure resources and infrastructure.
These strategies represent a shift in military medicine toward prolonged onsite stabilization rather than rapid evacuation. The combined use of underground facilities and repurposed infrastructure has helped maintain medical capacity under high threat conditions, improving survival among wounded individuals, and strengthening healthcare system resilience during conflict, according to US Army reports.
In addition to serving as a model for this shift in military medicine, the underground hospital project received the Partnership for Sustainability Award 2025 in Ukraine from the United Nations Global Compact in the “Rebuilding Ukraine” category. The award, presented by the United Nations network that promotes corporate sustainability and Sustainable Development Goals, recognizes private sector initiatives that support postwar reconstruction and strengthen social and institutional resilience.
The project was recognized for its contribution to saving lives and strengthening medical capacity in areas affected by active hostility.
This article was translated from El Médico Interactivo on Univadis, part of the Medscape Professional Network.
A version of this article appeared on Medscape.com.
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
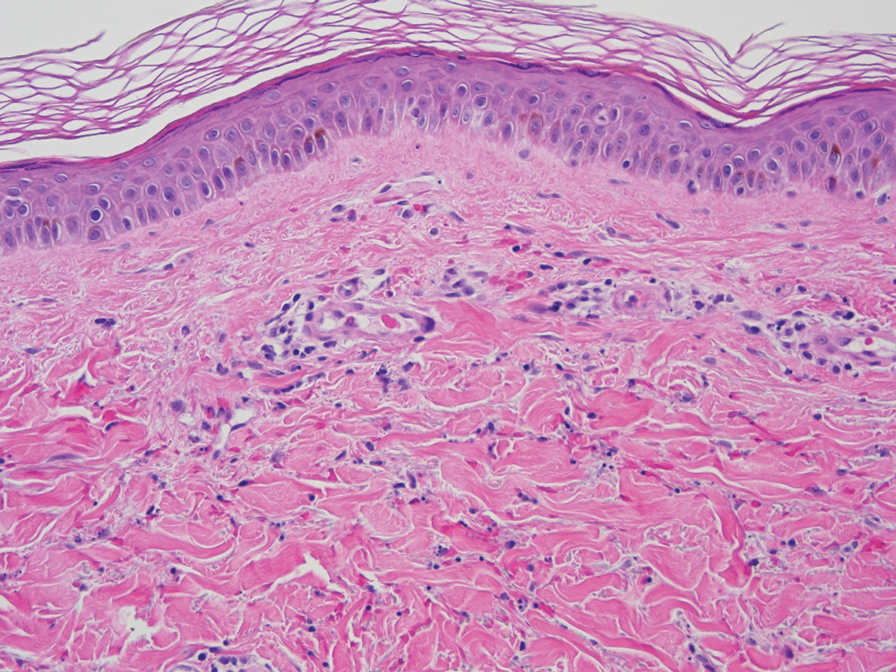
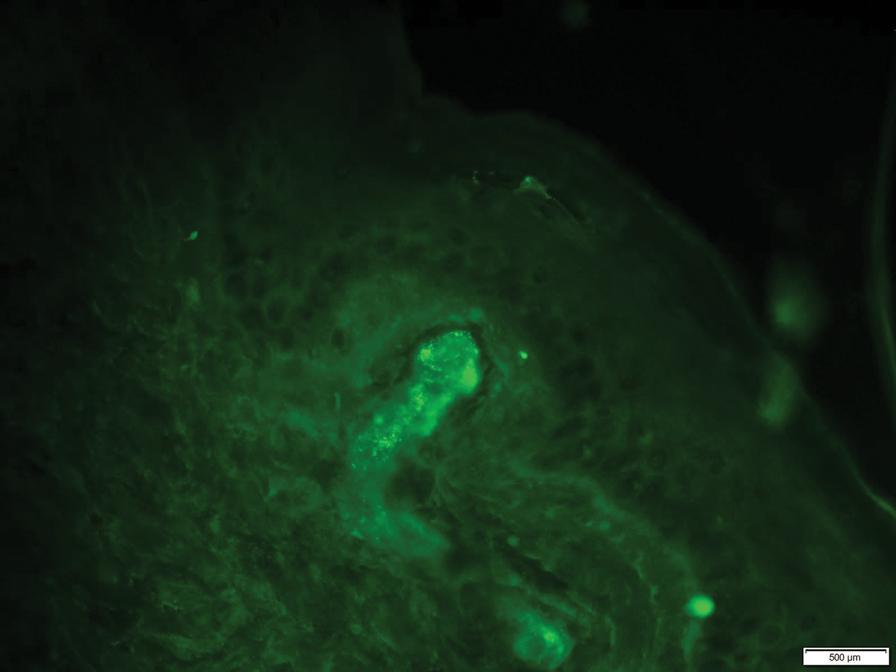
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
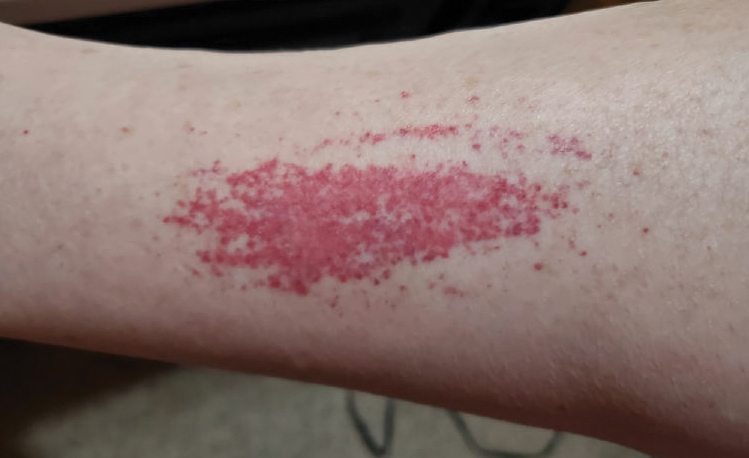
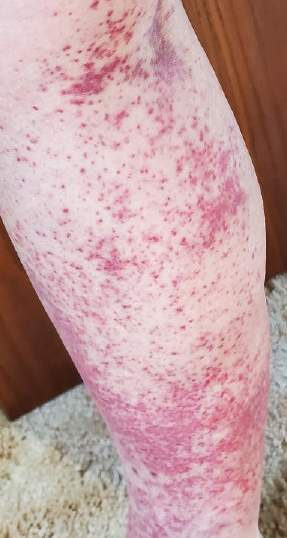
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
Hypergammaglobulinemic purpura of Waldenström (HGPW) is a rare chronic skin condition characterized by recurrent petechiae and purpura on the lower legs, elevated erythrocyte sedimentation rate (ESR), polyclonal hypergammaglobulinemia, and elevated titers of IgG and IgA rheumatoid factor (RF).1,2 This condition can be a primary (idiopathic) syndrome or secondary to an autoimmune connective tissue disease. We report 2 cases of patients with episodic skin eruptions that were consistent with HGPW.
Patient 1
A 41-year-old woman presented to our clinic with a rash on the legs of 20 years’ duration. She had first been evaluated at an outside dermatology clinic 5 years prior, and a biopsy performed at the time led to a diagnosis of leukocytoclastic vasculitis (LCV). The rash affected her ability to work, as her job involved standing for prolonged periods of time. If she stood for more than 2 hours, she experienced leg pain and worsening of the rash. The rash also was exacerbated by nonsteroidal anti-inflammatory drugs but improved with multiple days of rest. She had been on dapsone 75 mg daily, but the dose was reduced to 50 mg daily after elevated liver enzymes were noted. This regimen had improved her rash for 4 years until she experienced breakthrough symptoms, leading to her re-evaluation. Prior outside therapies included systemic steroids with limited response, then oral dapsone.
Upon our initial evaluation, laboratory tests were notable for an elevated ESR of 43 mm/h. Results of antinuclear antibody (ANA), anti–double-stranded DNA, extractable nuclear antigen, RF, HIV, cryoglobulin, hepatitis panel, serum protein electrophoresis, complete blood count, basic metabolic panel, urinalysis, and thyroid-stimulating hormone testing were within reference range. Physical examination revealed scattered pinpoint violaceous papules on the lower extremities. Photographs on the patient’s phone from 2 months prior showed a more robust manifestation with diffuse palpable purpura on the lower extremities.
At 3-year follow-up, laboratory evaluation including ESR, IgA, IgG, IgM, serum protein electrophoresis with reflex immunofixation, and Mycoplasma pneumoniae IgM/IgG showed elevated ESR (29 mm/h) and IgG (1654 mg), with otherwise unremarkable results. Because of the extended period of time since the previous biopsy, a repeat biopsy with hematoxylin and eosin staining and direct immunofluorescence was performed. Biopsy from the left calf demonstrated a perivascular and interstitial infiltrate with lymphocytes and neutrophils with nuclear debris and hemorrhage (Figure 1). Direct immunofluorescence was positive for IgA, C3, and fibrin within vessel walls (Figure 2).
Overall the features of recurrent dependent palpable purpura and the pathology findings were consistent with evolving LCV. Given the chronic nature of her symptoms; flares with prolonged standing; presence of polyclonol hypergammaglobulinemia; and negative evaluation for underling autoimmune disease, infection, and malignancy, the clinicopathologic correlation was most consistent with primary HGPW. The patient was treated with colchicine 0.6 mg twice daily and continued on dapsone 50 mg daily. The colchicine was reduced to once daily due to diarrhea. Nonetheless, the patient had less frequent and less intense flares. On follow-up examination 4 months later, she was satisfied with her current level of control and did not wish to escalate her treatment.
Patient 2
A 53-year-old woman with a 1-year history of sicca symptoms presented for evaluation of a transient rash on the legs and feet of 2 months’ duration. At that time, the heels began to feel swollen. The rash was painful on the feet and caused calf myalgias. She did not endorse pruritus or pain elsewhere. The rash was not associated with prolonged standing, walking, or wearing tight socks. She had no fevers, chills, or joint pain. Flares would come and go within a week.
Laboratory evaluation was notable for an ANA of 1:1280 (reference range, 1:80) with positive anti-Ro/SS-A and anti-La/SS-B. Rheumatology evaluation confirmed the diagnosis of Sjögren syndrome. Physical examination revealed minimal petechiae on the heel of the left foot. Photographs from the previous month provided by the patient revealed linear petechiae of the lower extremities with postinflammatory hyperpigmentation (Figure 3). An additional photograph from the prior week revealed more diffuse erythematous plaques without secondary changes on the feet up to the ankles (Figure 4).
The patient experienced a recurrence of the rash within a month and had an expedited visit for biopsies, which demonstrated mixed inflammation with neutrophils, nuclear debris, hemorrhage, and C3 and fibrin immunoreactants within vessel walls. As with patient 1, the features were consistent with LCV.
In the context of Sjögren syndrome and elevated IgG and RF, the patient’s symptoms were consistent with secondary HGPW. Rheumatology prescribed hydroxychloroquine 400 mg daily alternating every other day with 300 mg and 0.6 mg of colchicine. The rash cleared within approximately 1 month.
Comment
Also known as benign hypergammaglobulinemic purpura, HGPW is a rare purpuric eruption that is exacerbated with prolonged standing and increased hydrostatic pressure.3 First described in 1943, HGPW is characterized by recurrent petechiae, purpuric macules, or palpable purpura, depending on the degree of inflammation.1,4,5 It typically is distributed on the bilateral lower extremities or trunk. Chronic postinflammatory hyperpigmentation with hemosiderin deposition also can be observed. The lesions last for up to 1 week at a time and are frequently asymmetrically distributed.2
Patient 1 demonstrated the typical clinical manifestations and laboratory findings of HGPW. The eruption often is asymptomatic, and patients report that the skin worsens with prolonged immobilization, walking, and wearing of tight clothing.2,6-8 Increased hydrostatic pressure is thought to cause the erythrocyte extravasation, resulting in the purpuric lesions. However, patient 2 was less typical, presenting with prominent skin pain and myalgias. Some patients experience discomfort, burning dysesthesia, pruritus, and swelling of the affected area.1 Hypergammaglobulinemic purpura of Waldenström is a chronic condition. Recurrent episodes can occur yearly or as frequently as multiple times per week.8
Women are most commonly diagnosed with HGPW, but many cases have been reported in children.9,10 In spite of the “condition being considered largely benign,” women with a diagnosis of HGPW require preconception counseling due to risks for congenital heart block, neonatal lupus, intrauterine growth restriction, intrauterine demise, and preterm birth.7,9,11,12
The etiology of the rash remains undefined. It is hypothesized that it develops due to underlying immune dysregulation with associated immune complex formation and deposition in the blood vessel wall.1 Small circulating immune complexes containing IgG or IgA RF are a specific finding in patients with HGPW. These highly soluble autoantibodies are hypothesized to influence the rapid appearance and disappearance of lesions.1
The role of hypergammaglobulinemia in the pathogenesis of HGPW is unknown.13 Serum IgG levels do not correlate with the appearance and regression of lesions.13 Additionally, hypergammaglobulinemia can be found in autoimmune connective tissue diseases such as Sjögren syndrome without resulting cutaneous vasculitis.13
Characteristic laboratory abnormalities include polyclonal hypergammaglobulinemia, elevated ESR, and elevated IgA and IgG RF. Positive ANA and anti-Ro/SS-A and anti-La/SS-B indicate a potential to develop autoimmune connective tissue diseases, including Sjögren syndrome, systemic lupus erythematosus, and rheumatoid arthritis.1,14 Additional recommended workup includes complete blood counts, metabolic panel, complement levels, urinalysis, and urine protein/creatinine ratio.9 Repeat monitoring for antibodies, inflammatory markers, immunoglobulins, and RF should be completed 3 months after initial evaluation. Patients with symptoms of systemic disease should have laboratory evaluation repeated.
Erythrocyte sedimentation rate abnormalities are a defining feature of HGPW. Erythrocyte sedimentation rate is an inexpensive and commonly ordered inflammatory marker that measures settling of erythrocytes within 1 hour and can be elevated by plasma proteins such as gamma globulins. Erythrocyte sedimentation rate is nonspecific and is not sensitive as a general screening test. It can be elevated by autoimmune connective tissue disease, infection, and malignancy.15 Notably, ESR is not specific to inflammation. Confounding factors include red blood cell abnormalities, physiologic factors, and the quantity of plasma proteins such as fibrinogen.16 These positively charged plasma proteins neutralize the negative surface charge of erythrocytes, resulting in erythrocytes that are prone to rouleaux formation.17
The utility of the ESR is to expedite the diagnostic process and indicate the need for further workup.16 Patients with mild to moderate elevation in ESR without an identified etiology should have repeat testing to confirm the validity of the laboratory value. Patients with an ESR higher than 100 mm/h are more likely have an infectious cause, collagen vascular disease, or underlying malignancy.15 Elevation of ESR in HGPW is likely a result of increased immunoglobulins and acute phase proteins.17
The histopathology of HGPW is nonspecific and may show LCV or erythrocyte extravasation with mild perivascular lymphocytic infiltrates.1,9 Direct immunofluorescence testing may show immune-complex deposition.5 For patients with evidence of LCV, the biopsy of a fresh but well-developed lesion is important in confirming the presence of vasculitis.1 Incorrect sampling may lead to underreporting of LCV with HGPW.3
Associated underlying conditions include Sjögren syndrome, systemic lupus erythematosus, rheumatoid arthritis, hepatitis C, and hematologic malignancies.1,3 Our patients demonstrated primary and secondary causes of HGPW. Patient 1’s case was not associated with any autoimmune disease but demonstrated chronic recurrence. Patient 2’s case was secondary to Sjögren syndrome.
In patients with suspected HGPW, differential diagnoses to consider include IgA vasculitis, cutaneous small vessel vasculitis, pigmented purpuric dermatoses, idiopathic thrombocytopenic purpura, thrombotic thrombocytopenic purpura, and scurvy.1,4
For patients with primary disease, treatment is focused on symptom management with compression stockings and avoidance of triggers. Compression stockings may exacerbate purpura but can provide symptom relief in some individuals.14 Patients with frequent or painful episodes can benefit from systemic treatment. In patients with an underlying disease, systemic therapies include prednisone, hydroxychloroquine, indomethacin, colchicine, chlorambucil, mycophenolate mofetil, rituximab, and plasmapheresis. Dapsone, a treatment for LCV, has been reported to be beneficial in patients with a neutrophilic infiltrate.18
Hypergammaglobulinemic purpura of Waldenström requires a thorough evaluation due to its association with underlying systemic disease. Patients without evidence of systemic disease should receive long-term monitoring and coordination of care with rheumatology, as systemic manifestations can develop years after the initial cutaneous manifestation. Dermatologists should consider HGPW in the differential diagnosis for cutaneous vasculitides.
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
- Piette WW. Purpura: mechanisms and differential diagnosis.In: Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Elsevier Health Sciences; 2018:376-389.
- Finder KA, McCollough ML, Dixon SL, et al. Hypergammaglobulinemic purpura of Waldenström. J Am Acad Dermatol. 1990;23(4 Pt 1):669-676. doi:10.1016/0190-9622(90)70271-i
- Mathis J, Zirwas M, Elkins CT, et al. Persistent and progressive purpura in a patient with an elevated rheumatoid factor and polyclonal gammopathy (hypergammaglobulinemic purpura of Waldenström). J Am Acad Dermatol. 2015;72:374-376. doi:10.1016/j.jaad.2013.02.020
- 4. Alexandrescu DT, Levi M. The vascular purpuras. In: Kaushansky K, Prchal JT, Burns LJ, et al, eds. Williams Hematology. 10th ed. McGraw Hill; 2021:1-34.
- Lewin JM, Hunt R, Fischer M, et al. Hypergammaglobulinemic purpura of Waldenström. Dermatol Online J. 2012;18:2.
- Habib GS, Stimmer MM, Quismorio FP. Hypergammaglobulinemic purpura of Waldenstrom associated with systemic lupus erythematosus: report of a case and review of the literature. Lupus. 1995;4:19-22. doi:10.1177/096120339500400105
- Maeda-Tanaka M, Haruta S, Sado T, et al. Juvenile-onset hypergammaglobulinemic purpura and fetal congenital heart block.J Dermatol. 2006;33:714-718. doi:10.1111/j.1346-8138.2006.00166.x
- Malaviya AN, Kaushik P, Budhiraja S, et al. Hypergammaglobulinemic purpura of Waldenström: report of 3 cases with a short review. Clin Exp Rheumatol. 2000;18:518-522.
- Theisen E, Lee DE, Pei S, et al. Hypergammaglobulinemic purpura of Waldenström in children. Pediatr Dermatol. 2020;37:467-475. doi:10.1111/pde.14120
- Martini A, Ravelli A, Viola S, et al. Hypergammaglobulinemic purpura in childhood. Report of two cases and review of the literature. Helv Paediatr Acta. 1988;43:225-231.
- Jolly EC, Hunt BJ, Ellis S, et al. “Benign” hypergammaglobulinemic purpura is not benign in pregnancy. Clin Rheumatol. 2009;28(Suppl 1):S11-S15. doi:10.1007/s10067-008-1038-2
- Cheung VY, Bocking AD, Hollomby D, et al. Waldenström hypergammaglobulinemic purpura and pregnancy. Obstet Gynecol. 1993;82(4 Pt 2 Suppl):685-687.
- Kimura K, Miyabe C, Miyata R, et al. Hypergammaglobulinemic purpura: does hypergammaglobulinemia cause purpura? J Dermatol. 2021;48:e556-e557. doi:10.1111/1346-8138.16122
- Frankel A, Ingraffea A, Massé M, et al. Hypergammaglobulinemic purpura of Waldenström. Cutis. 2010;86:23-24.
- Brigden ML. Clinical utility of the erythrocyte sedimentation rate. Am Fam Physician. 1999;60:1443-1450.
- Solberg BL, Olson RJ. Clinical utility of the erythrocyte sedimentation rate: a case study. Clin Lab Sci. 2014;27:72-77.
- Tishkowski K, Gupta V. Erythrocyte sedimentation rate. In: StatPearls. StatPearls Publishing; May 9, 2021.
- Cheah J, Fields T. Hypergammaglobulinemic purpura of Waldenström. October 2018. Accessed November 14, 2021. https://www.hss.edu/files/HSS-Grand-Rounds-Complex-Cases-Vol7-Issue3.pdf
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Hypergammaglobulinemic Purpura of Waldenström With Primary and Autoimmune Associations
Practice Points
- Elevation of the erythrocyte sedimentation rate (ESR) is nonspecific for inflammation and may be observed in the setting of increased immunoglobulin levels.
- Patients with elevated ESR and clinical evidence of recurrent petechiae and purpura should be screened for monoclonal and polyclonal gammopathies.

A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
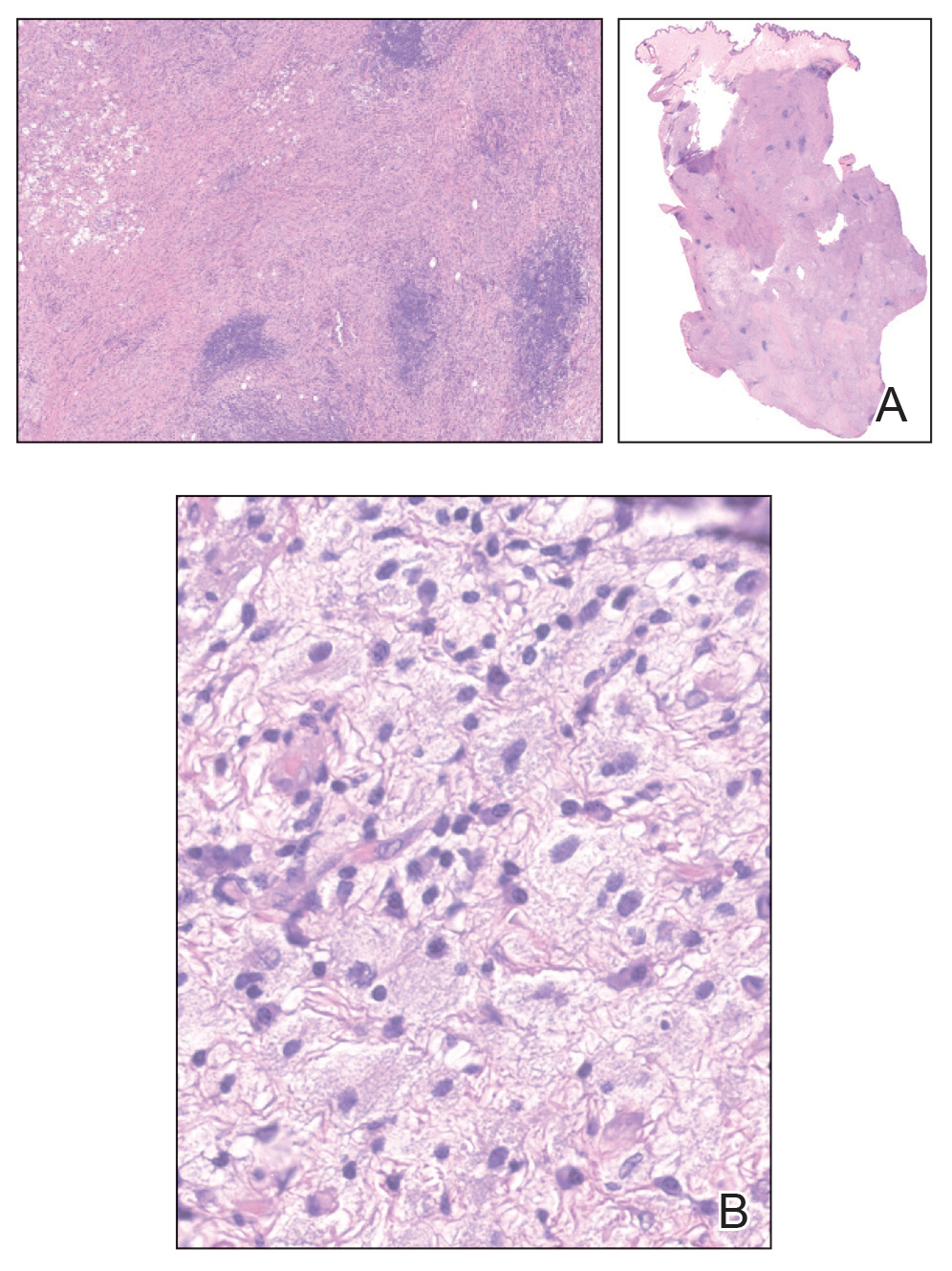
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
- Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Stefanato CM, Ellerin PS, Bhawan J. Cutaneous sinus histiocytosis (Rosai-Dorfman disease) presenting clinically as vasculitis. J Am Acad Dermatol. 2002;46:775-778.
- Dalia S, Sagatys E, Sokol L, et al. Rosai-Dorfman Disease: tumor biology, clinical features, pathology, and treatment. Cancer Control. 2014;21:322-327.
- Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73:697.
- Bolognia J, Jorizzo J, Schaffer J. Dermatology. 3rd ed. ed. Elsevier Saunders 2012.
- Salva KA, Stenstrom M, Breadon JY, et al. Possible association of cutaneous rosai-dorfman disease and chronic crohn disease: a case series report. JAMA Dermatol. 2014;150:177-181.
- Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002; 24:385-391.
- Betini N, Munger AM, Rottmann D, et al. Rare presentation of Rosai- Dorfman disease in soft tissue: diagnostic findings and surgical treatment. Case Rep Surg. 2022;2022:8440836.
- Cravero JC, Ibrahim S. Recurrent soft tissue rosai dorfman disease of right medial thigh lipoma with lymph node involvement. Fed Pract. 2024;41(suppl 2):S20-S23
- Tenny SO, McGinness M, Zhang D, et al. Rosai-Dorfman disease presenting as a breast mass and enlarged axillary lymph node mimicking malignancy: a case report and review of the literature. Breast J. 2011;17:516-520.
- Goyal G, Ravindran A, Young JR, et al. Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105:348-357.
- Li H, Li D, Xia J, et al. Radiological features of Rosai-Dorfman disease: case series and review of the literature. Clin Radiol. 2022;77:E799-E805.
- Elbaz Younes I, Sokol L, Zhang L. Rosai-Dorfman disease between proliferation and neoplasia. Cancers. 2022;14:5271.
- Ravindran A, Rech KL. How I diagnose Rosai-Dorfman disease. Am J Clin Pathol. 2023;160:1-10.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- Luppi M, Barozzi P, Garber R, et al. Expression of human herpesvirus 6 antigens in benign and malignant lymphoproliferative diseases. Am J Pathol. 1998;153:815-823.
- Wang KH, Chen WY, Liu HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
- Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer. 2014;61:1329-1335.
- Ravindran A, Goyal G, Go RS, et al. Rosai-Dorfman disease displays a unique monocyte-macrophage phenotype characterized by expression of OCT2. Am J Surg Pathol. 2021;45:35-44.
- Hoogewerf CJ, van Baar ME, Middelkoop E, et al. Impact of facial burns: relationship between depressive symptoms, self-esteem and scar severity. Gen Hosp Psychiatry. 2014;36:271-276.
- Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131:2877-2890.
- Al-Khateeb THH. Cutaneous Rosai-Dorfman disease of the face: a comprehensive literature review and case report. J Oral Maxillofacial Surg. 2016;74:528-540.
- Cheng SP, Jeng KS, Liu CL. Subcutaneous Rosai–Dorfman disease: is surgical excision justified? J Eur Acad Dermatol Venereol. 2005; 19:747-750.
- Garcia RA, DiCarlo EF. Rosai-Dorfman disease of bone and soft tissue. Arch Pathol Lab Med. 2021;146:40-46.
A Solitary Axillary Subcutaneous Mass
A Solitary Axillary Subcutaneous Mass
A 34-year-old man presented to our dermatology clinic for evaluation of a lesion in the right axilla of 1 year’s duration that had recently increased in size. The lesion was nontender and intermittently pruritic and was associated with focal hypohidrosis. The patient denied any fevers, chills, or recent weight change. His medical history was otherwise unremarkable. His only medications were daily ashwagandha and vitamin B and C supplements. On physical examination, a firm, 6-cm, subcutaneous nodule was noted in the right axilla with central alopecia and without a clear punctum. He had no palpable cervical, postauricular, or inguinal lymphadenopathy. The left axilla was clear, and there were no other relevant skin findings. Laboratory testing including a complete blood count, comprehensive metabolic panel, and sexually transmitted infections panel was unremarkable. Ultrasonography and subsequent magnetic resonance imaging of the right axilla showed a 4.9-cm nodule located in the subcutaneous fat with minimal deep infiltration and relatively smooth margins. An incisional biopsy of the lesion was performed.
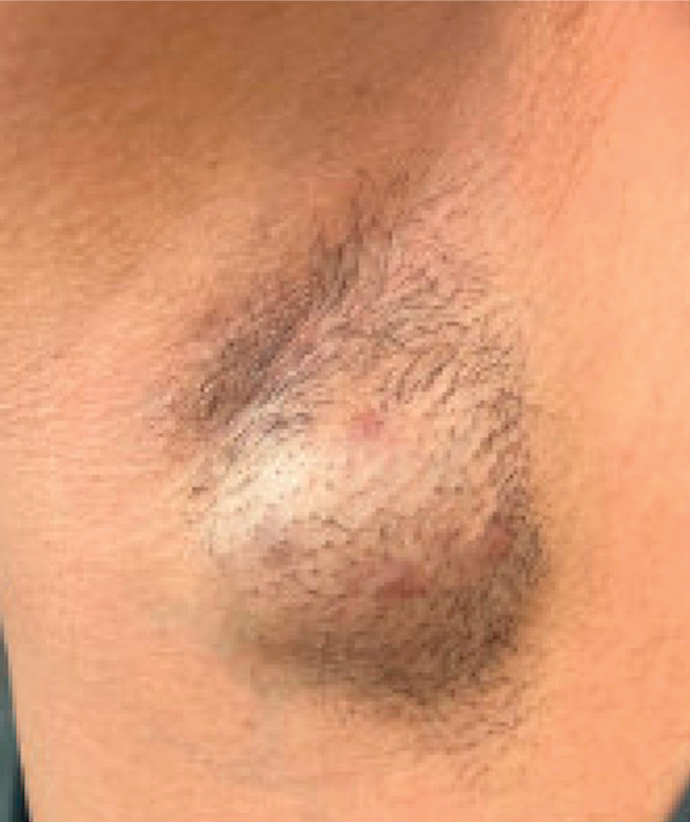

Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
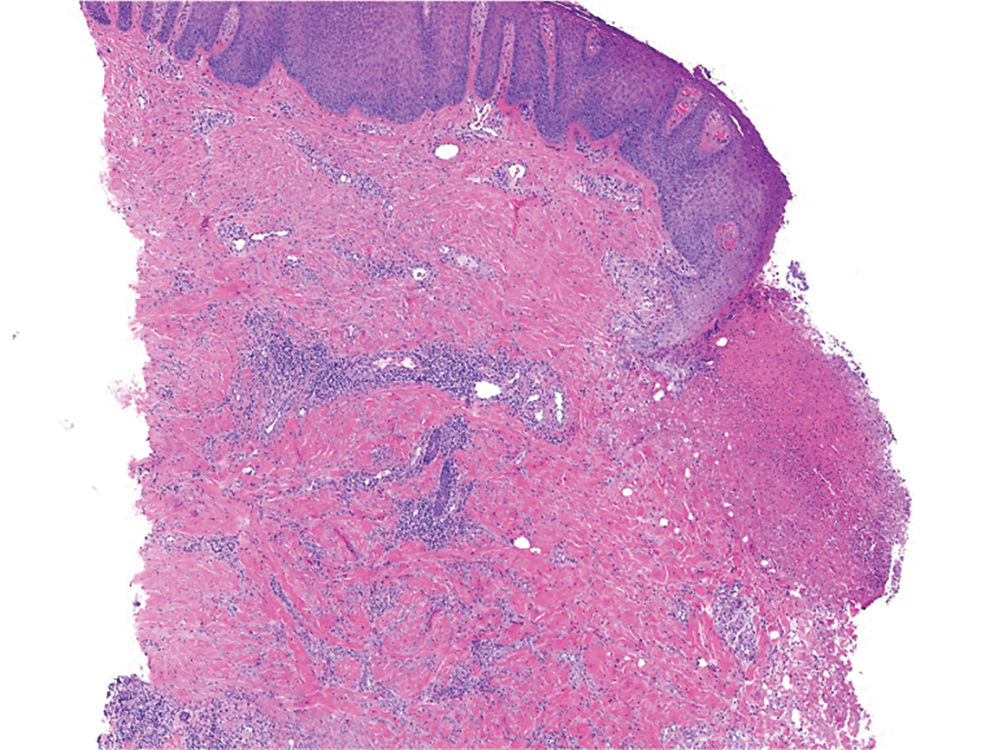
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
To the Editor:
Xylazine, commonly referred to by its street name tranq, is a veterinary tranquilizer that has recently gained attention due to its increasing misuse in human populations. It often is combined with recreational drugs like fentanyl to extend the duration of drug effects. As a partial α2 receptor agonist, xylazine acts by reducing dopamine and norepinephrine release, resulting in sedative effects. This case report highlights xylazine skin necrosis manifesting as wrist drop and chronic wounds in a patient with a history of intravenous (IV) drug use.
A 35-year-old man with a history of IV drug use presented to the emergency department with a nonprogressive right wrist drop that had persisted for 2 weeks, along with new-onset left wrist drop of 1 day’s duration. The patient did not report any sensory symptoms or pain. Physical examination revealed an ulcerated necrotic plaque with hemorrhagic crust and focal areas of scarring on the right posterior forearm (Figure 1). The left hand exhibited a well-healed pink scar symmetric to the ulcer on the right forearm. The patient reported a history of a similar ulcer on the left hand that had resolved after discontinuation of IV drug use in that arm. He denied any history of trauma to the area.
The patient’s laboratory results demonstrated elevated inflammatory markers, including an erythrocyte sedimentation rate of 105 mm/h (reference range, <15 mm/h in men younger than 50 years) and a C-reactive protein level of 7.7 mg/dL (reference range, <0.9 mg/dL). Additionally, antinuclear antibody and antineutrophil cytoplasmic antibody tests were positive. A urine drug screen returned positive results for various substances, including cocaine, cocaine metabolites, fentanyl, norfentanyl, β-hydroxyfentanyl or fentanyl metabolite, caffeine, caffeine metabolite or theophylline, nicotine metabolite, and xylazine. Magnetic resonance imaging of the right upper extremity excluded osteomyelitis but revealed multiple subepidermal abscesses.
A punch biopsy from the right forearm demonstrated an ulcer with a mixed infiltrate, dermal necrosis, and clusters of Gram-positive cocci, indicating a bacterial infection. There was no evidence of leukocytoclastic vasculitis (Figures 2 and 3). Electromyography confirmed mononeuritis multiplex as the cause of the right wrist drop. The patient was found to have cytoplasmic antineutrophil cytoplasmic antibody–positive vasculitis in the setting of levamisole-adulterated cocaine use. Since no vasculitis was identified on histopathology of the ulcer and xylazine was detected on drug screening, a diagnosis of xylazine-induced skin necrosis was made. In our case, the patient did not show evidence of active osteomyelitis or sepsis and left the hospital against medical advice without adequate wound debridement.
Our case highlights xylazine-induced skin necrosis that can occur in individuals who use IV drugs. The combination of xylazine with other recreational drugs such as fentanyl poses unique challenges for clinicians. Xylazine has been increasingly found in cases of overdose-related mortality1 and recently has been reported to induce skin ulcers.2 Xylazine intoxication, though uncommon, can result in distinct clinical presentations, including recalcitrant skin ulcers and deep necrotizing wounds.
The precise mechanism behind these wounds remains unclear. Xylazine is a partial α2 receptor agonist, and it is postulated that the necrotic wounds develop secondary to local vasoconstriction, leading to decreased skin perfusion.3 A recent study found that xylazine used in combination with cocaine or an active metabolite in heroin can cause cytotoxicity to vascular endothelial cells, which can lead to dysregulation of vascular tone.4 Decreased perfusion and impaired wound healing put patients at risk for secondary infections, infected ulcers, osteomyelitis, and sepsis.
In patients with known fentanyl use in conjunction with skin necrosis, a high degree of suspicion for xylazine intoxication should be employed. Ruling out vasculitis (via serologic markers and skin biopsy) as well as atypical skin infections is important in these patients to identify potential cases of xylazine-induced skin necrosis. Other IV drugs such as krokodil (desomorphine) can cause severe skin necrosis and therefore should be considered in these patients. Early detection of these skin ulcers is imperative, as delayed diagnosis increases the risk for osteomyelitis and/or the need for amputation.
This case emphasizes the importance of health care providers remaining vigilant about emerging trends in drug misuse. Early recognition of xylazine intoxication and its potential complications is crucial for timely intervention and appropriate management, which may include wound debridement and antibiotic therapy. In addition, proper counseling regarding discontinuation of drug use is important in wound healing, though this poses a challenging conversation with the patient. Increased awareness among health care professionals and continued research in illicit drug–induced skin necrosis will aid in better understanding and addressing the growing issue of xylazine misuse.
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
- Friedman J, Montero F, Bourgois P, et al. Xylazine spreads across the US: a growing component of the increasingly synthetic and polysubstance overdose crisis. Drug Alcohol Depend. 2022;233:109380. doi:10.1016/j.drugalcdep.2022.109380
- Malayala SV, Papudesi BN, Bobb R, et al. Xylazine-induced skin ulcers in a person who injects drugs in Philadelphia, Pennsylvania, USA. Cureus. 2022;14:E28160. doi:10.7759/cureus.28160
- McNinch J, Maguire M, Wallace L, et al. A case of skin necrosis caused by intravenous xylazine abuse. Abstract presented at: SHM Converge; May 3-7, 2021.
- Silva-Torres LA, Vélez C, Lyvia Alvarez J, et al. Toxic effects of xylazine on endothelial cells in combination with cocaine and 6-monoacetylmorphine. Toxicol In Vitro. 2014;28:1312-1319. doi:10.1016/j.tiv.2014.06.013
Xylazine-Induced Skin Necrosis
Xylazine-Induced Skin Necrosis
Practice Points
- Dermatologists should be aware of the potential for xylazine to cause ulcers in patients with a history of intravenous drug use.
- Early recognition of xylazine skin ulcers is imperative, as delayed diagnosis increases morbidity such as soft-tissue and bone infection, sepsis, and death.

Multiple Papules and Pustules on the Face and Neck
Multiple Papules and Pustules on the Face and Neck
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
THE DIAGNOSIS: Demodicosis
Direct microscopic examination of the purulent fluid revealed a considerable number of actively motile Demodex mites (Figure). Based on the microscopy results and the patient’s history of prolonged topical immunosuppressive therapy, a known risk factor for Demodex overgrowth, a diagnosis of demodicosis was made. The patient was prescribed a single dose of oral metronidazole 2 g as well as metronidazole solution 0.5% to be applied 3 times daily. The folliculitis gradually improved and eventually resolved completely.
Demodex is a parasitic mite inhabiting the pilosebaceous units of human skin. Evidence suggests the vast majority of adults carry these mites. Demodex mites maintain a balance with the human immune system in appropriate microenvironments, with the immune system controlling their numbers without eliciting an inflammatory response; however, immunosuppression, as induced by topical corticosteroids and other immunomodulators, can lead to an increase in Demodex mite populations on facial skin. Clinical manifestations and severity of demodicosis are highly variable, ranging from nonspecific dry, sensitive skin and papules to nodules or granulomas, depending on mite density, the cutaneous microenvironment, and the host immune response.1 Consequently, demodicosis often is mistaken for other dermatologic conditions with similar skin lesions.
High Demodex mite density is considered a pathogenic factor in demodicosis; therefore, determining Demodex mite density is essential to the diagnosis of demodicosis. Standard skin surface biopsy and direct microscopic examination commonly are used methods for measuring Demodex mite density; however, the accuracy of these methods is subject to the technical proficiency of the investigator. Noninvasive examination tools like dermoscopy and confocal laser scanning also offer advantages in diagnosing demodicosis. Dermoscopy, by direct contact with skin lesions, typically reveals gelatinous filaments extending from the follicular openings.
Importantly, Demodex mite density alone does not determine the severity of clinical symptoms. In addition, mites may migrate to the skin surface or reside deep within follicles, rendering them difficult to detect with standard examination methods.1 Therefore, diagnostic criteria should extend beyond mite proliferation to include characteristic clinical lesions, response to acaricidal therapy, and normalization of mite density.
Rosacea was included in the differential diagnosis for our patient, but it typically manifests in the central facial area (eg, forehead, nose, chin). Patients may have a history of facial flushing associated with alcohol consumption, heat exposure, or emotional stress.2 Additionally, rosacea typically has an insidious onset and does not erupt suddenly within a short period of time; however, our patient presented with a sudden onset of widespread papules and pustules on the face without facial flushing, and there was no exacerbation upon exposure to heat or emotional stress. Furthermore, rosacea tends to be recurrent and challenging to cure, whereas our patient responded rapidly to treatment without recurrence. Therefore, the likelihood of rosacea was minimal. Histopathologic examination also can differentiate between rosacea and demodicosis. Histologically, the features of rosacea include dilated blood and lymphatic vessels and infiltration of T lymphocytes, macrophages, and mast cells around blood vessels, often with increased solar elastosis and dermal edema.3 Demodicosis can reveal Demodex mites within the infundibulum of hair follicles, with dense neutrophil and monocyte infiltration around and between the infundibula.4
Bacterial folliculitis is primarily characterized by perifollicular erythema, papules, and pustules, often accompanied by pain. Positive bacterial culture of purulent fluid is indicative.5 Our patient’s lesions shared certain similarities with bacterial folliculitis but lacked the characteristic pain, instead exhibiting pronounced pruritus. Remarkable therapeutic efficacy was observed following topical acaricidal treatment, thus rendering the diagnosis of bacterial folliculitis less probable.
Acne vulgaris is a noninfectious folliculitis caused by follicular occlusion. Abnormal keratinization leads to the obstruction of follicles by keratin, hindering the outflow of sebum from the follicles. Sebum accumulation within the follicles provides a rich substrate for Propionibacterium acnes, which metabolizes sebum into proinflammatory free fatty acids, resulting in the formation of comedones, papules, and pustules.5 Our patient did not exhibit comedonal lesions on the face and lacked a seborrheic complexion, hence diminishing the likelihood of acne vulgaris.
Tinea corporis is another intensely pruritic condition, especially in areas subjected to prolonged use of topical immunosuppressants. It is caused by dermatophyte fungi and typically manifests as erythematous pruritic patches, often presenting as ring-shaped lesions with active margins and sometimes accompanied by scaling.6 While long-term use of immunosuppressants may be a risk factor for fungal infections and increase the probability of tinea corporis, our patient’s presentation of papules and pustules without a ring-shaped configuration or scaling diminished the likelihood of tinea corporis.
Our patient represents an intriguing case of an eruptive form of demodicosis induced by long-term intermittent and inconsistent application of topical immunosuppressive agents. Demodicosis encompasses a spectrum of clinical presentations, including pityriasis folliculorum, rosacealike, folliculitislike, and perioral dermatitis–like forms.1 It is prone to misdiagnosis, as it is clinically similar to other conditions, such as acne, rosacea, or bacterial folliculitis, and it also is susceptible to missed diagnosis. Demodicosis tends to erupt in immunocompromised individuals, and the use of topical immunosuppressive and corticosteroid medications can exacerbate Demodex activity. Dermatologists should be aware that demodicosis is not a rare skin disorder, and timely identification and diagnosis can reduce the incidence of disease and improve quality of life for affected patients. Conversely, the consequences of misdiagnosis can be severe, with inappropriate treatment potentially exacerbating the condition.
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
- Paichitrojjana A. Demodex: the worst enemies are the ones that used to be friends. Dermatol Reports. 2022;14:9339. doi:10.4081 /dr.2022.9339
- Del RJ, Baldwin H, Bhatia N, et al. A review of the diagnostic and therapeutic gaps in rosacea management: consensus opinion. Dermatol Ther (Heidelb). 2024;14:271-284. doi:10.1007/s13555-023-01087-8
- Powell FC. The histopathology of rosacea: ‘where’s the beef?’ Dermatology. 2004;209:173-174. doi:10.1159/000079884
- Helou W, Avitan-Hersh E, Bergman R. Demodex folliculitis of the scalp: clinicopathological study of an uncommon entity. Am J Dermatopathol. 2016;38:658-663. doi:10.1097/DAD.0000000000000512
- Laureano AC, Schwartz RA, Cohen PJ. Facial bacterial infections: folliculitis. Clin Dermatol. 2014;32:711-714. doi:10.1016 /j.clindermatol.2014.02.009
- Leung AK, Lam JM, Leong KF, et al. Tinea corporis: an updated review. Drugs Context. 2020;9. doi:10.7573/dic.2020-5-6
Multiple Papules and Pustules on the Face and Neck
Multiple Papules and Pustules on the Face and Neck
A 26-year-old woman presented to our clinic with multiple papules and pustules on the face and neck. One year prior, the patient had developed a pruritic rash on the face after using a new over-the-counter skin care product. An outside physician had diagnosed the rash as contact dermatitis and prescribed tacrolimus cream 0.1%. Initially, the patient noted improvement, but the rash recurred intermittently over the next year. She continued using the cream, but 2 months prior to the current presentation, the patient developed more papules and pustules on the face, prompting further evaluation.
Physical examination at the current presentation revealed widespread papules and pustules on the face and neck. Due to the patient’s aesthetic concerns, a more invasive biopsy was avoided, and purulent fluid from the lesions was collected for microscopic examination.

A Legacy in Dermatology: Dr. Vincent A. DeLeo Named AAD Master Dermatologist
A Legacy in Dermatology: Dr. Vincent A. DeLeo Named AAD Master Dermatologist
The Cutis editorial staff is proud to announce that Vincent A. DeLeo, MD, Editor-in-Chief, was honored with the Master Dermatologist Award at the 2026 Annual Meeting of the American Academy of Dermatology (AAD) in Denver, Colorado.
Presented as part of the AAD’s “Stars of the Academy” program, this award is reserved for physicians whose careers have advanced dermatology through leadership, service, and meaningful contributions to patient care, education, and research. The award reflects Dr. DeLeo’s impact across the specialty.

“Vince’s passion for dermatology has impacted all aspects of our specialty. He has been at the forefront of basic science research, clinical dermatology, education, mentoring, and leadership of specialty organizations and societies.” –Susan C. Taylor, MD
During the presentation, outgoing AAD president Susan C. Taylor, MD, emphasized Dr. DeLeo’s wide-ranging influence, noting his reputation as a researcher, compassionate physician, and skilled diagnostician. He is adept at managing complex cases and improving patient outcomes. Dr. DeLeo is widely recognized for his expertise in contact dermatitis, photomedicine, and photoprotection, as well as for his contributions to dermatologic education.
Beyond his clinical and editorial leadership of Cutis for the past 25 years, Dr. DeLeo is committed to mentorship and leadership by serving on the AAD Board of Directors as well as other specialty organizations such as the American Contact Dermatitis Society.
We congratulate Dr. DeLeo on this well-deserved distinction and thank him for his continued vision and dedication to our readers and the specialty at large.
The Cutis editorial staff is proud to announce that Vincent A. DeLeo, MD, Editor-in-Chief, was honored with the Master Dermatologist Award at the 2026 Annual Meeting of the American Academy of Dermatology (AAD) in Denver, Colorado.
Presented as part of the AAD’s “Stars of the Academy” program, this award is reserved for physicians whose careers have advanced dermatology through leadership, service, and meaningful contributions to patient care, education, and research. The award reflects Dr. DeLeo’s impact across the specialty.
“Vince’s passion for dermatology has impacted all aspects of our specialty. He has been at the forefront of basic science research, clinical dermatology, education, mentoring, and leadership of specialty organizations and societies.” –Susan C. Taylor, MD
During the presentation, outgoing AAD president Susan C. Taylor, MD, emphasized Dr. DeLeo’s wide-ranging influence, noting his reputation as a researcher, compassionate physician, and skilled diagnostician. He is adept at managing complex cases and improving patient outcomes. Dr. DeLeo is widely recognized for his expertise in contact dermatitis, photomedicine, and photoprotection, as well as for his contributions to dermatologic education.
Beyond his clinical and editorial leadership of Cutis for the past 25 years, Dr. DeLeo is committed to mentorship and leadership by serving on the AAD Board of Directors as well as other specialty organizations such as the American Contact Dermatitis Society.
We congratulate Dr. DeLeo on this well-deserved distinction and thank him for his continued vision and dedication to our readers and the specialty at large.
The Cutis editorial staff is proud to announce that Vincent A. DeLeo, MD, Editor-in-Chief, was honored with the Master Dermatologist Award at the 2026 Annual Meeting of the American Academy of Dermatology (AAD) in Denver, Colorado.
Presented as part of the AAD’s “Stars of the Academy” program, this award is reserved for physicians whose careers have advanced dermatology through leadership, service, and meaningful contributions to patient care, education, and research. The award reflects Dr. DeLeo’s impact across the specialty.
“Vince’s passion for dermatology has impacted all aspects of our specialty. He has been at the forefront of basic science research, clinical dermatology, education, mentoring, and leadership of specialty organizations and societies.” –Susan C. Taylor, MD
During the presentation, outgoing AAD president Susan C. Taylor, MD, emphasized Dr. DeLeo’s wide-ranging influence, noting his reputation as a researcher, compassionate physician, and skilled diagnostician. He is adept at managing complex cases and improving patient outcomes. Dr. DeLeo is widely recognized for his expertise in contact dermatitis, photomedicine, and photoprotection, as well as for his contributions to dermatologic education.
Beyond his clinical and editorial leadership of Cutis for the past 25 years, Dr. DeLeo is committed to mentorship and leadership by serving on the AAD Board of Directors as well as other specialty organizations such as the American Contact Dermatitis Society.
We congratulate Dr. DeLeo on this well-deserved distinction and thank him for his continued vision and dedication to our readers and the specialty at large.
A Legacy in Dermatology: Dr. Vincent A. DeLeo Named AAD Master Dermatologist
A Legacy in Dermatology: Dr. Vincent A. DeLeo Named AAD Master Dermatologist

Cutaneous Reactions to Triatomine (Kissing Bug) Bites and the Risk for Chagas Disease
Cutaneous Reactions to Triatomine (Kissing Bug) Bites and the Risk for Chagas Disease
Triatome bugs cause painful bites and serve as vectors for Chagas disease. In this article, we will address diagnosis and vector identification.
Key Morphologic Features
Insects from the subfamily Triatominae are identifiable by their long legs and a shieldlike abdomen behind a platelike pronotum that covers the thorax. Their half-membranous wings overlap, covering the central abdomen but leaving the lateral portions visible. Tigerlike stripes are characteristically prominent on the visible portions of the lateral abdomen. The stalklike head has an articulated beaklike mouth that can be retracted and used to deliver a powerful bite (Figure 1).

Feeding Mechanisms and Host Reactions
Triatome bugs are blood-feeding arthropods that hide in cracks and crevices in domestic structures by day and feed at night. They are shy feeders, and laboratory colonies have been known to die rather than feed in daylight. They are particularly common in thatched or wattle-and-daub dwellings, where they can be present in great numbers and descend on sleeping inhabitants at night. Triatome bugs require regular blood meals throughout the 5 developmental nymph stages in order to undergo successful molting.
In the wild, triatome bugs feed on a range of animals with little specificity, but in domestic settings they feed largely on humans. Thermosensors in the antennae help them locate blood vessels under the skin, which they penetrate easily due to their long mouthparts. Like other blood-sucking arthropods, they release an anticoagulant that facilitates continuous blood flow while feeding, which accounts for many of the cutaneous reactions observed after the host sustains a triatomine bite.1
Triatomine bugs have trouble feeding through clothing and seek out exposed skin, particularly the eyelids, producing the characteristic unilateral eyelid swelling known as the Romaña sign. Other bite reactions include purpura; macular erythema; and vesiculobullous, papular, and urticarial lesions (Figure 2).2 Associated lymphangitis or lymphadenopathy may be noted, and anaphylaxis has been reported. Similar to those of cockroaches, triatome antigens have been associated with atopic dermatitis and asthma.3
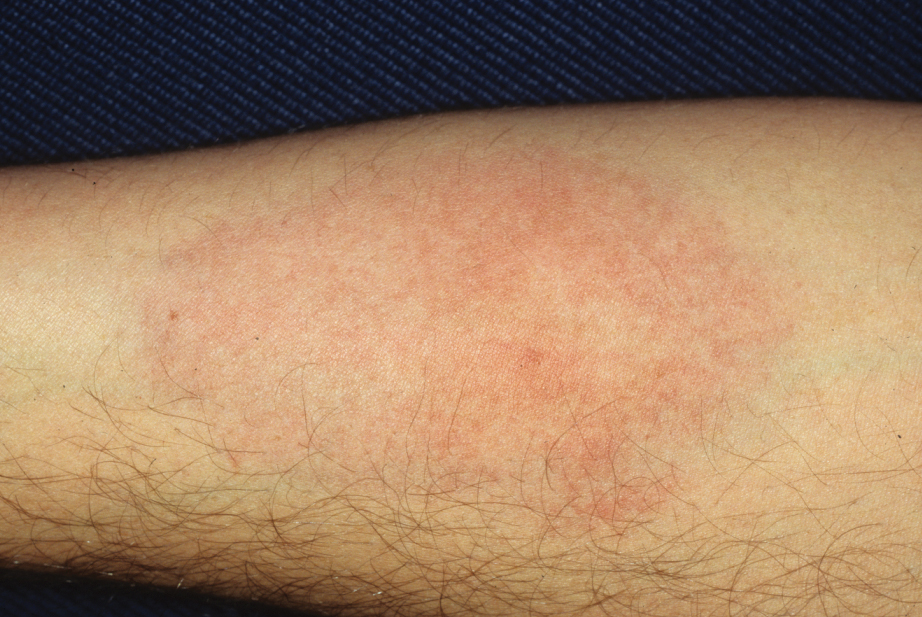
Chagas Disease Risk and Transmission
Triatomine reduviids are the primary vector of Chagas disease, and the geographic range of both continues to expand, particularly in North America. The disease remains endemic in Latin America, with the highest incidence now reported in Brazil.4 An estimated 240,000 to 350,000 individuals in the United States are infected, primarily immigrants from Mexico, Central America, and South America; approximately 30% of those infected will develop cardiac and/or gastrointestinal complications.4 If left untreated, Chagas disease leads to autonomic ganglion destruction and subsequent gastrointestinal and cardiac complications, including megacolon, dilated cardiomyopathy, and heart failure.5
Trypanosoma cruzi, the microorganism responsible for Chagas disease, is spread to humans through triatomine fecal matter scratched into the bite wound.6 Triatomine bugs have a highly developed gastrocolic reflex and defecate liberally as they feed. Fecal volume is heavily dependent on species and sex, with fifth-stage female nymphs producing the highest volume of excrement and thereby acting as particularly adept disease vectors.6 Triatoma infestans and members of the genus Mepraia are key vectors of T cruzi.1 In areas of South America where populations of T infestans are controlled through public health measures, Mepraia emerge as a largely uncontrolled disease vector.1,7 While endemic to the southern United States and South America, T cruzi has spread to much of North America and Europe by way of Triatominae as naturalized or invasive species.8
There are 3 phases of Chagas disease: acute, indeterminate, and chronic. A chagoma is a localized erythematous swelling at the site of the bite. The acute phase often lacks systemic symptoms but may include fever, myalgia, and headache. The intermediate phase may include fatigue and recurrent fevers. The most serious manifestations occur in the chronic phase and include cardiomyopathy with signs of congestive heart failure, irregular heartbeat, cardiac arrest, abdominal pain, constipation, and dysphagia.
Deforestation has been identified as a driving factor in the spread of Chagas disease, as the disease vectors shift from wilderness areas and animal hosts to inhabited areas where humans are the most readily available food source. Triatome bugs in areas experiencing higher levels of development or forest harvesting are forced into human-populated areas. As a result, instances of Chagas disease are on the rise in these communities.7 Salvador, Bahia, Brazil, has been identified as one such target of increased vector presence due to heavy deforestation, and the hottest months were identified as having the greatest threat of vector exposure.9 Brazil became the leading geographic area for the disease partly because of heavy loss of forested land.10
Vector Control and Prevention Strategies
Elimination of cracks and crevices in walls; replacement of wattle and daub with stucco, plaster, and other solid building materials; and the use of insecticides with durability in the environment have been used to reduce triatome bug infestation in homes. However, highly persistent insecticides carry greater environmental risk and may drive resistance as declining concentrations select for resistant arthropods. Repellents have less environmental impact and play an important role in vector control. Citronella essential oil has been observed to repel several species of triatome bugs that are common in Arizona; specifically, the component alcohols geraniol and citronellol were found to be effective at inhibiting triatome feeding.11
Early detection of Chagas disease is essential, as end-stage cardiomyopathy and megacolon are difficult to treat. Newly developed multiantigen testing has shown promising results, suggesting a potential for more accurate testing for Chagas disease.8 Geospatial tracking and mapping of T cruzi vectors now are employed to track seasonal vector changes and disease patterns.9 Researchers also have developed a dedicated dichotomous key for the identification of triatome bugs endemic in Brazil with the hope of better identification and mapping of disease vector presence and density.10 The key consists of a series of statements with 2 choices in each step. It uses observable features of the arthropod to lead users to the correct identification.
Final Thoughts
Identification of triatome bugs can help with public health efforts to control the spread of disease. Patients with unilateral eyelid swelling should be evaluated for possible bedbug or triatome exposure.
- Egaña C, Pinto R, Vergara F, et al. Fluctuations in Trypanosoma cruzi discrete typing unit composition in two naturally infected triatomines: Mepraia gajardoi and M. spinolai after laboratory feeding. Acta Trop. 2016;160:9-14. Erratum in: Acta Trop. 2016;162:248. doi:10.1016/j.actatropica.2016.04.008
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128.
- Alonso A, Potenza M, Mouchián K, et al. Proteinase and gelatinolytic properties of a Triatoma infestans extract. Allergol Immunopathol (Madr). 2004;32:223-227.
- Hochberg NS, Montgomery SP. Chagas disease. Ann Intern Med. 2023;176:ITC17-ITC32. doi:10.7326/AITC202302210
- Pless M, Juranek D, Kozarsky P, et al. The epidemiology of Chagas’ disease in a hyperendemic area of Cochabamba, Bolivia: a clinical study including electrocardiography, seroreactivity to Trypanosoma cruzi, xenodiagnosis, and domiciliary triatomine distribution. Am J Trop Med Hyg. 1992;47:539-546.
- Piesman J, Sherlock IA. Factors controlling the volume of feces produced by triatomine vectors of Chagas’ disease. Acta Trop. 1983;40:351-358.
- Steverding D. The history of Chagas disease. Parasit Vectors. 2014;10:317.
- Granjon E, Dichtel-Danjoy ML, Saba E, et al. Development of a novel multiplex immunoassay multi-cruzi for the serological confirmation of Chagas disease. PLoS Negl Trop Dis. 2016;10:e0004596.
- Santana Kde S, Bavia ME, Lima AD, et al. Spatial distribution of triatomines (Reduviidae: Triatominae) in urban areas of the city of Salvador, Bahia, Brazil. Geospat Health. 2011;5:199-203.
- de Mello DV, Nhapulo EF, Cesaretto LP, et al. Dichotomous keys based on cytogenetic data for triatomines reported in Brazilian regions with outbreaks of orally transmitted Chagas disease (Pernambuco and Rio Grande Do Norte). Trop Med Infect Dis. 2023;8:196.
- Zamora D, Klotz SA, Meister EA, et al. Repellency of the components of the essential oil, citronella, to Triatoma rubida, Triatoma protracta, and Triatoma recurva (Hemiptera: Reduviidae: Triatominae). J Med Entomol. 2015;52:719-721.
Triatome bugs cause painful bites and serve as vectors for Chagas disease. In this article, we will address diagnosis and vector identification.
Key Morphologic Features
Insects from the subfamily Triatominae are identifiable by their long legs and a shieldlike abdomen behind a platelike pronotum that covers the thorax. Their half-membranous wings overlap, covering the central abdomen but leaving the lateral portions visible. Tigerlike stripes are characteristically prominent on the visible portions of the lateral abdomen. The stalklike head has an articulated beaklike mouth that can be retracted and used to deliver a powerful bite (Figure 1).
Feeding Mechanisms and Host Reactions
Triatome bugs are blood-feeding arthropods that hide in cracks and crevices in domestic structures by day and feed at night. They are shy feeders, and laboratory colonies have been known to die rather than feed in daylight. They are particularly common in thatched or wattle-and-daub dwellings, where they can be present in great numbers and descend on sleeping inhabitants at night. Triatome bugs require regular blood meals throughout the 5 developmental nymph stages in order to undergo successful molting.
In the wild, triatome bugs feed on a range of animals with little specificity, but in domestic settings they feed largely on humans. Thermosensors in the antennae help them locate blood vessels under the skin, which they penetrate easily due to their long mouthparts. Like other blood-sucking arthropods, they release an anticoagulant that facilitates continuous blood flow while feeding, which accounts for many of the cutaneous reactions observed after the host sustains a triatomine bite.1
Triatomine bugs have trouble feeding through clothing and seek out exposed skin, particularly the eyelids, producing the characteristic unilateral eyelid swelling known as the Romaña sign. Other bite reactions include purpura; macular erythema; and vesiculobullous, papular, and urticarial lesions (Figure 2).2 Associated lymphangitis or lymphadenopathy may be noted, and anaphylaxis has been reported. Similar to those of cockroaches, triatome antigens have been associated with atopic dermatitis and asthma.3
Chagas Disease Risk and Transmission
Triatomine reduviids are the primary vector of Chagas disease, and the geographic range of both continues to expand, particularly in North America. The disease remains endemic in Latin America, with the highest incidence now reported in Brazil.4 An estimated 240,000 to 350,000 individuals in the United States are infected, primarily immigrants from Mexico, Central America, and South America; approximately 30% of those infected will develop cardiac and/or gastrointestinal complications.4 If left untreated, Chagas disease leads to autonomic ganglion destruction and subsequent gastrointestinal and cardiac complications, including megacolon, dilated cardiomyopathy, and heart failure.5
Trypanosoma cruzi, the microorganism responsible for Chagas disease, is spread to humans through triatomine fecal matter scratched into the bite wound.6 Triatomine bugs have a highly developed gastrocolic reflex and defecate liberally as they feed. Fecal volume is heavily dependent on species and sex, with fifth-stage female nymphs producing the highest volume of excrement and thereby acting as particularly adept disease vectors.6 Triatoma infestans and members of the genus Mepraia are key vectors of T cruzi.1 In areas of South America where populations of T infestans are controlled through public health measures, Mepraia emerge as a largely uncontrolled disease vector.1,7 While endemic to the southern United States and South America, T cruzi has spread to much of North America and Europe by way of Triatominae as naturalized or invasive species.8
There are 3 phases of Chagas disease: acute, indeterminate, and chronic. A chagoma is a localized erythematous swelling at the site of the bite. The acute phase often lacks systemic symptoms but may include fever, myalgia, and headache. The intermediate phase may include fatigue and recurrent fevers. The most serious manifestations occur in the chronic phase and include cardiomyopathy with signs of congestive heart failure, irregular heartbeat, cardiac arrest, abdominal pain, constipation, and dysphagia.
Deforestation has been identified as a driving factor in the spread of Chagas disease, as the disease vectors shift from wilderness areas and animal hosts to inhabited areas where humans are the most readily available food source. Triatome bugs in areas experiencing higher levels of development or forest harvesting are forced into human-populated areas. As a result, instances of Chagas disease are on the rise in these communities.7 Salvador, Bahia, Brazil, has been identified as one such target of increased vector presence due to heavy deforestation, and the hottest months were identified as having the greatest threat of vector exposure.9 Brazil became the leading geographic area for the disease partly because of heavy loss of forested land.10
Vector Control and Prevention Strategies
Elimination of cracks and crevices in walls; replacement of wattle and daub with stucco, plaster, and other solid building materials; and the use of insecticides with durability in the environment have been used to reduce triatome bug infestation in homes. However, highly persistent insecticides carry greater environmental risk and may drive resistance as declining concentrations select for resistant arthropods. Repellents have less environmental impact and play an important role in vector control. Citronella essential oil has been observed to repel several species of triatome bugs that are common in Arizona; specifically, the component alcohols geraniol and citronellol were found to be effective at inhibiting triatome feeding.11
Early detection of Chagas disease is essential, as end-stage cardiomyopathy and megacolon are difficult to treat. Newly developed multiantigen testing has shown promising results, suggesting a potential for more accurate testing for Chagas disease.8 Geospatial tracking and mapping of T cruzi vectors now are employed to track seasonal vector changes and disease patterns.9 Researchers also have developed a dedicated dichotomous key for the identification of triatome bugs endemic in Brazil with the hope of better identification and mapping of disease vector presence and density.10 The key consists of a series of statements with 2 choices in each step. It uses observable features of the arthropod to lead users to the correct identification.
Final Thoughts
Identification of triatome bugs can help with public health efforts to control the spread of disease. Patients with unilateral eyelid swelling should be evaluated for possible bedbug or triatome exposure.
Triatome bugs cause painful bites and serve as vectors for Chagas disease. In this article, we will address diagnosis and vector identification.
Key Morphologic Features
Insects from the subfamily Triatominae are identifiable by their long legs and a shieldlike abdomen behind a platelike pronotum that covers the thorax. Their half-membranous wings overlap, covering the central abdomen but leaving the lateral portions visible. Tigerlike stripes are characteristically prominent on the visible portions of the lateral abdomen. The stalklike head has an articulated beaklike mouth that can be retracted and used to deliver a powerful bite (Figure 1).
Feeding Mechanisms and Host Reactions
Triatome bugs are blood-feeding arthropods that hide in cracks and crevices in domestic structures by day and feed at night. They are shy feeders, and laboratory colonies have been known to die rather than feed in daylight. They are particularly common in thatched or wattle-and-daub dwellings, where they can be present in great numbers and descend on sleeping inhabitants at night. Triatome bugs require regular blood meals throughout the 5 developmental nymph stages in order to undergo successful molting.
In the wild, triatome bugs feed on a range of animals with little specificity, but in domestic settings they feed largely on humans. Thermosensors in the antennae help them locate blood vessels under the skin, which they penetrate easily due to their long mouthparts. Like other blood-sucking arthropods, they release an anticoagulant that facilitates continuous blood flow while feeding, which accounts for many of the cutaneous reactions observed after the host sustains a triatomine bite.1
Triatomine bugs have trouble feeding through clothing and seek out exposed skin, particularly the eyelids, producing the characteristic unilateral eyelid swelling known as the Romaña sign. Other bite reactions include purpura; macular erythema; and vesiculobullous, papular, and urticarial lesions (Figure 2).2 Associated lymphangitis or lymphadenopathy may be noted, and anaphylaxis has been reported. Similar to those of cockroaches, triatome antigens have been associated with atopic dermatitis and asthma.3
Chagas Disease Risk and Transmission
Triatomine reduviids are the primary vector of Chagas disease, and the geographic range of both continues to expand, particularly in North America. The disease remains endemic in Latin America, with the highest incidence now reported in Brazil.4 An estimated 240,000 to 350,000 individuals in the United States are infected, primarily immigrants from Mexico, Central America, and South America; approximately 30% of those infected will develop cardiac and/or gastrointestinal complications.4 If left untreated, Chagas disease leads to autonomic ganglion destruction and subsequent gastrointestinal and cardiac complications, including megacolon, dilated cardiomyopathy, and heart failure.5
Trypanosoma cruzi, the microorganism responsible for Chagas disease, is spread to humans through triatomine fecal matter scratched into the bite wound.6 Triatomine bugs have a highly developed gastrocolic reflex and defecate liberally as they feed. Fecal volume is heavily dependent on species and sex, with fifth-stage female nymphs producing the highest volume of excrement and thereby acting as particularly adept disease vectors.6 Triatoma infestans and members of the genus Mepraia are key vectors of T cruzi.1 In areas of South America where populations of T infestans are controlled through public health measures, Mepraia emerge as a largely uncontrolled disease vector.1,7 While endemic to the southern United States and South America, T cruzi has spread to much of North America and Europe by way of Triatominae as naturalized or invasive species.8
There are 3 phases of Chagas disease: acute, indeterminate, and chronic. A chagoma is a localized erythematous swelling at the site of the bite. The acute phase often lacks systemic symptoms but may include fever, myalgia, and headache. The intermediate phase may include fatigue and recurrent fevers. The most serious manifestations occur in the chronic phase and include cardiomyopathy with signs of congestive heart failure, irregular heartbeat, cardiac arrest, abdominal pain, constipation, and dysphagia.
Deforestation has been identified as a driving factor in the spread of Chagas disease, as the disease vectors shift from wilderness areas and animal hosts to inhabited areas where humans are the most readily available food source. Triatome bugs in areas experiencing higher levels of development or forest harvesting are forced into human-populated areas. As a result, instances of Chagas disease are on the rise in these communities.7 Salvador, Bahia, Brazil, has been identified as one such target of increased vector presence due to heavy deforestation, and the hottest months were identified as having the greatest threat of vector exposure.9 Brazil became the leading geographic area for the disease partly because of heavy loss of forested land.10
Vector Control and Prevention Strategies
Elimination of cracks and crevices in walls; replacement of wattle and daub with stucco, plaster, and other solid building materials; and the use of insecticides with durability in the environment have been used to reduce triatome bug infestation in homes. However, highly persistent insecticides carry greater environmental risk and may drive resistance as declining concentrations select for resistant arthropods. Repellents have less environmental impact and play an important role in vector control. Citronella essential oil has been observed to repel several species of triatome bugs that are common in Arizona; specifically, the component alcohols geraniol and citronellol were found to be effective at inhibiting triatome feeding.11
Early detection of Chagas disease is essential, as end-stage cardiomyopathy and megacolon are difficult to treat. Newly developed multiantigen testing has shown promising results, suggesting a potential for more accurate testing for Chagas disease.8 Geospatial tracking and mapping of T cruzi vectors now are employed to track seasonal vector changes and disease patterns.9 Researchers also have developed a dedicated dichotomous key for the identification of triatome bugs endemic in Brazil with the hope of better identification and mapping of disease vector presence and density.10 The key consists of a series of statements with 2 choices in each step. It uses observable features of the arthropod to lead users to the correct identification.
Final Thoughts
Identification of triatome bugs can help with public health efforts to control the spread of disease. Patients with unilateral eyelid swelling should be evaluated for possible bedbug or triatome exposure.
- Egaña C, Pinto R, Vergara F, et al. Fluctuations in Trypanosoma cruzi discrete typing unit composition in two naturally infected triatomines: Mepraia gajardoi and M. spinolai after laboratory feeding. Acta Trop. 2016;160:9-14. Erratum in: Acta Trop. 2016;162:248. doi:10.1016/j.actatropica.2016.04.008
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128.
- Alonso A, Potenza M, Mouchián K, et al. Proteinase and gelatinolytic properties of a Triatoma infestans extract. Allergol Immunopathol (Madr). 2004;32:223-227.
- Hochberg NS, Montgomery SP. Chagas disease. Ann Intern Med. 2023;176:ITC17-ITC32. doi:10.7326/AITC202302210
- Pless M, Juranek D, Kozarsky P, et al. The epidemiology of Chagas’ disease in a hyperendemic area of Cochabamba, Bolivia: a clinical study including electrocardiography, seroreactivity to Trypanosoma cruzi, xenodiagnosis, and domiciliary triatomine distribution. Am J Trop Med Hyg. 1992;47:539-546.
- Piesman J, Sherlock IA. Factors controlling the volume of feces produced by triatomine vectors of Chagas’ disease. Acta Trop. 1983;40:351-358.
- Steverding D. The history of Chagas disease. Parasit Vectors. 2014;10:317.
- Granjon E, Dichtel-Danjoy ML, Saba E, et al. Development of a novel multiplex immunoassay multi-cruzi for the serological confirmation of Chagas disease. PLoS Negl Trop Dis. 2016;10:e0004596.
- Santana Kde S, Bavia ME, Lima AD, et al. Spatial distribution of triatomines (Reduviidae: Triatominae) in urban areas of the city of Salvador, Bahia, Brazil. Geospat Health. 2011;5:199-203.
- de Mello DV, Nhapulo EF, Cesaretto LP, et al. Dichotomous keys based on cytogenetic data for triatomines reported in Brazilian regions with outbreaks of orally transmitted Chagas disease (Pernambuco and Rio Grande Do Norte). Trop Med Infect Dis. 2023;8:196.
- Zamora D, Klotz SA, Meister EA, et al. Repellency of the components of the essential oil, citronella, to Triatoma rubida, Triatoma protracta, and Triatoma recurva (Hemiptera: Reduviidae: Triatominae). J Med Entomol. 2015;52:719-721.
- Egaña C, Pinto R, Vergara F, et al. Fluctuations in Trypanosoma cruzi discrete typing unit composition in two naturally infected triatomines: Mepraia gajardoi and M. spinolai after laboratory feeding. Acta Trop. 2016;160:9-14. Erratum in: Acta Trop. 2016;162:248. doi:10.1016/j.actatropica.2016.04.008
- Moffitt JE, Venarske D, Goddard J, et al. Allergic reactions to Triatoma bites. Ann Allergy Asthma Immunol. 2003;91:122-128.
- Alonso A, Potenza M, Mouchián K, et al. Proteinase and gelatinolytic properties of a Triatoma infestans extract. Allergol Immunopathol (Madr). 2004;32:223-227.
- Hochberg NS, Montgomery SP. Chagas disease. Ann Intern Med. 2023;176:ITC17-ITC32. doi:10.7326/AITC202302210
- Pless M, Juranek D, Kozarsky P, et al. The epidemiology of Chagas’ disease in a hyperendemic area of Cochabamba, Bolivia: a clinical study including electrocardiography, seroreactivity to Trypanosoma cruzi, xenodiagnosis, and domiciliary triatomine distribution. Am J Trop Med Hyg. 1992;47:539-546.
- Piesman J, Sherlock IA. Factors controlling the volume of feces produced by triatomine vectors of Chagas’ disease. Acta Trop. 1983;40:351-358.
- Steverding D. The history of Chagas disease. Parasit Vectors. 2014;10:317.
- Granjon E, Dichtel-Danjoy ML, Saba E, et al. Development of a novel multiplex immunoassay multi-cruzi for the serological confirmation of Chagas disease. PLoS Negl Trop Dis. 2016;10:e0004596.
- Santana Kde S, Bavia ME, Lima AD, et al. Spatial distribution of triatomines (Reduviidae: Triatominae) in urban areas of the city of Salvador, Bahia, Brazil. Geospat Health. 2011;5:199-203.
- de Mello DV, Nhapulo EF, Cesaretto LP, et al. Dichotomous keys based on cytogenetic data for triatomines reported in Brazilian regions with outbreaks of orally transmitted Chagas disease (Pernambuco and Rio Grande Do Norte). Trop Med Infect Dis. 2023;8:196.
- Zamora D, Klotz SA, Meister EA, et al. Repellency of the components of the essential oil, citronella, to Triatoma rubida, Triatoma protracta, and Triatoma recurva (Hemiptera: Reduviidae: Triatominae). J Med Entomol. 2015;52:719-721.
Cutaneous Reactions to Triatomine (Kissing Bug) Bites and the Risk for Chagas Disease
Cutaneous Reactions to Triatomine (Kissing Bug) Bites and the Risk for Chagas Disease
Practice Points
- Triatomine bugs, commonly known as kissing bugs, are widespread, especially in warmer climates, and their geographic range is expanding.
- The Romaña sign, characterized by unilateral swelling of the eyelid, is common in triatomine bites.
- Triatomine bugs are the primary vector for transmission of the parasite Trypanosoma cruzi, the causative agent of Chagas disease.
- In recent years, T cruzi has been detected in triatomine reduviids in suburban areas of the southwestern United States.

Table Salt Method Following Cryotherapy for Recurrent Pyogenic Granuloma on the Fingertip
Table Salt Method Following Cryotherapy for Recurrent Pyogenic Granuloma on the Fingertip
Practice Gap
Pyogenic granulomas (PGs) are benign endothelial tumors of the skin or mucosae that frequently become ulcerated and may cause patients substantial discomfort or distress due to rapid enlargement and bleeding.1 These lesions often manifest as solitary red papules or polyps following localized trauma or irritation. They can grow up to 1 cm over a few weeks to several months. Pyogenic granulomas can develop at any age, but they most commonly are seen in children and young adults, with a slight male predominance.1,2 The differential diagnosis for PG includes amelanotic melanoma, bacillary angiomatosis, Kaposi sarcoma, glomus tumor, infantile hemangioma, and irritated melanocytic nevus.1 Histologically, PGs are well-circumscribed exophytic or pedunculated proliferations of small capillaries that often are arranged in a lobular pattern. Early lesions show packed endothelial cells, while advanced lesions display more ectatic vessels, erosion, and crusting.3 The term pyogenic granuloma is a misnomer, as these lesions display neither an infectious etiology nor granulomatous tissue on dermatopathologic examination.4 A more accurate clinical description for this lesion is a lobular capillary hemangioma.
Numerous surgical and laser techniques have been used to treat PGs, with varying degrees of success. Treatment often consists of either shave excision followed by electrosurgery at the base or full excision with suturing under local anesthesia for patients who can tolerate anesthetic injections.1 Pulsed dye laser has been proven to be a safe alternative treatment option, particularly in children who otherwise would not tolerate surgical procedures.5 Topical beta-blockers, silver nitrate cauterization, sclerotherapy, and liquid nitrogen all have been used as alternative treatment methods.1
Pyogenic granulomas often recur after first-line treatments, and patients may hesitate to try more invasive techniques when the first choice has failed. Children may not be amenable to any of these curative techniques, as they may not tolerate the pain associated with lidocaine injection and/or have a fear of needles or surgical intervention; even adults may be reluctant to have a procedure they perceive as painful. We present a less invasive technique for treatment of recurrent PGs using table salt and cryotherapy.
The Technique
A 51-year-old woman with no notable medical history presented to the emergency department for evaluation of a black dot on the pulp of the right third fingertip of 1 week’s duration. The patient reported rapid progression to an ulcerated red nodule with associated bleeding for the past 3 days (Figure 1). Direct pressure temporarily alleviated the bleeding, but it started again upon cessation of pressure. She denied any preceding trauma to the area or any associated systemic symptoms such as fever, chills, nausea, or vomiting.
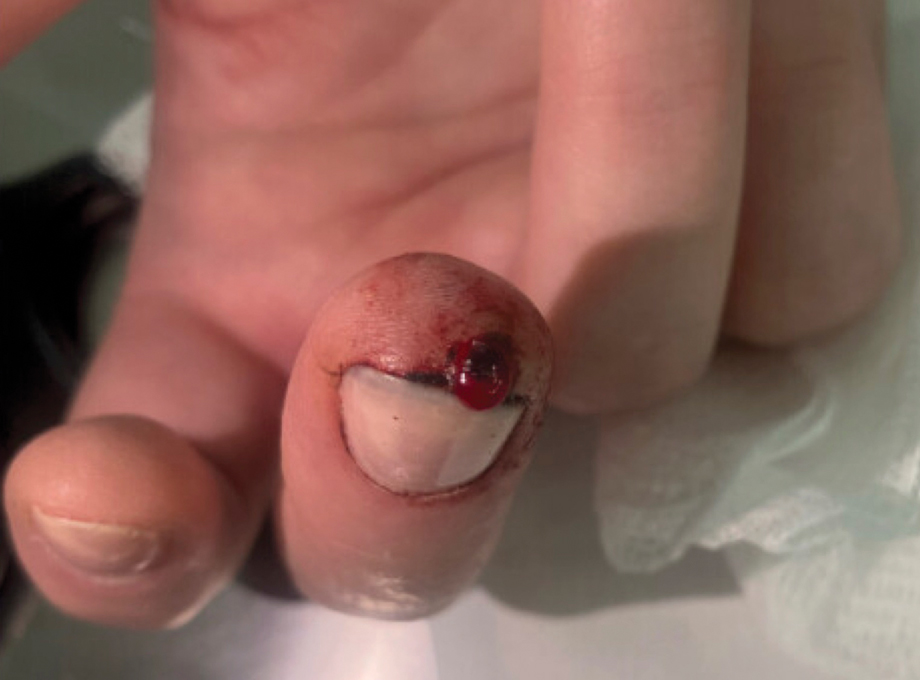
The inpatient dermatology team recommended that the patient be discharged following silver nitrate cautery, with a referral sent to outpatient dermatology; however, the patient returned to the dermatology clinic 4 days later, at which time physical examination revealed a well-circumscribed, 5-mm, bright-red, erosive papule with overlying hemorrhagic crust that was not actively bleeding, as well as fissuring of the surrounding skin. The entire lesion was removed using tangential excision followed by electrodesiccation at the base. Pathology revealed small capillaries arranged in a lobular pattern, confirming the diagnosis of PG. At a 2-week follow-up visit, the patient noted that the lesion had recurred within 24 hours after the procedure and was larger (Figure 2). At this visit, management was switched to a single treatment of cryotherapy (3 cycles for 5 seconds per cycle), and the table salt method was recommended based on a literature review for alternative nonpainful approaches for PG.6-11 We used this technique in our patient as an adjuvant to cryotherapy with the goal of reducing the need for additional painful procedures, but table salt also can be used as a standalone treatment without prior cryotherapy.
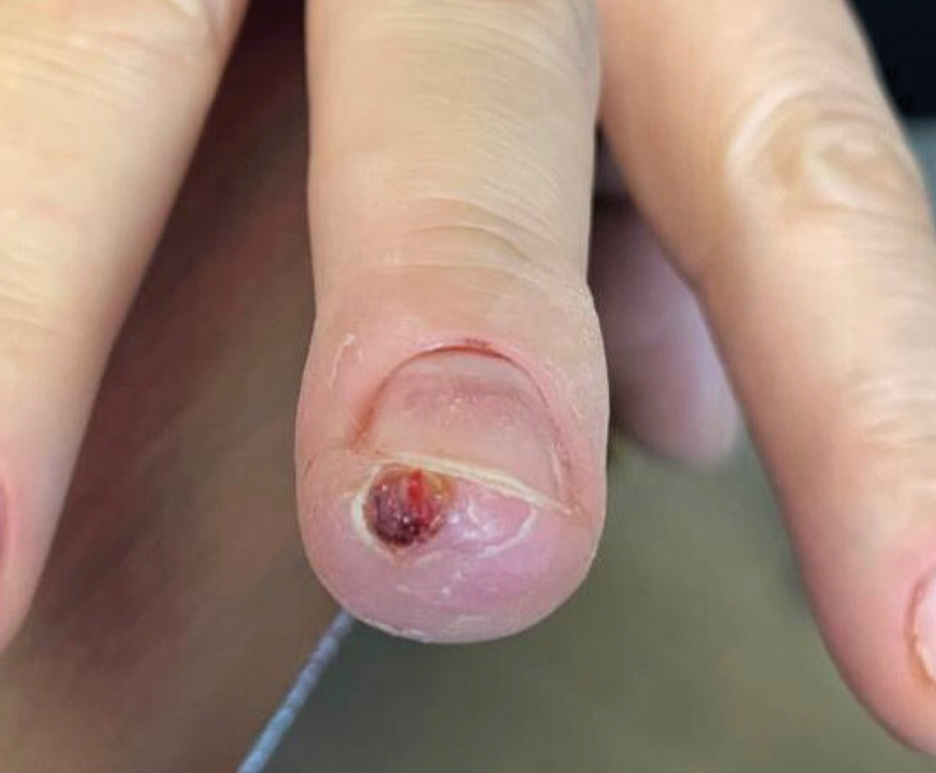
The patient was instructed to apply table salt to the lesion once daily for 2 weeks by pressing the lesion into a small amount of salt placed on a clean plate and then applying an occlusive dressing such as surgical or paper tape. She also was advised to apply petroleum jelly around the periphery of the lesion prior to salt application to protect the unaffected skin from irritation. Complete resolution of the lesion was seen when the patient followed up 2 weeks later (Figure 3). At the most recent follow-up 2 months after treatment, no further recurrence of the PG was reported.
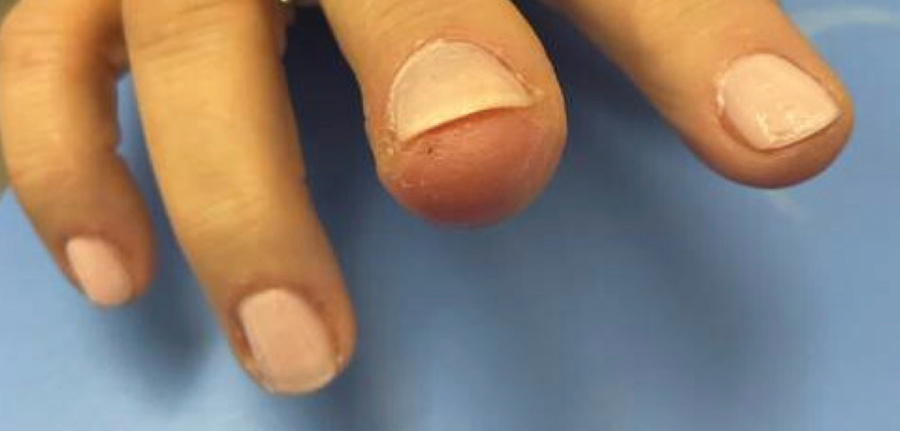
Practice Implication
Pyogenic granulomas can be distressing for both patients and providers because they are cosmetically bothersome and prone to spontaneous bleeding. Various medical and surgical options exist to treat PGs, but there is no clear consensus on a superior modality. A 2019 study by Daruwalla and Dhurat6 highlighted a less invasive treatment option for PGs using table salt applied once daily for 2 weeks under an occlusive dressing with good outcomes and without involving other treatments such as cryotherapy. Several other case reports have endorsed this approach, adding anecdotal evidence for its utility.7-11 Topical sodium chloride may treat PGs primarily through osmotic desiccation, drawing water out of the lesion and leading to endothelial cell shrinkage and collapse of its capillary network. This hyperosmolar environment also may induce microvascular thrombosis and ischemia, promoting lesion necrosis. Additionally, repeated application creates a dry, mildly irritative surface that may suppress angiogenesis and encourage regression of the vascular proliferation.
Consider topical application of table salt for treatment of PGs in certain subsets of patients; for example, patients who are not amenable to surgery or are too young for advanced surgical techniques may be good candidates for this method, as it does not require anesthetic injections and generally is pain free. Patients with resistant or recurrent PGs that did not respond to first-line treatments such as cryotherapy, tangential excision, or electrodesiccation may be more amenable to a less invasive secondary approach.
Importantly, we used a dual-therapy approach in our patient, initially using a single application of cryotherapy followed by the table salt method once daily for 2 weeks. This imposes limitations on the generalizability of table salt as a standalone approach for treating PGs. In this case, we did not have prior practical experience using table salt for this condition and only had small reports to justify its use. As a result, we attempted a more traditional treatment initially (cryotherapy) to avoid potential delays in resolution. The clinicians recommended table salt as an adjuvant prior to seeing the cryotherapy results because this treatment was benign and offered potential additive results, and therefore waiting was not necessary. However, various other cases have reported similar success using table salt as monotherapy.6-9,11 Patients should be advised of potential mild adverse events, such as irritation to the surrounding skin. Higher-level evidence studies are required to further vet the utility of the table salt method for treatment of PGs.
- Bolognia JL, Schaffer JV, Cerroni L. Vascular neoplasms and neoplastic‑like proliferations. In: Dermatology. Elsevier; 2018.
- Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: an epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
- Ferringer TK, DiCaudo DJ, Elston D, et al. Dermatopathology. W.B. Saunders; 2008.
- Gomes SR, Shakir QJ, Thaker PV, et al. Pyogenic granuloma of the gingiva: a misnomer? - a case report and review of literature. J Indian Soc Periodontol. 2013;17:514-519. doi:10.4103/0972-124X.118327
- Sud AR, Tan ST. Pyogenic granuloma-treatment by shave-excision and/or pulsed-dye laser. J Plast Reconstr Aesthet Surg. 2010;63:1364-1368. doi:10.1016/j.bjps.2009.06.031
- Daruwalla SB, Dhurat RS. A pinch of salt is all it takes! the novel use of table salt for the effective treatment of pyogenic granuloma. J Am Acad Dermatol. 2020;83:E107-E108. doi:10.1016/j.jaad.2019.12.013
- Alhammad G, Albaraka M, Alotaibi H, et al. The use of common salt for the treatment of pyogenic granuloma. JAAD Case Rep. 2024;53:40-42. doi:10.1016/j.jdcr.2024.08.016
- Weiss ES, Wood D. Simple, safe, and effective treatment for pyogenic granuloma. Can Fam Physician. 2023;69:479-480. doi:10.46747/cfp.6907479
- Bernales Salinas A, Toro Sepúlveda A, Meier Pincheira H, et al. Case report: pyogenic granuloma-just salt, a simple and pain-free treatment. Dermatol Ther. 2022;35:E15194. doi:10.1111/dth.15194
- Martín-Nieto González J, Rodríguez-Sánchez B, Berna-Rico E, et al. Pyogenic granuloma resolved with timolol and table salt. An Pediatr (Engl Ed). 2025;102:503706. doi:10.1016/j.anpede.2025.503706
- Bin Rubaian NF. Complete resolution of a refractory pyogenic granuloma following topical salt treatment. Open Access Emerg Med. 2021;13:445-448. doi:10.2147/OAEM.S323793
Practice Gap
Pyogenic granulomas (PGs) are benign endothelial tumors of the skin or mucosae that frequently become ulcerated and may cause patients substantial discomfort or distress due to rapid enlargement and bleeding.1 These lesions often manifest as solitary red papules or polyps following localized trauma or irritation. They can grow up to 1 cm over a few weeks to several months. Pyogenic granulomas can develop at any age, but they most commonly are seen in children and young adults, with a slight male predominance.1,2 The differential diagnosis for PG includes amelanotic melanoma, bacillary angiomatosis, Kaposi sarcoma, glomus tumor, infantile hemangioma, and irritated melanocytic nevus.1 Histologically, PGs are well-circumscribed exophytic or pedunculated proliferations of small capillaries that often are arranged in a lobular pattern. Early lesions show packed endothelial cells, while advanced lesions display more ectatic vessels, erosion, and crusting.3 The term pyogenic granuloma is a misnomer, as these lesions display neither an infectious etiology nor granulomatous tissue on dermatopathologic examination.4 A more accurate clinical description for this lesion is a lobular capillary hemangioma.
Numerous surgical and laser techniques have been used to treat PGs, with varying degrees of success. Treatment often consists of either shave excision followed by electrosurgery at the base or full excision with suturing under local anesthesia for patients who can tolerate anesthetic injections.1 Pulsed dye laser has been proven to be a safe alternative treatment option, particularly in children who otherwise would not tolerate surgical procedures.5 Topical beta-blockers, silver nitrate cauterization, sclerotherapy, and liquid nitrogen all have been used as alternative treatment methods.1
Pyogenic granulomas often recur after first-line treatments, and patients may hesitate to try more invasive techniques when the first choice has failed. Children may not be amenable to any of these curative techniques, as they may not tolerate the pain associated with lidocaine injection and/or have a fear of needles or surgical intervention; even adults may be reluctant to have a procedure they perceive as painful. We present a less invasive technique for treatment of recurrent PGs using table salt and cryotherapy.
The Technique
A 51-year-old woman with no notable medical history presented to the emergency department for evaluation of a black dot on the pulp of the right third fingertip of 1 week’s duration. The patient reported rapid progression to an ulcerated red nodule with associated bleeding for the past 3 days (Figure 1). Direct pressure temporarily alleviated the bleeding, but it started again upon cessation of pressure. She denied any preceding trauma to the area or any associated systemic symptoms such as fever, chills, nausea, or vomiting.
The inpatient dermatology team recommended that the patient be discharged following silver nitrate cautery, with a referral sent to outpatient dermatology; however, the patient returned to the dermatology clinic 4 days later, at which time physical examination revealed a well-circumscribed, 5-mm, bright-red, erosive papule with overlying hemorrhagic crust that was not actively bleeding, as well as fissuring of the surrounding skin. The entire lesion was removed using tangential excision followed by electrodesiccation at the base. Pathology revealed small capillaries arranged in a lobular pattern, confirming the diagnosis of PG. At a 2-week follow-up visit, the patient noted that the lesion had recurred within 24 hours after the procedure and was larger (Figure 2). At this visit, management was switched to a single treatment of cryotherapy (3 cycles for 5 seconds per cycle), and the table salt method was recommended based on a literature review for alternative nonpainful approaches for PG.6-11 We used this technique in our patient as an adjuvant to cryotherapy with the goal of reducing the need for additional painful procedures, but table salt also can be used as a standalone treatment without prior cryotherapy.
The patient was instructed to apply table salt to the lesion once daily for 2 weeks by pressing the lesion into a small amount of salt placed on a clean plate and then applying an occlusive dressing such as surgical or paper tape. She also was advised to apply petroleum jelly around the periphery of the lesion prior to salt application to protect the unaffected skin from irritation. Complete resolution of the lesion was seen when the patient followed up 2 weeks later (Figure 3). At the most recent follow-up 2 months after treatment, no further recurrence of the PG was reported.
Practice Implication
Pyogenic granulomas can be distressing for both patients and providers because they are cosmetically bothersome and prone to spontaneous bleeding. Various medical and surgical options exist to treat PGs, but there is no clear consensus on a superior modality. A 2019 study by Daruwalla and Dhurat6 highlighted a less invasive treatment option for PGs using table salt applied once daily for 2 weeks under an occlusive dressing with good outcomes and without involving other treatments such as cryotherapy. Several other case reports have endorsed this approach, adding anecdotal evidence for its utility.7-11 Topical sodium chloride may treat PGs primarily through osmotic desiccation, drawing water out of the lesion and leading to endothelial cell shrinkage and collapse of its capillary network. This hyperosmolar environment also may induce microvascular thrombosis and ischemia, promoting lesion necrosis. Additionally, repeated application creates a dry, mildly irritative surface that may suppress angiogenesis and encourage regression of the vascular proliferation.
Consider topical application of table salt for treatment of PGs in certain subsets of patients; for example, patients who are not amenable to surgery or are too young for advanced surgical techniques may be good candidates for this method, as it does not require anesthetic injections and generally is pain free. Patients with resistant or recurrent PGs that did not respond to first-line treatments such as cryotherapy, tangential excision, or electrodesiccation may be more amenable to a less invasive secondary approach.
Importantly, we used a dual-therapy approach in our patient, initially using a single application of cryotherapy followed by the table salt method once daily for 2 weeks. This imposes limitations on the generalizability of table salt as a standalone approach for treating PGs. In this case, we did not have prior practical experience using table salt for this condition and only had small reports to justify its use. As a result, we attempted a more traditional treatment initially (cryotherapy) to avoid potential delays in resolution. The clinicians recommended table salt as an adjuvant prior to seeing the cryotherapy results because this treatment was benign and offered potential additive results, and therefore waiting was not necessary. However, various other cases have reported similar success using table salt as monotherapy.6-9,11 Patients should be advised of potential mild adverse events, such as irritation to the surrounding skin. Higher-level evidence studies are required to further vet the utility of the table salt method for treatment of PGs.
Practice Gap
Pyogenic granulomas (PGs) are benign endothelial tumors of the skin or mucosae that frequently become ulcerated and may cause patients substantial discomfort or distress due to rapid enlargement and bleeding.1 These lesions often manifest as solitary red papules or polyps following localized trauma or irritation. They can grow up to 1 cm over a few weeks to several months. Pyogenic granulomas can develop at any age, but they most commonly are seen in children and young adults, with a slight male predominance.1,2 The differential diagnosis for PG includes amelanotic melanoma, bacillary angiomatosis, Kaposi sarcoma, glomus tumor, infantile hemangioma, and irritated melanocytic nevus.1 Histologically, PGs are well-circumscribed exophytic or pedunculated proliferations of small capillaries that often are arranged in a lobular pattern. Early lesions show packed endothelial cells, while advanced lesions display more ectatic vessels, erosion, and crusting.3 The term pyogenic granuloma is a misnomer, as these lesions display neither an infectious etiology nor granulomatous tissue on dermatopathologic examination.4 A more accurate clinical description for this lesion is a lobular capillary hemangioma.
Numerous surgical and laser techniques have been used to treat PGs, with varying degrees of success. Treatment often consists of either shave excision followed by electrosurgery at the base or full excision with suturing under local anesthesia for patients who can tolerate anesthetic injections.1 Pulsed dye laser has been proven to be a safe alternative treatment option, particularly in children who otherwise would not tolerate surgical procedures.5 Topical beta-blockers, silver nitrate cauterization, sclerotherapy, and liquid nitrogen all have been used as alternative treatment methods.1
Pyogenic granulomas often recur after first-line treatments, and patients may hesitate to try more invasive techniques when the first choice has failed. Children may not be amenable to any of these curative techniques, as they may not tolerate the pain associated with lidocaine injection and/or have a fear of needles or surgical intervention; even adults may be reluctant to have a procedure they perceive as painful. We present a less invasive technique for treatment of recurrent PGs using table salt and cryotherapy.
The Technique
A 51-year-old woman with no notable medical history presented to the emergency department for evaluation of a black dot on the pulp of the right third fingertip of 1 week’s duration. The patient reported rapid progression to an ulcerated red nodule with associated bleeding for the past 3 days (Figure 1). Direct pressure temporarily alleviated the bleeding, but it started again upon cessation of pressure. She denied any preceding trauma to the area or any associated systemic symptoms such as fever, chills, nausea, or vomiting.
The inpatient dermatology team recommended that the patient be discharged following silver nitrate cautery, with a referral sent to outpatient dermatology; however, the patient returned to the dermatology clinic 4 days later, at which time physical examination revealed a well-circumscribed, 5-mm, bright-red, erosive papule with overlying hemorrhagic crust that was not actively bleeding, as well as fissuring of the surrounding skin. The entire lesion was removed using tangential excision followed by electrodesiccation at the base. Pathology revealed small capillaries arranged in a lobular pattern, confirming the diagnosis of PG. At a 2-week follow-up visit, the patient noted that the lesion had recurred within 24 hours after the procedure and was larger (Figure 2). At this visit, management was switched to a single treatment of cryotherapy (3 cycles for 5 seconds per cycle), and the table salt method was recommended based on a literature review for alternative nonpainful approaches for PG.6-11 We used this technique in our patient as an adjuvant to cryotherapy with the goal of reducing the need for additional painful procedures, but table salt also can be used as a standalone treatment without prior cryotherapy.
The patient was instructed to apply table salt to the lesion once daily for 2 weeks by pressing the lesion into a small amount of salt placed on a clean plate and then applying an occlusive dressing such as surgical or paper tape. She also was advised to apply petroleum jelly around the periphery of the lesion prior to salt application to protect the unaffected skin from irritation. Complete resolution of the lesion was seen when the patient followed up 2 weeks later (Figure 3). At the most recent follow-up 2 months after treatment, no further recurrence of the PG was reported.
Practice Implication
Pyogenic granulomas can be distressing for both patients and providers because they are cosmetically bothersome and prone to spontaneous bleeding. Various medical and surgical options exist to treat PGs, but there is no clear consensus on a superior modality. A 2019 study by Daruwalla and Dhurat6 highlighted a less invasive treatment option for PGs using table salt applied once daily for 2 weeks under an occlusive dressing with good outcomes and without involving other treatments such as cryotherapy. Several other case reports have endorsed this approach, adding anecdotal evidence for its utility.7-11 Topical sodium chloride may treat PGs primarily through osmotic desiccation, drawing water out of the lesion and leading to endothelial cell shrinkage and collapse of its capillary network. This hyperosmolar environment also may induce microvascular thrombosis and ischemia, promoting lesion necrosis. Additionally, repeated application creates a dry, mildly irritative surface that may suppress angiogenesis and encourage regression of the vascular proliferation.
Consider topical application of table salt for treatment of PGs in certain subsets of patients; for example, patients who are not amenable to surgery or are too young for advanced surgical techniques may be good candidates for this method, as it does not require anesthetic injections and generally is pain free. Patients with resistant or recurrent PGs that did not respond to first-line treatments such as cryotherapy, tangential excision, or electrodesiccation may be more amenable to a less invasive secondary approach.
Importantly, we used a dual-therapy approach in our patient, initially using a single application of cryotherapy followed by the table salt method once daily for 2 weeks. This imposes limitations on the generalizability of table salt as a standalone approach for treating PGs. In this case, we did not have prior practical experience using table salt for this condition and only had small reports to justify its use. As a result, we attempted a more traditional treatment initially (cryotherapy) to avoid potential delays in resolution. The clinicians recommended table salt as an adjuvant prior to seeing the cryotherapy results because this treatment was benign and offered potential additive results, and therefore waiting was not necessary. However, various other cases have reported similar success using table salt as monotherapy.6-9,11 Patients should be advised of potential mild adverse events, such as irritation to the surrounding skin. Higher-level evidence studies are required to further vet the utility of the table salt method for treatment of PGs.
- Bolognia JL, Schaffer JV, Cerroni L. Vascular neoplasms and neoplastic‑like proliferations. In: Dermatology. Elsevier; 2018.
- Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: an epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
- Ferringer TK, DiCaudo DJ, Elston D, et al. Dermatopathology. W.B. Saunders; 2008.
- Gomes SR, Shakir QJ, Thaker PV, et al. Pyogenic granuloma of the gingiva: a misnomer? - a case report and review of literature. J Indian Soc Periodontol. 2013;17:514-519. doi:10.4103/0972-124X.118327
- Sud AR, Tan ST. Pyogenic granuloma-treatment by shave-excision and/or pulsed-dye laser. J Plast Reconstr Aesthet Surg. 2010;63:1364-1368. doi:10.1016/j.bjps.2009.06.031
- Daruwalla SB, Dhurat RS. A pinch of salt is all it takes! the novel use of table salt for the effective treatment of pyogenic granuloma. J Am Acad Dermatol. 2020;83:E107-E108. doi:10.1016/j.jaad.2019.12.013
- Alhammad G, Albaraka M, Alotaibi H, et al. The use of common salt for the treatment of pyogenic granuloma. JAAD Case Rep. 2024;53:40-42. doi:10.1016/j.jdcr.2024.08.016
- Weiss ES, Wood D. Simple, safe, and effective treatment for pyogenic granuloma. Can Fam Physician. 2023;69:479-480. doi:10.46747/cfp.6907479
- Bernales Salinas A, Toro Sepúlveda A, Meier Pincheira H, et al. Case report: pyogenic granuloma-just salt, a simple and pain-free treatment. Dermatol Ther. 2022;35:E15194. doi:10.1111/dth.15194
- Martín-Nieto González J, Rodríguez-Sánchez B, Berna-Rico E, et al. Pyogenic granuloma resolved with timolol and table salt. An Pediatr (Engl Ed). 2025;102:503706. doi:10.1016/j.anpede.2025.503706
- Bin Rubaian NF. Complete resolution of a refractory pyogenic granuloma following topical salt treatment. Open Access Emerg Med. 2021;13:445-448. doi:10.2147/OAEM.S323793
- Bolognia JL, Schaffer JV, Cerroni L. Vascular neoplasms and neoplastic‑like proliferations. In: Dermatology. Elsevier; 2018.
- Harris MN, Desai R, Chuang TY, et al. Lobular capillary hemangiomas: an epidemiologic report, with emphasis on cutaneous lesions. J Am Acad Dermatol. 2000;42:1012-1016.
- Ferringer TK, DiCaudo DJ, Elston D, et al. Dermatopathology. W.B. Saunders; 2008.
- Gomes SR, Shakir QJ, Thaker PV, et al. Pyogenic granuloma of the gingiva: a misnomer? - a case report and review of literature. J Indian Soc Periodontol. 2013;17:514-519. doi:10.4103/0972-124X.118327
- Sud AR, Tan ST. Pyogenic granuloma-treatment by shave-excision and/or pulsed-dye laser. J Plast Reconstr Aesthet Surg. 2010;63:1364-1368. doi:10.1016/j.bjps.2009.06.031
- Daruwalla SB, Dhurat RS. A pinch of salt is all it takes! the novel use of table salt for the effective treatment of pyogenic granuloma. J Am Acad Dermatol. 2020;83:E107-E108. doi:10.1016/j.jaad.2019.12.013
- Alhammad G, Albaraka M, Alotaibi H, et al. The use of common salt for the treatment of pyogenic granuloma. JAAD Case Rep. 2024;53:40-42. doi:10.1016/j.jdcr.2024.08.016
- Weiss ES, Wood D. Simple, safe, and effective treatment for pyogenic granuloma. Can Fam Physician. 2023;69:479-480. doi:10.46747/cfp.6907479
- Bernales Salinas A, Toro Sepúlveda A, Meier Pincheira H, et al. Case report: pyogenic granuloma-just salt, a simple and pain-free treatment. Dermatol Ther. 2022;35:E15194. doi:10.1111/dth.15194
- Martín-Nieto González J, Rodríguez-Sánchez B, Berna-Rico E, et al. Pyogenic granuloma resolved with timolol and table salt. An Pediatr (Engl Ed). 2025;102:503706. doi:10.1016/j.anpede.2025.503706
- Bin Rubaian NF. Complete resolution of a refractory pyogenic granuloma following topical salt treatment. Open Access Emerg Med. 2021;13:445-448. doi:10.2147/OAEM.S323793
Table Salt Method Following Cryotherapy for Recurrent Pyogenic Granuloma on the Fingertip
Table Salt Method Following Cryotherapy for Recurrent Pyogenic Granuloma on the Fingertip

AAD 2026 Annual Meeting Highlights
AAD 2026 Annual Meeting Highlights
The American Academy of Dermatology’s 2026 Annual Meeting in Denver, Colorado, showcased advances in clinical practice and dermatology research. Selected key updates are summarized here for concise review of emerging dermatology data relevant to clinical practice.
AI Holds Promise in Dermatology, Issues Remain to be Addressed
Artificial intelligence (AI) is rapidly advancing in dermatology, improving image analysis, clinical decision support, and workflow efficiency; however, concerns remain about ethical use, training gaps, and potential skill loss among clinicians. While AI may enhance productivity and care, experts emphasize the need for cautious implementation, education, and ongoing evaluation of real-world performance.
Phase 2b Findings Support Novel Agent to Treat Alopecia Areata
A phase 2b trial of rezpegaldesleukin for severe alopecia areata showed considerably greater reductions in SALT scores vs placebo over 36 weeks, with higher response rates and no treatment plateau. The biologic, which enhances regulatory T-cell activity, demonstrated a favorable safety profile, with mainly mild injection-site reactions and no new safety signals.
JAK Inhibitors: Identifying Ideal Candidates and Putting Real-World Risks in Context
Emerging evidence suggests Janus kinase (JAK) inhibitors are safer in dermatology than early rheumatoid arthritis data indicated. Risks for cardiovascular events, thrombosis, and malignancy appear low and largely driven by baseline patient factors. With appropriate screening and monitoring, these agents can be used safely in most patients with inflammatory skin diseases.
Nemolizumab Phase 2 Findings Positive for Children 2-11 Years Old With Atopic Dermatitis
A phase 2 open-label study of nemolizumab in children aged 2 to 11 years with moderate to severe atopic dermatitis showed notable improvements in skin clearance, disease severity, and itch with weight-based dosing. Responses were rapid, durable through 52 weeks, and consistent with prior data, with no new safety signals identified in this population.
Melasma: A New Era of Topical Treatment Options Galore
Melasma treatment is rapidly expanding beyond traditional agents such as hydroquinone and triple combination therapy, with newer topicals including tranexamic acid, cysteamine, azelaic acid, thiamidol, and emerging compounds showing variable efficacy. While promising, evidence is still evolving, and combination regimens plus strict photoprotection remain the cornerstone of management.
Weight-Loss Drug–Biologic Combination Boosts Relief in Psoriatic Arthritis
In a phase 3b trial, combining tirzepatide with ixekizumab significantly improved joint and skin outcomes in patients with psoriatic arthritis and overweight/obesity (P<.05) compared with ixekizumab alone (P<.001). The combination yielded higher American College of Rheumatology and Psoriasis Area and Severity Index response rates, early symptom improvement, and meaningful weight loss, with safety profiles consistent with known effects.
Tips on Using Biologics for Psoriasis in Context of HIV
Evidence for biologic use in HIV-positive patients with moderate to severe psoriasis is limited, but available case reports suggest tumor necrosis factor inhibitors and newer IL-targeted biologics are generally effective without major impacts on viral load or CD4 counts. Experts recommend prioritizing nonimmunosuppressive options and coordinating care with HIV specialists due to potential infection risks.
Upadacitinib Results in Significant Improvements in Nonsegmental Vitiligo in Phase 3 Studies
Two phase 3 trials showed that the Janus kinase 1 inhibitor upadacitinib significantly improved repigmentation outcomes in adolescents and adults with nonsegmental vitiligo vs placebo over 48 weeks (P<.0001 for both), with a higher proportion achieving clinically meaningful reductions in Vitiligo Area and Severity Index scores. Benefits increased over time without plateau, and no new safety signals were identified.
The American Academy of Dermatology’s 2026 Annual Meeting in Denver, Colorado, showcased advances in clinical practice and dermatology research. Selected key updates are summarized here for concise review of emerging dermatology data relevant to clinical practice.
AI Holds Promise in Dermatology, Issues Remain to be Addressed
Artificial intelligence (AI) is rapidly advancing in dermatology, improving image analysis, clinical decision support, and workflow efficiency; however, concerns remain about ethical use, training gaps, and potential skill loss among clinicians. While AI may enhance productivity and care, experts emphasize the need for cautious implementation, education, and ongoing evaluation of real-world performance.
Phase 2b Findings Support Novel Agent to Treat Alopecia Areata
A phase 2b trial of rezpegaldesleukin for severe alopecia areata showed considerably greater reductions in SALT scores vs placebo over 36 weeks, with higher response rates and no treatment plateau. The biologic, which enhances regulatory T-cell activity, demonstrated a favorable safety profile, with mainly mild injection-site reactions and no new safety signals.
JAK Inhibitors: Identifying Ideal Candidates and Putting Real-World Risks in Context
Emerging evidence suggests Janus kinase (JAK) inhibitors are safer in dermatology than early rheumatoid arthritis data indicated. Risks for cardiovascular events, thrombosis, and malignancy appear low and largely driven by baseline patient factors. With appropriate screening and monitoring, these agents can be used safely in most patients with inflammatory skin diseases.
Nemolizumab Phase 2 Findings Positive for Children 2-11 Years Old With Atopic Dermatitis
A phase 2 open-label study of nemolizumab in children aged 2 to 11 years with moderate to severe atopic dermatitis showed notable improvements in skin clearance, disease severity, and itch with weight-based dosing. Responses were rapid, durable through 52 weeks, and consistent with prior data, with no new safety signals identified in this population.
Melasma: A New Era of Topical Treatment Options Galore
Melasma treatment is rapidly expanding beyond traditional agents such as hydroquinone and triple combination therapy, with newer topicals including tranexamic acid, cysteamine, azelaic acid, thiamidol, and emerging compounds showing variable efficacy. While promising, evidence is still evolving, and combination regimens plus strict photoprotection remain the cornerstone of management.
Weight-Loss Drug–Biologic Combination Boosts Relief in Psoriatic Arthritis
In a phase 3b trial, combining tirzepatide with ixekizumab significantly improved joint and skin outcomes in patients with psoriatic arthritis and overweight/obesity (P<.05) compared with ixekizumab alone (P<.001). The combination yielded higher American College of Rheumatology and Psoriasis Area and Severity Index response rates, early symptom improvement, and meaningful weight loss, with safety profiles consistent with known effects.
Tips on Using Biologics for Psoriasis in Context of HIV
Evidence for biologic use in HIV-positive patients with moderate to severe psoriasis is limited, but available case reports suggest tumor necrosis factor inhibitors and newer IL-targeted biologics are generally effective without major impacts on viral load or CD4 counts. Experts recommend prioritizing nonimmunosuppressive options and coordinating care with HIV specialists due to potential infection risks.
Upadacitinib Results in Significant Improvements in Nonsegmental Vitiligo in Phase 3 Studies
Two phase 3 trials showed that the Janus kinase 1 inhibitor upadacitinib significantly improved repigmentation outcomes in adolescents and adults with nonsegmental vitiligo vs placebo over 48 weeks (P<.0001 for both), with a higher proportion achieving clinically meaningful reductions in Vitiligo Area and Severity Index scores. Benefits increased over time without plateau, and no new safety signals were identified.
The American Academy of Dermatology’s 2026 Annual Meeting in Denver, Colorado, showcased advances in clinical practice and dermatology research. Selected key updates are summarized here for concise review of emerging dermatology data relevant to clinical practice.
AI Holds Promise in Dermatology, Issues Remain to be Addressed
Artificial intelligence (AI) is rapidly advancing in dermatology, improving image analysis, clinical decision support, and workflow efficiency; however, concerns remain about ethical use, training gaps, and potential skill loss among clinicians. While AI may enhance productivity and care, experts emphasize the need for cautious implementation, education, and ongoing evaluation of real-world performance.
Phase 2b Findings Support Novel Agent to Treat Alopecia Areata
A phase 2b trial of rezpegaldesleukin for severe alopecia areata showed considerably greater reductions in SALT scores vs placebo over 36 weeks, with higher response rates and no treatment plateau. The biologic, which enhances regulatory T-cell activity, demonstrated a favorable safety profile, with mainly mild injection-site reactions and no new safety signals.
JAK Inhibitors: Identifying Ideal Candidates and Putting Real-World Risks in Context
Emerging evidence suggests Janus kinase (JAK) inhibitors are safer in dermatology than early rheumatoid arthritis data indicated. Risks for cardiovascular events, thrombosis, and malignancy appear low and largely driven by baseline patient factors. With appropriate screening and monitoring, these agents can be used safely in most patients with inflammatory skin diseases.
Nemolizumab Phase 2 Findings Positive for Children 2-11 Years Old With Atopic Dermatitis
A phase 2 open-label study of nemolizumab in children aged 2 to 11 years with moderate to severe atopic dermatitis showed notable improvements in skin clearance, disease severity, and itch with weight-based dosing. Responses were rapid, durable through 52 weeks, and consistent with prior data, with no new safety signals identified in this population.
Melasma: A New Era of Topical Treatment Options Galore
Melasma treatment is rapidly expanding beyond traditional agents such as hydroquinone and triple combination therapy, with newer topicals including tranexamic acid, cysteamine, azelaic acid, thiamidol, and emerging compounds showing variable efficacy. While promising, evidence is still evolving, and combination regimens plus strict photoprotection remain the cornerstone of management.
Weight-Loss Drug–Biologic Combination Boosts Relief in Psoriatic Arthritis
In a phase 3b trial, combining tirzepatide with ixekizumab significantly improved joint and skin outcomes in patients with psoriatic arthritis and overweight/obesity (P<.05) compared with ixekizumab alone (P<.001). The combination yielded higher American College of Rheumatology and Psoriasis Area and Severity Index response rates, early symptom improvement, and meaningful weight loss, with safety profiles consistent with known effects.
Tips on Using Biologics for Psoriasis in Context of HIV
Evidence for biologic use in HIV-positive patients with moderate to severe psoriasis is limited, but available case reports suggest tumor necrosis factor inhibitors and newer IL-targeted biologics are generally effective without major impacts on viral load or CD4 counts. Experts recommend prioritizing nonimmunosuppressive options and coordinating care with HIV specialists due to potential infection risks.
Upadacitinib Results in Significant Improvements in Nonsegmental Vitiligo in Phase 3 Studies
Two phase 3 trials showed that the Janus kinase 1 inhibitor upadacitinib significantly improved repigmentation outcomes in adolescents and adults with nonsegmental vitiligo vs placebo over 48 weeks (P<.0001 for both), with a higher proportion achieving clinically meaningful reductions in Vitiligo Area and Severity Index scores. Benefits increased over time without plateau, and no new safety signals were identified.
AAD 2026 Annual Meeting Highlights
AAD 2026 Annual Meeting Highlights