User login
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.

There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
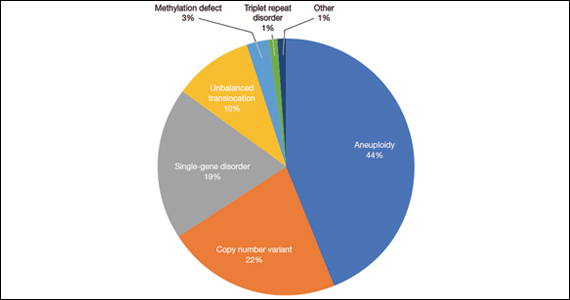
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
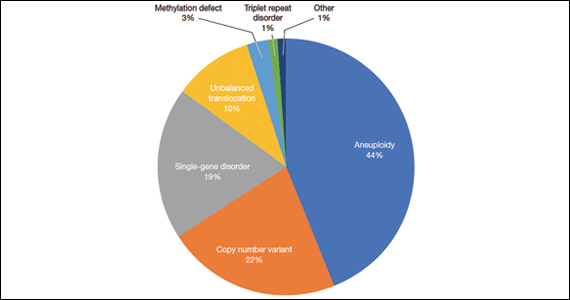
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.

Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD

Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD
Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Rebecca Reimers, MD
Dr. Reimers is a Clinical Fellow, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital and Boston Children’s Hospital, Massachusetts.
Stephanie Guseh, MD
Dr. Guseh is a Clinical Instructor, Maternal-Fetal Medicine and Clinical Genetics, Division of Maternal-Fetal Medicine, Brigham and Women’s Hospital.
The authors report no financial relationships relevant to this article.
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.
There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.
Last year, our Update focused on the expansion of sequencing in prenatal diagnosis. This year, we are taking a step sideways to remember the many diagnoses we may miss if we rely on exome sequencing alone. A recent case report in Prenatal Diagnosis describes a pregnancy affected by fetal akinesia sequence and polyhydramnios in which sequencing did not reveal a diagnosis. Expansion of the differential to include congenital myotonic dystrophy and subsequent triplet repeat testing led the clinicians to the diagnosis and identification of a triplet repeat expansion in the DMPK gene. This case serves as our first example of how complementary testing and technologies should continue to help us make critical diagnoses.
What is the yield of exome sequencing vs panels in nonimmune hydrops?
Rogers R, Moyer K, Moise KJ Jr. Congenital myotonic dystrophy: an overlooked diagnosis not amenable to detection by sequencing. Prenat Diagn. 2022;42:233-235. doi:10.1002/pd.6105.
Norton ME, Ziffle JV, Lianoglou BR, et al. Exome sequencing vs targeted gene panels for the evaluation of nonimmune hydrops fetalis. Am J Obstet Gynecol. 2021;28:S0002-9378(21)00828-0. doi:10.1016/j.ajog.2021.07.014.
We have had several illuminating discussions with our colleagues about the merits of exome sequencing (ES) versus panels and other modalities for fetal diagnosis. Many obstetricians practicing at the leading edge may feel like ES should be utilized uniformly for fetal anomalies with nondiagnostic karyotype or microarray. However, for well-defined phenotypes with clear and narrow lists of implicated genes (eg, skeletal dysplasias) or patients without insurance coverage, panel sequencing still has utility in prenatal diagnosis. The question of which phenotypes most benefit from ES versus panel sequencing is an area of interesting, ongoing research for several investigators.
Secondary analysis of nonimmune hydrops cohort
Norton and colleagues tackled one such cohort in a study presented in the American Journal of Obstetrics and Gynecology. They compared the proportion of diagnoses that would have been identified in commercial lab panels with their research of phenotype-driven ES in a cohort of 127 fetuses with features of nonimmune hydrops fetalis (NIHF). NIHF can be caused by a variety of single-gene disorders in addition to chromosomal disorders and copy number variants on chromosomal microarray. Patients were eligible for inclusion in the cohort if they had a nondiagnostic karyotype or microarray and any of the following features: nuchal translucency of 3.5 mm or greater, cystic hygroma, pleural effusion, pericardial effusion, ascites, or skin edema. Standard sequencing methods and variant analysis were performed. They assumed 100% analytical sensitivity and specificity of the panels for variant detection and collected cost information on the targeted gene panels.
Study outcomes
In the ES analysis of cases, 37 of 127 cases (29%) had a pathogenic or likely pathogenic variant in 1 of 29 genes, and another 12 of 127 cases (9%) had variants of uncertain significance that were strongly suspected to be the etiology during clinical analysis. The types of disorders that were identified are listed in the TABLE. In addition to a feature of NIHF, 50% of the cases had a structural anomaly.
There were 10 identified clinical panels from 7 clinical laboratories. These panels ranged in size from 11 to 128 genes. The highest simulated yield of any commercial panel was only 62% of the pathogenic variants identified by ES. The other commercial laboratory panels detection yield ranged from 11% to 62% of pathogenic variants detected by ES. For overall yield, the largest panel would have a diagnostic yield of 18% of diagnoses relative to the 29% diagnostic yield from ES.
The largest panel included 128 genes prior to the publication of the original cohort and was updated after publication to include 148 genes. The larger updated panel would have identified all of the patients in the ES cohort. However, many of the other panels listed would have identified a smaller fraction of the variants identified by ES (range, 11%-62%). At the time of publication, the cost of the panels ranged from $640 to $3,500, and the cost of prenatal ES ranged from $2,458 to $7,500.
Continue to: Strengths and limitations...
Strengths and limitations
Twenty-three percent of the patients who were sequenced had an increased fetal nuchal translucency or cystic hygroma, and another 17% had a single fetal effusion. This inclusivity makes this study more applicable to broader fetal anomaly populations. However, it is worth noting that only 61% of patients had NIHF by the definition of 2 or more fluid collections or skin thickening.
The authors assumed 100% sensitivity and specificity for the panel tests relative to diagnostic ES results, but this may not reflect real-life analysis. There is inherent subjectivity and subsequent differences in variant calling (deciding which genetic changes are pathogenic) between institutions and companies despite efforts to standardize this process. Due to the simulated nature of this study, these differences are not captured. Additionally, although the authors note that the research ES had at least 30 times the coverage (an adequate number of sequence reads for accurate testing) than did the commercial lab panels, some gene panels have additional sequencing of intronic regions, copy number analysis, and up to 10 times more coverage than ES, which could lead to more diagnoses.
This study illustrates that there is nuance involved in selecting which type of gene sequencing and which clinical laboratory to use for prenatal diagnosis. Labs with more updated literature searches and more inclusive gene panels may be excellent options for patients in whom ES is not covered by insurance or with phenotypes with a narrow range of suspected causative genes. However, there is a lag time in updating the genes offered on each panel, and new genedisease associations will not be captured by existing panels.
From a cost, speed-of-analysis, and depth-of-sequencing perspective, panel sequencing can have advantages that should be considered in some patients, particularly if the panels are large and regularly updated. However, the authors summarize our sentiments and their findings with the following:
“For disorders, such as NIHF with marked genetic heterogeneity and less clear in utero phenotypes of underlying genetic diseases, the broader coverage of exome sequencing makes it a superior option to targeted panel testing.”
We look forward to the publication of further anomaly-specific cohorts and secondary analyses of the utility of current panels and ES that may follow.
Frequency of Beckwith-Widemann syndrome in prenatally diagnosed omphaloceles
Abbasi N, Moore A, Chiu P, et al. Prenatally diagnosed omphaloceles: report of 92 cases and association with Beckwith-Wiedemann syndrome. Prenat Diagn. 2021;41:798-816. doi:10.1002/pd.5930.
An omphalocele is diagnosed prenatally on ultrasound when an anterior midline mass, often containing abdominal contents, is seen herniating into the base of the umbilical cord. Omphaloceles are often associated with additional structural abnormalities and underlying genetic syndromes, thus a thorough fetal assessment is required for accurate prenatal counseling and neonatal care.
Identification of Beckwith-Widemann syndrome (BWS) in the setting of a prenatally diagnosed omphalocele is difficult because of its wide range of clinical features and its unique genetic basis. Unlike many genetic disorders that are caused by specific genetic variants, or spelling changes in the genes, BWS results from a change in the expression of one or more of the genes in a specific region of chromosome 11. A high index of clinical suspicion as well as an understanding of the various genetic and epigenetics alterations that cause BWS is required for prenatal diagnosis.
Retrospective cohort at a single center
The authors in this study reviewed all pregnancies in which an omphalocele was diagnosed prenatally at a single center between 2010 and 2015. They describe a standard prenatal evaluation following identification of an omphalocele including echocardiogram, detailed anatomic survey, and availability of an amniocentesis to facilitate aneuploidy screening and testing for BWS. This review also includes an overview of perinatal and long-term outcomes for cases of BWS diagnosed at their center between 2000 and 2015.
Study outcomes
Results of prenatal genetic testing in this cohort were divided between cases of an isolated omphalocele (without other structural changes) and cases of nonisolated omphaloceles. In the group of pregnancies with an isolated omphalocele, 2 of 27 pregnancies (7.4%) were found to have an abnormal karyotype, and 6 of 16 of the remaining pregnancies (37.5%) were diagnosed with BWS. Among the group of pregnancies with a nonisolated omphalocele, 23 of 59 pregnancies (39%) were found to have an abnormal karyotype, and 1 of 20 pregnancies (5%) were diagnosed with BWS.
Prenatal sonographic features associated with cases of BWS included polyhydramnios in 12 of 19 cases (63%) and macrosomia in 8 of 19 cases (42%). Macroglossia is another characteristic feature of the disorder, which was identified in 4 of 19 cases (21%) prenatally and in an additional 10 of 19 cases (52.6%) postnatally. Interestingly, only 1 of the cases of BWS was caused by a microdeletion at 11p15.4—a change that was identified on microarray. The additional 6 cases of BWS were caused by imprinting changes in the region, which are only detectable with a specific methylation-analysis technique.
Among the 19 cases of BWS identified over a 15-year period, there was 1 intrauterine demise. Preterm birth occurred in 10 of 19 cases (52.6%), including 8 of 19 cases (42.1%) of spontaneous preterm labor. Respiratory distress (27.8%), hypoglycemia (61%), and gastrointestinal reflux (59%) were common neonatal complications. Embryonal tumors were diagnosed in 2 of 16 patients (12.5%). Although neurodevelopmental outcomes were incomplete, their data suggested normal development in 75% of children. There were 2 neonatal deaths in this cohort and 1 childhood death at age 2 years.
Study strengths and limitations
As with many studies investigating a rare disorder, this study is limited by its retrospective nature and small sample size. Nevertheless, it adds an important cohort of patients with a prenatally diagnosed omphalocele to the literature and illuminates the utility of a standardized approach to testing for BWS in this population.
In this cohort with prenatally diagnosed omphaloceles with standardized testing for BWS, the prevalence of the disorder was approximately 8% and more common in cases of an isolated omphalocele. The most common supporting sonographic features of BWS may not be detected until later in gestation, including polyhydramnios and macrosomia. This demonstrates the importance of both sonographic follow-up as well as universal testing for BWS in euploid cases of a prenatally diagnosed omphalocele. Almost all cases of BWS in this cohort required specialized molecular techniques for diagnosis, and the diagnosis would have been missed on karyotype, microarray, and ES.
Continue to: Genetic diagnoses that could have been identified by expanded carrier screening...
Genetic diagnoses that could have been identified by expanded carrier screening
Stevens BK, Nunley PB, Wagner C, et al. Utility of expanded carrier screening in pregnancies with ultrasound abnormalities. Prenat Diagn. 2022;42:60-78. doi:10.1002/pd.6069.
This series is a thorough retrospective review of patients evaluated in a pediatric genetics clinic from 2014 through 2017. Patients were included if they were evaluated in the first 6 months of life and had a structural abnormality that might be detected on prenatal ultrasonography. The genetic testing results were analyzed and categorized according to types of genetic disorders, with the goal of identifying how many patients might have been identified by expanded carrier screening (ECS) panels.
Study outcomes
A total of 931 charts were reviewed, and 85% (791 of 931) of patients evaluated in the first 6 months of life were determined to have a structural anomaly that might be detected on prenatal ultrasonography. Of those patients, 691 went on to have genetic testing and 32.1% (222 of 691) of them had a diagnostic (pathogenic) genetic testing result related to the phenotype. The types of diagnostic testing results are shown in the FIGURE. Notably, 42 single-gene disorders were detected.
FIGURE Diagnostic test result in pediatric patients evaluated under age 6 months
Of those 222 patients with diagnostic results, there were 8 patients with autosomal recessive and X-linked conditions that could be detected using a 500-gene ECS panel. Five patients could be detected with a 271-gene panel. After nondiagnostic microarray, 11.3% of patients had a condition that could be detected by using a 500-gene ECS panel. The identified conditions included cystic fibrosis, CYP21‐related congenital adrenal hyperplasia, autosomal recessive polycystic kidney disease, Antley‐Bixler syndrome, and Morquio syndrome type A.
Furthermore, the authors conducted a literature review of 271 conditions and found that 32% (88 of 271) of conditions may be associated with ultrasound findings.
Study strengths and limitations
When applying these data to prenatal populations, the authors acknowledge several notable limitations. There is a selection bias toward less-severe phenotypes for many patients choosing to continue rather than to interrupt a pregnancy. Additionally, only 23% of the patients in the study had a microarray and ES, which may lead to an underrepresentation of single-gene disorders and an underestimation of the utility of ECS. Finally, a retrospective classification of structural abnormalities that may be detectable by ultrasonography may not always reflect what is actually reported in prenatal imaging.
However, the work that the authors put forth to evaluate and categorize 931 participants by the results of genetic testing and structural anomalies is appreciated, and the level of detail is impressive for this retrospective chart review. Additionally, the tables itemizing the authors’ review of 271 ECS disorders that may have ultrasonography findings categorized by disorder and system are helpful and quick diagnostic references for clinicians providing prenatal care. ●
This study of potentially detectable prenatal findings from the lens of a pediatric genetics clinic lends an interesting perspective: Exome sequencing is not the primary route to establish a diagnosis; karyotype, microarray, methylation disorders, and triplet repeat disorders all have an established role in the diagnostic toolkit. Keeping in mind the contribution of these modalities to pediatric testing may shorten the diagnostic odyssey to continue pregnancies or help to fully counsel patients on expectations and decision-making after birth.
Carrier screening is not a substitute for diagnostic testing in pregnancy. However, in appropriately selected patients, a broad carrier screening panel may have added utility. ECS can be conducted while awaiting microarray results to help target testing and may be particularly useful for patients who decline diagnostic testing until the postnatal period. It is important to counsel patients that carrier screening is not a diagnostic test, and results will only report likely pathogenic or pathogenic variants, not variants of uncertain significance that may be of clinical relevance. However, our practice has had several insightful diagnoses reached through ECS, in conjunction with microarray testing that allowed for faster and more targeted sequencing and precise fetal diagnosis. This readily available molecular tool (often covered by insurance) deserves a spot in your fetal diagnosis tool belt based on available evidence.