User login
American Society of Hematology (ASH): ASH 2015
Statin may reduce vaso-occlusive pain in SCD

Photo courtesy of the CDC
ORLANDO, FL—In a small study, the cholesterol-lowering medication simvastatin reduced the frequency of vaso-occlusive pain in adults and children with sickle cell disease (SCD).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain after 3 months of treatment with simvastatin.
There was a slight overall decrease in the intensity of pain as well, but this was not statistically significant.
Still, investigators observed a decrease in biomarkers of inflammation and said the drug appeared to be safe for this patient population.
The team believes these preliminary data suggest the need to conduct a larger, randomized trial of simvastatin in SCD.
Carolyn C. Hoppe, MD, of UCSF Benioff Children’s Hospital Oakland in California, presented the data at the 2015 ASH Annual Meeting (abstract 545).*
“Vaso-occlusive pain is a clinical hallmark and major cause of morbidity in sickle cell disease,” Dr Hoppe said. “Triggered by polymerization and hemolysis, vaso-occlusion involves multiple pathways.”
Similarly, although statins are best known for their cholesterol-inhibiting ability, they also inhibit oxidative stress and inflammation.
With this in mind, Dr Hoppe and her colleagues previously tested simvastatin in a phase 1 study of SCD patients who were 13 years of age or older.
The investigators found the safety profile to be acceptable, and they observed an improvement in biomarkers of inflammation. So they decided to carry out the current study.
This was a single-center, uncontrolled trial that enrolled SCD patients ages 10 and older. They received once-daily oral simvastatin (40 mg) for 3 months.
The primary outcome measure was the frequency and intensity of vaso-occlusive pain, as recorded by daily electronic pain diaries, before and after simvastatin treatment.
Clinical laboratory studies and plasma biomarkers were evaluated at baseline, at 0.5, 1, 2, and 3 months during treatment, as well as 1 month after the discontinuation of simvastatin.
Results
Nineteen patients completed the study. They had a mean age of 19 (range, 10-34), and 13 were female. Seventeen had HbSS genotype, and 2 had S/β0 thalassemia. Ten patients were receiving hydroxyurea.
The simvastatin adherence rate was 85%, and the adherence to using the daily pain diary was 73%.
Dr Hoppe said there were no new safety issues or drug-related adverse events in this trial. There was no myalgia or myopathy. One subject did experience transient facial swelling that may have been drug-related.
The patients’ total cholesterol decreased by 20% from baseline. There was a significant decrease in both LDL and HDL cholesterol (P<0.001 for both).
Creatinine kinase remained stable during treatment, as did hemoglobin levels.
Dr Hoppe noted that the study was not designed to include an assessment of fetal hemoglobin, so she and her colleagues did not have data on that measure for all the patients, but the team did observe an increase in fetal hemoglobin levels from baseline among the patients who were receiving hydroxyurea.
The investigators observed a decrease from baseline in markers of hemolysis—absolute reticulocyte count (P=0.006) and total bilirubin (P=0.02).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain from baseline (P=0.005) and a 10% decrease in the intensity of pain (which was not significant).
There was a 59% decrease in hsCRP (P=0.003), an 18% decrease in sE-selectin (P=0.01), a 5% decrease in sICAM (P=0.03), and a 17% decrease in VEGF (P=0.05). There was no significant effect on plasma nitric oxide metabolites, sVCAM1, or P-selectin levels.
“These results are basically preliminary data to give clinical support for a larger, randomized trial of simvastatin to assess its clinical efficacy in SCD,” Dr Hoppe concluded.
She reported receiving research funding and consultancy payments from Eli Lilly and Company, and another investigator involved in this study is an employee of Pharmacyclics LLC. ![]()
*Data in the abstract differ from the data presented.
Photo courtesy of the CDC
ORLANDO, FL—In a small study, the cholesterol-lowering medication simvastatin reduced the frequency of vaso-occlusive pain in adults and children with sickle cell disease (SCD).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain after 3 months of treatment with simvastatin.
There was a slight overall decrease in the intensity of pain as well, but this was not statistically significant.
Still, investigators observed a decrease in biomarkers of inflammation and said the drug appeared to be safe for this patient population.
The team believes these preliminary data suggest the need to conduct a larger, randomized trial of simvastatin in SCD.
Carolyn C. Hoppe, MD, of UCSF Benioff Children’s Hospital Oakland in California, presented the data at the 2015 ASH Annual Meeting (abstract 545).*
“Vaso-occlusive pain is a clinical hallmark and major cause of morbidity in sickle cell disease,” Dr Hoppe said. “Triggered by polymerization and hemolysis, vaso-occlusion involves multiple pathways.”
Similarly, although statins are best known for their cholesterol-inhibiting ability, they also inhibit oxidative stress and inflammation.
With this in mind, Dr Hoppe and her colleagues previously tested simvastatin in a phase 1 study of SCD patients who were 13 years of age or older.
The investigators found the safety profile to be acceptable, and they observed an improvement in biomarkers of inflammation. So they decided to carry out the current study.
This was a single-center, uncontrolled trial that enrolled SCD patients ages 10 and older. They received once-daily oral simvastatin (40 mg) for 3 months.
The primary outcome measure was the frequency and intensity of vaso-occlusive pain, as recorded by daily electronic pain diaries, before and after simvastatin treatment.
Clinical laboratory studies and plasma biomarkers were evaluated at baseline, at 0.5, 1, 2, and 3 months during treatment, as well as 1 month after the discontinuation of simvastatin.
Results
Nineteen patients completed the study. They had a mean age of 19 (range, 10-34), and 13 were female. Seventeen had HbSS genotype, and 2 had S/β0 thalassemia. Ten patients were receiving hydroxyurea.
The simvastatin adherence rate was 85%, and the adherence to using the daily pain diary was 73%.
Dr Hoppe said there were no new safety issues or drug-related adverse events in this trial. There was no myalgia or myopathy. One subject did experience transient facial swelling that may have been drug-related.
The patients’ total cholesterol decreased by 20% from baseline. There was a significant decrease in both LDL and HDL cholesterol (P<0.001 for both).
Creatinine kinase remained stable during treatment, as did hemoglobin levels.
Dr Hoppe noted that the study was not designed to include an assessment of fetal hemoglobin, so she and her colleagues did not have data on that measure for all the patients, but the team did observe an increase in fetal hemoglobin levels from baseline among the patients who were receiving hydroxyurea.
The investigators observed a decrease from baseline in markers of hemolysis—absolute reticulocyte count (P=0.006) and total bilirubin (P=0.02).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain from baseline (P=0.005) and a 10% decrease in the intensity of pain (which was not significant).
There was a 59% decrease in hsCRP (P=0.003), an 18% decrease in sE-selectin (P=0.01), a 5% decrease in sICAM (P=0.03), and a 17% decrease in VEGF (P=0.05). There was no significant effect on plasma nitric oxide metabolites, sVCAM1, or P-selectin levels.
“These results are basically preliminary data to give clinical support for a larger, randomized trial of simvastatin to assess its clinical efficacy in SCD,” Dr Hoppe concluded.
She reported receiving research funding and consultancy payments from Eli Lilly and Company, and another investigator involved in this study is an employee of Pharmacyclics LLC. ![]()
*Data in the abstract differ from the data presented.
Photo courtesy of the CDC
ORLANDO, FL—In a small study, the cholesterol-lowering medication simvastatin reduced the frequency of vaso-occlusive pain in adults and children with sickle cell disease (SCD).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain after 3 months of treatment with simvastatin.
There was a slight overall decrease in the intensity of pain as well, but this was not statistically significant.
Still, investigators observed a decrease in biomarkers of inflammation and said the drug appeared to be safe for this patient population.
The team believes these preliminary data suggest the need to conduct a larger, randomized trial of simvastatin in SCD.
Carolyn C. Hoppe, MD, of UCSF Benioff Children’s Hospital Oakland in California, presented the data at the 2015 ASH Annual Meeting (abstract 545).*
“Vaso-occlusive pain is a clinical hallmark and major cause of morbidity in sickle cell disease,” Dr Hoppe said. “Triggered by polymerization and hemolysis, vaso-occlusion involves multiple pathways.”
Similarly, although statins are best known for their cholesterol-inhibiting ability, they also inhibit oxidative stress and inflammation.
With this in mind, Dr Hoppe and her colleagues previously tested simvastatin in a phase 1 study of SCD patients who were 13 years of age or older.
The investigators found the safety profile to be acceptable, and they observed an improvement in biomarkers of inflammation. So they decided to carry out the current study.
This was a single-center, uncontrolled trial that enrolled SCD patients ages 10 and older. They received once-daily oral simvastatin (40 mg) for 3 months.
The primary outcome measure was the frequency and intensity of vaso-occlusive pain, as recorded by daily electronic pain diaries, before and after simvastatin treatment.
Clinical laboratory studies and plasma biomarkers were evaluated at baseline, at 0.5, 1, 2, and 3 months during treatment, as well as 1 month after the discontinuation of simvastatin.
Results
Nineteen patients completed the study. They had a mean age of 19 (range, 10-34), and 13 were female. Seventeen had HbSS genotype, and 2 had S/β0 thalassemia. Ten patients were receiving hydroxyurea.
The simvastatin adherence rate was 85%, and the adherence to using the daily pain diary was 73%.
Dr Hoppe said there were no new safety issues or drug-related adverse events in this trial. There was no myalgia or myopathy. One subject did experience transient facial swelling that may have been drug-related.
The patients’ total cholesterol decreased by 20% from baseline. There was a significant decrease in both LDL and HDL cholesterol (P<0.001 for both).
Creatinine kinase remained stable during treatment, as did hemoglobin levels.
Dr Hoppe noted that the study was not designed to include an assessment of fetal hemoglobin, so she and her colleagues did not have data on that measure for all the patients, but the team did observe an increase in fetal hemoglobin levels from baseline among the patients who were receiving hydroxyurea.
The investigators observed a decrease from baseline in markers of hemolysis—absolute reticulocyte count (P=0.006) and total bilirubin (P=0.02).
Overall, there was a 46% decrease in the frequency of vaso-occlusive pain from baseline (P=0.005) and a 10% decrease in the intensity of pain (which was not significant).
There was a 59% decrease in hsCRP (P=0.003), an 18% decrease in sE-selectin (P=0.01), a 5% decrease in sICAM (P=0.03), and a 17% decrease in VEGF (P=0.05). There was no significant effect on plasma nitric oxide metabolites, sVCAM1, or P-selectin levels.
“These results are basically preliminary data to give clinical support for a larger, randomized trial of simvastatin to assess its clinical efficacy in SCD,” Dr Hoppe concluded.
She reported receiving research funding and consultancy payments from Eli Lilly and Company, and another investigator involved in this study is an employee of Pharmacyclics LLC. ![]()
*Data in the abstract differ from the data presented.
Potential new alternative in CML when TKI therapy fails

ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.
Chemo quadruples risk for myeloid cancers
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.

“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
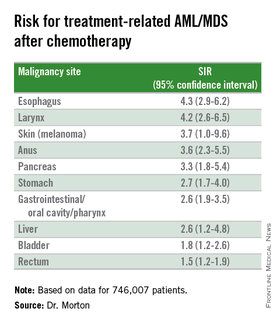
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
AT ASH 2015
Key clinical point: This study quantifies the risks for treatment-related myeloid cancers after chemotherapy.
Major finding: Chemotherapy is associated with a fourfold risk for treatment-related AML/MDS, compared with the general population.
Data source: Retrospective review of data on 746,007 adults treated with chemotherapy for a first primary malignancy.
Disclosures: The National Cancer Institute supported the study. Dr. Morton reported having no conflicts of interest to disclose.

More complete cytogenetic responses at 12 months with radotinib than imatinib
Radotinib was associated with significantly higher complete cytogenetic responses and major molecular responses than imatinib was at a minimum 12 months of follow-up in a randomized, open-label, phase III clinical trialof patients with newly diagnosed chronic myeloid leukemia-chronic phase (CML-CP).
Radotinib, an investigational BCR-ABL1 tyrosine kinase inhibitor developed by IL-YANG Pharmaceuticals, is approved in Korea for the treatment of CML-CP in patients who have failed prior TKIs.
Dr. Jae-Yong Kwak of Chonbuk National University Medical School and Hospital, Jeonju, South Korea, and his colleagues randomized 241 patients to either radotinib 300 mg twice daily (n = 79), radotinib 400 mg twice daily (n = 81), or imatinib 400 mg once daily (n = 81). All three study groups were balanced in regard to baseline age, gender, race, and Sokal risk score.
At a minimum follow-up of 12 months, the proportions of patients receiving a study drug were 86% (69/79) in the radotinib 300 mg twice-daily group, 72% (58/81) in the radotinib 400 mg twice-daily group, and 82% (66/81) in the imatinib 400 mg once-daily group.
The rates of major molecular response at 12 months were significantly higher in patients receiving radotinib 300 mg b.i.d. (52%, P = .0044) and radotinib 400 mg b.i.d. (46%, P = .0342), compared with imatinib (30%), Dr. Kwak reported at the annual meeting of the American Society of Hematology in Orlando.
Among responders, the median times to major molecular response were shorter on radotinib 300 mg b.i.d. (5.7 months) and radotinib 400 mg b.i.d. (5.6 months) than on imatinib (8.2 months). The MR4.5 rates by 12 months were also higher for both radotinib 300 mg b.i.d. (15%) and 400 mg b.i.d. (14%), compared with imatinib (9%). The complete cytogenetic response rates by 12 months were 91% for radotinib 300 mg b.i.d. (P = .0120), compared with imatinib (77%). None of the patients in the study had progressed to accelerated phase or blast crisis at 12 months.
Drug discontinuation due to adverse events (AEs) or laboratory abnormalities occurred in 9% of patients on radotinib 300 mg b.i.d., 20% on radotinib 400 mg b.i.d., and 6% on imatinib.
The major side effects included grade 3/4 thrombocytopenia in 16% of patients receiving radotinib 300 mg b.i.d., 14% on radotinib 400 mg b.i.d., and 20% receiving imatinib. Grade 3/4 neutropenia occurred in 19%, 24%, and 30% for radotinib 300 mg b.i.d., 400 mg b.i.d., and imatinib, respectively.
Overall, grade 3/4 nonlaboratory AEs were uncommon in all groups. The most common nonlaboratory AEs in the radotinib groups were skin rash (about 33% in both), nausea/vomiting (about 23% in both), headache (19% and 31%), and pruritus (19% and 30%). In the imatinib group, the most common adverse events were edema (35%), myalgia (28%), nausea/vomiting (27%), and skin rash (22%).
Dr. Kwak had no relevant disclosures. Some of his colleagues received research funding from IL-YANG Pharmaceutical Co. and Alexion Pharmaceuticals.
On Twitter @maryjodales
Radotinib was associated with significantly higher complete cytogenetic responses and major molecular responses than imatinib was at a minimum 12 months of follow-up in a randomized, open-label, phase III clinical trialof patients with newly diagnosed chronic myeloid leukemia-chronic phase (CML-CP).
Radotinib, an investigational BCR-ABL1 tyrosine kinase inhibitor developed by IL-YANG Pharmaceuticals, is approved in Korea for the treatment of CML-CP in patients who have failed prior TKIs.
Dr. Jae-Yong Kwak of Chonbuk National University Medical School and Hospital, Jeonju, South Korea, and his colleagues randomized 241 patients to either radotinib 300 mg twice daily (n = 79), radotinib 400 mg twice daily (n = 81), or imatinib 400 mg once daily (n = 81). All three study groups were balanced in regard to baseline age, gender, race, and Sokal risk score.
At a minimum follow-up of 12 months, the proportions of patients receiving a study drug were 86% (69/79) in the radotinib 300 mg twice-daily group, 72% (58/81) in the radotinib 400 mg twice-daily group, and 82% (66/81) in the imatinib 400 mg once-daily group.
The rates of major molecular response at 12 months were significantly higher in patients receiving radotinib 300 mg b.i.d. (52%, P = .0044) and radotinib 400 mg b.i.d. (46%, P = .0342), compared with imatinib (30%), Dr. Kwak reported at the annual meeting of the American Society of Hematology in Orlando.
Among responders, the median times to major molecular response were shorter on radotinib 300 mg b.i.d. (5.7 months) and radotinib 400 mg b.i.d. (5.6 months) than on imatinib (8.2 months). The MR4.5 rates by 12 months were also higher for both radotinib 300 mg b.i.d. (15%) and 400 mg b.i.d. (14%), compared with imatinib (9%). The complete cytogenetic response rates by 12 months were 91% for radotinib 300 mg b.i.d. (P = .0120), compared with imatinib (77%). None of the patients in the study had progressed to accelerated phase or blast crisis at 12 months.
Drug discontinuation due to adverse events (AEs) or laboratory abnormalities occurred in 9% of patients on radotinib 300 mg b.i.d., 20% on radotinib 400 mg b.i.d., and 6% on imatinib.
The major side effects included grade 3/4 thrombocytopenia in 16% of patients receiving radotinib 300 mg b.i.d., 14% on radotinib 400 mg b.i.d., and 20% receiving imatinib. Grade 3/4 neutropenia occurred in 19%, 24%, and 30% for radotinib 300 mg b.i.d., 400 mg b.i.d., and imatinib, respectively.
Overall, grade 3/4 nonlaboratory AEs were uncommon in all groups. The most common nonlaboratory AEs in the radotinib groups were skin rash (about 33% in both), nausea/vomiting (about 23% in both), headache (19% and 31%), and pruritus (19% and 30%). In the imatinib group, the most common adverse events were edema (35%), myalgia (28%), nausea/vomiting (27%), and skin rash (22%).
Dr. Kwak had no relevant disclosures. Some of his colleagues received research funding from IL-YANG Pharmaceutical Co. and Alexion Pharmaceuticals.
On Twitter @maryjodales
Radotinib was associated with significantly higher complete cytogenetic responses and major molecular responses than imatinib was at a minimum 12 months of follow-up in a randomized, open-label, phase III clinical trialof patients with newly diagnosed chronic myeloid leukemia-chronic phase (CML-CP).
Radotinib, an investigational BCR-ABL1 tyrosine kinase inhibitor developed by IL-YANG Pharmaceuticals, is approved in Korea for the treatment of CML-CP in patients who have failed prior TKIs.
Dr. Jae-Yong Kwak of Chonbuk National University Medical School and Hospital, Jeonju, South Korea, and his colleagues randomized 241 patients to either radotinib 300 mg twice daily (n = 79), radotinib 400 mg twice daily (n = 81), or imatinib 400 mg once daily (n = 81). All three study groups were balanced in regard to baseline age, gender, race, and Sokal risk score.
At a minimum follow-up of 12 months, the proportions of patients receiving a study drug were 86% (69/79) in the radotinib 300 mg twice-daily group, 72% (58/81) in the radotinib 400 mg twice-daily group, and 82% (66/81) in the imatinib 400 mg once-daily group.
The rates of major molecular response at 12 months were significantly higher in patients receiving radotinib 300 mg b.i.d. (52%, P = .0044) and radotinib 400 mg b.i.d. (46%, P = .0342), compared with imatinib (30%), Dr. Kwak reported at the annual meeting of the American Society of Hematology in Orlando.
Among responders, the median times to major molecular response were shorter on radotinib 300 mg b.i.d. (5.7 months) and radotinib 400 mg b.i.d. (5.6 months) than on imatinib (8.2 months). The MR4.5 rates by 12 months were also higher for both radotinib 300 mg b.i.d. (15%) and 400 mg b.i.d. (14%), compared with imatinib (9%). The complete cytogenetic response rates by 12 months were 91% for radotinib 300 mg b.i.d. (P = .0120), compared with imatinib (77%). None of the patients in the study had progressed to accelerated phase or blast crisis at 12 months.
Drug discontinuation due to adverse events (AEs) or laboratory abnormalities occurred in 9% of patients on radotinib 300 mg b.i.d., 20% on radotinib 400 mg b.i.d., and 6% on imatinib.
The major side effects included grade 3/4 thrombocytopenia in 16% of patients receiving radotinib 300 mg b.i.d., 14% on radotinib 400 mg b.i.d., and 20% receiving imatinib. Grade 3/4 neutropenia occurred in 19%, 24%, and 30% for radotinib 300 mg b.i.d., 400 mg b.i.d., and imatinib, respectively.
Overall, grade 3/4 nonlaboratory AEs were uncommon in all groups. The most common nonlaboratory AEs in the radotinib groups were skin rash (about 33% in both), nausea/vomiting (about 23% in both), headache (19% and 31%), and pruritus (19% and 30%). In the imatinib group, the most common adverse events were edema (35%), myalgia (28%), nausea/vomiting (27%), and skin rash (22%).
Dr. Kwak had no relevant disclosures. Some of his colleagues received research funding from IL-YANG Pharmaceutical Co. and Alexion Pharmaceuticals.
On Twitter @maryjodales
FROM ASH 2015
Key clinical point: Radotinib was associated with significantly higher complete cytogenetic responses and major molecular responses than was imatinib at a minimum 12 months of follow-up.
Major finding: By 12 months, the rates of major molecular response were significantly higher in patients receiving radotinib 300 mg b.i.d. (52%, P = .0044) and radotinib 400 mg b.i.d. (46%, P = .0342), compared with imatinib 400 mg/day (30%).
Data source: Randomized, open-label, phase III clinical trial involving 241 patients.
Disclosures: Dr. Kwak had no relevant disclosures. Some of his colleagues received research funding from IL-YANG Pharmaceutical Co. and Alexion Pharmaceuticals.
Nilotinib safe, effective as first-line therapy for CML-CP patients age 65 and older
ORLANDO – Age did not affect molecular response or the incidence of adverse reactions to nilotinib among patients with chronic myeloid leukemia in chronic phase (CML-CP), based on results from a subanalysis of the ENEST1st study.
The analysis of the ENEST1st study, reported by Dr. Francis J. Giles, compared outcomes for 1,089 newly diagnosed CML-CP patients, 19% were aged 65 years or older and 81% were younger than age 65 years. All patients had typical transcripts and were treated for 3 months or less with nilotinib 300 mg twice daily in the open-label study.

For those 65 years and older, Sokal risk scores were low in 4.5%, intermediate in 61.2%, and high in 23.4%, with missing data for 10.9%. For younger patients, Sokal risk scores were low in 42.1%, intermediate in 32%, and high in 16.9%, with missing data for 9.
At 18 months, there was an overall 38.4% rate (95% CI, 35.5%-41.3%) of MR4 grade molecular response, which was defined as BCR-ABL level of 0.01% or less on the International Scale or undetectable BCR-ABL in cDNA with at least 10,000 ABL transcripts.
The MR4 rate at 18 months did not significantly vary by age. For patients under age 65, the cumulative incidence of MR4 by 18 months was 48.8% (95% CI, 45.4% - 52.1%); among patients aged 65 and older, the incidence of MR4 was 48.3% (95% CI, 41.4% - 55.2%). The MR4.5 rate by 18 months was 32.5% in younger patients and 28.4% in older patients, reported Dr. Giles of the Institute for Drug Development, Cancer Therapy and Research Center, at the University of Texas Health Science Center at San Antonio, and his colleagues.
Based on Sokal score, the MR4 rate by 18 months in younger patients was 53.6% (low), 45.2% (intermediate), and 35.4% (high), respectively. For older patients, the MR4 rate by 18 months based on Sokal score was 44.4% (low), 49.6% (intermediate), and 44.7% (high).
Six patients (0.6%) progressed to accelerated phase/blast crisis (AP/BC) on study; 13 patients (1.2%) died by 24 months. The most common adverse events were rash (21.4%), pruritus (16.5%), and headache (15.2%).
Novartis is the sponsor of the ENEST1st study. Dr. Giles consults for and receives honoraria and research funding from Novartis.
On Twitter @maryjodales
ORLANDO – Age did not affect molecular response or the incidence of adverse reactions to nilotinib among patients with chronic myeloid leukemia in chronic phase (CML-CP), based on results from a subanalysis of the ENEST1st study.
The analysis of the ENEST1st study, reported by Dr. Francis J. Giles, compared outcomes for 1,089 newly diagnosed CML-CP patients, 19% were aged 65 years or older and 81% were younger than age 65 years. All patients had typical transcripts and were treated for 3 months or less with nilotinib 300 mg twice daily in the open-label study.
For those 65 years and older, Sokal risk scores were low in 4.5%, intermediate in 61.2%, and high in 23.4%, with missing data for 10.9%. For younger patients, Sokal risk scores were low in 42.1%, intermediate in 32%, and high in 16.9%, with missing data for 9.
At 18 months, there was an overall 38.4% rate (95% CI, 35.5%-41.3%) of MR4 grade molecular response, which was defined as BCR-ABL level of 0.01% or less on the International Scale or undetectable BCR-ABL in cDNA with at least 10,000 ABL transcripts.
The MR4 rate at 18 months did not significantly vary by age. For patients under age 65, the cumulative incidence of MR4 by 18 months was 48.8% (95% CI, 45.4% - 52.1%); among patients aged 65 and older, the incidence of MR4 was 48.3% (95% CI, 41.4% - 55.2%). The MR4.5 rate by 18 months was 32.5% in younger patients and 28.4% in older patients, reported Dr. Giles of the Institute for Drug Development, Cancer Therapy and Research Center, at the University of Texas Health Science Center at San Antonio, and his colleagues.
Based on Sokal score, the MR4 rate by 18 months in younger patients was 53.6% (low), 45.2% (intermediate), and 35.4% (high), respectively. For older patients, the MR4 rate by 18 months based on Sokal score was 44.4% (low), 49.6% (intermediate), and 44.7% (high).
Six patients (0.6%) progressed to accelerated phase/blast crisis (AP/BC) on study; 13 patients (1.2%) died by 24 months. The most common adverse events were rash (21.4%), pruritus (16.5%), and headache (15.2%).
Novartis is the sponsor of the ENEST1st study. Dr. Giles consults for and receives honoraria and research funding from Novartis.
On Twitter @maryjodales
ORLANDO – Age did not affect molecular response or the incidence of adverse reactions to nilotinib among patients with chronic myeloid leukemia in chronic phase (CML-CP), based on results from a subanalysis of the ENEST1st study.
The analysis of the ENEST1st study, reported by Dr. Francis J. Giles, compared outcomes for 1,089 newly diagnosed CML-CP patients, 19% were aged 65 years or older and 81% were younger than age 65 years. All patients had typical transcripts and were treated for 3 months or less with nilotinib 300 mg twice daily in the open-label study.
For those 65 years and older, Sokal risk scores were low in 4.5%, intermediate in 61.2%, and high in 23.4%, with missing data for 10.9%. For younger patients, Sokal risk scores were low in 42.1%, intermediate in 32%, and high in 16.9%, with missing data for 9.
At 18 months, there was an overall 38.4% rate (95% CI, 35.5%-41.3%) of MR4 grade molecular response, which was defined as BCR-ABL level of 0.01% or less on the International Scale or undetectable BCR-ABL in cDNA with at least 10,000 ABL transcripts.
The MR4 rate at 18 months did not significantly vary by age. For patients under age 65, the cumulative incidence of MR4 by 18 months was 48.8% (95% CI, 45.4% - 52.1%); among patients aged 65 and older, the incidence of MR4 was 48.3% (95% CI, 41.4% - 55.2%). The MR4.5 rate by 18 months was 32.5% in younger patients and 28.4% in older patients, reported Dr. Giles of the Institute for Drug Development, Cancer Therapy and Research Center, at the University of Texas Health Science Center at San Antonio, and his colleagues.
Based on Sokal score, the MR4 rate by 18 months in younger patients was 53.6% (low), 45.2% (intermediate), and 35.4% (high), respectively. For older patients, the MR4 rate by 18 months based on Sokal score was 44.4% (low), 49.6% (intermediate), and 44.7% (high).
Six patients (0.6%) progressed to accelerated phase/blast crisis (AP/BC) on study; 13 patients (1.2%) died by 24 months. The most common adverse events were rash (21.4%), pruritus (16.5%), and headache (15.2%).
Novartis is the sponsor of the ENEST1st study. Dr. Giles consults for and receives honoraria and research funding from Novartis.
On Twitter @maryjodales
FROM ASH 2015
Key clinical point: Age did not affect molecular response or the incidence of adverse reactions to nilotinib among patients with CML-CP.
Major finding: For patients younger than age 65 years, the cumulative incidence of MR4 by 18 months was 48.8% (95% CI, 45.4%-52.1%); among patients aged 65 years and older, the incidence of MR4 was 48.3% (95% CI, 41.4%-55.2%).
Data source: The analysis of the ENEST1st study compared outcomes for 1,089 newly diagnosed CML-CP patients.
Disclosures: Novartis is the sponsor of the ENEST1st study. Dr. Giles consults for and receives honoraria and research funding from Novartis.

Osteoarticular pain affects CML patients stopping TKI

Photo courtesy of ASH
ORLANDO, FL—Cases of musculoskeletal pain have been reported after patients stop taking tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML).
TKI discontinuation trials—notably, the STOP imatinib (STIM) trials and EURO-SKI trial—have been conducted to assess the feasibility of maintaining molecular remission once patients discontinue a TKI.
However, none of the studies collected low-grade events before or after patients discontinued TKI therapy.
So investigators collected data from the STIM2 study and EUROSKI trial and recorded all events from the time of TKI discontinuation.
They discovered that about 23% of patients who stopped TKI therapy experienced a withdrawal syndrome (WS) consisting largely of musculoskeletal pain, regardless of the TKI they were taking.
Philippe Rousselot, MD, PhD, of University of Versailles St-Quentin-en-Yvelines, Versailles, France, discussed this finding at the 2015 ASH Annual Meeting as abstract 137.*
Dr Rousselot noted that investigators first reported the TKI WS in 2014 in CML patients enrolled on the EURO-SKI trial who were discontinuing imatinib (Richter et al, JCO 2014).
A team of French investigators undertook the current observational study to estimate the prevalence of the WS and to identify clinical factors associated with it.
They collected, prospectively, the adverse events from all 428 French patients who were enrolled in the STIM2 (n=204) and EURO-SKI (n=224) trials. And they compared patients who stopped taking TKIs and had a painful WS to those who stopped TKIs and did not have a painful syndrome.
Patient characteristics
Patient characteristics were well balanced between the STIM2 and EURO-SKI groups, with the exception of the median time on TKI before discontinuation. In the STIM2 group, patients were a median of 77.4 months on TKI therapy. In the EURO-SKI group, the median time on a TKI was 100.4 months (P<0.001).
In all, there were 208 male and 220 female patients included. They were a median age of 64 (range, 53–73) and 63 (range, 53–70) years in the STIM2 and EURO-SKI groups, respectively.
Sokal scores were also comparable between the cohorts, with most patients falling in the low and intermediate ranges.
Prevalence and characteristics of WS
Overall, 326 patients (76.2%) were without WS and 102 (23.8%) had WS. In the STIM2 cohort, 193 patients (86.2%) were without WS and 31 (13.8%) had WS. In the EURO-SKI cohort, 133 patients (65.2%) were without WS and 71 (34.8%) had WS.
“And these differences [between cohorts] are significant,” Dr Rousselot pointed out.
Investigators analyzed clinical characteristics of WS in 40 patients and determined that the median time from TKI discontinuation to WS was 21 days, and the median duration of WS was 7 months (range, 3–30).
Pain was located in the shoulder and spine for 67% of the patients and elsewhere in 33%. About two-thirds of patients (62.5%) experienced grade 1–2 pain, and 37.5% experienced grade 3–4 pain.
Nineteen patients resumed TKI therapy, “because of loss of MMR [major molecular response] or loss of clinical response,” Dr Rousselot said.
And the pain disappeared in 52.6% of them when they resumed TKI therapy. The median duration of TKI therapy before WS pain disappeared was 3 weeks.
Risk factors for WS
Investigators determined that CML duration, time on a TKI, and previous history of osteoarticular symptoms were risk factors for WS.
Patients without WS had CML for a shorter time—a mean of 8.7 ± 3.1 months, compared to 9.7 ± 3.8 for those with WS (P=0.02).
Patients without WS were also on a TKI for a shorter time—a median of 81.2 months (range, 61.2–108.0), compared to 97.3 months (range, 73.7–122.9) for those with WS (P<0.001).
Patients with a previous history of osteoarticular symptoms were more likely to experience WS—22.9%, compared to 9.8% without a previous history (P=0.002).
Most patients were receiving imatinib—323 without WS and 100 with WS. The 1 patient receiving dasatinib had no WS. And of the 4 patients receiving nilotinib, 2 had WS and 2 didn’t.
And so the type of TKI therapy—dasatinib, imatinib, or nilotinib—was not significant (P=0.42).
Investigators performed a multivariate analysis adjusted for gender, CML duration, and Sokal score, and 2 risk factors emerged: previous history of osteoarticular symptoms (relative risk: 2.08) and time on TKI (relative risk: 2.23).
Discussion
Dr Rousselot compared the Richter trial (Richter et al, JCO 2014) to the current study and noted that the Richter trial, with an enrollment of 50 patients, had a WS prevalence of 30%. But the current trial had a prevalence of 24%.
The difference in WS may be due to time on TKI, Dr Rousselot said, as patients in the Richter trial were on TKI treatment for a longer period of time.
“The time of onset is the same [in both trials],” Dr Rousselot said, as are the TKI used, location of pain, and duration of pain.
“So what we can say is [with] shorter TKI treatment . . . , we have a higher risk of molecular relapse but a lower risk of withdrawal syndrome.”
And with longer TKI treatment, the converse appears to be true. It reduces the risk of molecular relapse but raises the risk of withdrawal syndrome. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of ASH
ORLANDO, FL—Cases of musculoskeletal pain have been reported after patients stop taking tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML).
TKI discontinuation trials—notably, the STOP imatinib (STIM) trials and EURO-SKI trial—have been conducted to assess the feasibility of maintaining molecular remission once patients discontinue a TKI.
However, none of the studies collected low-grade events before or after patients discontinued TKI therapy.
So investigators collected data from the STIM2 study and EUROSKI trial and recorded all events from the time of TKI discontinuation.
They discovered that about 23% of patients who stopped TKI therapy experienced a withdrawal syndrome (WS) consisting largely of musculoskeletal pain, regardless of the TKI they were taking.
Philippe Rousselot, MD, PhD, of University of Versailles St-Quentin-en-Yvelines, Versailles, France, discussed this finding at the 2015 ASH Annual Meeting as abstract 137.*
Dr Rousselot noted that investigators first reported the TKI WS in 2014 in CML patients enrolled on the EURO-SKI trial who were discontinuing imatinib (Richter et al, JCO 2014).
A team of French investigators undertook the current observational study to estimate the prevalence of the WS and to identify clinical factors associated with it.
They collected, prospectively, the adverse events from all 428 French patients who were enrolled in the STIM2 (n=204) and EURO-SKI (n=224) trials. And they compared patients who stopped taking TKIs and had a painful WS to those who stopped TKIs and did not have a painful syndrome.
Patient characteristics
Patient characteristics were well balanced between the STIM2 and EURO-SKI groups, with the exception of the median time on TKI before discontinuation. In the STIM2 group, patients were a median of 77.4 months on TKI therapy. In the EURO-SKI group, the median time on a TKI was 100.4 months (P<0.001).
In all, there were 208 male and 220 female patients included. They were a median age of 64 (range, 53–73) and 63 (range, 53–70) years in the STIM2 and EURO-SKI groups, respectively.
Sokal scores were also comparable between the cohorts, with most patients falling in the low and intermediate ranges.
Prevalence and characteristics of WS
Overall, 326 patients (76.2%) were without WS and 102 (23.8%) had WS. In the STIM2 cohort, 193 patients (86.2%) were without WS and 31 (13.8%) had WS. In the EURO-SKI cohort, 133 patients (65.2%) were without WS and 71 (34.8%) had WS.
“And these differences [between cohorts] are significant,” Dr Rousselot pointed out.
Investigators analyzed clinical characteristics of WS in 40 patients and determined that the median time from TKI discontinuation to WS was 21 days, and the median duration of WS was 7 months (range, 3–30).
Pain was located in the shoulder and spine for 67% of the patients and elsewhere in 33%. About two-thirds of patients (62.5%) experienced grade 1–2 pain, and 37.5% experienced grade 3–4 pain.
Nineteen patients resumed TKI therapy, “because of loss of MMR [major molecular response] or loss of clinical response,” Dr Rousselot said.
And the pain disappeared in 52.6% of them when they resumed TKI therapy. The median duration of TKI therapy before WS pain disappeared was 3 weeks.
Risk factors for WS
Investigators determined that CML duration, time on a TKI, and previous history of osteoarticular symptoms were risk factors for WS.
Patients without WS had CML for a shorter time—a mean of 8.7 ± 3.1 months, compared to 9.7 ± 3.8 for those with WS (P=0.02).
Patients without WS were also on a TKI for a shorter time—a median of 81.2 months (range, 61.2–108.0), compared to 97.3 months (range, 73.7–122.9) for those with WS (P<0.001).
Patients with a previous history of osteoarticular symptoms were more likely to experience WS—22.9%, compared to 9.8% without a previous history (P=0.002).
Most patients were receiving imatinib—323 without WS and 100 with WS. The 1 patient receiving dasatinib had no WS. And of the 4 patients receiving nilotinib, 2 had WS and 2 didn’t.
And so the type of TKI therapy—dasatinib, imatinib, or nilotinib—was not significant (P=0.42).
Investigators performed a multivariate analysis adjusted for gender, CML duration, and Sokal score, and 2 risk factors emerged: previous history of osteoarticular symptoms (relative risk: 2.08) and time on TKI (relative risk: 2.23).
Discussion
Dr Rousselot compared the Richter trial (Richter et al, JCO 2014) to the current study and noted that the Richter trial, with an enrollment of 50 patients, had a WS prevalence of 30%. But the current trial had a prevalence of 24%.
The difference in WS may be due to time on TKI, Dr Rousselot said, as patients in the Richter trial were on TKI treatment for a longer period of time.
“The time of onset is the same [in both trials],” Dr Rousselot said, as are the TKI used, location of pain, and duration of pain.
“So what we can say is [with] shorter TKI treatment . . . , we have a higher risk of molecular relapse but a lower risk of withdrawal syndrome.”
And with longer TKI treatment, the converse appears to be true. It reduces the risk of molecular relapse but raises the risk of withdrawal syndrome. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of ASH
ORLANDO, FL—Cases of musculoskeletal pain have been reported after patients stop taking tyrosine kinase inhibitors (TKIs) for chronic myeloid leukemia (CML).
TKI discontinuation trials—notably, the STOP imatinib (STIM) trials and EURO-SKI trial—have been conducted to assess the feasibility of maintaining molecular remission once patients discontinue a TKI.
However, none of the studies collected low-grade events before or after patients discontinued TKI therapy.
So investigators collected data from the STIM2 study and EUROSKI trial and recorded all events from the time of TKI discontinuation.
They discovered that about 23% of patients who stopped TKI therapy experienced a withdrawal syndrome (WS) consisting largely of musculoskeletal pain, regardless of the TKI they were taking.
Philippe Rousselot, MD, PhD, of University of Versailles St-Quentin-en-Yvelines, Versailles, France, discussed this finding at the 2015 ASH Annual Meeting as abstract 137.*
Dr Rousselot noted that investigators first reported the TKI WS in 2014 in CML patients enrolled on the EURO-SKI trial who were discontinuing imatinib (Richter et al, JCO 2014).
A team of French investigators undertook the current observational study to estimate the prevalence of the WS and to identify clinical factors associated with it.
They collected, prospectively, the adverse events from all 428 French patients who were enrolled in the STIM2 (n=204) and EURO-SKI (n=224) trials. And they compared patients who stopped taking TKIs and had a painful WS to those who stopped TKIs and did not have a painful syndrome.
Patient characteristics
Patient characteristics were well balanced between the STIM2 and EURO-SKI groups, with the exception of the median time on TKI before discontinuation. In the STIM2 group, patients were a median of 77.4 months on TKI therapy. In the EURO-SKI group, the median time on a TKI was 100.4 months (P<0.001).
In all, there were 208 male and 220 female patients included. They were a median age of 64 (range, 53–73) and 63 (range, 53–70) years in the STIM2 and EURO-SKI groups, respectively.
Sokal scores were also comparable between the cohorts, with most patients falling in the low and intermediate ranges.
Prevalence and characteristics of WS
Overall, 326 patients (76.2%) were without WS and 102 (23.8%) had WS. In the STIM2 cohort, 193 patients (86.2%) were without WS and 31 (13.8%) had WS. In the EURO-SKI cohort, 133 patients (65.2%) were without WS and 71 (34.8%) had WS.
“And these differences [between cohorts] are significant,” Dr Rousselot pointed out.
Investigators analyzed clinical characteristics of WS in 40 patients and determined that the median time from TKI discontinuation to WS was 21 days, and the median duration of WS was 7 months (range, 3–30).
Pain was located in the shoulder and spine for 67% of the patients and elsewhere in 33%. About two-thirds of patients (62.5%) experienced grade 1–2 pain, and 37.5% experienced grade 3–4 pain.
Nineteen patients resumed TKI therapy, “because of loss of MMR [major molecular response] or loss of clinical response,” Dr Rousselot said.
And the pain disappeared in 52.6% of them when they resumed TKI therapy. The median duration of TKI therapy before WS pain disappeared was 3 weeks.
Risk factors for WS
Investigators determined that CML duration, time on a TKI, and previous history of osteoarticular symptoms were risk factors for WS.
Patients without WS had CML for a shorter time—a mean of 8.7 ± 3.1 months, compared to 9.7 ± 3.8 for those with WS (P=0.02).
Patients without WS were also on a TKI for a shorter time—a median of 81.2 months (range, 61.2–108.0), compared to 97.3 months (range, 73.7–122.9) for those with WS (P<0.001).
Patients with a previous history of osteoarticular symptoms were more likely to experience WS—22.9%, compared to 9.8% without a previous history (P=0.002).
Most patients were receiving imatinib—323 without WS and 100 with WS. The 1 patient receiving dasatinib had no WS. And of the 4 patients receiving nilotinib, 2 had WS and 2 didn’t.
And so the type of TKI therapy—dasatinib, imatinib, or nilotinib—was not significant (P=0.42).
Investigators performed a multivariate analysis adjusted for gender, CML duration, and Sokal score, and 2 risk factors emerged: previous history of osteoarticular symptoms (relative risk: 2.08) and time on TKI (relative risk: 2.23).
Discussion
Dr Rousselot compared the Richter trial (Richter et al, JCO 2014) to the current study and noted that the Richter trial, with an enrollment of 50 patients, had a WS prevalence of 30%. But the current trial had a prevalence of 24%.
The difference in WS may be due to time on TKI, Dr Rousselot said, as patients in the Richter trial were on TKI treatment for a longer period of time.
“The time of onset is the same [in both trials],” Dr Rousselot said, as are the TKI used, location of pain, and duration of pain.
“So what we can say is [with] shorter TKI treatment . . . , we have a higher risk of molecular relapse but a lower risk of withdrawal syndrome.”
And with longer TKI treatment, the converse appears to be true. It reduces the risk of molecular relapse but raises the risk of withdrawal syndrome. ![]()
*Data in the abstract differ from the presentation.
ASH: First shot across the bow for CAR T cells in multiple myeloma
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.

CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.
CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Chimeric antigen receptor (CAR) T cells targeting BCMA eradicated myeloma cells in a patient with a heavy burden of chemotherapy-resistant multiple myeloma.
The patient had received three prior myeloma therapies and relapsed with myeloma making up more than 90% of his bone marrow cells just 3 months after autologous transplant.
CD138-positive plasma cells were completely absent, however, one month after infusion of CAR T cells directed against the B-cell maturation antigen (BCMA), and remain undetectable 14 weeks after infusion.
“We have demonstrated for the first time that CAR T cells can have powerful activity against measurable multiple myeloma,” Dr. James N. Kochenderfer of the National Cancer Institute in Bethesda, Md., said at the annual meeting of the American Society of Hematology.
A second patient, who had five prior lines of myeloma therapy and myeloma in 80% of his bone marrow cells, also experienced a dramatic reduction in myeloma, resulting so far in a partial response. The patient has returned to full-time work and myeloma cells continue to decrease 6 weeks after infusion.
The two ongoing responses, however, were associated with the most severe clinical signs of cytokine release syndrome in the late-breaking abstract study (LBA-1), Dr. Kochenderfer said.
The complete responder experienced cytokine release toxicities including fever, tachycardia, hypotension, elevated liver enzymes, and elevated creatinine kinase that resolved. The patient was also platelet transfusion-dependent for 9 weeks after infusion and a baseline absolute neutrophil count of less than 500 microliters remained at that level for 40 days after infusion. All symptoms resolved within two weeks, he said.
The ongoing partial responder experienced fever, tachycardia, hypotension, acute kidney injury, dyspnea, delirium, and prolonged thrombocytopenia and all completely resolved.
Both patients had much higher serum levels of interleukin-6 than the other patients, he noted.
Both patients also received the highest dose level of anti-BCMA CAR T cells. Going forward, that dose will now only be administered to patients with less than 50% bone marrow plasma cells, Dr. Kochenderfer said.
Other responses among the 12 patients treated thus far include 1 transient very good partial response of 8-week duration, 1 transient partial response lasting 2 weeks, and 8 responses of stable disease.
While three new drugs (elotuzumab, daratumumab, ixazomib) were approved for the treatment of multiple myeloma in the month of November alone, this is early days for CAR T-cell therapy in multiple myeloma.
“Almost all of the CAR T-cell work has been in B-cell malignancies, acute lymphocytic leukemia, and chronic lymphocytic leukemia, so is this the right antigen, we don’t know? Is this the right construct, we don’t know? But there’s clearly activity and that is very exciting,” session comoderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston, said in an interview.
Dana-Farber is conducting a CAR T cell trial in myeloma using a different target and NKG2D ligands. Other centers are also using CAR T cells with different targets. “So maybe other targets would have different results and a better safety profile,” he added.
BCMA is an appropriate target for CAR T cell therapy for multiple reasons, Dr. Kochenderfer said. Multiple myeloma is still an incurable disease and BCMA, a member of the tumor necrosis factor superfamily, is uniformly expressed in 60% to 70% of cases.
Preclinical studies by the team also show that BCMA is not detectable in normal human tissues, but is selectively expressed only in bone marrow, lymphoid organs, and organs known to have plasma cells in their lamina propria and by plasma cells and a small fraction of B cells.
The investigators genetically modified autologous T cells to express an anti-BCMA CAR and ligated it into a replication-incompetent gamma retrovirus. The T cells were stimulated with the anti-CD3 monoclonal antibody OKT3 before transduction, with the entire culture process taking 9 days from start to finish. T cells expressing this CAR recognize BCMA with great specificity, Dr. Kochenderfer observed.
The 12 patients received 300 mg/m2 of cyclophosphamide and 30 mg/m2 of fludarabine (Fludara) daily for 3 days before a single infusion of anti-BCMA CAR at dose levels of 0.3 x 106 to 9 x 106 T cells/kg. All patients had at least 3 prior therapies and normal organ function. Five had amyloid light chain only, 4 had immunoglobulin gamma disease, and 4 patients, including the complete responder, had immunoglobulin alpha disease.
It’s unclear why only a few patients responded to the CAR T cells, but dose level was likely a big factor and there is a lot of patient variability with T-cell therapies, Dr. Kochenderfer said at a press briefing.
“I wish we knew exactly why some patients respond and some don’t,” he added. “The CAR T cells have shown remarkable activity against really previously refractory diseases like acute lymphocytic leukemia,” noted Dr. George Daley of Harvard Medical School, Boston, in an interview at the meeting. “There’s been a lot of excitement that maybe we can extend the principles that work in leukemia to other types of diseases.”
The study is still ongoing and accruing patients, but a multicenter trial is also being initiated in myeloma with Bluebird Bio using “a closely related, but slightly different CAR,” Dr. Kochenderfer said.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2015
Key clinical point: The first clinical trial of a CAR targeting BCMA demonstrated strong antimyeloma activity in patients with at least 3 prior lines of myeloma therapy.
Major finding: Myeloma in plasma cells was reduced from 90% to 0% in one patient.
Data source: Phase I study in 12 patients with at least 3 prior lines of multiple myeloma therapy.
Disclosures: Dr. Kochenderfer reported consultancy with Bluebird Bio and off-label use of cyclophosphamide and fludarabine.

Anti-BCL2, CD20 combo safe, effective in untreated CLL
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.

“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
AT ASH 2015
Key clinical point: Patients with CLL and comorbidities were able to tolerate an experimental regimen of obinutuzumab and venetoclax.
Major finding: Two of 12 evaluable patients had evidence of laboratory-defined but not clinical tumor lysis syndrome.
Data source: Safety run-in phase of a randomized phase 3 trial with 13 patients with chronic lymphocytic leukemia and comorbidities.
Disclosures: The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.

Venetoclax produces deep responses in ultra-high-risk CLL

Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
Photo courtesy of ASH
ORLANDO, FL—The pivotal phase 2 study of venetoclax monotherapy in patients with relapsed/refractory 17p-deleted chronic lymphocytic leukemia (CLL) has achieved unprecedented deep responses, according to investigators.
More than 10% of patients had a complete response (CR), complete response with incomplete blood count recovery (CRi), or near partial response (nPR), as confirmed by an independent review committee (IRC).
And more than 20% of responders became negative for minimal residual disease (MRD).
Venetoclax is an orally bioavailable, selective BCL-2 inhibitor that directly induces apoptosis in CLL cells independent of p53.
The US Food and Drug Administration granted venetoclax breakthrough therapy designation for relapsed/refractory CLL earlier this year.
“Patients with a 17p deletion in CLL have very poor prognosis,” said Stephan Stilgenbauer, MD, of University of Ulm in Germany, “and limited treatment options.”
The median progression-free survival (PFS) with frontline chemoimmunotherapy in this population is less than 12 months.
The first-in-human study of venetoclax, which was recently published in NEJM, showed a 79% overall response rate (ORR) in relapsed/refractory CLL patients.
Dr Stilgenbauer presented the pivotal phase 2 results at the 2015 ASH Annual Meeting as LBA-6.
Study overview
The primary objective of the trial was ORR by independent review committee. The secondary endpoints were CR/PR rates, time to first response, duration of response, PFS, overall survival (OS), and safety.
Investigators also included the exploratory endpoint of MRD as determined by flow cytometry with a sensitivity of less than 10-4.
Patients had to have an ECOG score of 2 or less, an absolute neutrophil count of 1000/μL or greater, a platelet count of 40,000/mm3 or higher, and a hemoglobin count of at least 8 g/dL. They also had to have a creatinine clearance of 50 mL/min or more.
“With regard to performance status, blood counts, and creatinine clearance,” Dr Stilgenbauer said, “inclusion criteria were relatively liberal, allowing patients with comorbidity on the trial.”
Patients were excluded if they had prior allogeneic stem cell transplantation, Richter’s transformation, uncontrolled autoimmune cytopenia, other malignancy, or major organ dysfunction.
Trial design
Patients received an oral dose of venetoclax once daily continuously until disease progression or discontinuation for another reason.
Because tumor lysis syndrome (TLS) was a concern, investigators devised a step-wise weekly ramp-up with risk-based prophylaxis to mitigate TLS.
Patients started on a dose of 20 mg on day 1. If they did not experience any electrolyte abnormalities, they received a 50 mg daily dose for the rest of the first week, escalating to 100 mg, 200 mg, and to the target dose of 400 mg daily on subsequent weeks. Patients continued on 400 mg daily for the remainder of the study.
The investigators assessed response using iwCLL 2008 criteria with monthly physical exams and blood counts, CT scans to confirm clinical response at week 36, and a bone marrow biopsy to confirm CR.
Patient population and disposition
Investigators enrolled 107 patients with a median age of 67 (range, 37–85). Seventy (65%) were male.
Patients had a median of 2 prior therapies (range, 1–10): 54 (50%) had prior bendamustine and 38 (70%) were refractory to it; 78 (73%) had prior fludarabine and 34 (44%) were refractory to it; and 90 (84%) had a prior CD20 monoclonal antibody.
About half (52%) were ECOG grade 1, 53% had 1 or more nodes 5 cm or larger, and 51% had absolute lymphocyte (ALC) levels 25 x 109/L or higher.
Eighty-two percent of patients were in the medium and high TLS risk categories, slightly less than half were Rai stage III or IV, and 81% were IGHV unmutated.
As of the data lock on April 30, 2015, patients remained a median of 12.1 months on study (range, 0.03–21.5). Seventy are still active on venetoclax, and 37 discontinued the treatment.
Eleven patients discontinued due to Richter’s transformation, 11 due to CLL progression, and 9 due to adverse events. Three patients proceeded to stem cell transplant, 2 withdrew consent, and 1 was noncompliant.
Eighteen patients died, 14 due to disease progression.
Response
Eighty-five patients responded, for an ORR of 79.4% by IRC. Eight patients (7.5%) achieved a CR or CRi, 3 (2.8%) had an nPR, and 74 (69.2%) had a PR. Twenty-two patients (20.6%) had no response.
Twenty-five of 48 patients had no evidence of CLL in their bone marrow by immunohistochemistry, and 18 of 45 patients assessed were MRD-negative in the peripheral blood.
Reduction in lymphocytosis “was quite a universal phenomenon across this trial,” Dr Stilgenbauer said. Only 4 patients of 87 with baseline lymphocytosis failed to reduce their lymphocyte count to below 4 x 109/L, the usual threshold for a CR. And the median time to normalization was 22 days (range, 2–122).
Eighty-nine of 96 patients had 50% or more reduction in their nodal size in a median of 2.7 months (range, 0.7–8.4).
The median time to first response was 0.8 months (range, 0.1–8.1), and the median time to CR/CRi was 8.2 months (range, 3.0–16.3).
“And this number still appears to evolve over the duration of the trial,” Dr Stilgenbauer said.
The median duration of response has not yet been reached. But investigators estimated that of the 85 responders, 84.7% would maintain their response at 12 months, 100% of patients in the CR/CRi and nPR groups would maintain their response, and 94.4% of patients who were MRD-negative would maintain their response.
The median PFS and OS have not been reached. The PFS estimate for 12 months was 72.0%, and the OS estimate was 86.7%.
Adverse events
Treatment-emergent adverse events of any grade occurred in 96% of patients. The most frequent were neutropenia (43%), diarrhea (29%), nausea (29%), anemia (27%), fatigue (22%), pyrexia (20%), thrombocytopenia (19%), hyperphosphatemia (16%), vomiting (15%), and upper respiratory tract infection (15%).
The most frequent grade 3/4 adverse events were neutropenia (40%), anemia (18%), and thrombocytopenia (15%).
Dr Stilgenbauer pointed out that 22.4% of patients had neutropenia at baseline. Neutropenia was managed with dose interruption or reduction, G-CSF, and/or antibiotics.
Infections occurred in 72% of patients, with 20% of patients experiencing grade 3 or higher.
“The types of infections were the usual expected ones,” Dr Stilgenbauer said.
Laboratory TLS occurred in 5 patients exclusively during the ramp-up period. Two required a dose interruption of 1 day each. There were no clinical TLS events.
Serious adverse events occurred in 55% of patients, the most common being pyrexia (7%), autoimmune hemolytic anemia (7%), pneumonia (6%), and febrile neutropenia (5%).
The investigators concluded that venetoclax offers a favorable risk-benefit profile. The risk of TLS can be effectively mitigated with no clinical TLS, and the incidence of neutropenia and infection are similar to frontline chemoimmunotherapy.
“Venetoclax may provide an attractive treatment option for 17p-deleted CLL as monotherapy or as a component of novel combination strategies,” Dr Stilgenbauer said.
AbbVie and Genentech, collaborators in the development of venetoclax, provided financial support for the study design, study conduct, analysis, data interpretation, writing, and review. ![]()
CAR T cells persist for 3 years in young ALL patients
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.
Photo courtesy of Penn Medicine
ORLANDO, FL—CTL019, a CD19 chimeric antigen receptor (CAR) T-cell therapy, can persist for 3 years or longer in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL), according to the latest results of a pilot study.
This suggests CTL019 can offer long-term disease control without subsequent therapy, such as stem cell transplant, said study author Stephan Grupp, MD, PhD, of the University of Pennsylvania in Philadelphia.
Dr Grupp presented these results at the 2015 ASH Annual Meeting (abstract 681*).
Previously, Dr Grupp and his colleagues reported that CTL019 led to durable antitumor activity, including sustained complete responses (CRs) in adults and children with ALL (ASH 2012, NEJM 2013, ASH 2013, NEJM 2014, ASH 2014).
At this year’s ASH meeting, he reported on outcomes and longer follow-up of the first 59 patients with relapsed/refractory ALL treated in a pilot trial. The patients had a median age of 11 years.
The median follow-up was 12 months. Fifty-five patients (93%) achieved a CR at 1 month. Six patients went on to receive a transplant, and 1 patient went on to receive donor lymphocyte infusion.
The relapse-free survival was 76% at 6 months and 55% at 12 months. Overall survival was 79% at 12 months.
“There were no relapses past 1 year,” Dr Grupp said, noting that 18 patients beyond 1 year are in remission, and 13 patients have not received further therapy.
“We are able to get patients into remission,” he said, adding that response is similar at high and low disease burden.
“We see massive proliferation of CAR T cells. Prolonged CTL019 persistence is detected by flow cytometry. About 70% of patients maintain CAR T cells.”
Resistance was seen in 7% of patients due to failure of T cells to proliferate. One-third of recurrences occurred in those with CD19-positive relapse and two-thirds with CD19-negative relapse.
Dr Grupp noted that CTL019 has an impact on central nervous system (CNS) disease.
“We are able to control CNS disease,” he said. “We found no CNS relapses in patients who were treated, and 98% of them still have CTL019 in the cerebral spinal fluid.”
B-cell aplasia persists beyond 3.5 years in all responding patients. This is managed by immunoglobulin replacement.
“Patients in remission and with B-cell aplasia at 1 year are still alive at 2 to 3 years,” Dr Grupp said. “We do not know how long this will last and if B cells will recover.”
Severe cytokine release syndrome (CRS), observed in 88% of patients, is the principal toxicity. This is controlled with anti-IL6 therapy. Patients who are less likely to have CRS are those with a lower disease burden.
“IL-6 is a major player but does not predict CRS; it only correlates strongly with it,” Dr Grupp said.
Some toxicity is also associated with macrophage activation syndrome and neurotoxicity.
CTL019 was invented at The University of Pennsylvania but has been licensed to Novartis. Most of the researchers involved in this study reported research funding and/or consultancy payments from Novartis, and 1 researcher is employed by the company. ![]()
*Data in the abstract differ from the presentation.