User login
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
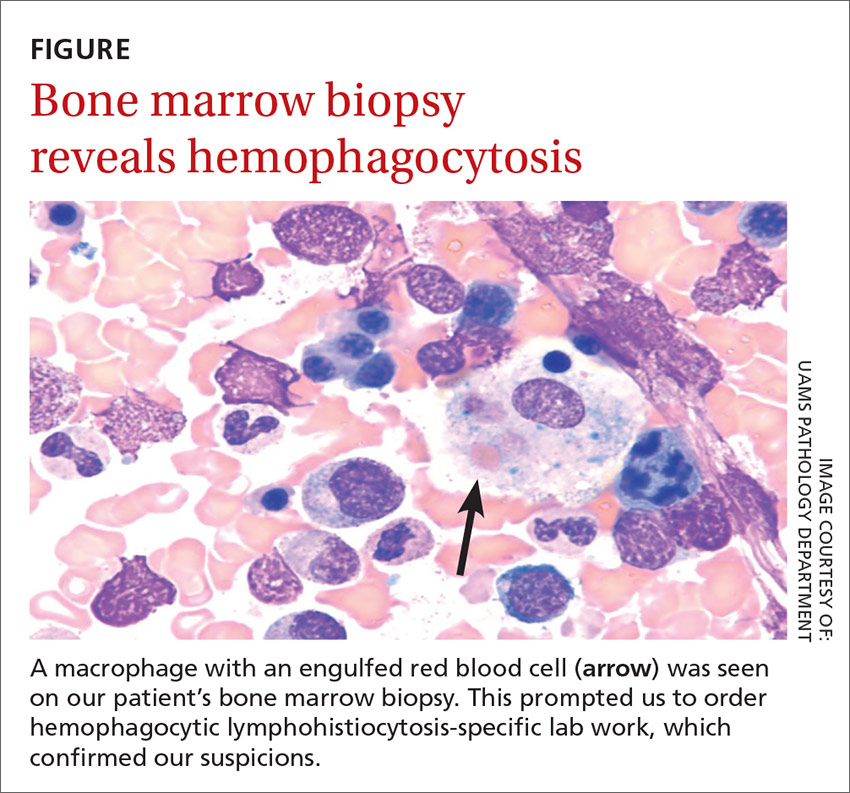
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
THE CASE
A 47-year-old African American woman was admitted to the hospital with pulmonary edema revealed on a computed tomography (CT) scan. She had a history of systemic lupus erythematosus (SLE), hypertension, and end-stage renal disease (ESRD). The patient had been hospitalized one month earlier for lupus nephritis with a hypertensive emergency that led to a seizure. During this earlier hospitalization, she was given a diagnosis of posterior reversible encephalopathy syndrome.
Two weeks into her more recent hospitalization, the patient developed a fever that was accompanied by cough and fatigue. By the third week, there was no identified cause of the fever, and the patient met the criteria for fever of unknown origin (FUO).
Her medications included cyclophosphamide, prednisone, nebivolol, clonidine, phenytoin, and epoetin alfa. The patient was also receiving dialysis every other day. Chest x-ray findings suggested pneumonia, and the patient was treated with vancomycin and piperacillin/tazobactam. However, her fever persisted after completing the antibiotics. Central line sepsis was high in the differential, as the patient was on dialysis, but blood and catheter tip cultures were negative. Chest and abdominal CT scans showed no new disease process. Urine and sputum cultures were collected and were negative for infection. Drug-induced fever was then suspected, but was ruled out when the fever persisted after the removal of potential offending agents (phenytoin, nebivolol, and cyclophosphamide).
THE DIAGNOSIS
We then followed the American Academy of Family Physicians’ diagnostic protocol for FUO.1
Initial labs included a complete blood count (CBC), 2 blood cultures, a urine culture, erythrocyte sedimentation rate (ESR), a purified protein derivative skin test, chest and abdominal CT scans, and double-stranded DNA (dsDNA) levels (since this patient had known SLE). The patient’s hemoglobin level and mean corpuscular volume were consistent with normocytic anemia, which was attributed to the ESRD. The ESR was mildly elevated at 46 mm/hr, but dsDNA was not, ruling out a lupus flare. Thrombocytopenia (platelet count, 82 K/mcL) and lymphocytopenia (absolute lymphocyte count, 0.2 K/mcL) were assumed to be secondary to cyclophosphamide use.
Because the initial labs were non-diagnostic, we proceeded with a sputum stain and culture, human immunodeficiency virus testing, a hepatitis panel, and a peripheral blood smear.1 All were negative except for the peripheral blood smear, which showed hemophagocytic cells. This was the first finding that brought hemophagocytic lymphohistiocytosis (HLH) into the differential.
We then performed a bone marrow biopsy (FIGURE), which also revealed hemophagocytic cells, so we ordered HLH-specific labs (more on those in a bit). Liver enzymes were elevated to 3 times their normal value. Triglycerides (414 mg/dL), ferritin (>15,000 ng/mL), and interleukin-2 (IL-2) receptor levels (>20,000 pg/m) were also elevated.
The patient was tested for herpes simplex virus, Epstein-Barr virus (EBV), and cytomegalovirus (CMV), since these viruses are associated with HLH. She had 3.1 million copies/mL of CMV, leading to the diagnosis of secondary HLH. This diagnosis might not have been made if not for a persistent fever investigation.
DISCUSSION
HLH is a life-threatening syndrome of excessive immune activation that results in tissue damage.2 There are primary and secondary forms, but they share the same mechanism of impaired regulation of cytotoxic granules and cytokines. Primary HLH results from a congenital gene mutation,3 while secondary HLH is triggered by an autoimmune or inflammatory disease or an infection.4 EBV is the most common viral etiology, followed closely by CMV.5
The diagnosis may be established genetically (based on mutations of the genes loci PRF1, UNC13D, or STX11) or by fulfillment of 5 out of 8 criteria: fever; splenomegaly; cytopenia; hypertriglyceridemia; hypofibrinogenemia; hemophagocytosis in the bone marrow, spleen, or lymph nodes; low or absent natural killer cell activity; and an elevated ferritin level (>500 ng/mL). Elevated soluble CD25 and IL-2 receptor markers are HLH-specific markers.3 This patient had fever, cytopenia, hypertriglyceridemia, hemophagocytosis, and elevated ferritin with elevated IL-2, meeting the criteria for secondary HLH.
First treat the underlying condition, then the HLH
Treatment for HLH includes treating the underlying condition (such as EBV or CMV) with antiretroviral medications, and using immunosuppressive agents such as chemotherapy drugs and steroids for the HLH.
Our patient was treated with valganciclovir 900 mg/d for 2 weeks for the CMV and an etoposide/prednisone taper for 3 months for HLH chemotherapy and suppression. Within one month, her CMV viral load decreased to <300 copies/mL and her fever resolved. Ferritin, triglycerides, and liver enzyme levels returned to normal within 3 months.
THE TAKEAWAY
FUO can be frustrating for both the physician and the patient. Not only is the differential large, but testing is extensive. It is important to get a thorough history and to consider medications as the cause. Testing should be patient-specific and systematic. Persistent investigation is critical to saving the patient’s life.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.
1. Roth AR, Basello GM. Approach to the adult patient with fever of unknown origin. Am Fam Physician. 2003;68:2223-2228.
2. Filipovich A, McClain K, Grom A. Histiocytic disorders: recentinsights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82-S89.
3. Larroche C. Hemophagocytic lymphohistiocytosis in adults: diagnosis and treatment. Joint Bone Spine. 2012;79:356-361.
4. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-822.
5. Janka GE, Lehmberg K. Hemophagocytic syndromes—an update. Blood Rev. 2014;28:135-142.
