User login
The link between suicide and sleep
According to the Centers for Disease Control and Prevention, suicide is the 10th leading cause of mortality in the United States, with rates of suicide rising over the past 2 decades. In 2016, completed suicides accounted for approximately 45,000 deaths in the United States (Ivey-Stephenson AZ, et al. MMWR Surveill Summ. 2017;66[18]:1). While progress has been made to lower mortality rates of other leading causes of death, very little progress has been made on reducing the rates of suicide. The term “suicide,” as referred to in this article, encompasses suicidal ideation, suicidal behavior, and suicide death.

Researchers have been investigating potential risk factors and prevention strategies for suicide. The relationship between suicide and sleep disturbances, specifically insomnia and nightmares, has been well documented in the literature. Given that insomnia and nightmares are potentially modifiable risk factors, it continues to be an area of active exploration for suicide rate reduction. While there are many different types of sleep disorders, including excessive daytime sleepiness, parasomnias, obstructive sleep apnea, and restless legs syndrome, this article will focus on the relationship between insomnia and nightmares with suicide.
Insomnia
Insomnia disorder, according to the American Psychiatric Association’s DSM-5, is a dissatisfaction of sleep quantity or quality that occurs at least three nights per week for a minimum of 3 months despite adequate opportunity for sleep. This may present as difficulty with falling asleep, staying asleep, or early morning awakenings. The sleep disturbance results in functional impairment or significant distress in at least one area of life (American Psychiatric Association. Arlington, Virginia: APA; 2013). While insomnia is often a symptom of many psychiatric disorders, research has shown that insomnia is an independent risk factor for suicide, even when controlling for mental illness. Studies have shown that there is up to a 2.4 relative risk of suicide death with insomnia after adjusting for depression severity (McCall W, et al. J Clin Sleep Med. 2013;32[9]:135).
Nightmares
Nightmares, as defined by the American Psychiatric Association’s DSM-5, are “typically lengthy, elaborate, story-like sequences of dream imagery that seem real and incite anxiety, fear, or other dysphoric emotions” (American Psychiatric Association. Arlington, Virginia: APA; 2013). They are common symptoms in posttraumatic stress disorder (PTSD), with up to 90% of individuals with PTSD experiencing nightmares following a traumatic event (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393). Nightmares have also been shown to be an independent risk factor for suicide when controlling for mental illness. Studies have shown that nightmares are associated with an elevated risk factor of 1.5 to 3 times for suicidal ideation and 3 to 4 times for suicide attempts. The data suggest that nightmares may be a stronger risk factor for suicide than insomnia (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Proposed Mechanism
The mechanism linking insomnia and nightmares with suicide has been theorized and studied by researchers. A couple of the most noteworthy proposed psychological mechanisms involve dysfunctional beliefs and attitudes about sleep, as well as deficits in problem solving capability. Dysfunctional beliefs and attitudes about sleep (DBAS) are negative cognitions pertaining to sleep, and they have been shown to be related to the intensity of suicidal ideations. Many of the DBAS are pessimistic thoughts that contain a “hopelessness flavor” to them, which lead to the perpetuation of insomnia. Hopelessness has been found to be a strong risk factor for suicide. In addition to DBAS, insomnia has also shown to lead to impairments in complex problem solving. The lack of problem solving skills in these patients may lead to fewer quantity and quality of solutions during stressful situations and leave suicide as the perceived best or only option.
The biological theories focus on serotonin and hyperarousal mediated by the hypothalamic-pituitary-adrenal (HPA) axis. Serotonin is a neurotransmitter that is involved in the induction and maintenance of sleep. Of interesting note, low levels of serotonin’s main metabolite, 5-hydroxyindoleacetic acid (5-HIAA) have been found in the cerebrospinal fluid of suicide victims. Evidence has also shown that sleep and the HPA axis are closely related. The HPA axis is activated by stress leading to a cascade of hormones that can cause susceptibility of hyperarousal, REM alterations, and suicide. Hyperarousal, shared in context with PTSD and insomnia, can lead to hyperactivation of the noradrenergic systems in the medial prefrontal cortex, which can lead to decrease in executive decision making (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Treatment Strategies
The benefit of treating insomnia and nightmares, in regards to reducing suicidality, continues to be an area of active research. Many of the previous studies have theorized that treating symptoms of insomnia and nightmares may indirectly reduce suicide. Pharmaceutical and nonpharmaceutical treatments are currently being used to help treat patients with insomnia and nightmares, but the benefit for reducing suicidality is still unknown.

One of the main treatment modalities for insomnia is hypnotic medication; however, these medications carry their own potential risk for suicide. Reports of suicide death in conjunction with hypnotic medication has led the FDA to add warnings about the increased risk of suicide with these medications. Some of these medications include zolpidem, zaleplon, eszopiclone, doxepin, ramelteon, and suvorexant. A review of research studies and case reports was completed in 2017 and showed that there was an odds ratio of 2 to 3 for hypnotic use in suicide deaths. However, most of the studies that were reviewed reported a potential confounding bias of the individual’s current mental health state. Furthermore, many of the suicide case reports that involved hypnotics also had additional substances detected, such as alcohol. Hypnotic medication has been shown to be an effective treatment for insomnia, but caution needs to be used when prescribing these medications. Strategies that may be beneficial when using hypnotic medication to reduce the risk of an adverse outcome include using the lowest effective dose and educating the patient of not combining the medication with alcohol or other sedative/hypnotics (McCall W, et al. Am J Psychiatry. 2017;174[1]:18).
For patients who have recurrent nightmares in the context of PTSD, the alpha-1 adrenergic receptor antagonist, prazosin, may provide some benefit; however, the literature is divided. There have been several randomized, placebo-controlled clinical trials with prazosin, which has shown a moderate to large effect for alleviating trauma-related nightmares and improving sleep quality. Some of the limitations of these studies were that the trials were small to moderate in size, and the length of the trials was 15 weeks or less. In 2018, Raskin and colleagues completed a follow-up randomized, placebo-controlled study for 26 weeks with 304 participants and did not find a significant difference between prazosin and placebo in regards to nightmares and sleep quality (Raskind MA, et al. N Engl J Med. 2018;378[6]:507).
Cognitive behavioral therapy for insomnia (CBT-I) and image rehearsal therapy (IRT) are two sleep-targeted therapy modalities that are evidence based. CBT-I targets dysfunctional beliefs and attitudes regarding sleep (McCall W, et al. J Clin Sleep Med. 2013;9[2]:135). IRT, on the other hand, specifically targets nightmares by having the patient write out a narrative of the nightmare, followed by re-scripting an alternative ending to something that is less distressing. The patient will rehearse the new dream narrative before going to sleep. There is still insufficient evidence to determine if these therapies have benefit in reducing suicide (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393).
While the jury is still out on how best to target and treat the risk factors of insomnia and nightmares in regards to suicide, there are still steps that health-care providers can take to help keep their patients safe. During the patient interview, new or worsening insomnia and nightmares should prompt further investigation of suicidal thoughts and behaviors. After a thorough interview, treatment options, with a discussion of risks and benefits, can be tailored to the individual’s needs. Managing insomnia and nightmares may be one avenue of suicide prevention.
Drs. Locrotondo and McCall are with the Department of Psychiatry and Health Behavior at the Medical College of Georgia, Augusta University, Augusta, Georgia.
According to the Centers for Disease Control and Prevention, suicide is the 10th leading cause of mortality in the United States, with rates of suicide rising over the past 2 decades. In 2016, completed suicides accounted for approximately 45,000 deaths in the United States (Ivey-Stephenson AZ, et al. MMWR Surveill Summ. 2017;66[18]:1). While progress has been made to lower mortality rates of other leading causes of death, very little progress has been made on reducing the rates of suicide. The term “suicide,” as referred to in this article, encompasses suicidal ideation, suicidal behavior, and suicide death.
Researchers have been investigating potential risk factors and prevention strategies for suicide. The relationship between suicide and sleep disturbances, specifically insomnia and nightmares, has been well documented in the literature. Given that insomnia and nightmares are potentially modifiable risk factors, it continues to be an area of active exploration for suicide rate reduction. While there are many different types of sleep disorders, including excessive daytime sleepiness, parasomnias, obstructive sleep apnea, and restless legs syndrome, this article will focus on the relationship between insomnia and nightmares with suicide.
Insomnia
Insomnia disorder, according to the American Psychiatric Association’s DSM-5, is a dissatisfaction of sleep quantity or quality that occurs at least three nights per week for a minimum of 3 months despite adequate opportunity for sleep. This may present as difficulty with falling asleep, staying asleep, or early morning awakenings. The sleep disturbance results in functional impairment or significant distress in at least one area of life (American Psychiatric Association. Arlington, Virginia: APA; 2013). While insomnia is often a symptom of many psychiatric disorders, research has shown that insomnia is an independent risk factor for suicide, even when controlling for mental illness. Studies have shown that there is up to a 2.4 relative risk of suicide death with insomnia after adjusting for depression severity (McCall W, et al. J Clin Sleep Med. 2013;32[9]:135).
Nightmares
Nightmares, as defined by the American Psychiatric Association’s DSM-5, are “typically lengthy, elaborate, story-like sequences of dream imagery that seem real and incite anxiety, fear, or other dysphoric emotions” (American Psychiatric Association. Arlington, Virginia: APA; 2013). They are common symptoms in posttraumatic stress disorder (PTSD), with up to 90% of individuals with PTSD experiencing nightmares following a traumatic event (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393). Nightmares have also been shown to be an independent risk factor for suicide when controlling for mental illness. Studies have shown that nightmares are associated with an elevated risk factor of 1.5 to 3 times for suicidal ideation and 3 to 4 times for suicide attempts. The data suggest that nightmares may be a stronger risk factor for suicide than insomnia (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Proposed Mechanism
The mechanism linking insomnia and nightmares with suicide has been theorized and studied by researchers. A couple of the most noteworthy proposed psychological mechanisms involve dysfunctional beliefs and attitudes about sleep, as well as deficits in problem solving capability. Dysfunctional beliefs and attitudes about sleep (DBAS) are negative cognitions pertaining to sleep, and they have been shown to be related to the intensity of suicidal ideations. Many of the DBAS are pessimistic thoughts that contain a “hopelessness flavor” to them, which lead to the perpetuation of insomnia. Hopelessness has been found to be a strong risk factor for suicide. In addition to DBAS, insomnia has also shown to lead to impairments in complex problem solving. The lack of problem solving skills in these patients may lead to fewer quantity and quality of solutions during stressful situations and leave suicide as the perceived best or only option.
The biological theories focus on serotonin and hyperarousal mediated by the hypothalamic-pituitary-adrenal (HPA) axis. Serotonin is a neurotransmitter that is involved in the induction and maintenance of sleep. Of interesting note, low levels of serotonin’s main metabolite, 5-hydroxyindoleacetic acid (5-HIAA) have been found in the cerebrospinal fluid of suicide victims. Evidence has also shown that sleep and the HPA axis are closely related. The HPA axis is activated by stress leading to a cascade of hormones that can cause susceptibility of hyperarousal, REM alterations, and suicide. Hyperarousal, shared in context with PTSD and insomnia, can lead to hyperactivation of the noradrenergic systems in the medial prefrontal cortex, which can lead to decrease in executive decision making (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Treatment Strategies
The benefit of treating insomnia and nightmares, in regards to reducing suicidality, continues to be an area of active research. Many of the previous studies have theorized that treating symptoms of insomnia and nightmares may indirectly reduce suicide. Pharmaceutical and nonpharmaceutical treatments are currently being used to help treat patients with insomnia and nightmares, but the benefit for reducing suicidality is still unknown.
One of the main treatment modalities for insomnia is hypnotic medication; however, these medications carry their own potential risk for suicide. Reports of suicide death in conjunction with hypnotic medication has led the FDA to add warnings about the increased risk of suicide with these medications. Some of these medications include zolpidem, zaleplon, eszopiclone, doxepin, ramelteon, and suvorexant. A review of research studies and case reports was completed in 2017 and showed that there was an odds ratio of 2 to 3 for hypnotic use in suicide deaths. However, most of the studies that were reviewed reported a potential confounding bias of the individual’s current mental health state. Furthermore, many of the suicide case reports that involved hypnotics also had additional substances detected, such as alcohol. Hypnotic medication has been shown to be an effective treatment for insomnia, but caution needs to be used when prescribing these medications. Strategies that may be beneficial when using hypnotic medication to reduce the risk of an adverse outcome include using the lowest effective dose and educating the patient of not combining the medication with alcohol or other sedative/hypnotics (McCall W, et al. Am J Psychiatry. 2017;174[1]:18).
For patients who have recurrent nightmares in the context of PTSD, the alpha-1 adrenergic receptor antagonist, prazosin, may provide some benefit; however, the literature is divided. There have been several randomized, placebo-controlled clinical trials with prazosin, which has shown a moderate to large effect for alleviating trauma-related nightmares and improving sleep quality. Some of the limitations of these studies were that the trials were small to moderate in size, and the length of the trials was 15 weeks or less. In 2018, Raskin and colleagues completed a follow-up randomized, placebo-controlled study for 26 weeks with 304 participants and did not find a significant difference between prazosin and placebo in regards to nightmares and sleep quality (Raskind MA, et al. N Engl J Med. 2018;378[6]:507).
Cognitive behavioral therapy for insomnia (CBT-I) and image rehearsal therapy (IRT) are two sleep-targeted therapy modalities that are evidence based. CBT-I targets dysfunctional beliefs and attitudes regarding sleep (McCall W, et al. J Clin Sleep Med. 2013;9[2]:135). IRT, on the other hand, specifically targets nightmares by having the patient write out a narrative of the nightmare, followed by re-scripting an alternative ending to something that is less distressing. The patient will rehearse the new dream narrative before going to sleep. There is still insufficient evidence to determine if these therapies have benefit in reducing suicide (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393).
While the jury is still out on how best to target and treat the risk factors of insomnia and nightmares in regards to suicide, there are still steps that health-care providers can take to help keep their patients safe. During the patient interview, new or worsening insomnia and nightmares should prompt further investigation of suicidal thoughts and behaviors. After a thorough interview, treatment options, with a discussion of risks and benefits, can be tailored to the individual’s needs. Managing insomnia and nightmares may be one avenue of suicide prevention.
Drs. Locrotondo and McCall are with the Department of Psychiatry and Health Behavior at the Medical College of Georgia, Augusta University, Augusta, Georgia.
According to the Centers for Disease Control and Prevention, suicide is the 10th leading cause of mortality in the United States, with rates of suicide rising over the past 2 decades. In 2016, completed suicides accounted for approximately 45,000 deaths in the United States (Ivey-Stephenson AZ, et al. MMWR Surveill Summ. 2017;66[18]:1). While progress has been made to lower mortality rates of other leading causes of death, very little progress has been made on reducing the rates of suicide. The term “suicide,” as referred to in this article, encompasses suicidal ideation, suicidal behavior, and suicide death.
Researchers have been investigating potential risk factors and prevention strategies for suicide. The relationship between suicide and sleep disturbances, specifically insomnia and nightmares, has been well documented in the literature. Given that insomnia and nightmares are potentially modifiable risk factors, it continues to be an area of active exploration for suicide rate reduction. While there are many different types of sleep disorders, including excessive daytime sleepiness, parasomnias, obstructive sleep apnea, and restless legs syndrome, this article will focus on the relationship between insomnia and nightmares with suicide.
Insomnia
Insomnia disorder, according to the American Psychiatric Association’s DSM-5, is a dissatisfaction of sleep quantity or quality that occurs at least three nights per week for a minimum of 3 months despite adequate opportunity for sleep. This may present as difficulty with falling asleep, staying asleep, or early morning awakenings. The sleep disturbance results in functional impairment or significant distress in at least one area of life (American Psychiatric Association. Arlington, Virginia: APA; 2013). While insomnia is often a symptom of many psychiatric disorders, research has shown that insomnia is an independent risk factor for suicide, even when controlling for mental illness. Studies have shown that there is up to a 2.4 relative risk of suicide death with insomnia after adjusting for depression severity (McCall W, et al. J Clin Sleep Med. 2013;32[9]:135).
Nightmares
Nightmares, as defined by the American Psychiatric Association’s DSM-5, are “typically lengthy, elaborate, story-like sequences of dream imagery that seem real and incite anxiety, fear, or other dysphoric emotions” (American Psychiatric Association. Arlington, Virginia: APA; 2013). They are common symptoms in posttraumatic stress disorder (PTSD), with up to 90% of individuals with PTSD experiencing nightmares following a traumatic event (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393). Nightmares have also been shown to be an independent risk factor for suicide when controlling for mental illness. Studies have shown that nightmares are associated with an elevated risk factor of 1.5 to 3 times for suicidal ideation and 3 to 4 times for suicide attempts. The data suggest that nightmares may be a stronger risk factor for suicide than insomnia (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Proposed Mechanism
The mechanism linking insomnia and nightmares with suicide has been theorized and studied by researchers. A couple of the most noteworthy proposed psychological mechanisms involve dysfunctional beliefs and attitudes about sleep, as well as deficits in problem solving capability. Dysfunctional beliefs and attitudes about sleep (DBAS) are negative cognitions pertaining to sleep, and they have been shown to be related to the intensity of suicidal ideations. Many of the DBAS are pessimistic thoughts that contain a “hopelessness flavor” to them, which lead to the perpetuation of insomnia. Hopelessness has been found to be a strong risk factor for suicide. In addition to DBAS, insomnia has also shown to lead to impairments in complex problem solving. The lack of problem solving skills in these patients may lead to fewer quantity and quality of solutions during stressful situations and leave suicide as the perceived best or only option.
The biological theories focus on serotonin and hyperarousal mediated by the hypothalamic-pituitary-adrenal (HPA) axis. Serotonin is a neurotransmitter that is involved in the induction and maintenance of sleep. Of interesting note, low levels of serotonin’s main metabolite, 5-hydroxyindoleacetic acid (5-HIAA) have been found in the cerebrospinal fluid of suicide victims. Evidence has also shown that sleep and the HPA axis are closely related. The HPA axis is activated by stress leading to a cascade of hormones that can cause susceptibility of hyperarousal, REM alterations, and suicide. Hyperarousal, shared in context with PTSD and insomnia, can lead to hyperactivation of the noradrenergic systems in the medial prefrontal cortex, which can lead to decrease in executive decision making (McCall W, et al. Curr Psychiatr Rep. 2013;15[9]:389).
Treatment Strategies
The benefit of treating insomnia and nightmares, in regards to reducing suicidality, continues to be an area of active research. Many of the previous studies have theorized that treating symptoms of insomnia and nightmares may indirectly reduce suicide. Pharmaceutical and nonpharmaceutical treatments are currently being used to help treat patients with insomnia and nightmares, but the benefit for reducing suicidality is still unknown.
One of the main treatment modalities for insomnia is hypnotic medication; however, these medications carry their own potential risk for suicide. Reports of suicide death in conjunction with hypnotic medication has led the FDA to add warnings about the increased risk of suicide with these medications. Some of these medications include zolpidem, zaleplon, eszopiclone, doxepin, ramelteon, and suvorexant. A review of research studies and case reports was completed in 2017 and showed that there was an odds ratio of 2 to 3 for hypnotic use in suicide deaths. However, most of the studies that were reviewed reported a potential confounding bias of the individual’s current mental health state. Furthermore, many of the suicide case reports that involved hypnotics also had additional substances detected, such as alcohol. Hypnotic medication has been shown to be an effective treatment for insomnia, but caution needs to be used when prescribing these medications. Strategies that may be beneficial when using hypnotic medication to reduce the risk of an adverse outcome include using the lowest effective dose and educating the patient of not combining the medication with alcohol or other sedative/hypnotics (McCall W, et al. Am J Psychiatry. 2017;174[1]:18).
For patients who have recurrent nightmares in the context of PTSD, the alpha-1 adrenergic receptor antagonist, prazosin, may provide some benefit; however, the literature is divided. There have been several randomized, placebo-controlled clinical trials with prazosin, which has shown a moderate to large effect for alleviating trauma-related nightmares and improving sleep quality. Some of the limitations of these studies were that the trials were small to moderate in size, and the length of the trials was 15 weeks or less. In 2018, Raskin and colleagues completed a follow-up randomized, placebo-controlled study for 26 weeks with 304 participants and did not find a significant difference between prazosin and placebo in regards to nightmares and sleep quality (Raskind MA, et al. N Engl J Med. 2018;378[6]:507).
Cognitive behavioral therapy for insomnia (CBT-I) and image rehearsal therapy (IRT) are two sleep-targeted therapy modalities that are evidence based. CBT-I targets dysfunctional beliefs and attitudes regarding sleep (McCall W, et al. J Clin Sleep Med. 2013;9[2]:135). IRT, on the other hand, specifically targets nightmares by having the patient write out a narrative of the nightmare, followed by re-scripting an alternative ending to something that is less distressing. The patient will rehearse the new dream narrative before going to sleep. There is still insufficient evidence to determine if these therapies have benefit in reducing suicide (Littlewood DL, et al. J Clin Sleep Med. 2016;12[3]:393).
While the jury is still out on how best to target and treat the risk factors of insomnia and nightmares in regards to suicide, there are still steps that health-care providers can take to help keep their patients safe. During the patient interview, new or worsening insomnia and nightmares should prompt further investigation of suicidal thoughts and behaviors. After a thorough interview, treatment options, with a discussion of risks and benefits, can be tailored to the individual’s needs. Managing insomnia and nightmares may be one avenue of suicide prevention.
Drs. Locrotondo and McCall are with the Department of Psychiatry and Health Behavior at the Medical College of Georgia, Augusta University, Augusta, Georgia.
ECMO for ARDS in the modern era
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
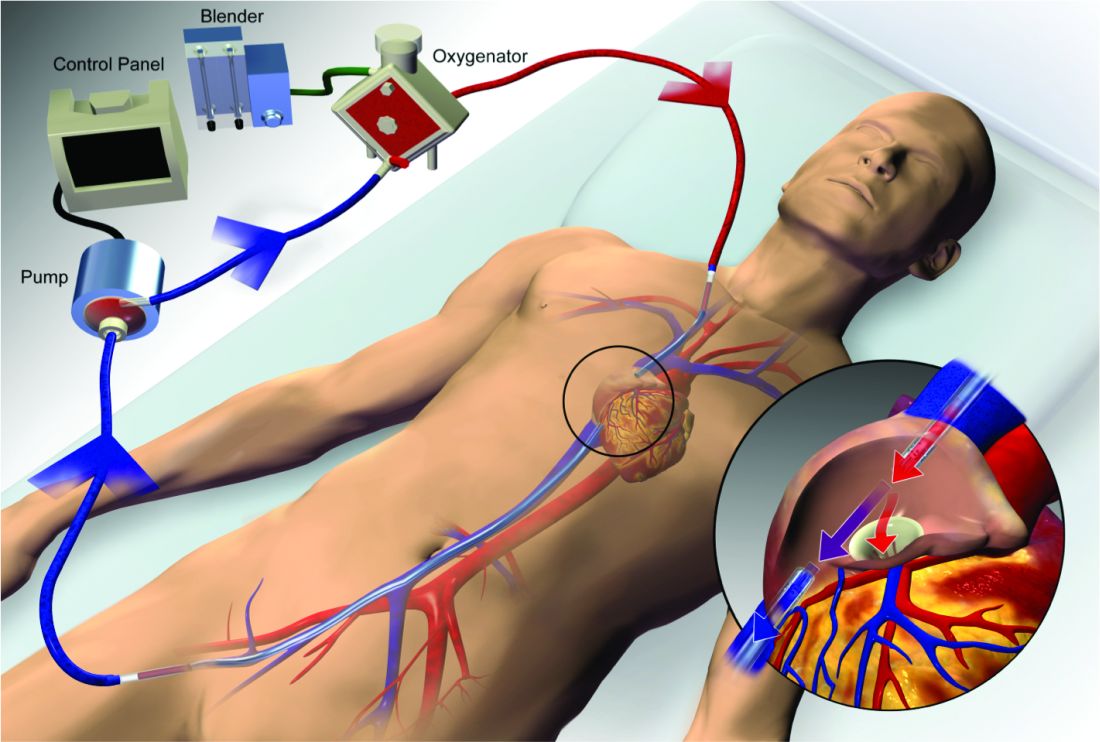
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.

Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.
Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
Extracorporeal membrane oxygenation (ECMO) has become increasingly accepted as a rescue therapy for severe respiratory failure from a variety of conditions, though most commonly, the acute respiratory distress syndrome (ARDS) (Thiagarajan R, et al. ASAIO. 2017;63[1]:60). ECMO can provide respiratory or cardiorespiratory support for failing lungs, heart, or both. The most common ECMO configuration used in ARDS is venovenous ECMO, in which blood is withdrawn from a catheter placed in a central vein, pumped through a gas exchange device known as an oxygenator, and returned to the venous system via another catheter. The blood flowing through the oxygenator is separated from a continuous supply of oxygen-rich sweep gas by a semipermeable membrane, across which diffusion-mediated gas exchange occurs, so that the blood exiting it is rich in oxygen and low in carbon dioxide. As venovenous ECMO functions in series with the native circulation, the well-oxygenated blood exiting the ECMO circuit mixes with poorly oxygenated blood flowing through the lungs. Therefore, oxygenation is dependent on native cardiac output to achieve systemic oxygen delivery (Figure 1).
ECMO been used successfully in adults with ARDS since the early 1970s (Hill JD, et al. N Engl J Med. 1972;286[12]:629-34) but, until recently, was limited to small numbers of patients at select global centers and associated with a high-risk profile. In the last decade, however, driven by improvements in ECMO circuit components making the device safer and easier to use, encouraging worldwide experience during the 2009 influenza A (H1N1) pandemic (Davies A, et al. JAMA. 2009;302[17]1888-95), and publication of the Efficacy and Economic Assessment of Conventional Ventilatory Support versus Extracorporeal Membrane Oxygenation for Severe Adult Respiratory Failure (CESAR) trial (Peek GJ, et al. Lancet. 2009;374[9698]:1351-63), ECMO use has markedly increased.
Despite its rapid growth, however, rigorous evidence supporting the use of ECMO has been lacking. The CESAR trial, while impressive in execution, had methodological issues that limited the strength of its conclusions. CESAR was a pragmatic trial that randomized 180 adults with severe respiratory failure from multiple etiologies to conventional management or transfer to an experienced, ECMO-capable center. CESAR met its primary outcome of improved survival without disability in the ECMO-referred group (63% vs 47%, relative risk [RR] 0.69; 95% confidence interval [CI] 0.05 to 0.97, P=.03), but not all patients in that group ultimately received ECMO. In addition, the use of lung protective ventilation was significantly higher in the ECMO-referred group, making it difficult to separate its benefit from that of ECMO. A conservative interpretation is that CESAR showed the clinical benefit of treatment at an ECMO-capable center, experienced in the management of patients with severe respiratory failure.
Not until the release of the Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome (EOLIA) trial earlier this year (Combes A, et al. N Engl J Med. 2018;378[21]:1965-75), did a modern, randomized controlled trial evaluating the use of ECMO itself exist. The EOLIA trial addressed the limitations of CESAR and randomized adult patients with early, severe ARDS to conventional, standard of care management that included a protocolized lung protective strategy in the control group vs immediate initiation of ECMO combined with an ultra-lung protective strategy (targeting end-inspiratory plateau pressure ≤24 cmH2O) in the intervention group. The primary outcome was all-cause mortality at 60 days. Of note, patients enrolled in EOLIA met entry criteria despite greater than 90% of patients receiving neuromuscular blockade and around 60% treated with prone positioning at the time of randomization (importantly, 90% of control group patients ultimately underwent prone positioning).
EOLIA was powered to detect a 20% decrease in mortality in the ECMO group. Based on trial design and the results of the fourth interim analysis, the trial was stopped for futility to reach that endpoint after enrollment of 249 of a maximum 331 patients. Although a 20% mortality reduction was not achieved, 60-day mortality was notably lower in the ECMO-treated group (35% vs 46%, RR 0.76, 95% CI 0.55 to 1.04, P=.09). The key secondary outcome of risk of treatment failure (defined as death in the ECMO group and death or crossover to ECMO in the control group) favored the ECMO group with a RR for mortality of 0.62 (95% CI, 0.47 to 0.82; P<.001), as did other secondary endpoints such as days free of renal and other organ failure.
A major limitation of the trial was that 35 (28%) of control group patients ultimately crossed over to ECMO, which diluted the effect of ECMO observed in the intention-to-treat analysis. Crossover occurred at clinician discretion an average of 6.5 days after randomization and after stringent criteria for crossover was met. These patients were incredibly ill, with a median oxygen saturation of 77%, rapidly worsening inotropic scores, and lactic acidosis; nine individuals had already suffered cardiac arrest, and six had received ECMO as part of extracorporeal cardiopulmonary resuscitation (ECPR), the initiation of venoarterial ECMO during cardiac arrest in attempt to restore spontaneous circulation. Mortality was considerably worse in the crossover group than in conventionally managed cohort overall, and, notably, 33% of patients crossed over to ECMO still survived.
In order to estimate the effect of ECMO on survival times if crossover had not occurred, the authors performed a post-hoc, rank-preserving structural failure time analysis. Though this relies on some assessment regarding the effect of the treatment itself, it showed a hazard ratio for mortality in the ECMO group of 0.51 (95% CI 0.24 to 1.02, P=.055). Although the EOLIA trial was not positive by traditional interpretation, all three major analyses and all secondary endpoints suggest some degree of benefit in patients with severe ARDS managed with ECMO.
Importantly, ECMO was well tolerated (at least when performed at expert centers, as done in this trial). There were significantly more bleeding events and cases of severe thrombocytopenia in the ECMO-treated group, but massive hemorrhage, ischemic and hemorrhagic stroke, arrhythmias, and other complications were similar.
Where do we go from here? Based on the totality of information, it is reasonable to consider ECMO for cases of severe ARDS not responsive to conventional measures, such as a lung protective ventilator strategy, neuromuscular blockade, and prone positioning. Initiation of ECMO may be reasonable prior to implementation of standard of care therapies, in order to permit safe transfer to an experienced center from a center not able to provide them.
Two take-away points: First, it is important to recognize that much of the clinical benefit derived from ECMO may be beyond its ability to normalize gas exchange and be due, at least in part, to the fact that ECMO allows the enhancement of proven lung protective ventilatory strategies. Initiation of ECMO and the “lung rest” it permits reduce the mechanical power applied to the injured alveoli and may attenuate ventilator-induced lung injury, cytokine release, and multiorgan failure that portend poor clinical outcomes in ARDS. Second, ECMO in EOLIA was conducted at expert centers with relatively low rates of complications.
It is too early to know how the critical care community will view ECMO for ARDS in light of EOLIA as well as a growing body of global ECMO experience, or how its wider application may impact the distribution and organization of ECMO centers. Regardless, of paramount importance in using ECMO as a treatment modality is optimizing patient management both prior to and after its initiation.
Dr. Agerstrand is Assistant Professor of Medicine, Director of the Medical ECMO Program, Columbia University College of Physicians and Surgeons, New York-Presbyterian Hospital.
The importance of diversity and inclusion in medicine
Diversity
There is growing appreciation for diversity and inclusion (DI) as drivers of excellence in medicine. CHEST also promotes excellence in medicine. Therefore, it is intuitive that CHEST promote DI. Diversity encompasses differences in gender, race/ethnicity, vocational training, age, sexual orientation, thought processes, etc.
Academic medicine is rich with examples of how diversity is critical to the health of our nation:
– Diverse student populations have been shown to improve our learners’ satisfaction with their educational experience.
– Diverse teams have been shown to be more capable of solving complex problems than homogenous teams.
– Health care is moving toward a team-based, interprofessional model that values the contributions of a range of providers’ perspectives in improving patient outcomes.
– In biomedical research, investigators ask different research questions based on their own background and experiences. This implies that finding solutions to diseases that affect specific populations will require a diverse pool of biomedical researchers.
– Faculty diversity as a key component of excellence for medical education and research has been documented.
Diversity alone doesn’t drive inclusion. Noted diversity advocate, Verna Myers, stated, “Diversity is being invited to the party. Inclusion is being asked to dance.” In my opinion, diversity is the commencement of work, but inclusion helps complete the task.
Inclusion

An inclusive environment values the unique contributions all members bring. Teams with diversity of thought are more innovative as individual members with different backgrounds and points of view bring an extensive range of ideas and creativity to scientific discovery and decision-making processes. Inclusion leverages the power of our unique differences to accomplish our mutual goals. By valuing everyone’s perspective, we demonstrate excellence.
I recommend an article from the Harvard Business Review (HBR Feb 2017). The authors suggest several ways to promote inclusiveness: (1) ensuring team members speak up and are heard; (2) making it safe to propose novel ideas; (3) empowering team members to make decisions; (4) taking advice and implementing feedback; (5) giving actionable feedback; and ( 6) sharing credit for team success. If the team leader possesses at least three of these traits, 87% of team members say they feel welcome and included in their team; 87% say they feel free to express their views and opinions; and 74% say they feel that their ideas are heard and recognized. If the team leader possessed none of these traits, those percentages dropped to 51%, 46%, and 37%, respectively. I believe this concept is applicable in medicine also.
Sponsors
What can we do to advance diversity and inclusion individually and in our individual institutions? A sponsor is a senior level leader who advocates for key assignments, promotes for and puts his or her reputation on the line for the protégé’s advancement. This invigorates and drives engagement. One key to rising above the playing field for women and people of color is sponsorship. Being a sponsor does not mean one would recommend someone who is not qualified. It means one recommends or supports those who are capable of doing the job but would not otherwise be given the opportunity.
Ask yourself: Have I served as a sponsor? What would prevent me from being a sponsor? Do I believe in this concept?
Cause for Alarm
Numerous publications have recently discussed the crisis of the decline of black men entering medicine. In 1978, there were 1,410 black male applicants to medical school, and in 2014, there were 1,337. Additionally, the number of black male matriculants to medical school over more than 35 years has not surpassed the 1978 numbers. In 1978, there were 542 black male matriculants, and in 2014, there were 515 (J of Racial and Ethnic Health Disparities. 2017, 4:317-321). This report is thorough and insightful and illustrates the work that we must do to help improve this situation.
Dr. Marc Nivet, Association of American Medical Colleges (AAMC) Chief Diversity Officer, stated “No other minority group has experienced such declines. The inability to find, engage, and develop candidates for careers in medicine from all members of our society limits our ability to improve health care for all.” I recommend you read the 2015 AAMC publication entitled: Altering the Course: Black Males in Medicine.
Health-care Disparities
Research suggests that the overall health of Americans has improved; however, disparities continue to persist among many populations within the United States. Racial and ethnic minority populations have poorer access to care and worse outcomes than their white counterparts. Approximately 20% of the nation living in rural areas is less likely than those living in urban areas to receive preventive care and more likely to experience language barriers.
Individuals identifying as lesbian, gay, bisexual, or transgender are likely to experience discrimination in health-care settings. These individuals often face insurance-based barriers and are less likely to have a usual source of care than patients who identify as straight.
A 2002 report by the Institute of Medicine entitled: Unequal Treatment: What Healthcare Providers Need to Know about Racial and Ethnic Disparities in Healthcare is revealing. Salient information reported is: It is generally accepted that a diverse workforce is a key component in the delivery of quality, competent care throughout the nation. Physicians from racial and ethnic backgrounds typically underrepresented in medicine are significantly more likely to practice primary care than white physicians and are more likely to practice in impoverished and medically underserved areas. Diversity in the physician workforce impacts the quality of care received by patients. Race concordance between patient and physician results in longer visits and increased patient satisfaction, and language concordance is positively associated with adherence to treatment among certain racial or ethnic groups.
Improving the patient experience or quality of care received also requires attention to education and training on cultural competence. By weaving together a diverse and culturally responsive pool of physicians working collaboratively with other health-care professionals, access and quality of care can improve throughout the nation.
CHEST cannot attain more racial diversity in our organization if we don’t have this diversity in medical education and training. This is why CHEST must be actively involved in addressing these issues.
Unconscious Bias
Despite many examples of how diversity enriches the quality of health care and health research, there is still much work to be done to address the human biases that impede our ability to benefit from diversity in medicine. While academic medicine has made progress toward addressing overt discrimination, unconscious bias (implicit bias) represents another threat. Unconscious bias describes the prejudices we don’t know we have. While unconscious biases vary from person to person, we all possess them. The existence of unconscious bias in academic medicine, while uncomfortable and unsettling, is a reality. The AAMC developed an unconscious bias learning lab for the health professions and produced an oft-cited video about addressing unconscious bias in the faculty advancement, promotion, and tenure process. We must consider this and other ways in which we can help promote the acknowledgment of unconscious bias. The CHEST staff have undergone unconscious bias training, and I recommend it for all faculty in academic medicine.
Summary
Diversity and inclusion in medicine is of paramount importance. It leads to better patient care and better trainee education and will decrease health-care disparities. Progress has been made, but there is more work to be done.
CHEST is supportive of these efforts and has worked on this previously and with a renewed push in the past 2 years with the DI Task Force initially and, now, the DI Roundtable, which has representatives from each of the standing committees, including the Board of Regents. This roundtable group will help advance the DI initiatives of the organization. I ask that each person reading this article consider what we as individuals can do in helping make DI in medicine a priority.
Dr. Haynes is Professor of Medicine at The University of Mississippi Medical Center in Jackson, MS. He is also the Executive Vice Chair of the Department of Medicine. At CHEST, he is a member of the training and transitions committee, executive scientific program committee, former chair of the diversity and inclusion task force, and is the current chair of the diversity and inclusion roundtable.
Diversity
There is growing appreciation for diversity and inclusion (DI) as drivers of excellence in medicine. CHEST also promotes excellence in medicine. Therefore, it is intuitive that CHEST promote DI. Diversity encompasses differences in gender, race/ethnicity, vocational training, age, sexual orientation, thought processes, etc.
Academic medicine is rich with examples of how diversity is critical to the health of our nation:
– Diverse student populations have been shown to improve our learners’ satisfaction with their educational experience.
– Diverse teams have been shown to be more capable of solving complex problems than homogenous teams.
– Health care is moving toward a team-based, interprofessional model that values the contributions of a range of providers’ perspectives in improving patient outcomes.
– In biomedical research, investigators ask different research questions based on their own background and experiences. This implies that finding solutions to diseases that affect specific populations will require a diverse pool of biomedical researchers.
– Faculty diversity as a key component of excellence for medical education and research has been documented.
Diversity alone doesn’t drive inclusion. Noted diversity advocate, Verna Myers, stated, “Diversity is being invited to the party. Inclusion is being asked to dance.” In my opinion, diversity is the commencement of work, but inclusion helps complete the task.
Inclusion
An inclusive environment values the unique contributions all members bring. Teams with diversity of thought are more innovative as individual members with different backgrounds and points of view bring an extensive range of ideas and creativity to scientific discovery and decision-making processes. Inclusion leverages the power of our unique differences to accomplish our mutual goals. By valuing everyone’s perspective, we demonstrate excellence.
I recommend an article from the Harvard Business Review (HBR Feb 2017). The authors suggest several ways to promote inclusiveness: (1) ensuring team members speak up and are heard; (2) making it safe to propose novel ideas; (3) empowering team members to make decisions; (4) taking advice and implementing feedback; (5) giving actionable feedback; and ( 6) sharing credit for team success. If the team leader possesses at least three of these traits, 87% of team members say they feel welcome and included in their team; 87% say they feel free to express their views and opinions; and 74% say they feel that their ideas are heard and recognized. If the team leader possessed none of these traits, those percentages dropped to 51%, 46%, and 37%, respectively. I believe this concept is applicable in medicine also.
Sponsors
What can we do to advance diversity and inclusion individually and in our individual institutions? A sponsor is a senior level leader who advocates for key assignments, promotes for and puts his or her reputation on the line for the protégé’s advancement. This invigorates and drives engagement. One key to rising above the playing field for women and people of color is sponsorship. Being a sponsor does not mean one would recommend someone who is not qualified. It means one recommends or supports those who are capable of doing the job but would not otherwise be given the opportunity.
Ask yourself: Have I served as a sponsor? What would prevent me from being a sponsor? Do I believe in this concept?
Cause for Alarm
Numerous publications have recently discussed the crisis of the decline of black men entering medicine. In 1978, there were 1,410 black male applicants to medical school, and in 2014, there were 1,337. Additionally, the number of black male matriculants to medical school over more than 35 years has not surpassed the 1978 numbers. In 1978, there were 542 black male matriculants, and in 2014, there were 515 (J of Racial and Ethnic Health Disparities. 2017, 4:317-321). This report is thorough and insightful and illustrates the work that we must do to help improve this situation.
Dr. Marc Nivet, Association of American Medical Colleges (AAMC) Chief Diversity Officer, stated “No other minority group has experienced such declines. The inability to find, engage, and develop candidates for careers in medicine from all members of our society limits our ability to improve health care for all.” I recommend you read the 2015 AAMC publication entitled: Altering the Course: Black Males in Medicine.
Health-care Disparities
Research suggests that the overall health of Americans has improved; however, disparities continue to persist among many populations within the United States. Racial and ethnic minority populations have poorer access to care and worse outcomes than their white counterparts. Approximately 20% of the nation living in rural areas is less likely than those living in urban areas to receive preventive care and more likely to experience language barriers.
Individuals identifying as lesbian, gay, bisexual, or transgender are likely to experience discrimination in health-care settings. These individuals often face insurance-based barriers and are less likely to have a usual source of care than patients who identify as straight.
A 2002 report by the Institute of Medicine entitled: Unequal Treatment: What Healthcare Providers Need to Know about Racial and Ethnic Disparities in Healthcare is revealing. Salient information reported is: It is generally accepted that a diverse workforce is a key component in the delivery of quality, competent care throughout the nation. Physicians from racial and ethnic backgrounds typically underrepresented in medicine are significantly more likely to practice primary care than white physicians and are more likely to practice in impoverished and medically underserved areas. Diversity in the physician workforce impacts the quality of care received by patients. Race concordance between patient and physician results in longer visits and increased patient satisfaction, and language concordance is positively associated with adherence to treatment among certain racial or ethnic groups.
Improving the patient experience or quality of care received also requires attention to education and training on cultural competence. By weaving together a diverse and culturally responsive pool of physicians working collaboratively with other health-care professionals, access and quality of care can improve throughout the nation.
CHEST cannot attain more racial diversity in our organization if we don’t have this diversity in medical education and training. This is why CHEST must be actively involved in addressing these issues.
Unconscious Bias
Despite many examples of how diversity enriches the quality of health care and health research, there is still much work to be done to address the human biases that impede our ability to benefit from diversity in medicine. While academic medicine has made progress toward addressing overt discrimination, unconscious bias (implicit bias) represents another threat. Unconscious bias describes the prejudices we don’t know we have. While unconscious biases vary from person to person, we all possess them. The existence of unconscious bias in academic medicine, while uncomfortable and unsettling, is a reality. The AAMC developed an unconscious bias learning lab for the health professions and produced an oft-cited video about addressing unconscious bias in the faculty advancement, promotion, and tenure process. We must consider this and other ways in which we can help promote the acknowledgment of unconscious bias. The CHEST staff have undergone unconscious bias training, and I recommend it for all faculty in academic medicine.
Summary
Diversity and inclusion in medicine is of paramount importance. It leads to better patient care and better trainee education and will decrease health-care disparities. Progress has been made, but there is more work to be done.
CHEST is supportive of these efforts and has worked on this previously and with a renewed push in the past 2 years with the DI Task Force initially and, now, the DI Roundtable, which has representatives from each of the standing committees, including the Board of Regents. This roundtable group will help advance the DI initiatives of the organization. I ask that each person reading this article consider what we as individuals can do in helping make DI in medicine a priority.
Dr. Haynes is Professor of Medicine at The University of Mississippi Medical Center in Jackson, MS. He is also the Executive Vice Chair of the Department of Medicine. At CHEST, he is a member of the training and transitions committee, executive scientific program committee, former chair of the diversity and inclusion task force, and is the current chair of the diversity and inclusion roundtable.
Diversity
There is growing appreciation for diversity and inclusion (DI) as drivers of excellence in medicine. CHEST also promotes excellence in medicine. Therefore, it is intuitive that CHEST promote DI. Diversity encompasses differences in gender, race/ethnicity, vocational training, age, sexual orientation, thought processes, etc.
Academic medicine is rich with examples of how diversity is critical to the health of our nation:
– Diverse student populations have been shown to improve our learners’ satisfaction with their educational experience.
– Diverse teams have been shown to be more capable of solving complex problems than homogenous teams.
– Health care is moving toward a team-based, interprofessional model that values the contributions of a range of providers’ perspectives in improving patient outcomes.
– In biomedical research, investigators ask different research questions based on their own background and experiences. This implies that finding solutions to diseases that affect specific populations will require a diverse pool of biomedical researchers.
– Faculty diversity as a key component of excellence for medical education and research has been documented.
Diversity alone doesn’t drive inclusion. Noted diversity advocate, Verna Myers, stated, “Diversity is being invited to the party. Inclusion is being asked to dance.” In my opinion, diversity is the commencement of work, but inclusion helps complete the task.
Inclusion
An inclusive environment values the unique contributions all members bring. Teams with diversity of thought are more innovative as individual members with different backgrounds and points of view bring an extensive range of ideas and creativity to scientific discovery and decision-making processes. Inclusion leverages the power of our unique differences to accomplish our mutual goals. By valuing everyone’s perspective, we demonstrate excellence.
I recommend an article from the Harvard Business Review (HBR Feb 2017). The authors suggest several ways to promote inclusiveness: (1) ensuring team members speak up and are heard; (2) making it safe to propose novel ideas; (3) empowering team members to make decisions; (4) taking advice and implementing feedback; (5) giving actionable feedback; and ( 6) sharing credit for team success. If the team leader possesses at least three of these traits, 87% of team members say they feel welcome and included in their team; 87% say they feel free to express their views and opinions; and 74% say they feel that their ideas are heard and recognized. If the team leader possessed none of these traits, those percentages dropped to 51%, 46%, and 37%, respectively. I believe this concept is applicable in medicine also.
Sponsors
What can we do to advance diversity and inclusion individually and in our individual institutions? A sponsor is a senior level leader who advocates for key assignments, promotes for and puts his or her reputation on the line for the protégé’s advancement. This invigorates and drives engagement. One key to rising above the playing field for women and people of color is sponsorship. Being a sponsor does not mean one would recommend someone who is not qualified. It means one recommends or supports those who are capable of doing the job but would not otherwise be given the opportunity.
Ask yourself: Have I served as a sponsor? What would prevent me from being a sponsor? Do I believe in this concept?
Cause for Alarm
Numerous publications have recently discussed the crisis of the decline of black men entering medicine. In 1978, there were 1,410 black male applicants to medical school, and in 2014, there were 1,337. Additionally, the number of black male matriculants to medical school over more than 35 years has not surpassed the 1978 numbers. In 1978, there were 542 black male matriculants, and in 2014, there were 515 (J of Racial and Ethnic Health Disparities. 2017, 4:317-321). This report is thorough and insightful and illustrates the work that we must do to help improve this situation.
Dr. Marc Nivet, Association of American Medical Colleges (AAMC) Chief Diversity Officer, stated “No other minority group has experienced such declines. The inability to find, engage, and develop candidates for careers in medicine from all members of our society limits our ability to improve health care for all.” I recommend you read the 2015 AAMC publication entitled: Altering the Course: Black Males in Medicine.
Health-care Disparities
Research suggests that the overall health of Americans has improved; however, disparities continue to persist among many populations within the United States. Racial and ethnic minority populations have poorer access to care and worse outcomes than their white counterparts. Approximately 20% of the nation living in rural areas is less likely than those living in urban areas to receive preventive care and more likely to experience language barriers.
Individuals identifying as lesbian, gay, bisexual, or transgender are likely to experience discrimination in health-care settings. These individuals often face insurance-based barriers and are less likely to have a usual source of care than patients who identify as straight.
A 2002 report by the Institute of Medicine entitled: Unequal Treatment: What Healthcare Providers Need to Know about Racial and Ethnic Disparities in Healthcare is revealing. Salient information reported is: It is generally accepted that a diverse workforce is a key component in the delivery of quality, competent care throughout the nation. Physicians from racial and ethnic backgrounds typically underrepresented in medicine are significantly more likely to practice primary care than white physicians and are more likely to practice in impoverished and medically underserved areas. Diversity in the physician workforce impacts the quality of care received by patients. Race concordance between patient and physician results in longer visits and increased patient satisfaction, and language concordance is positively associated with adherence to treatment among certain racial or ethnic groups.
Improving the patient experience or quality of care received also requires attention to education and training on cultural competence. By weaving together a diverse and culturally responsive pool of physicians working collaboratively with other health-care professionals, access and quality of care can improve throughout the nation.
CHEST cannot attain more racial diversity in our organization if we don’t have this diversity in medical education and training. This is why CHEST must be actively involved in addressing these issues.
Unconscious Bias
Despite many examples of how diversity enriches the quality of health care and health research, there is still much work to be done to address the human biases that impede our ability to benefit from diversity in medicine. While academic medicine has made progress toward addressing overt discrimination, unconscious bias (implicit bias) represents another threat. Unconscious bias describes the prejudices we don’t know we have. While unconscious biases vary from person to person, we all possess them. The existence of unconscious bias in academic medicine, while uncomfortable and unsettling, is a reality. The AAMC developed an unconscious bias learning lab for the health professions and produced an oft-cited video about addressing unconscious bias in the faculty advancement, promotion, and tenure process. We must consider this and other ways in which we can help promote the acknowledgment of unconscious bias. The CHEST staff have undergone unconscious bias training, and I recommend it for all faculty in academic medicine.
Summary
Diversity and inclusion in medicine is of paramount importance. It leads to better patient care and better trainee education and will decrease health-care disparities. Progress has been made, but there is more work to be done.
CHEST is supportive of these efforts and has worked on this previously and with a renewed push in the past 2 years with the DI Task Force initially and, now, the DI Roundtable, which has representatives from each of the standing committees, including the Board of Regents. This roundtable group will help advance the DI initiatives of the organization. I ask that each person reading this article consider what we as individuals can do in helping make DI in medicine a priority.
Dr. Haynes is Professor of Medicine at The University of Mississippi Medical Center in Jackson, MS. He is also the Executive Vice Chair of the Department of Medicine. At CHEST, he is a member of the training and transitions committee, executive scientific program committee, former chair of the diversity and inclusion task force, and is the current chair of the diversity and inclusion roundtable.
Value-based sleep: understanding and maximizing value in sleep medicine care
In addition to well-documented health consequences, obstructive sleep apnea (OSA) is associated with substantial economic costs borne by patients, payers, employers, and society at large. For example, in a recent white paper commissioned by the American Academy of Sleep Medicine, the total societal-level costs of OSA were estimated to exceed $150 billion per year in the United States alone. In addition to direct costs associated with OSA diagnosis and treatment, indirect costs were estimated at $86.9 billion for lost workplace productivity; $30 billion for increased health-care utilization (HCU); $26.2 billion for motor vehicle crashes (MVC); and $6.5 billion for workplace accidents and injuries.1
More important, evidence suggests that OSA treatments provide positive economic impact, for example reducing health-care utilization and reducing days missed from work. Our group at the University of Maryland is currently heavily involved in related research examining the health economic impact of sleep disorders and their treatments.
Value-based sleep is a concept that I created several years ago to guide a greater emphasis on health economic outcomes in order to advance our field. In addition to working with payers, industry partners, employers, and forward-thinking startups, we are investing much effort into provider education regarding the health economic aspects of sleep. This article examines what value-based sleep is, how to increase the value of sleep in your practice setting, and steps to prepare for payment models of the future.
Value is in the eye of the beholder
Unlike sleep medicine providers (and some patients), the majority of society views sleep as means to an end and not as an end-in-itself. That is, people only value sleep insofar as sleep will help them achieve their primary objectives, whatever they might be. In health economic terms, these distinct viewpoints are referred to as perspectives. For example, from the patient perspective, sleep is valued to the extent that it helps to increase quality of life. From the payer perspective, sleep is valued to the extent that reduces health-care utilization. From the employer perspective, sleep is valued to the extent that it increases workplace productivity and reduces health-care expenses. Table 1 summarizes common stakeholders and perspectives in sleep medicine.
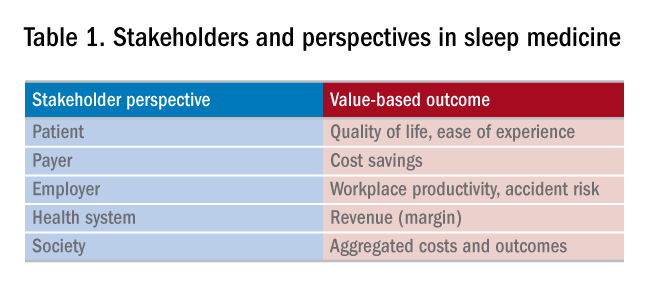
Speaking the language of value
In order to define, demonstrate, and maximize the perceived value of sleep medicine services, sleep physicians must understand and clearly articulate the values of these multiple constituents. Most important, this means that sleep physicians must move beyond discussing the apnea-hypopnea index (AHI). To be clear, no one other than sleep medicine insiders care about the AHI! Of course, the AHI is an important (albeit imperfect) measure of OSA disease severity and treatment outcomes. However, when was the last time that a patient told you they woke up one morning dreaming about a lower AHI? It simply does not happen. Instead, stakeholders care about outcomes that matters to them, from their own unique perspectives. To speak directly to these interests and frame the value of sleep, sleep medicine providers must methodically develop value propositions with each unique target constituency in mind. Speak the language of your audience, and use terms that matter to them.
Adopting value-based payments
Much has been spoken about a transition from fee-for-service to value-based care in medicine. New health-care business models will soon impact patients, providers, payers, and health systems. To guide and ensure sustainable change, multi-stakeholder organizations, such as the Health Care Payment & Learning Action Network, are heavily engaged in the development and implementation of alternate payment models (APMs) to facilitate the transition from fee-for-service to population health. As depicted in Figure 1, sequential steps toward value-based care include increased fees corresponding to improved outcomes. A reimbursement model that is fully value-based centers on shared financial risks. Although private practitioners may be ill-equipped to provide population-level services or negotiate fully value-based models, sleep medicine providers should do well to increase familiarity with APMs and their impact on primary and specialty care services. 
Five steps to a value-based approach
In the modern health-care climate of increasing costs on the one hand and limited resources on the other, sleep medicine providers must embrace a value-based perspective to survive, thrive, and grow in a new world of value-based care. This will require sleep medicine providers to learn, adapt, and adjust. The good news is that regardless of your practice or organizational setting, these strategies and tactics will help guide you:
1. Know thyself. What are your personal and organization objectives? Where are you, career-wise? Where do you want to be in 2, 3, and 5 years?
2. Know your customer. Whom do you serve? More broadly, whom does sleep serve? Listen carefully and identify the outcomes that matter to your constituents. Make these your endpoints.
3. Develop customer-centric language. Develop scripts. Rehearse them.
4. Understand trends in payments and technology. Is your region adopting bundled payments or paying more for improved outcomes? How might telemedicine or preauthorization for PAP impact your practice?
5. Know your numbers. To negotiate with confidence, you need to know your numbers. What are your costs per patient, per test, per outcome, and lifetime value of the patient?
Summary and next steps
To survive and thrive in a value-based future, you need to define, demonstrate, and maximize your perceived value. This will require greater attention to the language that you use, the results that you emphasize, and the data that you use to make decisions, all while attending to the perspectives of diverse stakeholders. The need for sleep medicine services has never been greater. Adopt a value-based sleep approach to ensure your bright future.
References
1. American Academy of Sleep M. Hidden health crisis costing America billions. Underdiagnosing and undertreating obstructive sleep apnea draining healthcare system. Mountain View, CA: Frost & Sullivan; 2016.
2. Wickwire EM, Verma T. Value and payment in sleep medicine. J Clin Sleep Med. 2018;14(5):881-884.
Dr. Wickwire is Associate Professor of Psychiatry and Medicine at the University of Maryland School of Medicine, where he directs the insomnia program. His current research interests include health and economic consequences of sleep disorders and their treatments and targeting sleep treatments for specific populations.
In addition to well-documented health consequences, obstructive sleep apnea (OSA) is associated with substantial economic costs borne by patients, payers, employers, and society at large. For example, in a recent white paper commissioned by the American Academy of Sleep Medicine, the total societal-level costs of OSA were estimated to exceed $150 billion per year in the United States alone. In addition to direct costs associated with OSA diagnosis and treatment, indirect costs were estimated at $86.9 billion for lost workplace productivity; $30 billion for increased health-care utilization (HCU); $26.2 billion for motor vehicle crashes (MVC); and $6.5 billion for workplace accidents and injuries.1
More important, evidence suggests that OSA treatments provide positive economic impact, for example reducing health-care utilization and reducing days missed from work. Our group at the University of Maryland is currently heavily involved in related research examining the health economic impact of sleep disorders and their treatments.
Value-based sleep is a concept that I created several years ago to guide a greater emphasis on health economic outcomes in order to advance our field. In addition to working with payers, industry partners, employers, and forward-thinking startups, we are investing much effort into provider education regarding the health economic aspects of sleep. This article examines what value-based sleep is, how to increase the value of sleep in your practice setting, and steps to prepare for payment models of the future.
Value is in the eye of the beholder
Unlike sleep medicine providers (and some patients), the majority of society views sleep as means to an end and not as an end-in-itself. That is, people only value sleep insofar as sleep will help them achieve their primary objectives, whatever they might be. In health economic terms, these distinct viewpoints are referred to as perspectives. For example, from the patient perspective, sleep is valued to the extent that it helps to increase quality of life. From the payer perspective, sleep is valued to the extent that reduces health-care utilization. From the employer perspective, sleep is valued to the extent that it increases workplace productivity and reduces health-care expenses. Table 1 summarizes common stakeholders and perspectives in sleep medicine.
Speaking the language of value
In order to define, demonstrate, and maximize the perceived value of sleep medicine services, sleep physicians must understand and clearly articulate the values of these multiple constituents. Most important, this means that sleep physicians must move beyond discussing the apnea-hypopnea index (AHI). To be clear, no one other than sleep medicine insiders care about the AHI! Of course, the AHI is an important (albeit imperfect) measure of OSA disease severity and treatment outcomes. However, when was the last time that a patient told you they woke up one morning dreaming about a lower AHI? It simply does not happen. Instead, stakeholders care about outcomes that matters to them, from their own unique perspectives. To speak directly to these interests and frame the value of sleep, sleep medicine providers must methodically develop value propositions with each unique target constituency in mind. Speak the language of your audience, and use terms that matter to them.
Adopting value-based payments
Much has been spoken about a transition from fee-for-service to value-based care in medicine. New health-care business models will soon impact patients, providers, payers, and health systems. To guide and ensure sustainable change, multi-stakeholder organizations, such as the Health Care Payment & Learning Action Network, are heavily engaged in the development and implementation of alternate payment models (APMs) to facilitate the transition from fee-for-service to population health. As depicted in Figure 1, sequential steps toward value-based care include increased fees corresponding to improved outcomes. A reimbursement model that is fully value-based centers on shared financial risks. Although private practitioners may be ill-equipped to provide population-level services or negotiate fully value-based models, sleep medicine providers should do well to increase familiarity with APMs and their impact on primary and specialty care services.
Five steps to a value-based approach
In the modern health-care climate of increasing costs on the one hand and limited resources on the other, sleep medicine providers must embrace a value-based perspective to survive, thrive, and grow in a new world of value-based care. This will require sleep medicine providers to learn, adapt, and adjust. The good news is that regardless of your practice or organizational setting, these strategies and tactics will help guide you:
1. Know thyself. What are your personal and organization objectives? Where are you, career-wise? Where do you want to be in 2, 3, and 5 years?
2. Know your customer. Whom do you serve? More broadly, whom does sleep serve? Listen carefully and identify the outcomes that matter to your constituents. Make these your endpoints.
3. Develop customer-centric language. Develop scripts. Rehearse them.
4. Understand trends in payments and technology. Is your region adopting bundled payments or paying more for improved outcomes? How might telemedicine or preauthorization for PAP impact your practice?
5. Know your numbers. To negotiate with confidence, you need to know your numbers. What are your costs per patient, per test, per outcome, and lifetime value of the patient?
Summary and next steps
To survive and thrive in a value-based future, you need to define, demonstrate, and maximize your perceived value. This will require greater attention to the language that you use, the results that you emphasize, and the data that you use to make decisions, all while attending to the perspectives of diverse stakeholders. The need for sleep medicine services has never been greater. Adopt a value-based sleep approach to ensure your bright future.
References
1. American Academy of Sleep M. Hidden health crisis costing America billions. Underdiagnosing and undertreating obstructive sleep apnea draining healthcare system. Mountain View, CA: Frost & Sullivan; 2016.
2. Wickwire EM, Verma T. Value and payment in sleep medicine. J Clin Sleep Med. 2018;14(5):881-884.
Dr. Wickwire is Associate Professor of Psychiatry and Medicine at the University of Maryland School of Medicine, where he directs the insomnia program. His current research interests include health and economic consequences of sleep disorders and their treatments and targeting sleep treatments for specific populations.
In addition to well-documented health consequences, obstructive sleep apnea (OSA) is associated with substantial economic costs borne by patients, payers, employers, and society at large. For example, in a recent white paper commissioned by the American Academy of Sleep Medicine, the total societal-level costs of OSA were estimated to exceed $150 billion per year in the United States alone. In addition to direct costs associated with OSA diagnosis and treatment, indirect costs were estimated at $86.9 billion for lost workplace productivity; $30 billion for increased health-care utilization (HCU); $26.2 billion for motor vehicle crashes (MVC); and $6.5 billion for workplace accidents and injuries.1
More important, evidence suggests that OSA treatments provide positive economic impact, for example reducing health-care utilization and reducing days missed from work. Our group at the University of Maryland is currently heavily involved in related research examining the health economic impact of sleep disorders and their treatments.
Value-based sleep is a concept that I created several years ago to guide a greater emphasis on health economic outcomes in order to advance our field. In addition to working with payers, industry partners, employers, and forward-thinking startups, we are investing much effort into provider education regarding the health economic aspects of sleep. This article examines what value-based sleep is, how to increase the value of sleep in your practice setting, and steps to prepare for payment models of the future.
Value is in the eye of the beholder
Unlike sleep medicine providers (and some patients), the majority of society views sleep as means to an end and not as an end-in-itself. That is, people only value sleep insofar as sleep will help them achieve their primary objectives, whatever they might be. In health economic terms, these distinct viewpoints are referred to as perspectives. For example, from the patient perspective, sleep is valued to the extent that it helps to increase quality of life. From the payer perspective, sleep is valued to the extent that reduces health-care utilization. From the employer perspective, sleep is valued to the extent that it increases workplace productivity and reduces health-care expenses. Table 1 summarizes common stakeholders and perspectives in sleep medicine.
Speaking the language of value
In order to define, demonstrate, and maximize the perceived value of sleep medicine services, sleep physicians must understand and clearly articulate the values of these multiple constituents. Most important, this means that sleep physicians must move beyond discussing the apnea-hypopnea index (AHI). To be clear, no one other than sleep medicine insiders care about the AHI! Of course, the AHI is an important (albeit imperfect) measure of OSA disease severity and treatment outcomes. However, when was the last time that a patient told you they woke up one morning dreaming about a lower AHI? It simply does not happen. Instead, stakeholders care about outcomes that matters to them, from their own unique perspectives. To speak directly to these interests and frame the value of sleep, sleep medicine providers must methodically develop value propositions with each unique target constituency in mind. Speak the language of your audience, and use terms that matter to them.
Adopting value-based payments
Much has been spoken about a transition from fee-for-service to value-based care in medicine. New health-care business models will soon impact patients, providers, payers, and health systems. To guide and ensure sustainable change, multi-stakeholder organizations, such as the Health Care Payment & Learning Action Network, are heavily engaged in the development and implementation of alternate payment models (APMs) to facilitate the transition from fee-for-service to population health. As depicted in Figure 1, sequential steps toward value-based care include increased fees corresponding to improved outcomes. A reimbursement model that is fully value-based centers on shared financial risks. Although private practitioners may be ill-equipped to provide population-level services or negotiate fully value-based models, sleep medicine providers should do well to increase familiarity with APMs and their impact on primary and specialty care services.
Five steps to a value-based approach
In the modern health-care climate of increasing costs on the one hand and limited resources on the other, sleep medicine providers must embrace a value-based perspective to survive, thrive, and grow in a new world of value-based care. This will require sleep medicine providers to learn, adapt, and adjust. The good news is that regardless of your practice or organizational setting, these strategies and tactics will help guide you:
1. Know thyself. What are your personal and organization objectives? Where are you, career-wise? Where do you want to be in 2, 3, and 5 years?
2. Know your customer. Whom do you serve? More broadly, whom does sleep serve? Listen carefully and identify the outcomes that matter to your constituents. Make these your endpoints.
3. Develop customer-centric language. Develop scripts. Rehearse them.
4. Understand trends in payments and technology. Is your region adopting bundled payments or paying more for improved outcomes? How might telemedicine or preauthorization for PAP impact your practice?
5. Know your numbers. To negotiate with confidence, you need to know your numbers. What are your costs per patient, per test, per outcome, and lifetime value of the patient?
Summary and next steps
To survive and thrive in a value-based future, you need to define, demonstrate, and maximize your perceived value. This will require greater attention to the language that you use, the results that you emphasize, and the data that you use to make decisions, all while attending to the perspectives of diverse stakeholders. The need for sleep medicine services has never been greater. Adopt a value-based sleep approach to ensure your bright future.
References
1. American Academy of Sleep M. Hidden health crisis costing America billions. Underdiagnosing and undertreating obstructive sleep apnea draining healthcare system. Mountain View, CA: Frost & Sullivan; 2016.
2. Wickwire EM, Verma T. Value and payment in sleep medicine. J Clin Sleep Med. 2018;14(5):881-884.
Dr. Wickwire is Associate Professor of Psychiatry and Medicine at the University of Maryland School of Medicine, where he directs the insomnia program. His current research interests include health and economic consequences of sleep disorders and their treatments and targeting sleep treatments for specific populations.
Diagnosis and Management of Critical Illness-Related Corticosteroid Insufficiency (CIRCI): Updated Guidelines 2017
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines

Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines
Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Critical illness-related corticosteroid insufficiency (CIRCI) was first introduced in 2008 by a task force convened by the Society of Critical Care Medicine (SCCM) to describe the impairment of the hypothalamic-pituitary-adrenal (HPA) axis during critical illness (Marik PE, et al. Crit Care Med. 2008;36(6):1937).
CIRCI is characterized by dysregulated systemic inflammation resulting from inadequate cellular corticosteroid activity for the severity of the patient’s critical illness. Signs and symptoms of CIRCI include hypotension poorly responsive to fluids, decreased sensitivity to catecholamines, fever, altered mental status, hypoxemia, and laboratory abnormalities such as hyponatremia and hypoglycemia. CIRCI can occur in a variety of acute conditions, such as sepsis and septic shock, acute respiratory distress syndrome (ARDS), severe community-acquired pneumonia, and non-septic systemic inflammatory response syndrome (SIRS) states associated with shock, such as trauma, cardiac arrest, and cardiopulmonary bypass surgery. Three major pathophysiologic events are considered to constitute CIRCI: dysregulation of the HPA axis, altered cortisol metabolism, and tissue resistance to glucocorticoids (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2089; Intensive Care Med. 2017;43(12):1781). Plasma clearance of cortisol is markedly reduced during critical illness, due to suppressed expression and activity of the primary cortisol-metabolizing enzymes in the liver and kidney. Furthermore, despite the elevated cortisol levels during critical illness, tissue resistance to glucocorticoids is believed to occur because of insufficient glucocorticoid receptor alpha-mediated anti-inflammatory activity.
Reviewing the Updated Guidelines
Against this background of recent insights into the understanding of CIRCI and the widespread use of corticosteroids in critically ill patients, an international panel of experts of the SCCM and the European Society of Intensive Care Medicine (ESICM) recently updated the guidelines for the diagnosis and management of CIRCI in a two-part guideline document (Annane D, Pastores SM, et al. Crit Care Med. 2017;45(12):2078; Intensive Care Med. 2017;43(12):1751; Pastores SM, Annane D, et al. Crit Care Med. 2018;46(1):146; Pastores SM, Annane D, et al. Intensive Care Med. 2018;44(4):474). For this update, the multidisciplinary task force used the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) methodology to formulate actionable recommendations for the diagnosis and treatment of CIRCI. The recommendations and their strength (strong or conditional) required the agreement of at least 80% of the task force members. The task force spent considerable time and spirited discussions on the diagnosis of CIRCI and the use of corticosteroids for clinical disorders that most clinicians associate with CIRCI: sepsis/septic shock, ARDS, and major trauma.
Diagnosis
The task force was unable to reach agreement on a single test that can reliably diagnose CIRCI. However, they acknowledged that a delta cortisol less than 9 µg/dL at 60 minutes after administration of 250 µg of cosyntropin and a random plasma cortisol level of less than 10 µg/dL may be used by clinicians. They also suggested against the use of plasma-free cortisol or salivary cortisol level over plasma total cortisol. Unequivocally, the panel acknowledged the limitations of the current diagnostic tools to identify patients at risk for CIRCI and how this may impact the way corticosteroids are used in clinical practice.
Sepsis and Septic Shock
Despite dozens of observational studies and randomized controlled trials (RCTs) over several decades, the benefit-to-risk ratio of corticosteroids to treat sepsis and septic shock remains controversial with systematic reviews and meta-analyses either confirming (Annane D, et al. Cochrane Database Syst Rev. 2015;12:CD002243) or refuting (Volbeda M, et al. Intensive Care Med. 2015;41:1220) the survival benefit of corticosteroids. Based on the best available data, the task force recommended the use of corticosteroids in adult patients with septic shock that is not responsive to fluids and moderate-to-high vasopressor therapy but not for patients with sepsis who are not in shock. Intravenous hydrocortisone less than 400 mg/day for at least greater than or equal to 3 days at full dose was recommended rather than high dose and short course. The panel emphasized the consistent benefit of corticosteroids on shock reversal and the low risk for superinfection with low dose corticosteroids.
Since the publication of the updated CIRCI guidelines, two large RCTs (more than 5,000 combined patients) of low-dose corticosteroids in patients with septic shock were reported: The Adjunctive Corticosteroid Treatment in Critically Ill Patients with Septic Shock (ADRENAL) trial (Venkatesh B, et al. N Engl J Med. 2018;378:797) and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial (Annane D, et al. N Engl J Med. 2018;378:809). The ADRENAL trial included 3,800 patients in five countries and did not show a significant difference in 90-day mortality between the hydrocortisone group and the placebo group (27.9% vs 28.8%, respectively, P=.50). In contrast, the APROCCHSS trial, involving 1,241 patients in France, reported a lower 90-day mortality in the hydrocortisone-fludrocortisone group compared with the placebo group (43% vs 49.1%, P=.03). Both trials showed a beneficial effect of hydrocortisone in the number of vasopressor-free and mechanical ventilation-free days. Blood transfusions were less common in the in the hydrocortisone group than among those who received placebo in the ADRENAL trial. Besides hyperglycemia, which was more common in the hydrocortisone group in both trials, the overall rates of adverse events were relatively low.
It is important to highlight the key differences in study design between these two RCTs. First, in the APROCCHSS trial, oral fludrocortisone (50-μg once daily for 7 days) was added to hydrocortisone to provide additional mineralocorticoid potency, although a previous study had shown no added benefit (Annane D, et al. JAMA. 2010;303:341). Second, hydrocortisone was administered as a 50-mg IV bolus every 6 hours in APROCCHSS and given as a continuous infusion of 200 mg/day for 7 days or until death or ICU discharge in ADRENAL. It is noteworthy that the subjects in the ADRENAL trial had a higher rate of surgical admissions (31.5% vs 18.3%), a lower rate of renal-replacement therapy (12.7% vs 27.6%), lower rates of lung infection (35.2% vs 59.4%) and urinary tract infection (7.5% vs 17.7%), and a higher rate of abdominal infection (25.5% vs 11.5%). Patients in the APROCCHSS trial had high Sequential Organ Failure Assessment (SOFA) scores and Simplified Acute Physiology Score (SAPS) II values suggesting a sicker population and probably accounting for the higher mortality rates in both hydrocortisone and placebo groups compared with ADRENAL. In view of the current evidence, the author believes that survival benefit with corticosteroids in septic shock is dependent on several factors: dose (hydrocortisone greater than 400 mg/day), longer duration (at least 3 or more days), and severity of sepsis. “The more severe the sepsis, the more septic shock the patient is in, the more likely it is for corticosteroids to help these patients get off vasopressors and mechanical ventilation. I consider the addition of fludrocortisone as optional.”
ARDS
In patients with early moderate-to-severe ARDS (PaO2/FIO2 of less than 200 and within 14 days of onset), the task force recommended the use of IV methylprednisolone in a dose of 1 mg/kg/day followed by slow tapering over 2 weeks to prevent the development of a rebound inflammatory response, and adherence to infection surveillance. In patients with major trauma and influenza, the panel suggested against the use of corticosteroids. Corticosteroids were recommended for patients with severe community-acquired pneumonia (less than 400 mg/day of IV hydrocortisone or equivalent for 5 to 7 days), meningitis, adults undergoing cardiopulmonary bypass surgery, and adults who suffer a cardiac arrest. The task force highlighted that the quality of evidence for the use of corticosteroids in these disease states was often low and that additional well-designed RCTs with carefully selected patients were warranted.
To conclude, as with any clinical practice guideline, the task force reiterated that the updated CIRCI guidelines were not intended to define a standard of care and should not be interpreted as prescribing an exclusive course of management. Good clinical judgment should always prevail!
Dr. Pastores is Program Director, Critical Care Medicine, Vice-Chair of Education, Department of Anesthesiology and Critical Care Medicine, Memorial Sloan Kettering Cancer Center; Professor of Medicine and Anesthesiology, Weill Cornell Medical College, New York, NY.
Hurricane relief and patient care
In October 2017, in support of the Federal Emergency Management Agency’s response to assist the Governor and people of Puerto Rico, three Department of Defense (DOD) military hospital platforms were deployed; one each, by the US Army, Navy, and Air Force. They arrived on the island at different times with predominantly wartime surgical capabilities and augmented the Federal Emergency Management Agency (FEMA), US Public Health Service, National Guard, and Puerto Rico Department of Health efforts. My perspective is that of patient care and transport between the Centro Medico hospital complex in San Juan, the larger regional hospitals, the Veterans Administration hospital, the DOD response, FEMA Disaster Medical Assistance Teams (DMAT), and FEMA Federal Medical Shelters about 4 to 6 weeks after Hurricanes Maria and Irma struck. Based upon this experience, I would like to offer the following.
Pre-Disaster: All clinicians have a few patients that teeter “on the edge.” When basic services go away, these patients fall over that edge and become inpatients. Establish a list of patients who require oxygen and devices such as vests, cough-assist, or ventilation. If evacuation before the disaster is possible, those patients need to leave. If they refuse, or are unable to leave, they need to be able to supply their own generated power for a prolonged period of time, as batteries will run out prior to power restoration. They must be able to use oxygen concentrators, as tank re-supply may not be readily available. By law, FEMA cannot give generators to individuals, so individuals must prepare for themselves. In a hurricane-prone area where seasonal risk can be established, planning medication refills at the beginning of the season or giving a larger than normal supply may prove useful. In an area prone to sudden disaster, such as earthquake or tornado, then counseling patients to request refills at least 2 weeks early may be adequate.

Post-Disaster: The most reliable form of communication will be text. You likely already have text contacts for your staff and family members; add other providers, responders, planners, pharmacists, and oxygen suppliers to your text contacts. While you may wish to share a text point of contact with patients, understand that your ability to actually help during the initial disaster will likely be limited. Identify possible language translation needs and possible translators among your staff and/or friends as telephone services will be limited or absent following the disaster. Finally, identify your local emergency response planners on Facebook, Twitter, or other social media feeds. This will allow you to direct others to these sites for accurate information after the disaster.
Responder Recommendations: A single social media post can DESTROY your plans and hamper your efforts. Advertise a single contact point and an information resource (eg, bulletin board, webpage) early and often. Publicly and accurately declare the means by which people will access health care and health-care services, such as medications, dialysis, and oxygen. There will be nongovernment organizations (NGOs), friends, and other well-meaning individuals who will try to assist people in need through unconventional channels. Yet, by requesting assistance through nonroutine channels, those efforts tend to delay assistance, cause confusion, and/or squander resources. Continue to direct those requests through the established response channels, ie, the local 911 equivalent.
Plan to use cellular texts to communicate. While satellite telephones are great in concept, in execution, they are difficult to utilize when transmitting complex medical information. If you have an expansive budget, there are now devices available that allow for Iridium satellite-based text communications that require batteries but not intact cellular towers.
Facilities with electricity, water, oxygen, medications, laboratory testing, and CT scanners need to be identified and advertised within the responder community. If FEMA is involved, these resources will be identified and updated on a routine basis. The information will be distributed to their DMAT teams. Those DMAT teams will be distributed throughout the response area. Additionally, if the resources and budgeting are approved, then FEMA will also help re-establish medical transport, as well as Federal Medical Shelters (FMS). The FMS can temporarily house patients who can perform basic activities of daily living but require power, oxygen, or medication administration. For those patients in need of medications without insurance, FEMA may activate medication assistance through the Emergency Prescription Assistance Program. This will allow up to 30 days of medication to be distributed at no cost to the individual through participating pharmacies.
External responders will obviously need to pair with local providers/professionals who can navigate the system and, if necessary, can translate medical terms and care plans. Additionally, external responders will be targets for individuals looking to obtain resources for secondary gain or profit. Establishing a plan or consistently redirecting people to the appropriate resources for those needs may limit the inevitable damage these individuals will cause. Additionally, understand that the efficiencies of the modern society will be gone, and tasks will take much longer than expected. Even if you can communicate by text, the transporting of patients, delivering supplies, meeting with groups, and assessing sites will take far longer than you are used to when none of the stoplights are functional or if gasoline is in limited supply.
Finally, there will be patients for whom no solution, short of an intact, well-resourced medical system, exists—those with severe congenital issues, patients with advanced dementia, patients with advanced cancer, and those with multiple-antibiotic-resistant osteomyelitis are a few of the patients that this response encountered. If transport out of the area is unavailable, NGOs and other charities may be the best, and at times, the only resource for these patients. During this response, I observed NGO and charities helping individual patients and their families with their power, shelter, and medical needs that could not be legally provided by federal government response.
While I hope you may never need to use them, preparations for evacuation, medication, power, and communications before a potential disaster occurs will prove helpful to your patients. After the disaster, consistent and simple communications to the public will be necessary to limit the damage from the social media rumor mill. Working within the organized response framework and leveraging local knowledge and targeted NGO involvement will maximize the effect of your efforts.
In October 2017, in support of the Federal Emergency Management Agency’s response to assist the Governor and people of Puerto Rico, three Department of Defense (DOD) military hospital platforms were deployed; one each, by the US Army, Navy, and Air Force. They arrived on the island at different times with predominantly wartime surgical capabilities and augmented the Federal Emergency Management Agency (FEMA), US Public Health Service, National Guard, and Puerto Rico Department of Health efforts. My perspective is that of patient care and transport between the Centro Medico hospital complex in San Juan, the larger regional hospitals, the Veterans Administration hospital, the DOD response, FEMA Disaster Medical Assistance Teams (DMAT), and FEMA Federal Medical Shelters about 4 to 6 weeks after Hurricanes Maria and Irma struck. Based upon this experience, I would like to offer the following.
Pre-Disaster: All clinicians have a few patients that teeter “on the edge.” When basic services go away, these patients fall over that edge and become inpatients. Establish a list of patients who require oxygen and devices such as vests, cough-assist, or ventilation. If evacuation before the disaster is possible, those patients need to leave. If they refuse, or are unable to leave, they need to be able to supply their own generated power for a prolonged period of time, as batteries will run out prior to power restoration. They must be able to use oxygen concentrators, as tank re-supply may not be readily available. By law, FEMA cannot give generators to individuals, so individuals must prepare for themselves. In a hurricane-prone area where seasonal risk can be established, planning medication refills at the beginning of the season or giving a larger than normal supply may prove useful. In an area prone to sudden disaster, such as earthquake or tornado, then counseling patients to request refills at least 2 weeks early may be adequate.
Post-Disaster: The most reliable form of communication will be text. You likely already have text contacts for your staff and family members; add other providers, responders, planners, pharmacists, and oxygen suppliers to your text contacts. While you may wish to share a text point of contact with patients, understand that your ability to actually help during the initial disaster will likely be limited. Identify possible language translation needs and possible translators among your staff and/or friends as telephone services will be limited or absent following the disaster. Finally, identify your local emergency response planners on Facebook, Twitter, or other social media feeds. This will allow you to direct others to these sites for accurate information after the disaster.
Responder Recommendations: A single social media post can DESTROY your plans and hamper your efforts. Advertise a single contact point and an information resource (eg, bulletin board, webpage) early and often. Publicly and accurately declare the means by which people will access health care and health-care services, such as medications, dialysis, and oxygen. There will be nongovernment organizations (NGOs), friends, and other well-meaning individuals who will try to assist people in need through unconventional channels. Yet, by requesting assistance through nonroutine channels, those efforts tend to delay assistance, cause confusion, and/or squander resources. Continue to direct those requests through the established response channels, ie, the local 911 equivalent.
Plan to use cellular texts to communicate. While satellite telephones are great in concept, in execution, they are difficult to utilize when transmitting complex medical information. If you have an expansive budget, there are now devices available that allow for Iridium satellite-based text communications that require batteries but not intact cellular towers.
Facilities with electricity, water, oxygen, medications, laboratory testing, and CT scanners need to be identified and advertised within the responder community. If FEMA is involved, these resources will be identified and updated on a routine basis. The information will be distributed to their DMAT teams. Those DMAT teams will be distributed throughout the response area. Additionally, if the resources and budgeting are approved, then FEMA will also help re-establish medical transport, as well as Federal Medical Shelters (FMS). The FMS can temporarily house patients who can perform basic activities of daily living but require power, oxygen, or medication administration. For those patients in need of medications without insurance, FEMA may activate medication assistance through the Emergency Prescription Assistance Program. This will allow up to 30 days of medication to be distributed at no cost to the individual through participating pharmacies.
External responders will obviously need to pair with local providers/professionals who can navigate the system and, if necessary, can translate medical terms and care plans. Additionally, external responders will be targets for individuals looking to obtain resources for secondary gain or profit. Establishing a plan or consistently redirecting people to the appropriate resources for those needs may limit the inevitable damage these individuals will cause. Additionally, understand that the efficiencies of the modern society will be gone, and tasks will take much longer than expected. Even if you can communicate by text, the transporting of patients, delivering supplies, meeting with groups, and assessing sites will take far longer than you are used to when none of the stoplights are functional or if gasoline is in limited supply.
Finally, there will be patients for whom no solution, short of an intact, well-resourced medical system, exists—those with severe congenital issues, patients with advanced dementia, patients with advanced cancer, and those with multiple-antibiotic-resistant osteomyelitis are a few of the patients that this response encountered. If transport out of the area is unavailable, NGOs and other charities may be the best, and at times, the only resource for these patients. During this response, I observed NGO and charities helping individual patients and their families with their power, shelter, and medical needs that could not be legally provided by federal government response.
While I hope you may never need to use them, preparations for evacuation, medication, power, and communications before a potential disaster occurs will prove helpful to your patients. After the disaster, consistent and simple communications to the public will be necessary to limit the damage from the social media rumor mill. Working within the organized response framework and leveraging local knowledge and targeted NGO involvement will maximize the effect of your efforts.
In October 2017, in support of the Federal Emergency Management Agency’s response to assist the Governor and people of Puerto Rico, three Department of Defense (DOD) military hospital platforms were deployed; one each, by the US Army, Navy, and Air Force. They arrived on the island at different times with predominantly wartime surgical capabilities and augmented the Federal Emergency Management Agency (FEMA), US Public Health Service, National Guard, and Puerto Rico Department of Health efforts. My perspective is that of patient care and transport between the Centro Medico hospital complex in San Juan, the larger regional hospitals, the Veterans Administration hospital, the DOD response, FEMA Disaster Medical Assistance Teams (DMAT), and FEMA Federal Medical Shelters about 4 to 6 weeks after Hurricanes Maria and Irma struck. Based upon this experience, I would like to offer the following.
Pre-Disaster: All clinicians have a few patients that teeter “on the edge.” When basic services go away, these patients fall over that edge and become inpatients. Establish a list of patients who require oxygen and devices such as vests, cough-assist, or ventilation. If evacuation before the disaster is possible, those patients need to leave. If they refuse, or are unable to leave, they need to be able to supply their own generated power for a prolonged period of time, as batteries will run out prior to power restoration. They must be able to use oxygen concentrators, as tank re-supply may not be readily available. By law, FEMA cannot give generators to individuals, so individuals must prepare for themselves. In a hurricane-prone area where seasonal risk can be established, planning medication refills at the beginning of the season or giving a larger than normal supply may prove useful. In an area prone to sudden disaster, such as earthquake or tornado, then counseling patients to request refills at least 2 weeks early may be adequate.
Post-Disaster: The most reliable form of communication will be text. You likely already have text contacts for your staff and family members; add other providers, responders, planners, pharmacists, and oxygen suppliers to your text contacts. While you may wish to share a text point of contact with patients, understand that your ability to actually help during the initial disaster will likely be limited. Identify possible language translation needs and possible translators among your staff and/or friends as telephone services will be limited or absent following the disaster. Finally, identify your local emergency response planners on Facebook, Twitter, or other social media feeds. This will allow you to direct others to these sites for accurate information after the disaster.
Responder Recommendations: A single social media post can DESTROY your plans and hamper your efforts. Advertise a single contact point and an information resource (eg, bulletin board, webpage) early and often. Publicly and accurately declare the means by which people will access health care and health-care services, such as medications, dialysis, and oxygen. There will be nongovernment organizations (NGOs), friends, and other well-meaning individuals who will try to assist people in need through unconventional channels. Yet, by requesting assistance through nonroutine channels, those efforts tend to delay assistance, cause confusion, and/or squander resources. Continue to direct those requests through the established response channels, ie, the local 911 equivalent.
Plan to use cellular texts to communicate. While satellite telephones are great in concept, in execution, they are difficult to utilize when transmitting complex medical information. If you have an expansive budget, there are now devices available that allow for Iridium satellite-based text communications that require batteries but not intact cellular towers.
Facilities with electricity, water, oxygen, medications, laboratory testing, and CT scanners need to be identified and advertised within the responder community. If FEMA is involved, these resources will be identified and updated on a routine basis. The information will be distributed to their DMAT teams. Those DMAT teams will be distributed throughout the response area. Additionally, if the resources and budgeting are approved, then FEMA will also help re-establish medical transport, as well as Federal Medical Shelters (FMS). The FMS can temporarily house patients who can perform basic activities of daily living but require power, oxygen, or medication administration. For those patients in need of medications without insurance, FEMA may activate medication assistance through the Emergency Prescription Assistance Program. This will allow up to 30 days of medication to be distributed at no cost to the individual through participating pharmacies.
External responders will obviously need to pair with local providers/professionals who can navigate the system and, if necessary, can translate medical terms and care plans. Additionally, external responders will be targets for individuals looking to obtain resources for secondary gain or profit. Establishing a plan or consistently redirecting people to the appropriate resources for those needs may limit the inevitable damage these individuals will cause. Additionally, understand that the efficiencies of the modern society will be gone, and tasks will take much longer than expected. Even if you can communicate by text, the transporting of patients, delivering supplies, meeting with groups, and assessing sites will take far longer than you are used to when none of the stoplights are functional or if gasoline is in limited supply.
Finally, there will be patients for whom no solution, short of an intact, well-resourced medical system, exists—those with severe congenital issues, patients with advanced dementia, patients with advanced cancer, and those with multiple-antibiotic-resistant osteomyelitis are a few of the patients that this response encountered. If transport out of the area is unavailable, NGOs and other charities may be the best, and at times, the only resource for these patients. During this response, I observed NGO and charities helping individual patients and their families with their power, shelter, and medical needs that could not be legally provided by federal government response.
While I hope you may never need to use them, preparations for evacuation, medication, power, and communications before a potential disaster occurs will prove helpful to your patients. After the disaster, consistent and simple communications to the public will be necessary to limit the damage from the social media rumor mill. Working within the organized response framework and leveraging local knowledge and targeted NGO involvement will maximize the effect of your efforts.
Life after angiotensin II
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.

Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).

Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.
Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).
Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
Hypotension is an often-underestimated adversary. Even brief periods of intraoperative mean arterial pressure (MAP) <65 mm Hg increase the odds of both myocardial ischemia and acute kidney injury in the postoperative period. The threshold may be even higher in the postoperative critically ill population (Khanna, et al. Crit Care Med. 2018;46(1):71). Hypotension that is refractory to high-dose vasopressors is associated with an all-cause mortality of 50% to 80%.
The vasopressor toolbox centers around escalating doses of catecholamines with or without the addition of vasopressin. High-dose catecholamines, albeit a frequent choice, is associated with adverse cardiac events (Schmittinger, et al. Intensive Care Med. 2012;38[6]:950) and is an independent predictor of ICU mortality (Sviri, et al. J Crit Care. 2014;29[1]:157).
The evidence behind angiotensin II
Angiotensin II (AT II) is a naturally occurring hormone in the renin-angiotensin-aldosterone (RAA) system that modulates blood pressure through direct arterial vasoconstriction and direct stimulation of the kidneys and adrenal cortex to release vasopressin and aldosterone, respectively.
Positive results from the recent phase 3 trial for AT II have offered hope that this agent would add the needed balance to the current scarcity of vasopressor options (Khanna, et al. N Engl J Med. 2017;377[5]:419). AT II would provide the missing piece in the jigsaw that would allow the intensivist to manage refractory hypotension, while keeping a multimodal vasopressor dosing regimen within therapeutic limits.
Irvine Page and coworkers are credited with most of the initial work on AT II, which they did nearly 70 years ago. Anecdotal use in humans has been reported since the early 1960s (Del Greco, et al. JAMA 1961;178:994). After a prolonged period of quiescence, the Angiotensin II in High-Output Shock (ATHOS) pilot study, which was done in 2014 as a single-center “proof of concept” study of 20 patients, reinvigorated clinical enthusiasm for this agent (Chawla, et al. Crit Care. 2014;18[5]:534). ATHOS demonstrated the effectiveness of AT II at decreasing norepinephrine (NE) requirements of patients in vasodilatory shock (mean NE dose in AT II group 7.4 ug/min vs 27.6 ug/min in placebo, P=.06). These promising results were followed by ATHOS-3, a phase 3, double-blind, multicenter randomized controlled trial of stable human synthetic AT II. This trial was conducted under a special protocol assessment agreement with the US Food and Drug Administration (FDA). A total of 344 patients with predefined criteria for vasodilatory shock were randomized to AT II or placebo as the intention-to-treat population. The primary end-point was a response in MAP by hour 3 of AT II initiation; response was defined as either a MAP rise to 75 mm Hg or an increase in MAP ≥ 10 mm Hg. The primary end-point was reached more frequently in the AT II group than in the placebo group (69.9% AT II vs 23.4% placebo, OR 7.95, 95% CI 4.76-13.3, P<.001). The AT II group had significantly lower cardiovascular sequential organ failure assessment (SOFA) scores at 48 hours and achieved a consistent decrease in background vasopressor doses. Post-hoc data analysis found that the highest benefit was in patients who were AT II deficient (high ratio of AT I:AT II) (Wunderink, et al. Intensive Care Med Exp. 2017;5(Suppl 2):44). The patients who were AT II depleted and received placebo had a higher hazard ratio of death (HR 1.77, 95% CI 1.10-2.85, P=.019), while those who were AT II depleted and received AT II had a decreased risk of mortality (HR 0.64, 95% CI 0.41-1.00, P=.047). The data suggest not only that AT II levels may be predictive of mortality in vasodilatory shock but also that exogenous AT II administration may favorably modulate mortality in this population. Further, a subset data analysis of severely ill patients (APACHE II scores > 30) showed that those who received AT II and standard vasopressors had a significantly lower 28-day mortality compared with patients who only received standard vasopressors (Szerlip, et al. Crit Care Med. 2018;46[1]:3). Considering that the endothelial cells in the lungs and kidneys are locations where AT I is hydrolyzed by angiotensin-converting enzyme (ACE) into AT II, patients receiving ACE-inhibitors and individuals with pulmonary or renal disease are at greatest risk for AT II deficiency. As such, the use of AT II in the extra-corporeal membrane oxygenation (ECMO), post cardiopulmonary bypass, acute respiratory distress syndrome (ARDS), and renal failure populations are of future interest.
Is there a downside?
Appropriate caution is necessary when interpreting these outcomes. One criticism that ATHOS-3 received was the use of a MAP goal of 75 mm Hg, a higher value than currently recommended by clinical guidelines, in the first 3 hours of AT II administration. Because this was a phase 3 trial, both the safety and efficacy of the drug were examined. These goals are difficult to accomplish if simultaneously manipulating other variables. Therefore, to isolate the effects of drug efficacy and safety, a higher MAP goal (75 mm Hg) was established to minimize any effect from varying background vasopressor doses during the first 3 hours of the study.
Furthermore, ATHOS-3 did find an increase in venous and arterial thromboembolic events in patients who received AT II (13% AT II vs 5% placebo). Previously, a systematic review of over 30,000 patients did not report this increased thromboembolic risk (Busse, et al. Crit Care. 2017;21[1]:324). According to the package insert, all patients receiving AT II should receive appropriate thromboembolic prophylaxis if medically indicated.
Where does AT II fit in our algorithm for resuscitation and the vasopressor toolbox?
Data from Wunderink et al indicate a potential mortality benefit in populations who are AT II depleted. However, we can only infer who these patients may be, as no commonly available assay can measure AT I and AT II levels. ATHOS and ATHOS-3 used AT II late during resuscitation, as did the Expanded Access Program (EAP) of the FDA, which gave physicians preliminary access to AT II while it was undergoing FDA review. Using similar inclusion criteria as ATHOS-3, the EAP did not permit patients to receive AT II until doses greater than or equal to 0.2 ug/kg/min of NE-equivalents were reached. In a recently published case report, AT II was successfully used in a patient with septic shock secondary to a colonic perforation (Chow, et al. Accepted for e-publication: A&A Practice. April 2018.). This individual was in vasodilatory shock despite standard resuscitation, 0.48 ug/kg/min of NE, and 0.04 units/min of vasopressin. Methylene blue and hydroxocobalamin had failed to relieve the vasoplegia, and only after the initiation of AT II at 40 ng/kg/min, the patient could be relieved of vasopressors and survived to be discharged from the hospital. In our opinion, best clinical practices would allow for an early multimodal vasopressor regimen that should include AT II at the earliest sign of rapid clinical decline (Jentzer, et al. Chest. 2018. Jan 9. pii: S0012-3692(18)30072-2. doi: 10.1016/j.chest.2017.12.021. [Epub ahead of print]).
Angiotensin II was recently approved by the FDA in December 2017 and is now available on the market for management of vasodilatory shock. This will undoubtedly have a profound impact on the way clinicians treat vasodilatory shock. Previously, we were confined to agents such methylene blue and hydroxocobalamin to rescue patients from profound vasoplegia. However, none of these agents are supported by robust evidence from randomized control trials.
Now, we can openly welcome a new challenger to the campaign, a new hue to the palette of vasopressor colors. This new class of vasopressor makes complete physiological sense and will provide an invaluable tool in our daily battle against sepsis and vasodilatory shock.
Dr. Chow is Assistant Professor, Division of Critical Care Medicine, Department of Anesthesiology, University of Maryland School of Medicine, Baltimore, MD; Dr. Khana is Assistant Professor of Anesthesiology, Staff Intensivist, Vice-Chief for Research, Center for Critical Care, Department of Outcomes Research & General Anesthesiology, Anesthesiology Institute, Cleveland Clinic, Cleveland, OH
Editor’s note
For decades, our options to treat patients with profound vasoplegia have been limited to high-dose catecholamines and vasopressin. Clinicians are often faced with the need to initiate multiple catecholamine agents knowing that these drugs stimulate similar receptors. The recent ATHOS-3 trial introduces AT II as a new option for the management of patients with refractory vasodilatory shock. This drug has a distinct mechanism of action that complements the effect of other vasopressors. Moreover, recent data suggest that this new agent is most beneficial in patients who are AT II deficient. Just like cancer therapies have evolved to precision medicine, will we perhaps face the need to better understand and promptly identify patients with AT II deficiency? For now, we have a new player on our vasopressor team.
Angel Coz, MD, FCCP
Section Editor
Postoperative pulmonary complications of cardiac surgery
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
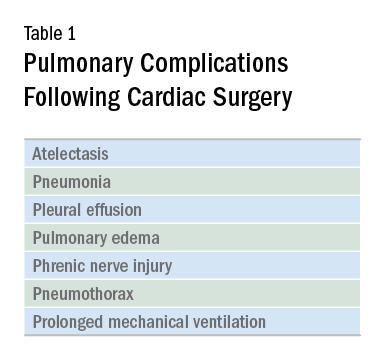
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).

Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).
Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
Cardiac surgery patients are sicker today than in previous decades due to an aging population and a rising complexity in medical care. There is an increasing reliance on noncardiac surgeons to care for these patients. The optimal postoperative providers and structure of the ICU where patients are cared for remain unclear, but what is irrefutable is patients’ increased postoperative morbidity. Pulmonary complications are a leading cause of morbidity in these patients, occurring in up to one-fifth of cases (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Common pulmonary complications of cardiac surgery are listed in Table 1. Those complications, captured by The Society of Thoracic Surgeons (STS) Cardiac Surgery Database, include receiving ventilation longer than 24 hours, pneumonia, pulmonary embolism, and pleural effusion requiring drainage (The Society of Thoracic Surgeons. STS National Database. https://www.sts.org/registries-research-center/sts-national-database. Accessed January 9, 2018).
It should come as no surprise that cardiac surgery can have pronounced effects on lung function. The anesthetic agents, chest wall alteration, and direct lung manipulation can all affect pulmonary parameters. Functional residual capacity (FRC) can decrease by up to 20% with anesthesia (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531), and the thoracic manipulation and alteration of rib cage mechanics with a classic median sternotomy approach can lead to decreases in forced vital capacity (FVC) and expiratory volume in the first second of forced expiration (FEV1) that can last for months after surgery. Use of the cardiopulmonary bypass circuit can also lead to bronchoconstriction. These changes in pulmonary function are less pronounced in alternative surgical approaches, such as partial sternotomies (Weissman C. Seminars in Cardiothoracic and Vascular Anesthesia: Pulmonary Complications After Cardiac Surgery. Glen Head, NY: Westminister Publications; 2004).
The most frequent pulmonary consequence of cardiac surgery is atelectasis, seen on postoperative chest radiographs in approximately 50% to 90% of patients (Szelowski LA, et al. Curr Probl Surg. 2015;52[1]:531). Induction, apnea during cardiopulmonary bypass, manual compression of the lungs for surgical exposure, internal mammary harvesting, and pleurotomy can lead to atelectasis in the intraoperative setting while weak cough, poor inspiratory efforts, interstitial edema, and immobility further contribute postoperatively (Weissman 2004). While frequently seen, clinically significant pulmonary consequences from this radiographic finding alone are rare (Weissman 2004).
Pleural effusions are seen on immediate postoperative chest radiographs in the majority of patients. Additionally, 10% to 40% of patients develop pleural effusions 2 to 3 weeks after surgery secondary to postpericardiotomy syndrome. While some effusions require drainage and further intervention (eg, hemothorax), most effusions require no specific treatment and resolve over time (Weissman 2004).
The prevalence of pneumonia following cardiac surgery varies based on differences in study populations and diagnostic criteria, but it remains an important source of morbidity and mortality. In one series, postoperative pneumonia occurred in 3.1% of patients, with higher rates observed in patients who were older, had worse left ventricular ejection fraction, had COPD, experienced longer bypass times, and received more red blood cell transfusions in the operating room (Allou N, et al. Crit Care Med. 2014;42[5]:1150). A meta-analysis found that an average of 6.37% of patients developed ventilator-associated pneumonia (VAP), and this rose to 35.2% in those receiving ventilation for greater than 48 hours. Those who developed VAP had an odds ratio of dying of 15.18 (95% CI 5.81-39.68) compared with those who did not (He S, et al. J Thorac Cardiovasc Surg. 2014;148[6]:3148).
A small proportion of patients go on to develop ARDS. While relatively uncommon, ARDS carries a high mortality rate. Many possible etiologies for ARDS in cardiac surgery patients have been proposed, including an inflammatory response related to the cardiopulmonary bypass circuit, reperfusion injury secondary to reduced pulmonary blood flow during bypass, protamine administration, transfusion, hypothermia, and lack of ventilation during bypass (Weissman 2004); (Stephens RS, et al. Ann Thorac Surg. 2013;95[3]:1122). Type of surgery may also play a role, as patients who undergo aortic surgery are at an even greater risk (Stephens 2013). As with other cases of ARDS, treatment is supportive: low tidal volume ventilation and careful management of fluid balance, as well as paralysis, prone positioning, and consideration for extracorporeal membrane oxygenation (ECMO), as appropriate (Stephens 2013).
Therapies to prevent postoperative pulmonary complications have included early extubation, aggressive pain control, deep breathing, physical therapy, early mobilization, and noninvasive ventilation in the form of CPAP and intermittent positive pressure breathing. A meta-analysis of 18 trials looking at the use of various forms of prophylactic postoperative physiotherapy did not show a difference in any measured clinical outcome (Pasquina P, Walder B. Br Med J. 2003;327[7428]:1379).
However, the heterogeneity, short follow-up, and low quality of included studies made it difficult to draw meaningful conclusions on the benefit or lack thereof for these therapies. More recent studies have shown promise for chest physiotherapy started several weeks prior to elective coronary bypass graft surgery and extended CPAP via nasal CPAP mask immediately following extubation (Hulzebos EH. JAMA. 2006;296[15]:1851), (Stephens 2013).
Ongoing areas for improvement include further clarification and standardization of best practices for postcardiac surgery patients, including blood product transfusion, optimal tidal volumes for surgical and postsurgical ventilation, timing of extubation, and the use of preventive therapies in the pre- and postsurgical periods. As providers who care for these patients, understanding how we can improve their postoperative pulmonary recovery will allow us to enhance our patient’s experience.
Dr. Noel is a Critical Care Fellow, Cooper Medical School of Rowan University, Camden, New Jersey.
On Diagnosing Sepsis
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.

However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.
However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Two years ago, a panel appointed by the Society of Critical Care Medicine and the European Society of Intensive Care Medicine, referred to as a consensus conference, proposed a new definition for sepsis and new diagnostic criteria for sepsis and septic shock, known as Sepsis-3 (Singer M, et al. JAMA. 2016;315[8]:801). The panel proposed that sepsis be defined as life-threatening organ dysfunction due to a dysregulated host response to infection. Upon reflection, one could see that what we had called definitions of sepsis, severe sepsis, and septic shock for over 2 decades actually represented diagnostic criteria more than concise definitions. In that regard, a concise definition is a useful addition in the tool kit for training all health-care professionals to recognize sepsis and to treat it early and aggressively.
However, the diagnostic criteria leave something to be desired, in terms of both practicality and sensitivity for detecting patients whose infection has made them seriously ill. Those who participate in quality improvement efforts in their own hospitals will recognize that to promote change and to achieve a goal of better, higher quality care, it is important to remove obstacles in the system and to structure it so that doing the right thing is easier than not doing it. For sepsis, the first step in the process, recognizing that sepsis is present, has always been complex enough that it has been the bane of the enterprise. As many as two-thirds of patients with sepsis presenting to the ED with severe sepsis never receive that diagnosis while in the hospital. (Deis AS, et al. Chest. 2018;153[1]:39). As any sepsis core measure coordinator can attest, diagnostic criteria that are readily visible on retrospective examination are often unnoticed or misinterpreted in real time.
The crux of this issue is that the very entity of sepsis is not a definite thing but a not-quite-focused idea. Much is known of pathophysiologic features that seem to be important, but there is no one unifying pathologic condition. Contrast that with another critical illness, myocardial infarction. The very name states the unifying pathology. Our predecessors were able to work backward from an understanding that acute blockage of a small artery led to ischemia and infarction, in order to identify methods to detect it while it is happening—measuring enzymes and evaluating an ECG. For sepsis, we don’t even understand why patients are sick or why they die. There is a complex interaction of inflammation, microcirculatory thrombosis, mitochondrial dysfunction, immune suppression, but there is no one combination of those things that is yet understood in a way that lends itself to diagnostic testing. The best we can say is that the patient reacted to their infection in a way that was detrimental to their own body’s functioning. Rather than recognizing a few symptoms and sending a confirmatory test, with sepsis, we must tote up the signs and symptoms in the domains of recognizing infection and recognizing organ dysfunction, then determine whether they are present in sufficient amounts; it is an exercise that requires mental discipline.
If the diagnostic criteria we use, whether Sepsis-1, 2, or 3, are all gross descriptions of complex internal interactions that are not specific, then the syndrome that any of these criteria identifies is also not specific for anything particular. It falls to the medical community, as a whole, to determine exactly what it is that we desire a given syndrome to be indicative of. The Sepsis-3 authors decided that the appropriate syndrome should predict death or prolonged ICU stay. They used several large data sets to develop and validate infection-associated variables that would have good predictive ability for that outcome, and they compared what they found with sepsis by the Sepsis-1 definition, infection plus SIRS (Seymour C, et al. JAMA. 2016;315[8]:762). Infection + SIRS is a strawman in this comparison, because they tested its predictive ability for the outcome against that of the Sequential Organ Failure Assessment (SOFA) and the Logistic Organ Dysfunction Score (LODS). These two scoring systems were developed as severity of injury scales and validated as mortality predictors; the higher the score, the likelier mortality, whereas SIRS clearly contains no information about organ dysfunction. The comparator of interest for this outcome is actually severe sepsis, infection plus SIRS plus organ dysfunction.
Although the criteria the Sepsis-3 investigators used for defining patients with suspected infection were novel and reasonable, we lack additional important information about the patients they studied. They did not report the spectrum of treatments for sepsis in their cohort, whether early or late, adequate or inadequate, so it is impossible to determine whether the criteria address patients who are undertreated, patients who are treated late, patients who will die regardless of adequate therapy, or some combination. In other words, there is no way to tell whether patients who were recognized early in their course via Sepsis-1 criteria and treated aggressively and effectively may have avoided shock, ICU admission, and death. It is, of course, the business of physicians and nurses to help patients avoid exactly those things. Multiple studies have now demonstrated that SIRS criteria are more sensitive than SOFA-based screens, specifically qSOFA, for identifying infection with organ dysfunction, and that qSOFA is more specific for mortality (Serafim, et al. Chest. 2017; http://dx.doi.org/10.1016/j.chest.2017.12.015).
In contrast, the Sepsis-1 authors proposed infection plus SIRS as a sensitive screening tool that could warn of the possibility of an associated organ dysfunction (Sprung, et al. Crit Care Med. 2017;45[9]:1564). Previous to the Sepsis-1 conference, Bone and colleagues had defined the sepsis syndrome, which incorporated both SIRS and organ dysfunction (Bone, et al. Crit Care Med. 1989;17[5]:389). It was the collective insight of the Sepsis-1 participants to recognize that SIRS induced by infection could be a harbinger of organ failure. The Sepsis-3 authors believe that SIRS is a “normal and adaptive” part of infection and that it is “not useful” in the diagnosis of sepsis. That analysis neglects a couple of important things about SIRS. First, numerous studies demonstrate that infection with SIRS is associated with a mortality rate of 7% to 9%, which is by no means trivial (Rangel-Frausto MS, et al. JAMA. 1995;273[2]:117). Second, the components of SIRS have been recognized as representative of serious illness for millennia; the assertion that the Sepsis-1 definitions are not evidence-based is mistaken and discounts the collective experience of the medical profession.
Finally, SIRS is criticized on the basis of being nonspecific. “If I climb a flight of stairs, I get SIRS.” This is clearly a true statement. In fact, one could propose that the name could more accurately be Systemic Stress Response Syndrome, though “scissors” is certainly less catchy than “sirs” when one says it aloud. However, the critique neglects an important concept, encapsulated in Bayes’ Theorem. The value of any positive test result is largely dependent on the prevalence of the disease being tested for in the population being tested. It is unlikely that the prevalence of sepsis is very high among patients whose SIRS is induced by climbing a flight of stairs. On the other hand, tachycardia and tachypnea in a patient who is indulging in no activity while lying on a bed feeling miserable should prompt a search for both the infection that could be causing it and the organ dysfunction that could be associated with it. The specificity of SIRS derives from the population in which it is witnessed, and its sensitivity is to be respected.
To quote a friend, the remarkable CEO of a small Kansas hospital, “If a patient with an infection feels bad enough that they climb up on that gurney and place themselves at our mercy, we owe it to them to prove why they don’t have sepsis, rather than why they do.”
Editor’s Comment
The progress made in the last several years emphasizes the importance of early identification and aggressive treatment of sepsis. The Third International Consensus Definitions (Sepsis-3) have sparked great controversy in the sepsis community, because they delay the recognition of sepsis until organ damage occurs. In this Critical Care Commentary, Dr. Steven Q. Simpson asserts with solid arguments that the use of a screening tool with higher specificity for mortality, at the expense of sensitivity, is not a step in the right direction. Moving away from criteria that have been widely adopted in clinical trials and quality improvement initiatives throughout the world can be a setback in the battle to improve sepsis outcomes. Until prospectively validated criteria that allow earlier identification of sepsis are developed, there is no compelling reason for change.
Angel Coz, MD, FCCP
Section Editor
Dr. Simpson is Professor, Interim Director; Division of Pulmonary and Critical Care Medicine, University of Kansas, Kansas City, Kansas.
Role of Obstructive Sleep Apnea in HTN
Heart disease and stroke are leading causes of death and disability. High blood pressure (BP) is a major risk factor for both.
The 2017 guidelines regarding “Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure” (JNC 7) were recently published, which is an update incorporating new information from studies regarding BP-related risk of cardiovascular disease (CVD) and strategies to improve hypertension (HTN) treatment and control.
Screening for secondary causes of HTN is necessary for new-onset or uncontrolled HTN in adults, including drug-resistant HTN. Screening includes testing for obstructive sleep apnea, which is highly prevalent in this population.
Obstructive sleep apnea is a common chronic condition characterized by recurrent collapse of upper airways during sleep, inducing intermittent episodes of apnea/hypopnea, hypoxemia, and sleep disruption (Pedrosa RP, et al. Chest. 2013;144[5]:1487).
It is estimated to affect 17% of US adults but is overwhelmingly underrecognized and untreated (JAMA. 2012;307[20]:2169). The prevalence is higher in men than women. The major risk factors for OSA are obesity, male sex, and advancing age. Since these conditions oftentimes predispose to and are concomitant with HTN, it can be challenging to determine the independent effects of OSA on the development of HTN.
The relationship between obstructive sleep apnea (OSA) and HTN has been a point of interest for decades, with untreated OSA being associated with an increased risk for developing new-onset HTN (JAMA. 2012;307[20]:2169).
There have been several landmark trials that have sought to determine the extent of a causal relationship between OSAS and HTN. Sleep Heart Health Study (Sleep. 2006;29;1009) was one such study, which was limited by the inability to prove that OSA preceded the onset of HTN.
Wisconsin Sleep Cohort (N Engl J Med. 2000;342:1378) was another landmark prospective longitudinal study that implicates OSA as a possible causal factor in HTN. The notable limitation of the study was the presence of HTN after initial assessment was found to be dependent upon the severity of OSA at baseline.
While these two cohort studies found an association between OSA and HTN, the Vitoria Sleep Cohort out of Spain (Am J Respir Crit Care Med. 2011;184[11]:1299), the third and most recent longitudinal cohort study, looked at younger and thinner patients than the SHHS and the Wisconsin Sleep Cohort, failed to show a significant association between OSA and incident HTN. Methodologic differences may help to explain the disparity in results.
NREM sleep has normal circadian variation of BP, causing “dipping” of both systolic and diastolic BP at night due to decreased sympathetic and increased parasympathetic activity. REM sleep has predominant sympathetic activity and transient nocturnal BP surges.

OSA results in hypoxemia, which causes nocturnal catecholamine surges, resulting in nocturnal increase in heart rate and BP that is most prominent during post-apneic hyperventilation.
Reduced nocturnal BP (nondipping) or even higher nocturnal BP than daytime BP is an undoubted risk factor for hypertensive patients due to the end-organ damage and subsequent cardiovascular events. With sleep apnea, sleep quality is decreased due to frequent arousal from sleep (Hypertension. 2006;47[5]:833).
Sleep duration of less than or equal to 5 hours per night was shown to significantly increase risk for HTN in patients less than or equal to 60 years of age, even after controlling for obesity and diabetes.
Sleep Heart Health Study suggests that sleep duration above or below a median of 7 to 8 hours per night is associated with a higher prevalence of HTN (Sleep. 2006;29:1009). Thus, improving duration and quality of sleep in sleep apnea patients may help decrease the risk of developing HTN.
Key question: Will treatment of OSA appreciably alter BP?
Continuous positive airway pressure (CPAP) is an efficacious treatment of choice for OSA. Interventional trials, though limited by issues related to compliance, have shown CPAP to acutely reduce sympathetic drive and BP during sleep. However, this improvement in BP control is not entirely consistent in all patients with the data being less clear-cut regarding nighttime CPAP therapy and impact on daytime BP.
A randomized controlled trial from Barbe et al suggests that normotensive subjects with severe OSA but without demonstrable daytime sleepiness are immune to the BP-reducing effects of CPAP (Ann Intern Med. 2001;134:1015); those who were objectively sleepy had a more robust response to the BP lowering effects of CPAP with better cardiovascular outcomes among patients who were adherent to CPAP therapy (≥4 hours per night).
Sleep Apnea Cardiovascular Endpoints (SAVE) study looked at CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea (N Engl J Med. 2016;375:919). CPAP significantly reduced snoring and daytime sleepiness and improved health-related quality of life and mood, but the risk of serious cardiovascular events was not lower among patients who received treatment with CPAP in addition to usual care compared with usual care alone. This study was not powered to provide definitive answers regarding the effects of CPAP on secondary cardiovascular end points, and the use of PAP was less than 4 hours.
A recent systematic review and meta-analysis looked at “Association of Positive Airway Pressure with Cardiovascular Events and Death in Adults with Sleep Apnea” (JAMA. 2017;318(2):156). No significant associations between PAP treatment and a range of cardiovascular events were noted in this meta-analysis.

It is possible that the limited adherence to therapy in many trials was insufficient to drive protection, along with short follow-up duration of most trials that may have given insufficient time for PAP to have affected vascular outcomes.
In a cross-over study of valsartan and CPAP, combining drug treatment with CPAP appeared to have a more synergistic effect in reducing BP than either agent alone (Am J Respir Crit Care Med. 2010;182:954).
The beneficial effect of CPAP remains an open question. Considering the multifactorial pathophysiology of OSA-associated HTN, proven therapies, such as BP lowering, lipid lowering, and antiplatelet therapy, along with PAP therapy, should be utilized. This combination strategy is likely to be more effective in improving both nocturnal and daytime BP control in OSA.
Dr. Singh is Director, Sleep Disorder and Research Center, Michael E. DeBakey VA Medical Center; and Dr. Singh is Assistant Professor and Dr. Velamuri is Associate Professor, Pulmonary, Critical Care and Sleep Medicine, Baylor College of Medicine. Houston, Texas.
Heart disease and stroke are leading causes of death and disability. High blood pressure (BP) is a major risk factor for both.
The 2017 guidelines regarding “Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure” (JNC 7) were recently published, which is an update incorporating new information from studies regarding BP-related risk of cardiovascular disease (CVD) and strategies to improve hypertension (HTN) treatment and control.
Screening for secondary causes of HTN is necessary for new-onset or uncontrolled HTN in adults, including drug-resistant HTN. Screening includes testing for obstructive sleep apnea, which is highly prevalent in this population.
Obstructive sleep apnea is a common chronic condition characterized by recurrent collapse of upper airways during sleep, inducing intermittent episodes of apnea/hypopnea, hypoxemia, and sleep disruption (Pedrosa RP, et al. Chest. 2013;144[5]:1487).
It is estimated to affect 17% of US adults but is overwhelmingly underrecognized and untreated (JAMA. 2012;307[20]:2169). The prevalence is higher in men than women. The major risk factors for OSA are obesity, male sex, and advancing age. Since these conditions oftentimes predispose to and are concomitant with HTN, it can be challenging to determine the independent effects of OSA on the development of HTN.
The relationship between obstructive sleep apnea (OSA) and HTN has been a point of interest for decades, with untreated OSA being associated with an increased risk for developing new-onset HTN (JAMA. 2012;307[20]:2169).
There have been several landmark trials that have sought to determine the extent of a causal relationship between OSAS and HTN. Sleep Heart Health Study (Sleep. 2006;29;1009) was one such study, which was limited by the inability to prove that OSA preceded the onset of HTN.
Wisconsin Sleep Cohort (N Engl J Med. 2000;342:1378) was another landmark prospective longitudinal study that implicates OSA as a possible causal factor in HTN. The notable limitation of the study was the presence of HTN after initial assessment was found to be dependent upon the severity of OSA at baseline.
While these two cohort studies found an association between OSA and HTN, the Vitoria Sleep Cohort out of Spain (Am J Respir Crit Care Med. 2011;184[11]:1299), the third and most recent longitudinal cohort study, looked at younger and thinner patients than the SHHS and the Wisconsin Sleep Cohort, failed to show a significant association between OSA and incident HTN. Methodologic differences may help to explain the disparity in results.
NREM sleep has normal circadian variation of BP, causing “dipping” of both systolic and diastolic BP at night due to decreased sympathetic and increased parasympathetic activity. REM sleep has predominant sympathetic activity and transient nocturnal BP surges.
OSA results in hypoxemia, which causes nocturnal catecholamine surges, resulting in nocturnal increase in heart rate and BP that is most prominent during post-apneic hyperventilation.
Reduced nocturnal BP (nondipping) or even higher nocturnal BP than daytime BP is an undoubted risk factor for hypertensive patients due to the end-organ damage and subsequent cardiovascular events. With sleep apnea, sleep quality is decreased due to frequent arousal from sleep (Hypertension. 2006;47[5]:833).
Sleep duration of less than or equal to 5 hours per night was shown to significantly increase risk for HTN in patients less than or equal to 60 years of age, even after controlling for obesity and diabetes.
Sleep Heart Health Study suggests that sleep duration above or below a median of 7 to 8 hours per night is associated with a higher prevalence of HTN (Sleep. 2006;29:1009). Thus, improving duration and quality of sleep in sleep apnea patients may help decrease the risk of developing HTN.
Key question: Will treatment of OSA appreciably alter BP?
Continuous positive airway pressure (CPAP) is an efficacious treatment of choice for OSA. Interventional trials, though limited by issues related to compliance, have shown CPAP to acutely reduce sympathetic drive and BP during sleep. However, this improvement in BP control is not entirely consistent in all patients with the data being less clear-cut regarding nighttime CPAP therapy and impact on daytime BP.
A randomized controlled trial from Barbe et al suggests that normotensive subjects with severe OSA but without demonstrable daytime sleepiness are immune to the BP-reducing effects of CPAP (Ann Intern Med. 2001;134:1015); those who were objectively sleepy had a more robust response to the BP lowering effects of CPAP with better cardiovascular outcomes among patients who were adherent to CPAP therapy (≥4 hours per night).
Sleep Apnea Cardiovascular Endpoints (SAVE) study looked at CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea (N Engl J Med. 2016;375:919). CPAP significantly reduced snoring and daytime sleepiness and improved health-related quality of life and mood, but the risk of serious cardiovascular events was not lower among patients who received treatment with CPAP in addition to usual care compared with usual care alone. This study was not powered to provide definitive answers regarding the effects of CPAP on secondary cardiovascular end points, and the use of PAP was less than 4 hours.
A recent systematic review and meta-analysis looked at “Association of Positive Airway Pressure with Cardiovascular Events and Death in Adults with Sleep Apnea” (JAMA. 2017;318(2):156). No significant associations between PAP treatment and a range of cardiovascular events were noted in this meta-analysis.
It is possible that the limited adherence to therapy in many trials was insufficient to drive protection, along with short follow-up duration of most trials that may have given insufficient time for PAP to have affected vascular outcomes.
In a cross-over study of valsartan and CPAP, combining drug treatment with CPAP appeared to have a more synergistic effect in reducing BP than either agent alone (Am J Respir Crit Care Med. 2010;182:954).
The beneficial effect of CPAP remains an open question. Considering the multifactorial pathophysiology of OSA-associated HTN, proven therapies, such as BP lowering, lipid lowering, and antiplatelet therapy, along with PAP therapy, should be utilized. This combination strategy is likely to be more effective in improving both nocturnal and daytime BP control in OSA.
Dr. Singh is Director, Sleep Disorder and Research Center, Michael E. DeBakey VA Medical Center; and Dr. Singh is Assistant Professor and Dr. Velamuri is Associate Professor, Pulmonary, Critical Care and Sleep Medicine, Baylor College of Medicine. Houston, Texas.
Heart disease and stroke are leading causes of death and disability. High blood pressure (BP) is a major risk factor for both.
The 2017 guidelines regarding “Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure” (JNC 7) were recently published, which is an update incorporating new information from studies regarding BP-related risk of cardiovascular disease (CVD) and strategies to improve hypertension (HTN) treatment and control.
Screening for secondary causes of HTN is necessary for new-onset or uncontrolled HTN in adults, including drug-resistant HTN. Screening includes testing for obstructive sleep apnea, which is highly prevalent in this population.
Obstructive sleep apnea is a common chronic condition characterized by recurrent collapse of upper airways during sleep, inducing intermittent episodes of apnea/hypopnea, hypoxemia, and sleep disruption (Pedrosa RP, et al. Chest. 2013;144[5]:1487).
It is estimated to affect 17% of US adults but is overwhelmingly underrecognized and untreated (JAMA. 2012;307[20]:2169). The prevalence is higher in men than women. The major risk factors for OSA are obesity, male sex, and advancing age. Since these conditions oftentimes predispose to and are concomitant with HTN, it can be challenging to determine the independent effects of OSA on the development of HTN.
The relationship between obstructive sleep apnea (OSA) and HTN has been a point of interest for decades, with untreated OSA being associated with an increased risk for developing new-onset HTN (JAMA. 2012;307[20]:2169).
There have been several landmark trials that have sought to determine the extent of a causal relationship between OSAS and HTN. Sleep Heart Health Study (Sleep. 2006;29;1009) was one such study, which was limited by the inability to prove that OSA preceded the onset of HTN.
Wisconsin Sleep Cohort (N Engl J Med. 2000;342:1378) was another landmark prospective longitudinal study that implicates OSA as a possible causal factor in HTN. The notable limitation of the study was the presence of HTN after initial assessment was found to be dependent upon the severity of OSA at baseline.
While these two cohort studies found an association between OSA and HTN, the Vitoria Sleep Cohort out of Spain (Am J Respir Crit Care Med. 2011;184[11]:1299), the third and most recent longitudinal cohort study, looked at younger and thinner patients than the SHHS and the Wisconsin Sleep Cohort, failed to show a significant association between OSA and incident HTN. Methodologic differences may help to explain the disparity in results.
NREM sleep has normal circadian variation of BP, causing “dipping” of both systolic and diastolic BP at night due to decreased sympathetic and increased parasympathetic activity. REM sleep has predominant sympathetic activity and transient nocturnal BP surges.
OSA results in hypoxemia, which causes nocturnal catecholamine surges, resulting in nocturnal increase in heart rate and BP that is most prominent during post-apneic hyperventilation.
Reduced nocturnal BP (nondipping) or even higher nocturnal BP than daytime BP is an undoubted risk factor for hypertensive patients due to the end-organ damage and subsequent cardiovascular events. With sleep apnea, sleep quality is decreased due to frequent arousal from sleep (Hypertension. 2006;47[5]:833).
Sleep duration of less than or equal to 5 hours per night was shown to significantly increase risk for HTN in patients less than or equal to 60 years of age, even after controlling for obesity and diabetes.
Sleep Heart Health Study suggests that sleep duration above or below a median of 7 to 8 hours per night is associated with a higher prevalence of HTN (Sleep. 2006;29:1009). Thus, improving duration and quality of sleep in sleep apnea patients may help decrease the risk of developing HTN.
Key question: Will treatment of OSA appreciably alter BP?
Continuous positive airway pressure (CPAP) is an efficacious treatment of choice for OSA. Interventional trials, though limited by issues related to compliance, have shown CPAP to acutely reduce sympathetic drive and BP during sleep. However, this improvement in BP control is not entirely consistent in all patients with the data being less clear-cut regarding nighttime CPAP therapy and impact on daytime BP.
A randomized controlled trial from Barbe et al suggests that normotensive subjects with severe OSA but without demonstrable daytime sleepiness are immune to the BP-reducing effects of CPAP (Ann Intern Med. 2001;134:1015); those who were objectively sleepy had a more robust response to the BP lowering effects of CPAP with better cardiovascular outcomes among patients who were adherent to CPAP therapy (≥4 hours per night).
Sleep Apnea Cardiovascular Endpoints (SAVE) study looked at CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea (N Engl J Med. 2016;375:919). CPAP significantly reduced snoring and daytime sleepiness and improved health-related quality of life and mood, but the risk of serious cardiovascular events was not lower among patients who received treatment with CPAP in addition to usual care compared with usual care alone. This study was not powered to provide definitive answers regarding the effects of CPAP on secondary cardiovascular end points, and the use of PAP was less than 4 hours.
A recent systematic review and meta-analysis looked at “Association of Positive Airway Pressure with Cardiovascular Events and Death in Adults with Sleep Apnea” (JAMA. 2017;318(2):156). No significant associations between PAP treatment and a range of cardiovascular events were noted in this meta-analysis.
It is possible that the limited adherence to therapy in many trials was insufficient to drive protection, along with short follow-up duration of most trials that may have given insufficient time for PAP to have affected vascular outcomes.
In a cross-over study of valsartan and CPAP, combining drug treatment with CPAP appeared to have a more synergistic effect in reducing BP than either agent alone (Am J Respir Crit Care Med. 2010;182:954).
The beneficial effect of CPAP remains an open question. Considering the multifactorial pathophysiology of OSA-associated HTN, proven therapies, such as BP lowering, lipid lowering, and antiplatelet therapy, along with PAP therapy, should be utilized. This combination strategy is likely to be more effective in improving both nocturnal and daytime BP control in OSA.
Dr. Singh is Director, Sleep Disorder and Research Center, Michael E. DeBakey VA Medical Center; and Dr. Singh is Assistant Professor and Dr. Velamuri is Associate Professor, Pulmonary, Critical Care and Sleep Medicine, Baylor College of Medicine. Houston, Texas.