User login
BRUISE CONTROL: Warfarin bests heparin bridging
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
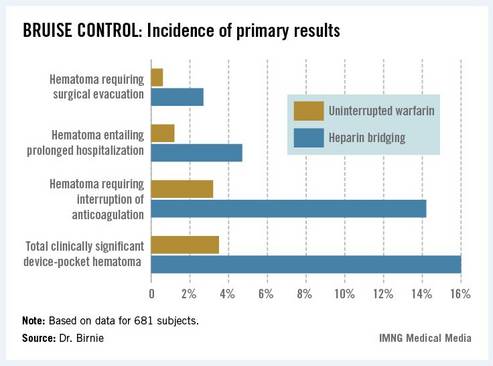
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
DENVER – Uninterrupted warfarin therapy during pacemaker or implantable cardioverter-defibrillator surgery in patients at high thromboembolic risk proved superior to the guideline-recommended practice of discontinuing warfarin and bridging with heparin, according to a large, multicenter, randomized clinical trial.
The primary outcome in the 17-center, 681-patient BRUISE CONTROL (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial) was the incidence of clinically significant device-pocket hematoma. The rate was 16% in patients randomized to heparin bridging, compared with 3.5% with uninterrupted warfarin, Dr. David H. Birnie reported at the annual meeting of the Heart Rhythm Society.
These results are clearly practice changing. Heparin bridging has been the standard of care. It is recommended in this common clinical scenario in all of the major guidelines, but that’s bound to change as a result of BRUISE CONTROL, predicted Dr. Birnie of the University of Ottawa Heart Institute.
"This trial was a home run. It was unequivocally positive," he commented. "For sure, our clinical practice changed as soon as we saw those results."
Device-pocket hematoma is a "very nasty" complication of cardiac device surgery, Dr. Birnie noted. It is quite painful, can cause device infection, and is difficult to treat. Clinically significant device-pocket hematoma was defined in this trial as a hematoma resulting in prolonged hospitalization for an additional day or more, or interruption of oral anticoagulation for at least 24 hours, and/or requiring additional surgery. All three components of the primary endpoint were significantly less frequent in the uninterrupted warfarin group (see chart).
Performing device surgery in patients on uninterrupted warfarin with a median international normalized ratio (INR) of 2.3 was not associated with any increase in major perioperative bleeding or other surgical or thromboembolic complications. And patient satisfaction surveys indicated subjects greatly preferred having their procedure without stopping their warfarin.
BRUISE CONTROL was planned as a definitive 1,000-patient clinical trial. However, the Data and Safety Monitoring Board halted the study after an interim analysis involving the first 681 subjects.
Of note, this was a study restricted to patients at high stroke risk – greater than 5% annually – as defined by the presence of atrial fibrillation and a CHADS2 score of 3 or more or the presence of a mechanical heart valve.
Dr. Birnie said that although it seems counterintuitive to have less bleeding when cardiac device surgery is performed on a fully anticoagulated patient, as occurred in BRUISE CONTROL, he and his coinvestigators have an explanatory hypothesis: "When bleeding occurs in a fully anticoagulated patient, the operator can readily see it and address it. On the contrary, with bridging you get hemostasis at the time of surgery, but you may have missed a tiny little thing, and then 24 hours later when you start up your heparin bridging again, that’s when the bleeding occurs. It’s a physiologically plausible explanation," he said.
In a multivariate analysis, neither the use of pressure dressings, nor the use of sandbags, nor injection of antibleeding agents into the device pocket before closure had any significant impact on the incidence of clinically significant hematomas. Only three factors did: uninterrupted warfarin therapy, which reduced the risk by 84%; aspirin therapy, which doubled the risk; and the presence of diabetes mellitus, which was inexplicably associated with a 52% reduction in hematoma risk.
Each year roughly 1.6 million people worldwide undergo pacemaker or implantable cardioverter-defibrillator (ICD) implantation. Up to one-third of them are on long-term oral anticoagulation, most commonly with warfarin.
Dr. Birnie stressed that the BRUISE CONTROL findings have no relevance to patients on one of the new oral anticoagulants.
"The whole risk/benefit ratio of the new agents is completely different from warfarin. Their onset and offset of action is hours as opposed to the 5 days for warfarin," he noted.
With that point in mind, he and his coinvestigators have just started the BRUISE CONTROL-2 trial, which is examining whether it’s better to stop the new agents around the time of device surgery or continue the medication uninterrupted.
Simultaneous with Dr. Birnie’s presentation of the BRUISE CONTROL data in Denver, the study results were published online (N. Engl. J. Med. 2013 May 9 [doi: 10.1056/NEJMoa1302946]).
BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
Major finding: The incidence of clinically significant device-pocket hematoma in patients at high thromboembolic risk who underwent pacemaker or implantable cardioverter-defibrillator surgery was 3.5% if they remained on full-dose warfarin, compared with 16% with the guideline-recommended, standard-of-care practice of interrupting warfarin in favor of heparin bridging.
Data source: BRUISE CONTROL was a 681-patient, 17-center randomized clinical trial.
Disclosures: BRUISE CONTROL was funded by the Canadian Institutes of Health Research and the Ministry of Health and Long-Term Care of Ontario. Dr. Birnie reported having no conflicts of interest.
Study: Reablate, don't medicate, after failed AF ablation
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
DENVER – After a failed first ablation procedure for paroxysmal atrial fibrillation, redo ablation proved more effective than did antiarrhythmic drug therapy in a randomized trial, Dr. Jonathan Steinberg reported at the annual meeting of the Heart Rhythm Society.
The success rate of a first ablation procedure in patients with symptomatic paroxysmal AF is typically about 60%. The question of what to do for the 40% who are nonresponders has been unclear, with no prior randomized clinical trial evidence available to guide decisions, noted Dr. Steinberg of Columbia University, New York.
The study comprised 154 patients with recurrent symptomatic paroxysmal AF 3 months after an initial ablation procedure involving only pulmonary vein isolation. All participants received an implantable loop recorder to track atrial arrhythmic events.
They were then randomized to redo ablation limited to reisolation of the pulmonary vein, which was successfully accomplished in all instances, or to guideline-based antiarrhythmic drug therapy. The choice of drug was left to individual investigator discretion. The three options were propafenone at 450-900 mg/day, sotalol at 160-320 mg/day, or flecainide at 200-400 mg/day. Propafenone was selected in the majority of cases, at an average dose of 579 mg/day.
The average AF burden as measured by implantable loop recorder at randomization was 15%. The primary study endpoint was AF burden at 36 months of follow-up, which was 5.6% in the redo-ablation group compared with 18.8% in the antiarrhythmic drug group.
Secondary endpoints uniformly favored redo ablation as well (see graphic).
Data from the implantable loop recorders was evaluated every 3 months during 3 years of follow-up. As early as 3 months into the study, the group given antiarrhythmic drugs had an AF burden of 3.3%, significantly higher than the 1.9% rate seen in the redo-ablation group.
Thereafter, the drug therapy group experienced a gradual increase in AF burden throughout the first 12-15 months, followed by a much more substantial increase during the remainder of the study.
"The redo-ablation group had a different pattern" of AF burden, Dr. Steinberg observed. "It was low throughout the first 12-15 months, with just a slight increase, and it then rose only gradually over time until the 36-month end of the study."
Freedom from any atrial tachy-arrhythmia at 1 year was 30% in the antiarrhythmic drug therapy group and 75% in the redo-ablation group. By 3 years, 12% of those in the drug therapy group were free of atrial tachyarrhythmias as were 58% in the redo-ablation group.
Complications in the redo-ablation group consisted of two cases of cardiac tamponade. In contrast, 49 patients, or 64%, in the antiarrhythmic drug therapy group discontinued medication because of intolerance or ineffectiveness.
Session cochair Dr. Gordon Tomaselli of Johns Hopkins University, Baltimore, said that in light of the potential proarrhythmic effects of virtually all antiarrhythmic drugs, it would have been useful to include a no-antiarrhythmic drug control group in the study.
Dr. Steinberg reported having no conflicts of interest.
Major Finding: At 3 years after a first ablation procedure had failed for symptomatic paroxysmal atrial fibrillation, 12% of those randomized to drug therapy and 58% of those in the redo-ablation group were free of atrial tachyarrhythmias.
Data Source: A randomized, prospective, multicenter clinical trial involving 154 patients whose atrial arrhythmia status was monitored via implantable loop recorder.
Disclosures: The presenter reported having no conflicts of interest.
Erythropoietin cuts heart surgery transfusions
MINNEAPOLIS – A single high dose of human recombinant erythropoietin given 2 days before heart surgery reduced blood transfusions by 65%, without substantially increasing mortality or morbidity in the prospective, randomized SHOT trial.
Patients who received 80,000 IU of erythropoietin (Eprex) in a single bolus plus iron supplementation until discharge needed 0.39 blood units/patient, compared with 1.12 units/patient for those receiving standard care involving thromboelastography and tranexamic acid (P less than .001; risk ratio, 0.338).

All-cause mortality at 45 days postoperative was 3.0% with erythropoietin and 3.33%
without it (P = .26), Dr. Luca Weltert said at the annual meeting of the American Association for Thoracic Surgery.
The investigators undertook the SHOT (Blood Sparing Strategies: Single Shot High Dose Erythropoietin Two Days Before Heart Surgery) trial because their original erythropoietin protocol proved too compli- cated for everyday use. The protocol was incorporated
into the 2012 Society of
Thoracic Surgeons and Society of Cardiovascular Anesthesiologists blood conservation clinical practice guidelines, but consisted of five different doses of erythropoietin in 5 days, totaling 52,000 IU, he explained.
Invited discussant Dr. Victor Ferraris, of the Lexington (Ky.) Veterans Affairs Medical Center, questioned whether the high dose of erythropoietin increased thromboembolic events, in light of the black box warnings added to erythropoiesis-stimulating agents due to an increased risk for thromboembolic events in patients with malignancy and an increased risk of serious cardiovascular events in patients with chronic kidney disease when the hemoglobin level exceeds 12 g/dL.
Thromboembolic events were not significantly different between groups, and the trial did not include cancer patients, said Dr. Weltert of the European Hospital in Rome.
Major nonfatal adverse events at 45 days were reported in 4.33% of erythropoietin and 5.67% of control patients (P = .13). Specifically, there were no differences in neurologic complications (4 vs. 5 events, respectively), long-term wound infection (4 in both groups), deep vein thrombosis (2 vs. 5), onset of acute hypertension (1 vs. 2), and new-onset renal failure (2 vs. 1).
Attendees continued this line of questioning, asking how reliable the negative finding is and whether the investigators could have missed a population with a positive result. The efficacy endpoint was based on a minimal sample size of 400 patients, and the trial enrolled 600 consecutive "all comers," Dr. Weltert said, but he added that 3,000 - 4,000 patients would be needed for the safety endpoint, "so the samples are ridiculously small to really state strongly that there is no harm for a subgroup."
The two study arms were well matched, with 35% of all patients undergoing coronary artery bypass grafting, 31% valve surgery, roughly 20% repair of the ascending aorta, and other in about 14%. The mean logistic EuroScore was 9.94 in the erythropoietin group and 10.12 in the control group, and the mean ages were 72.9 and 70.4 years, respectively.
Mean hemoglobin levels on day 4 postoperative was significantly higher in the erythropoietin group than in controls (10.21 vs. 9.01 g/dL; P = .02), Dr. Weltert said.
There were no significant differences between the erythropoietin and control groups in intensive care unit stay (2.71 vs. 2.60 days, respectively), perioperative myocardial infarction (7 vs. 7), cardiac tamponade (3 vs. 5), or need for reintubation (13 vs. 15).
An attendee asked whether hemoglobin levels drifted between days 3 and 4 postoperatively, as this can confound the decision to transfuse after cardiac surgery, and whether the investigators looked at another potential confounder, hemoglobin trigger, since no less than five randomized controlled trials have shown that a hemoglobin level of 7-8 g/dL should be used as a transfusion trigger.
Dr. Weltert said the team picked day 4 to check hemoglobin levels for practical reasons because that’s the day when you see the biggest drift and that the erythropoietin group fared better. He went on to say, "The trigger is extremely important, and I think it will be the very next step in bloodless surgery."
The investigators did compare costs for the two strategies, with standard care ringing up at 336 euros (U.S.$434), vs. 297 euros (U.S.$383) for the erythropoietin protocol (P = .05).
The European Hospital sponsored the study. Dr. Weltert reported no relevant disclosures.
MINNEAPOLIS – A single high dose of human recombinant erythropoietin given 2 days before heart surgery reduced blood transfusions by 65%, without substantially increasing mortality or morbidity in the prospective, randomized SHOT trial.
Patients who received 80,000 IU of erythropoietin (Eprex) in a single bolus plus iron supplementation until discharge needed 0.39 blood units/patient, compared with 1.12 units/patient for those receiving standard care involving thromboelastography and tranexamic acid (P less than .001; risk ratio, 0.338).
All-cause mortality at 45 days postoperative was 3.0% with erythropoietin and 3.33%
without it (P = .26), Dr. Luca Weltert said at the annual meeting of the American Association for Thoracic Surgery.
The investigators undertook the SHOT (Blood Sparing Strategies: Single Shot High Dose Erythropoietin Two Days Before Heart Surgery) trial because their original erythropoietin protocol proved too compli- cated for everyday use. The protocol was incorporated
into the 2012 Society of
Thoracic Surgeons and Society of Cardiovascular Anesthesiologists blood conservation clinical practice guidelines, but consisted of five different doses of erythropoietin in 5 days, totaling 52,000 IU, he explained.
Invited discussant Dr. Victor Ferraris, of the Lexington (Ky.) Veterans Affairs Medical Center, questioned whether the high dose of erythropoietin increased thromboembolic events, in light of the black box warnings added to erythropoiesis-stimulating agents due to an increased risk for thromboembolic events in patients with malignancy and an increased risk of serious cardiovascular events in patients with chronic kidney disease when the hemoglobin level exceeds 12 g/dL.
Thromboembolic events were not significantly different between groups, and the trial did not include cancer patients, said Dr. Weltert of the European Hospital in Rome.
Major nonfatal adverse events at 45 days were reported in 4.33% of erythropoietin and 5.67% of control patients (P = .13). Specifically, there were no differences in neurologic complications (4 vs. 5 events, respectively), long-term wound infection (4 in both groups), deep vein thrombosis (2 vs. 5), onset of acute hypertension (1 vs. 2), and new-onset renal failure (2 vs. 1).
Attendees continued this line of questioning, asking how reliable the negative finding is and whether the investigators could have missed a population with a positive result. The efficacy endpoint was based on a minimal sample size of 400 patients, and the trial enrolled 600 consecutive "all comers," Dr. Weltert said, but he added that 3,000 - 4,000 patients would be needed for the safety endpoint, "so the samples are ridiculously small to really state strongly that there is no harm for a subgroup."
The two study arms were well matched, with 35% of all patients undergoing coronary artery bypass grafting, 31% valve surgery, roughly 20% repair of the ascending aorta, and other in about 14%. The mean logistic EuroScore was 9.94 in the erythropoietin group and 10.12 in the control group, and the mean ages were 72.9 and 70.4 years, respectively.
Mean hemoglobin levels on day 4 postoperative was significantly higher in the erythropoietin group than in controls (10.21 vs. 9.01 g/dL; P = .02), Dr. Weltert said.
There were no significant differences between the erythropoietin and control groups in intensive care unit stay (2.71 vs. 2.60 days, respectively), perioperative myocardial infarction (7 vs. 7), cardiac tamponade (3 vs. 5), or need for reintubation (13 vs. 15).
An attendee asked whether hemoglobin levels drifted between days 3 and 4 postoperatively, as this can confound the decision to transfuse after cardiac surgery, and whether the investigators looked at another potential confounder, hemoglobin trigger, since no less than five randomized controlled trials have shown that a hemoglobin level of 7-8 g/dL should be used as a transfusion trigger.
Dr. Weltert said the team picked day 4 to check hemoglobin levels for practical reasons because that’s the day when you see the biggest drift and that the erythropoietin group fared better. He went on to say, "The trigger is extremely important, and I think it will be the very next step in bloodless surgery."
The investigators did compare costs for the two strategies, with standard care ringing up at 336 euros (U.S.$434), vs. 297 euros (U.S.$383) for the erythropoietin protocol (P = .05).
The European Hospital sponsored the study. Dr. Weltert reported no relevant disclosures.
MINNEAPOLIS – A single high dose of human recombinant erythropoietin given 2 days before heart surgery reduced blood transfusions by 65%, without substantially increasing mortality or morbidity in the prospective, randomized SHOT trial.
Patients who received 80,000 IU of erythropoietin (Eprex) in a single bolus plus iron supplementation until discharge needed 0.39 blood units/patient, compared with 1.12 units/patient for those receiving standard care involving thromboelastography and tranexamic acid (P less than .001; risk ratio, 0.338).
All-cause mortality at 45 days postoperative was 3.0% with erythropoietin and 3.33%
without it (P = .26), Dr. Luca Weltert said at the annual meeting of the American Association for Thoracic Surgery.
The investigators undertook the SHOT (Blood Sparing Strategies: Single Shot High Dose Erythropoietin Two Days Before Heart Surgery) trial because their original erythropoietin protocol proved too compli- cated for everyday use. The protocol was incorporated
into the 2012 Society of
Thoracic Surgeons and Society of Cardiovascular Anesthesiologists blood conservation clinical practice guidelines, but consisted of five different doses of erythropoietin in 5 days, totaling 52,000 IU, he explained.
Invited discussant Dr. Victor Ferraris, of the Lexington (Ky.) Veterans Affairs Medical Center, questioned whether the high dose of erythropoietin increased thromboembolic events, in light of the black box warnings added to erythropoiesis-stimulating agents due to an increased risk for thromboembolic events in patients with malignancy and an increased risk of serious cardiovascular events in patients with chronic kidney disease when the hemoglobin level exceeds 12 g/dL.
Thromboembolic events were not significantly different between groups, and the trial did not include cancer patients, said Dr. Weltert of the European Hospital in Rome.
Major nonfatal adverse events at 45 days were reported in 4.33% of erythropoietin and 5.67% of control patients (P = .13). Specifically, there were no differences in neurologic complications (4 vs. 5 events, respectively), long-term wound infection (4 in both groups), deep vein thrombosis (2 vs. 5), onset of acute hypertension (1 vs. 2), and new-onset renal failure (2 vs. 1).
Attendees continued this line of questioning, asking how reliable the negative finding is and whether the investigators could have missed a population with a positive result. The efficacy endpoint was based on a minimal sample size of 400 patients, and the trial enrolled 600 consecutive "all comers," Dr. Weltert said, but he added that 3,000 - 4,000 patients would be needed for the safety endpoint, "so the samples are ridiculously small to really state strongly that there is no harm for a subgroup."
The two study arms were well matched, with 35% of all patients undergoing coronary artery bypass grafting, 31% valve surgery, roughly 20% repair of the ascending aorta, and other in about 14%. The mean logistic EuroScore was 9.94 in the erythropoietin group and 10.12 in the control group, and the mean ages were 72.9 and 70.4 years, respectively.
Mean hemoglobin levels on day 4 postoperative was significantly higher in the erythropoietin group than in controls (10.21 vs. 9.01 g/dL; P = .02), Dr. Weltert said.
There were no significant differences between the erythropoietin and control groups in intensive care unit stay (2.71 vs. 2.60 days, respectively), perioperative myocardial infarction (7 vs. 7), cardiac tamponade (3 vs. 5), or need for reintubation (13 vs. 15).
An attendee asked whether hemoglobin levels drifted between days 3 and 4 postoperatively, as this can confound the decision to transfuse after cardiac surgery, and whether the investigators looked at another potential confounder, hemoglobin trigger, since no less than five randomized controlled trials have shown that a hemoglobin level of 7-8 g/dL should be used as a transfusion trigger.
Dr. Weltert said the team picked day 4 to check hemoglobin levels for practical reasons because that’s the day when you see the biggest drift and that the erythropoietin group fared better. He went on to say, "The trigger is extremely important, and I think it will be the very next step in bloodless surgery."
The investigators did compare costs for the two strategies, with standard care ringing up at 336 euros (U.S.$434), vs. 297 euros (U.S.$383) for the erythropoietin protocol (P = .05).
The European Hospital sponsored the study. Dr. Weltert reported no relevant disclosures.
AT THE AATS ANNUAL MEETING

Mitral valve bioprosthetic shows durability
NEW YORK – After following more than 400 patients who underwent mitral valve replacement for almost 25 years, French investigators have found that the Carpentier-Edwards Perimount Pericardial prosthetic has an expected durability of more than 16 years, with a low incidence of valve-related complications. The findings were presented by Dr. Thierry Bourguignon as one of the Plenary "Top 10" abstracts at the AATS Mitral Conclave.
"This is a great study. This very-long-term data has been missing for the Carpentier-Edwards Perimount bioprosthetic," said session moderator Dr. David H. Adams of Mount Sinai Medical Center in New York.
In this prospective study, investigators followed 404 patients who underwent mitral valve replacement between August 1984 and March 2011; 46 of these patients eventually needed a second bioprosthesis. Patients were asked to complete yearly clinical questionnaires and undergo an echocardiographic study. Their mean age was 68 years, but the range was 22-89 years. Almost one-fifth of the group was aged 60 years or younger. The mean follow-up time was 7.2 years, although it ranged from 0 to 24.8 years. Ten patients were lost during follow-up, yielding almost a 98% completion rate. Fifty-seven percent were New York Heart Association (NYHA) class III or IV.
The operative mortality rate was 3.3%. A total of 188 patients had late death (5.8%/valve-year). Forty of the deaths were valve related, including 5 due to thromboembolism, 4 to hemorrhage, 4 to endocarditis, 4 to structural valve dysfunction (SVD), and 23 to sudden death. Valve-related survival was more than 60% at 20 years post surgery. Risk factors affecting late survival were age at implant (hazard ratio, 1.06; P less than .001) and preoperative NYHA class III or IV (HR, 1.86; P less than .001).
"Valve-related events, including endocarditis, thromboembolism, and bleeding, were rare," said Dr. Bourguignon, a cardiovascular surgeon at Trousseau Hospital in Chambray Les Tours, France. There were no cases of valve thrombosis.
Seventy-six patients had an SVD, which was defined by echocardiography as severe mitral regurgitation and/or having a mean gradient of more than 8 mm Hg, even if patients were asymptomatic. Of these, 63 were reoperated and 13 died before reoperation. Three-quarters of the valves failed due to calcification, while 20% had late leaflet tears and 4% had mixed problems.
For the entire group, it took an average of 16.6 years before an SVD occurred, although freedom from SVD differed according to age at surgery. Older patients fared better. After 16.6 years, 75% of those over age 70 were expected to be free from SVD, while the rates were lower for those between 60 and 70 years (52%) and those under age 60 (40%). Older patients were also less likely to need the valve removed due to SVD.
What should a surgeon tell a patient about the risk of needing another operation to replace a failing mitral valve bioprosthetic? Using competing risk analysis, the authors predict that, for example, a 60-year-old patient at time of surgery will have a 20% chance of requiring reoperation due to an SVD after 11.9 years.
"In our experience, the CE Perimount valve is a reliable choice for patients older than 60, depending on the accepted risk of reoperation," said Dr. Bourguignon. Addressing a question from the audience, Dr. Bourguignon suggested that although the 25-year data were not available as yet for aortic valve replacements, preliminary findings indicate aortic valve replacement with bioprosthetics lasts longer than mitral valve replacement. At his hospital, patients under age 60 generally receive mechanical valves.
The conclave was sponsored by the American Association for Thoracic Surgery.
Dr. Bourguignon has a financial relationship with Edwards Lifesciences.
NEW YORK – After following more than 400 patients who underwent mitral valve replacement for almost 25 years, French investigators have found that the Carpentier-Edwards Perimount Pericardial prosthetic has an expected durability of more than 16 years, with a low incidence of valve-related complications. The findings were presented by Dr. Thierry Bourguignon as one of the Plenary "Top 10" abstracts at the AATS Mitral Conclave.
"This is a great study. This very-long-term data has been missing for the Carpentier-Edwards Perimount bioprosthetic," said session moderator Dr. David H. Adams of Mount Sinai Medical Center in New York.
In this prospective study, investigators followed 404 patients who underwent mitral valve replacement between August 1984 and March 2011; 46 of these patients eventually needed a second bioprosthesis. Patients were asked to complete yearly clinical questionnaires and undergo an echocardiographic study. Their mean age was 68 years, but the range was 22-89 years. Almost one-fifth of the group was aged 60 years or younger. The mean follow-up time was 7.2 years, although it ranged from 0 to 24.8 years. Ten patients were lost during follow-up, yielding almost a 98% completion rate. Fifty-seven percent were New York Heart Association (NYHA) class III or IV.
The operative mortality rate was 3.3%. A total of 188 patients had late death (5.8%/valve-year). Forty of the deaths were valve related, including 5 due to thromboembolism, 4 to hemorrhage, 4 to endocarditis, 4 to structural valve dysfunction (SVD), and 23 to sudden death. Valve-related survival was more than 60% at 20 years post surgery. Risk factors affecting late survival were age at implant (hazard ratio, 1.06; P less than .001) and preoperative NYHA class III or IV (HR, 1.86; P less than .001).
"Valve-related events, including endocarditis, thromboembolism, and bleeding, were rare," said Dr. Bourguignon, a cardiovascular surgeon at Trousseau Hospital in Chambray Les Tours, France. There were no cases of valve thrombosis.
Seventy-six patients had an SVD, which was defined by echocardiography as severe mitral regurgitation and/or having a mean gradient of more than 8 mm Hg, even if patients were asymptomatic. Of these, 63 were reoperated and 13 died before reoperation. Three-quarters of the valves failed due to calcification, while 20% had late leaflet tears and 4% had mixed problems.
For the entire group, it took an average of 16.6 years before an SVD occurred, although freedom from SVD differed according to age at surgery. Older patients fared better. After 16.6 years, 75% of those over age 70 were expected to be free from SVD, while the rates were lower for those between 60 and 70 years (52%) and those under age 60 (40%). Older patients were also less likely to need the valve removed due to SVD.
What should a surgeon tell a patient about the risk of needing another operation to replace a failing mitral valve bioprosthetic? Using competing risk analysis, the authors predict that, for example, a 60-year-old patient at time of surgery will have a 20% chance of requiring reoperation due to an SVD after 11.9 years.
"In our experience, the CE Perimount valve is a reliable choice for patients older than 60, depending on the accepted risk of reoperation," said Dr. Bourguignon. Addressing a question from the audience, Dr. Bourguignon suggested that although the 25-year data were not available as yet for aortic valve replacements, preliminary findings indicate aortic valve replacement with bioprosthetics lasts longer than mitral valve replacement. At his hospital, patients under age 60 generally receive mechanical valves.
The conclave was sponsored by the American Association for Thoracic Surgery.
Dr. Bourguignon has a financial relationship with Edwards Lifesciences.
NEW YORK – After following more than 400 patients who underwent mitral valve replacement for almost 25 years, French investigators have found that the Carpentier-Edwards Perimount Pericardial prosthetic has an expected durability of more than 16 years, with a low incidence of valve-related complications. The findings were presented by Dr. Thierry Bourguignon as one of the Plenary "Top 10" abstracts at the AATS Mitral Conclave.
"This is a great study. This very-long-term data has been missing for the Carpentier-Edwards Perimount bioprosthetic," said session moderator Dr. David H. Adams of Mount Sinai Medical Center in New York.
In this prospective study, investigators followed 404 patients who underwent mitral valve replacement between August 1984 and March 2011; 46 of these patients eventually needed a second bioprosthesis. Patients were asked to complete yearly clinical questionnaires and undergo an echocardiographic study. Their mean age was 68 years, but the range was 22-89 years. Almost one-fifth of the group was aged 60 years or younger. The mean follow-up time was 7.2 years, although it ranged from 0 to 24.8 years. Ten patients were lost during follow-up, yielding almost a 98% completion rate. Fifty-seven percent were New York Heart Association (NYHA) class III or IV.
The operative mortality rate was 3.3%. A total of 188 patients had late death (5.8%/valve-year). Forty of the deaths were valve related, including 5 due to thromboembolism, 4 to hemorrhage, 4 to endocarditis, 4 to structural valve dysfunction (SVD), and 23 to sudden death. Valve-related survival was more than 60% at 20 years post surgery. Risk factors affecting late survival were age at implant (hazard ratio, 1.06; P less than .001) and preoperative NYHA class III or IV (HR, 1.86; P less than .001).
"Valve-related events, including endocarditis, thromboembolism, and bleeding, were rare," said Dr. Bourguignon, a cardiovascular surgeon at Trousseau Hospital in Chambray Les Tours, France. There were no cases of valve thrombosis.
Seventy-six patients had an SVD, which was defined by echocardiography as severe mitral regurgitation and/or having a mean gradient of more than 8 mm Hg, even if patients were asymptomatic. Of these, 63 were reoperated and 13 died before reoperation. Three-quarters of the valves failed due to calcification, while 20% had late leaflet tears and 4% had mixed problems.
For the entire group, it took an average of 16.6 years before an SVD occurred, although freedom from SVD differed according to age at surgery. Older patients fared better. After 16.6 years, 75% of those over age 70 were expected to be free from SVD, while the rates were lower for those between 60 and 70 years (52%) and those under age 60 (40%). Older patients were also less likely to need the valve removed due to SVD.
What should a surgeon tell a patient about the risk of needing another operation to replace a failing mitral valve bioprosthetic? Using competing risk analysis, the authors predict that, for example, a 60-year-old patient at time of surgery will have a 20% chance of requiring reoperation due to an SVD after 11.9 years.
"In our experience, the CE Perimount valve is a reliable choice for patients older than 60, depending on the accepted risk of reoperation," said Dr. Bourguignon. Addressing a question from the audience, Dr. Bourguignon suggested that although the 25-year data were not available as yet for aortic valve replacements, preliminary findings indicate aortic valve replacement with bioprosthetics lasts longer than mitral valve replacement. At his hospital, patients under age 60 generally receive mechanical valves.
The conclave was sponsored by the American Association for Thoracic Surgery.
Dr. Bourguignon has a financial relationship with Edwards Lifesciences.
AT THE 2013 MITRAL CONCLAVE
Major finding: The mean durability of the Carpentier-Edwards Perimount Pericardial prosthetic was 16.6 years before a structural valve dysfunction occurred.
Data source: Prospective study of 404 patients who underwent mitral valve replacement using the Perimount prosthetic.
Disclosures: Dr. Bourguignon has a financial relationship with Edwards Lifesciences.
Coronary-intervention caseload for competency cut
The panel of cardiologists who revised U.S. competency standards for coronary interventionalists acknowledged prevailing case-volume realities and cut the minimum number of cases recommended for active operators to 50 per year, down from a long-standing recommendation of 75 coronary interventions annually.
The Clinical Competence and Training Task Force, assembled by the American College of Cardiology, the American Heart Association, and the Society for Cardiovascular Angiography and Interventions (SCAI), also made the linked change of halving prior recommended coronary case volumes for catheterization-laboratory centers, with a new recommended minimum target of 200 cases per year per site, down from the 400 annual cases called for in the 2007 version of the clinical competence statement for cardiac interventions.
The 2013 revision, with a focus specifically on coronary-artery interventions, appeared online on the websites of all three organizations (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc.2013.05.002]).
Although the revised case numbers will likely be what first captures the attention, these changes were not the most central to the new revision, said Dr. Theodore A. Bass, professor and chief of cardiology at the University of Florida, Jacksonville, and vice chair of the task force. He focused on the diverse, 35-item list of core competency components that is the backbone of the new revision.
"It’s a much broader view of what competency involves," he said in an interview. "In the past, competency was taking an exam, or having a certain knowledge base. But now we realize that other skills are also extremely important," such as appropriate patient selection, using technologies in a safe and appropriate manner, and delivering patient-centered care, said Dr. Bass, president-elect of SCAI.
The new statement "is the first cardiovascular competency statement to fully utilize the six-domains structure promulgated by the Accreditation Council of Graduate Medical Education and adopted and endorsed by the American Board of Internal Medicine," Dr. John Gordon Harold, chair of the writing committee, said in a written statement.
"It goes beyond medical knowledge and procedure performance to include the important issues of leading an interdisciplinary team, working in a complex system, communicating effectively, engaging in continuous quality improvement at individual and system levels, adhering to evidence-based medicine, and demonstrating the highest levels of professionalism," said Dr. Harold, who is also ACC president and a cardiologist at Cedars-Sinai Heart Institute in Los Angeles.
Although the new statement is wide ranging, the case-number issue stands out as something the task force worked on at length, with a quarter of the statement devoted to various aspects of the issue for both individual operators and for cath labs.
"Volume has been used as a surrogate for quality because it measurable, but there has never been clear data that there is a strong correlation," said Dr. Bass. Plus, the original individual volume number, the venerable figure of 75 cases per year that has been around since at least 1990, when it appeared in the first clinical competency statement, "was not data based; it was judgment based," he noted. "Volume is not the be all and end all. We thought that 100 cases over 2 years seemed in the sweet spot for all considerations."
"Low-volume operators can self- restrict what they do and get very good outcomes with less volume," said Dr. Christopher J. White, professor and chair for cardiovascular diseases at the Ochsner Clinic, New Orleans, and a member of the task force. "Rather than use an arbitrary volume as a surrogate for quality, it is more useful to actually measure quality, with a tool like the NCDR [National Cardiovascular Data Registry] for CathPCI."
Another issue is the feasibility of calling for annual rates of 75 cases individually and 400 per center, given recent trends with substantially fewer U.S. percutaneous coronary interventions (PCI), compared with the mid-2000s, and the growing number of interventionalists. The statement notes that "a majority of interventional cardiologists in the United States are not achieving the previously recommended threshold of 75 PCIs annually."
Also, these days most interventional cardiologists perform other types of procedures that may not count as PCIs but still keep their skills sharp – things like peripheral vascular procedures, carotid stenting, and trans- catheter aortic valve replacements, Dr. Bass said. And there is the issue of society’s need to have important acute care services like primary PCI for myocardial infarctions available in even remote areas, where higher case volumes are hard to maintain.
"Cath labs with fewer than 200 cases per year should examine what they do and have stringent quality assurance measures in place," he said.
Two other notable changes in the 2013 competency revision are the inclusion for the first time of radial-artery access as an identified competency. Radial access "is still just under 20% of all U.S. PCI, but that’s up exponentially from a few years ago," Dr. Bass said. "There is a huge amount of patient preference for it, and the next generation is now trained in it. Radial access is the future."
The new revision focused exclusively on coronary artery interventions, with other common cardiac percutaneous procedures like valvuloplasty on hold for a different competency task force that will deal with structural and noncoronary interventions, Dr. Bass said.
Dr. Bass, Dr. Harold, and Dr. White had no relevant disclosures.
The panel of cardiologists who revised U.S. competency standards for coronary interventionalists acknowledged prevailing case-volume realities and cut the minimum number of cases recommended for active operators to 50 per year, down from a long-standing recommendation of 75 coronary interventions annually.
The Clinical Competence and Training Task Force, assembled by the American College of Cardiology, the American Heart Association, and the Society for Cardiovascular Angiography and Interventions (SCAI), also made the linked change of halving prior recommended coronary case volumes for catheterization-laboratory centers, with a new recommended minimum target of 200 cases per year per site, down from the 400 annual cases called for in the 2007 version of the clinical competence statement for cardiac interventions.
The 2013 revision, with a focus specifically on coronary-artery interventions, appeared online on the websites of all three organizations (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc.2013.05.002]).
Although the revised case numbers will likely be what first captures the attention, these changes were not the most central to the new revision, said Dr. Theodore A. Bass, professor and chief of cardiology at the University of Florida, Jacksonville, and vice chair of the task force. He focused on the diverse, 35-item list of core competency components that is the backbone of the new revision.
"It’s a much broader view of what competency involves," he said in an interview. "In the past, competency was taking an exam, or having a certain knowledge base. But now we realize that other skills are also extremely important," such as appropriate patient selection, using technologies in a safe and appropriate manner, and delivering patient-centered care, said Dr. Bass, president-elect of SCAI.
The new statement "is the first cardiovascular competency statement to fully utilize the six-domains structure promulgated by the Accreditation Council of Graduate Medical Education and adopted and endorsed by the American Board of Internal Medicine," Dr. John Gordon Harold, chair of the writing committee, said in a written statement.
"It goes beyond medical knowledge and procedure performance to include the important issues of leading an interdisciplinary team, working in a complex system, communicating effectively, engaging in continuous quality improvement at individual and system levels, adhering to evidence-based medicine, and demonstrating the highest levels of professionalism," said Dr. Harold, who is also ACC president and a cardiologist at Cedars-Sinai Heart Institute in Los Angeles.
Although the new statement is wide ranging, the case-number issue stands out as something the task force worked on at length, with a quarter of the statement devoted to various aspects of the issue for both individual operators and for cath labs.
"Volume has been used as a surrogate for quality because it measurable, but there has never been clear data that there is a strong correlation," said Dr. Bass. Plus, the original individual volume number, the venerable figure of 75 cases per year that has been around since at least 1990, when it appeared in the first clinical competency statement, "was not data based; it was judgment based," he noted. "Volume is not the be all and end all. We thought that 100 cases over 2 years seemed in the sweet spot for all considerations."
"Low-volume operators can self- restrict what they do and get very good outcomes with less volume," said Dr. Christopher J. White, professor and chair for cardiovascular diseases at the Ochsner Clinic, New Orleans, and a member of the task force. "Rather than use an arbitrary volume as a surrogate for quality, it is more useful to actually measure quality, with a tool like the NCDR [National Cardiovascular Data Registry] for CathPCI."
Another issue is the feasibility of calling for annual rates of 75 cases individually and 400 per center, given recent trends with substantially fewer U.S. percutaneous coronary interventions (PCI), compared with the mid-2000s, and the growing number of interventionalists. The statement notes that "a majority of interventional cardiologists in the United States are not achieving the previously recommended threshold of 75 PCIs annually."
Also, these days most interventional cardiologists perform other types of procedures that may not count as PCIs but still keep their skills sharp – things like peripheral vascular procedures, carotid stenting, and trans- catheter aortic valve replacements, Dr. Bass said. And there is the issue of society’s need to have important acute care services like primary PCI for myocardial infarctions available in even remote areas, where higher case volumes are hard to maintain.
"Cath labs with fewer than 200 cases per year should examine what they do and have stringent quality assurance measures in place," he said.
Two other notable changes in the 2013 competency revision are the inclusion for the first time of radial-artery access as an identified competency. Radial access "is still just under 20% of all U.S. PCI, but that’s up exponentially from a few years ago," Dr. Bass said. "There is a huge amount of patient preference for it, and the next generation is now trained in it. Radial access is the future."
The new revision focused exclusively on coronary artery interventions, with other common cardiac percutaneous procedures like valvuloplasty on hold for a different competency task force that will deal with structural and noncoronary interventions, Dr. Bass said.
Dr. Bass, Dr. Harold, and Dr. White had no relevant disclosures.
The panel of cardiologists who revised U.S. competency standards for coronary interventionalists acknowledged prevailing case-volume realities and cut the minimum number of cases recommended for active operators to 50 per year, down from a long-standing recommendation of 75 coronary interventions annually.
The Clinical Competence and Training Task Force, assembled by the American College of Cardiology, the American Heart Association, and the Society for Cardiovascular Angiography and Interventions (SCAI), also made the linked change of halving prior recommended coronary case volumes for catheterization-laboratory centers, with a new recommended minimum target of 200 cases per year per site, down from the 400 annual cases called for in the 2007 version of the clinical competence statement for cardiac interventions.
The 2013 revision, with a focus specifically on coronary-artery interventions, appeared online on the websites of all three organizations (J. Am. Coll. Cardiol. 2013 [doi:10.1016/j.jacc.2013.05.002]).
Although the revised case numbers will likely be what first captures the attention, these changes were not the most central to the new revision, said Dr. Theodore A. Bass, professor and chief of cardiology at the University of Florida, Jacksonville, and vice chair of the task force. He focused on the diverse, 35-item list of core competency components that is the backbone of the new revision.
"It’s a much broader view of what competency involves," he said in an interview. "In the past, competency was taking an exam, or having a certain knowledge base. But now we realize that other skills are also extremely important," such as appropriate patient selection, using technologies in a safe and appropriate manner, and delivering patient-centered care, said Dr. Bass, president-elect of SCAI.
The new statement "is the first cardiovascular competency statement to fully utilize the six-domains structure promulgated by the Accreditation Council of Graduate Medical Education and adopted and endorsed by the American Board of Internal Medicine," Dr. John Gordon Harold, chair of the writing committee, said in a written statement.
"It goes beyond medical knowledge and procedure performance to include the important issues of leading an interdisciplinary team, working in a complex system, communicating effectively, engaging in continuous quality improvement at individual and system levels, adhering to evidence-based medicine, and demonstrating the highest levels of professionalism," said Dr. Harold, who is also ACC president and a cardiologist at Cedars-Sinai Heart Institute in Los Angeles.
Although the new statement is wide ranging, the case-number issue stands out as something the task force worked on at length, with a quarter of the statement devoted to various aspects of the issue for both individual operators and for cath labs.
"Volume has been used as a surrogate for quality because it measurable, but there has never been clear data that there is a strong correlation," said Dr. Bass. Plus, the original individual volume number, the venerable figure of 75 cases per year that has been around since at least 1990, when it appeared in the first clinical competency statement, "was not data based; it was judgment based," he noted. "Volume is not the be all and end all. We thought that 100 cases over 2 years seemed in the sweet spot for all considerations."
"Low-volume operators can self- restrict what they do and get very good outcomes with less volume," said Dr. Christopher J. White, professor and chair for cardiovascular diseases at the Ochsner Clinic, New Orleans, and a member of the task force. "Rather than use an arbitrary volume as a surrogate for quality, it is more useful to actually measure quality, with a tool like the NCDR [National Cardiovascular Data Registry] for CathPCI."
Another issue is the feasibility of calling for annual rates of 75 cases individually and 400 per center, given recent trends with substantially fewer U.S. percutaneous coronary interventions (PCI), compared with the mid-2000s, and the growing number of interventionalists. The statement notes that "a majority of interventional cardiologists in the United States are not achieving the previously recommended threshold of 75 PCIs annually."
Also, these days most interventional cardiologists perform other types of procedures that may not count as PCIs but still keep their skills sharp – things like peripheral vascular procedures, carotid stenting, and trans- catheter aortic valve replacements, Dr. Bass said. And there is the issue of society’s need to have important acute care services like primary PCI for myocardial infarctions available in even remote areas, where higher case volumes are hard to maintain.
"Cath labs with fewer than 200 cases per year should examine what they do and have stringent quality assurance measures in place," he said.
Two other notable changes in the 2013 competency revision are the inclusion for the first time of radial-artery access as an identified competency. Radial access "is still just under 20% of all U.S. PCI, but that’s up exponentially from a few years ago," Dr. Bass said. "There is a huge amount of patient preference for it, and the next generation is now trained in it. Radial access is the future."
The new revision focused exclusively on coronary artery interventions, with other common cardiac percutaneous procedures like valvuloplasty on hold for a different competency task force that will deal with structural and noncoronary interventions, Dr. Bass said.
Dr. Bass, Dr. Harold, and Dr. White had no relevant disclosures.
Big trials show no advantage to using off-pump CABG
SAN FRANCISCO - Off-pump coronary artery bypass graft surgery offered no advantages over on-pump CABG in any major end points at 1 year of follow-up in two major prospective randomized trials totaling more than 7,000 patients.
Among the key 1-year outcomes - which didn't differ between off- and on-pump CABG patients in the GOPCABE and CORONARY trials - were death, MI, stroke, neurocognitive function, quality of life, renal failure, and repeat revascularization, investigators reported at the annual meeting of the American College of Cardiology.

A third randomized trial presented at the same session of the ACC meeting did find a significant outcome advantage favoring off-pump CABG in high-operative-risk patients at 1 year. However, experts discounted this Czech study because it was small, single center, reported only 30-day results, and the advantage found for off-pump surgery hinged on an outdated and inadequate definition of MI.
The new study findings signal a striking fall from grace for off-pump CABG. Not long ago, this technique, while controversial, was viewed by many as a progressive development within heart surgery, one that would revitalize a mature operation whose annual case numbers were declining in the face of stiff competition from percutaneous coronary intervention by cardiologists. Off-pump CABG was an innovation designed to avoid the perioperative complications related to aortic cross-clamping and the heart-lung machine, including the lingering neurocognitive dysfunction known informally in surgical circles as "pump head."
However, the resounding lack of any demonstrable advantages for off-pump CABG in the two large trials presented in San Francisco left analysts scratching their heads as to the role remaining for this beating heart surgical technique, which is more difficult to learn and perform skillfully than on-pump bypass.
Discussant Dr. Michael J. Mack, a cardiac surgeon, voiced a similar sentiment. "I was an early advocate of off-pump surgery. But as a card-carrying off-pump bypass surgeon, it's getting harder and harder for me to maintain enthusiasm for a potential benefit from this," declared Dr. Mack, medical director of cardiovascular surgery for the Baylor Health Care System and director of cardiovascular research at the Heart Hospital in Plano, Tex. He noted that the CORONARY and GOPCABE trials follow upon the earlier ROOBY (Randomized On/Off Bypass) trial, which actually showed worse outcomes in the off-pump group. ROOBY enrolled 2,203 Veterans Affairs patients, with the off-pump CABG group having a significantly higher 1-year rate of the primary composite endpoint comprising death, nonfatal MI, or repeat revascularization, along with worse graft patency (N. Engl. J. Med. 2009;361:1827-37).
GOPCABE and CORONARY were designed in part to answer critics of ROOBY, who have argued that the VA trial used insufficiently experienced off-pump CABG surgeons and featured a patient population at too low an operative risk to detect a signal of benefit favoring off-pump surgery. The GOPCABE (German Off-Pump Coronary Artery Bypass Grafting in Elderly Patients) study involved 2,539 patients aged 75 years or older randomized at 12 German centers. A total of 60% of patients had triple-vessel disease, and no one was excluded from the trial because of left ventricular function or coronary artery anatomy. Participating surgeons were highly experienced. Those who performed off-pump CABG in the study had previously done an average of 514 of them, while the on-pump surgeons had done an average of 1,378 of those operations.
"We wanted to have the best off-pump vs. the best on-pump surgeons, like in a competition," explained Dr. Anno Diegeler, a surgeon at the Bad Neustadt (Germany) Heart Center.

He and his coinvestigators conducted GOPCABE because they believed it would be easier to show advantages for off-pump CABG in a population at high operative risk, such as elderly patients with many comorbidities. Indeed, the study hypothesis was that the off-pump group would show a robust 30% reduction in the primary endpoint, a composite of death, stroke, myocardial infarction, repeat revascularization, or new renal-replacement therapy at 1 year.
That didn't happen. The 30-day rate of the primary endpoint was 7.8% in the off-pump group and 8.2% with on-pump CABG, while the 1-year rates were 13.1% and 14.0%, respectively. None of the individual components of the composite endpoint differed significantly between the groups, either.
Neurocognitive function wasn't measured in GOPCABE, but it was in CORONARY (the CABG Off or On Pump Revascularization Study), which involved 4,752 randomized patients in 19 countries.
Dr. Andre Lamy presented the 1-year results. The primary endpoint was a composite of death, MI, stroke, or new renal failure requiring dialysis. The rate was 12.1% in patients in the off-pump group and similar at 13.3% in the on-pump group. As in GOPCABE, the surgeons participating in CORONARY were highly proficient. They had to have at least 2 years' experience as a staff cardiac surgeon and at least 100 prior cases of whichever operation they were assigned to. The vast majority met that standard for both procedures.
Quality of life and neurocognitive tests showed significant difference between the two groups.
However, neurocognitive testing was declared optional because it's so time consuming, and many patients opted out. For example, only 1,273 of the original 4,752 patients returned to take the Montreal Cognitive Assessment at 1 year, noted Dr. Lamy, a heart surgeon at the Population Health Research Institute at McMaster University in Hamilton, Ont.
Discussant Dr. Bernard Gersh zeroed in on the incomplete neurocognitive testing. "I think this is really a significant limitation. There's a huge bias. If there's any advantage to off-pump CABG, it may be in neurocognitive dysfunction," commented Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.
In response to questioning as to where off-pump CABG fits into clinical practice in light of the disappointing CORONARY and GOPCABE findings, Dr. Diegeler said he remains convinced that some high-operative-risk patients - those with aortic calcification or other evidence of generalized vascular disease - do benefit preferentially from off-pump surgery when performed by expert surgeons.
Dr. Lamy said a post hoc analysis of the CORONARY data showed that low-operative-risk patients as defined by a EuroScore of 0-2 tended to do better with on- than off-pump CABG, while the converse was true in those with moderate- or high-risk scores.
"In my personal practice now, my low-risk patients go on-pump and my moderate- and high-risk patients go off-pump," he added.
Dr. Jan Hlavicka presented the results of the PRAGUE-6 trial, in which 206 patients at high operative risk - a EuroScore of 6 or greater - were randomized to off- or on-pump CABG at Charles University, Prague. The operations were performed by five surgeons proficient in both procedures. The 30-day primary composite endpoint comprising death, MI, stroke, or new renal failure requiring dialysis occurred in 20.6% of the on-pump group, compared with 9.2% of off-pump patients.
The off-pump group required significantly fewer RBC transfusions. There were no significant differences between the two groups in terms of average hospital length of stay, wound infection rates, or total hospital costs.
Dr. Gersh noted that the only significant difference between the two groups in the individual components of the primary endpoint was in acute MI rates: 12.1% in the on- vs. 4.1% in the off-pump CABG group. He took issue with the Czech investigators? use of the 2004 Society of Thoracic Surgeons definition of acute MI. That's not sufficiently stringent. It surely captures many patients who don't really have an acute MI. The data should be reanalyzed using a contemporary definition which requires new Q waves, he added.
Dr. Hlavicka, Dr. Lamy, and Dr. Diegeler declared no conflicts.
SAN FRANCISCO - Off-pump coronary artery bypass graft surgery offered no advantages over on-pump CABG in any major end points at 1 year of follow-up in two major prospective randomized trials totaling more than 7,000 patients.
Among the key 1-year outcomes - which didn't differ between off- and on-pump CABG patients in the GOPCABE and CORONARY trials - were death, MI, stroke, neurocognitive function, quality of life, renal failure, and repeat revascularization, investigators reported at the annual meeting of the American College of Cardiology.
A third randomized trial presented at the same session of the ACC meeting did find a significant outcome advantage favoring off-pump CABG in high-operative-risk patients at 1 year. However, experts discounted this Czech study because it was small, single center, reported only 30-day results, and the advantage found for off-pump surgery hinged on an outdated and inadequate definition of MI.
The new study findings signal a striking fall from grace for off-pump CABG. Not long ago, this technique, while controversial, was viewed by many as a progressive development within heart surgery, one that would revitalize a mature operation whose annual case numbers were declining in the face of stiff competition from percutaneous coronary intervention by cardiologists. Off-pump CABG was an innovation designed to avoid the perioperative complications related to aortic cross-clamping and the heart-lung machine, including the lingering neurocognitive dysfunction known informally in surgical circles as "pump head."
However, the resounding lack of any demonstrable advantages for off-pump CABG in the two large trials presented in San Francisco left analysts scratching their heads as to the role remaining for this beating heart surgical technique, which is more difficult to learn and perform skillfully than on-pump bypass.
Discussant Dr. Michael J. Mack, a cardiac surgeon, voiced a similar sentiment. "I was an early advocate of off-pump surgery. But as a card-carrying off-pump bypass surgeon, it's getting harder and harder for me to maintain enthusiasm for a potential benefit from this," declared Dr. Mack, medical director of cardiovascular surgery for the Baylor Health Care System and director of cardiovascular research at the Heart Hospital in Plano, Tex. He noted that the CORONARY and GOPCABE trials follow upon the earlier ROOBY (Randomized On/Off Bypass) trial, which actually showed worse outcomes in the off-pump group. ROOBY enrolled 2,203 Veterans Affairs patients, with the off-pump CABG group having a significantly higher 1-year rate of the primary composite endpoint comprising death, nonfatal MI, or repeat revascularization, along with worse graft patency (N. Engl. J. Med. 2009;361:1827-37).
GOPCABE and CORONARY were designed in part to answer critics of ROOBY, who have argued that the VA trial used insufficiently experienced off-pump CABG surgeons and featured a patient population at too low an operative risk to detect a signal of benefit favoring off-pump surgery. The GOPCABE (German Off-Pump Coronary Artery Bypass Grafting in Elderly Patients) study involved 2,539 patients aged 75 years or older randomized at 12 German centers. A total of 60% of patients had triple-vessel disease, and no one was excluded from the trial because of left ventricular function or coronary artery anatomy. Participating surgeons were highly experienced. Those who performed off-pump CABG in the study had previously done an average of 514 of them, while the on-pump surgeons had done an average of 1,378 of those operations.
"We wanted to have the best off-pump vs. the best on-pump surgeons, like in a competition," explained Dr. Anno Diegeler, a surgeon at the Bad Neustadt (Germany) Heart Center.
He and his coinvestigators conducted GOPCABE because they believed it would be easier to show advantages for off-pump CABG in a population at high operative risk, such as elderly patients with many comorbidities. Indeed, the study hypothesis was that the off-pump group would show a robust 30% reduction in the primary endpoint, a composite of death, stroke, myocardial infarction, repeat revascularization, or new renal-replacement therapy at 1 year.
That didn't happen. The 30-day rate of the primary endpoint was 7.8% in the off-pump group and 8.2% with on-pump CABG, while the 1-year rates were 13.1% and 14.0%, respectively. None of the individual components of the composite endpoint differed significantly between the groups, either.
Neurocognitive function wasn't measured in GOPCABE, but it was in CORONARY (the CABG Off or On Pump Revascularization Study), which involved 4,752 randomized patients in 19 countries.
Dr. Andre Lamy presented the 1-year results. The primary endpoint was a composite of death, MI, stroke, or new renal failure requiring dialysis. The rate was 12.1% in patients in the off-pump group and similar at 13.3% in the on-pump group. As in GOPCABE, the surgeons participating in CORONARY were highly proficient. They had to have at least 2 years' experience as a staff cardiac surgeon and at least 100 prior cases of whichever operation they were assigned to. The vast majority met that standard for both procedures.
Quality of life and neurocognitive tests showed significant difference between the two groups.
However, neurocognitive testing was declared optional because it's so time consuming, and many patients opted out. For example, only 1,273 of the original 4,752 patients returned to take the Montreal Cognitive Assessment at 1 year, noted Dr. Lamy, a heart surgeon at the Population Health Research Institute at McMaster University in Hamilton, Ont.
Discussant Dr. Bernard Gersh zeroed in on the incomplete neurocognitive testing. "I think this is really a significant limitation. There's a huge bias. If there's any advantage to off-pump CABG, it may be in neurocognitive dysfunction," commented Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.
In response to questioning as to where off-pump CABG fits into clinical practice in light of the disappointing CORONARY and GOPCABE findings, Dr. Diegeler said he remains convinced that some high-operative-risk patients - those with aortic calcification or other evidence of generalized vascular disease - do benefit preferentially from off-pump surgery when performed by expert surgeons.
Dr. Lamy said a post hoc analysis of the CORONARY data showed that low-operative-risk patients as defined by a EuroScore of 0-2 tended to do better with on- than off-pump CABG, while the converse was true in those with moderate- or high-risk scores.
"In my personal practice now, my low-risk patients go on-pump and my moderate- and high-risk patients go off-pump," he added.
Dr. Jan Hlavicka presented the results of the PRAGUE-6 trial, in which 206 patients at high operative risk - a EuroScore of 6 or greater - were randomized to off- or on-pump CABG at Charles University, Prague. The operations were performed by five surgeons proficient in both procedures. The 30-day primary composite endpoint comprising death, MI, stroke, or new renal failure requiring dialysis occurred in 20.6% of the on-pump group, compared with 9.2% of off-pump patients.
The off-pump group required significantly fewer RBC transfusions. There were no significant differences between the two groups in terms of average hospital length of stay, wound infection rates, or total hospital costs.
Dr. Gersh noted that the only significant difference between the two groups in the individual components of the primary endpoint was in acute MI rates: 12.1% in the on- vs. 4.1% in the off-pump CABG group. He took issue with the Czech investigators? use of the 2004 Society of Thoracic Surgeons definition of acute MI. That's not sufficiently stringent. It surely captures many patients who don't really have an acute MI. The data should be reanalyzed using a contemporary definition which requires new Q waves, he added.
Dr. Hlavicka, Dr. Lamy, and Dr. Diegeler declared no conflicts.
SAN FRANCISCO - Off-pump coronary artery bypass graft surgery offered no advantages over on-pump CABG in any major end points at 1 year of follow-up in two major prospective randomized trials totaling more than 7,000 patients.
Among the key 1-year outcomes - which didn't differ between off- and on-pump CABG patients in the GOPCABE and CORONARY trials - were death, MI, stroke, neurocognitive function, quality of life, renal failure, and repeat revascularization, investigators reported at the annual meeting of the American College of Cardiology.
A third randomized trial presented at the same session of the ACC meeting did find a significant outcome advantage favoring off-pump CABG in high-operative-risk patients at 1 year. However, experts discounted this Czech study because it was small, single center, reported only 30-day results, and the advantage found for off-pump surgery hinged on an outdated and inadequate definition of MI.
The new study findings signal a striking fall from grace for off-pump CABG. Not long ago, this technique, while controversial, was viewed by many as a progressive development within heart surgery, one that would revitalize a mature operation whose annual case numbers were declining in the face of stiff competition from percutaneous coronary intervention by cardiologists. Off-pump CABG was an innovation designed to avoid the perioperative complications related to aortic cross-clamping and the heart-lung machine, including the lingering neurocognitive dysfunction known informally in surgical circles as "pump head."
However, the resounding lack of any demonstrable advantages for off-pump CABG in the two large trials presented in San Francisco left analysts scratching their heads as to the role remaining for this beating heart surgical technique, which is more difficult to learn and perform skillfully than on-pump bypass.
Discussant Dr. Michael J. Mack, a cardiac surgeon, voiced a similar sentiment. "I was an early advocate of off-pump surgery. But as a card-carrying off-pump bypass surgeon, it's getting harder and harder for me to maintain enthusiasm for a potential benefit from this," declared Dr. Mack, medical director of cardiovascular surgery for the Baylor Health Care System and director of cardiovascular research at the Heart Hospital in Plano, Tex. He noted that the CORONARY and GOPCABE trials follow upon the earlier ROOBY (Randomized On/Off Bypass) trial, which actually showed worse outcomes in the off-pump group. ROOBY enrolled 2,203 Veterans Affairs patients, with the off-pump CABG group having a significantly higher 1-year rate of the primary composite endpoint comprising death, nonfatal MI, or repeat revascularization, along with worse graft patency (N. Engl. J. Med. 2009;361:1827-37).
GOPCABE and CORONARY were designed in part to answer critics of ROOBY, who have argued that the VA trial used insufficiently experienced off-pump CABG surgeons and featured a patient population at too low an operative risk to detect a signal of benefit favoring off-pump surgery. The GOPCABE (German Off-Pump Coronary Artery Bypass Grafting in Elderly Patients) study involved 2,539 patients aged 75 years or older randomized at 12 German centers. A total of 60% of patients had triple-vessel disease, and no one was excluded from the trial because of left ventricular function or coronary artery anatomy. Participating surgeons were highly experienced. Those who performed off-pump CABG in the study had previously done an average of 514 of them, while the on-pump surgeons had done an average of 1,378 of those operations.
"We wanted to have the best off-pump vs. the best on-pump surgeons, like in a competition," explained Dr. Anno Diegeler, a surgeon at the Bad Neustadt (Germany) Heart Center.
He and his coinvestigators conducted GOPCABE because they believed it would be easier to show advantages for off-pump CABG in a population at high operative risk, such as elderly patients with many comorbidities. Indeed, the study hypothesis was that the off-pump group would show a robust 30% reduction in the primary endpoint, a composite of death, stroke, myocardial infarction, repeat revascularization, or new renal-replacement therapy at 1 year.
That didn't happen. The 30-day rate of the primary endpoint was 7.8% in the off-pump group and 8.2% with on-pump CABG, while the 1-year rates were 13.1% and 14.0%, respectively. None of the individual components of the composite endpoint differed significantly between the groups, either.
Neurocognitive function wasn't measured in GOPCABE, but it was in CORONARY (the CABG Off or On Pump Revascularization Study), which involved 4,752 randomized patients in 19 countries.
Dr. Andre Lamy presented the 1-year results. The primary endpoint was a composite of death, MI, stroke, or new renal failure requiring dialysis. The rate was 12.1% in patients in the off-pump group and similar at 13.3% in the on-pump group. As in GOPCABE, the surgeons participating in CORONARY were highly proficient. They had to have at least 2 years' experience as a staff cardiac surgeon and at least 100 prior cases of whichever operation they were assigned to. The vast majority met that standard for both procedures.
Quality of life and neurocognitive tests showed significant difference between the two groups.
However, neurocognitive testing was declared optional because it's so time consuming, and many patients opted out. For example, only 1,273 of the original 4,752 patients returned to take the Montreal Cognitive Assessment at 1 year, noted Dr. Lamy, a heart surgeon at the Population Health Research Institute at McMaster University in Hamilton, Ont.
Discussant Dr. Bernard Gersh zeroed in on the incomplete neurocognitive testing. "I think this is really a significant limitation. There's a huge bias. If there's any advantage to off-pump CABG, it may be in neurocognitive dysfunction," commented Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.
In response to questioning as to where off-pump CABG fits into clinical practice in light of the disappointing CORONARY and GOPCABE findings, Dr. Diegeler said he remains convinced that some high-operative-risk patients - those with aortic calcification or other evidence of generalized vascular disease - do benefit preferentially from off-pump surgery when performed by expert surgeons.
Dr. Lamy said a post hoc analysis of the CORONARY data showed that low-operative-risk patients as defined by a EuroScore of 0-2 tended to do better with on- than off-pump CABG, while the converse was true in those with moderate- or high-risk scores.
"In my personal practice now, my low-risk patients go on-pump and my moderate- and high-risk patients go off-pump," he added.
Dr. Jan Hlavicka presented the results of the PRAGUE-6 trial, in which 206 patients at high operative risk - a EuroScore of 6 or greater - were randomized to off- or on-pump CABG at Charles University, Prague. The operations were performed by five surgeons proficient in both procedures. The 30-day primary composite endpoint comprising death, MI, stroke, or new renal failure requiring dialysis occurred in 20.6% of the on-pump group, compared with 9.2% of off-pump patients.
The off-pump group required significantly fewer RBC transfusions. There were no significant differences between the two groups in terms of average hospital length of stay, wound infection rates, or total hospital costs.
Dr. Gersh noted that the only significant difference between the two groups in the individual components of the primary endpoint was in acute MI rates: 12.1% in the on- vs. 4.1% in the off-pump CABG group. He took issue with the Czech investigators? use of the 2004 Society of Thoracic Surgeons definition of acute MI. That's not sufficiently stringent. It surely captures many patients who don't really have an acute MI. The data should be reanalyzed using a contemporary definition which requires new Q waves, he added.
Dr. Hlavicka, Dr. Lamy, and Dr. Diegeler declared no conflicts.

PROACT Trial: Lower INR goal acceptable
MINNEAPOLIS – High-risk patients receiving the On-X mechanical aortic valve can be safely managed with less aggressive anticoagulation than currently recommended, interim results of the PROACT trial suggest.*
The lower target international normalized ratio (INR) in the trial resulted in a decline of more than 50% in bleeding events and did not increase the risk of thromboembolism, reported Dr. John Puskas, international principal investigator for PROACT (Prospective Randomized On-X Anticoagulation Clinical Trial) and associate chief of cardiothoracic surgery at Emory University in Atlanta.

"This aortic bileaflet mechanical valve may be safely managed in these select patients at an INR of 1.5 to 2.0, with daily low-dose aspirin," he said at the annual meeting of the American Association for Thoracic Surgery.
Current American College of Cardiology and American Heart Association guidelines recommend that warfarin be dosed to achieve an INR of 2.0 to 3.0 after implantation of a bileaflet mechanical valve, and that once-daily aspirin 75-100 mg be added for all patients with mechanical heart valves.
Dr. Puskas and his associates analyzed data from 375 high-risk patients randomly assigned to lower-dose warfarin (INR 1.5-2.0) or to continue standard-dose warfarin (INR 2.0-3.0), 3 months after implantation with the On-X bileaflet mechanical heart valve. All patients received aspirin 81 mg daily. INR was adjusted by rigorous home self-monitoring, with an average of 9 days between readings and at least 96% compliance.
High-risk patients included those with chronic atrial fibrillation, left ventricular ejection fraction less than 30%, ventricular aneurysm, left atrium diameter greater than 50 mm, prior neurological events, on estrogen replacement therapy, hypercoagulability, or inadequate platelet response to aspirin or clopidogrel (Plavix). There were 185 patients in the experimental, test arm and 190 in the control arm. After randomization, 11 test patients had a neurological event (5 strokes, 6 transient ischemic attacks) and crossed over to the control group, per protocol.
After an average follow-up of 3.82 years, patients managed with a lower target INR had a significant benefit compared with controls with respect to number of major bleeding events (10 vs. 25, respectively), minor bleeds (8 vs. 25), total bleeds (18 vs. 50), and all bleeding and thrombus (38 vs. 64), Dr. Puskas said. The corresponding rate ratios (RRs) were 0.45, 0.36, 0.40, and 0.66.
There was no difference between groups in the composite primary endpoint of major bleed, stroke, transient ischemic attack, thromboembolic events and thrombosis (30 events vs. 39 events; RR, 0.86; P = .54), he said.
Specifically, hemorrhagic stroke occurred in 1 test patient and 2 controls (RR, 0.56) and ischemic stroke in 5 patients in each group (RR, 1.12), he said.
Valve-related mortality was also similar in the test and control groups (5 deaths vs. 4 deaths), as was total mortality (10 vs. 11), Dr. Puskas said.
Invited discussant Dr. A. Pieter Kappetein, a member of the RE-ALIGN trial steering committee and professor of thoracic surgery at Erasmus Medical Center in Rotterdam, the Netherlands, said the number of patients in the analysis was extremely low and questioned the validity of combining bleeding and thromboembolic events in the primary endpoint.
"In this study, you mix the efficacy endpoint with the safety endpoints," he said, observing that they move in opposite directions.
In light of such large-scale trials as ARISTOTLE and RELY, he asked whether PROACT should be considered a pilot trial and whether a new trial, designed with roughly 8,000 patients, should be performed that would also include newer anticoagulation agents to adequately evaluate reduced anticoagulation in mechanical valves.
"Is it not potentially dangerous if we do not know what the increase is for thrombosis and follow your conclusions?" he added.
Dr. Puskas said he shared Dr. Kappetein’s concern about the noninferiority design of the trial and that it was a topic of great discussion with the Food and Drug Administration (FDA). He also agreed that thrombotic events and bleeding events move in the opposite direction.
"What we are really looking for is to determine the sweet spot where those two curves intersect," he said. "While it is theoretically and intellectually correct to say that thrombosis is the efficacy issue and hemorrhage is the safety issue, we are obliged to combine those for two reasons.
"The first is practical; no company will sponsor an 8,000-patient trial, and second, this is, in fact, a trade-off in the minds of patients and clinicians. So, it is a relevant clinical endpoint – the unholy composite, if you will – of thrombosis and hemorrhage."
Finally, a member of the audience asked whether the results would hold up with standard management because universal point-of-care home testing is not the "real world" in the United States.
Dr. Puskas replied that it is in Scandinavia and other parts of the world, and admonished American clinicians, including himself, "to catch up to what should be standard of care." He noted that, based on the roughly 53,000 INR readings in PROACT, controlling INR within your range was more important in terms of adverse events, particularly hemorrhagic events, than what arm patients were assigned to.
"Home monitoring is available, it’s not high tech and it’s much easier for patients," he said. "To be perfectly blunt, there’s really no excuse for us not using it uniformly in America. Quite frankly, it is a conflict of interest between local caregivers and their patients’ well-being.
"There is a small revenue stream to cardiology offices and primary care doctors running Coumadin clinics, and that is keeping us in the system that we have now rather than home monitoring through bigger, centralized Coumadin clinics."
Dr. Puskas did not report data on PROACT’s low-risk arm managed with clopidogrel 75 mg/day plus aspirin 325 mg/day, or a third arm managed on warfarin at an INR of 2.0-2.5 plus aspirin 81 mg/day. The low-risk data will not be available for at least one more year, although the investigators are in discussion with the FDA about a possible interim analysis, he said in an interview.
The evaluable high-risk patients were 79% male, 93% were in sinus rhythm preoperatively, and concomitant procedures included coronary artery bypass grafting in 27%, aortic aneurysm repair in 14%, and other procedures in 25%. Their average age was 55 years.
Life Technologies sponsored the study. Dr. Puskas reported having no financial relationship with Life Technologies.
Correction, 6/18/2013: An earlier version of this article stated that the On-X aortic valve is investigational. This was a misstatement. The valve itself has been approved in the United States since 2001. The PROACT trial applications of lowered INR and an aspirin/Plavix regimen are not approved.
MINNEAPOLIS – High-risk patients receiving the On-X mechanical aortic valve can be safely managed with less aggressive anticoagulation than currently recommended, interim results of the PROACT trial suggest.*
The lower target international normalized ratio (INR) in the trial resulted in a decline of more than 50% in bleeding events and did not increase the risk of thromboembolism, reported Dr. John Puskas, international principal investigator for PROACT (Prospective Randomized On-X Anticoagulation Clinical Trial) and associate chief of cardiothoracic surgery at Emory University in Atlanta.
"This aortic bileaflet mechanical valve may be safely managed in these select patients at an INR of 1.5 to 2.0, with daily low-dose aspirin," he said at the annual meeting of the American Association for Thoracic Surgery.
Current American College of Cardiology and American Heart Association guidelines recommend that warfarin be dosed to achieve an INR of 2.0 to 3.0 after implantation of a bileaflet mechanical valve, and that once-daily aspirin 75-100 mg be added for all patients with mechanical heart valves.
Dr. Puskas and his associates analyzed data from 375 high-risk patients randomly assigned to lower-dose warfarin (INR 1.5-2.0) or to continue standard-dose warfarin (INR 2.0-3.0), 3 months after implantation with the On-X bileaflet mechanical heart valve. All patients received aspirin 81 mg daily. INR was adjusted by rigorous home self-monitoring, with an average of 9 days between readings and at least 96% compliance.
High-risk patients included those with chronic atrial fibrillation, left ventricular ejection fraction less than 30%, ventricular aneurysm, left atrium diameter greater than 50 mm, prior neurological events, on estrogen replacement therapy, hypercoagulability, or inadequate platelet response to aspirin or clopidogrel (Plavix). There were 185 patients in the experimental, test arm and 190 in the control arm. After randomization, 11 test patients had a neurological event (5 strokes, 6 transient ischemic attacks) and crossed over to the control group, per protocol.
After an average follow-up of 3.82 years, patients managed with a lower target INR had a significant benefit compared with controls with respect to number of major bleeding events (10 vs. 25, respectively), minor bleeds (8 vs. 25), total bleeds (18 vs. 50), and all bleeding and thrombus (38 vs. 64), Dr. Puskas said. The corresponding rate ratios (RRs) were 0.45, 0.36, 0.40, and 0.66.
There was no difference between groups in the composite primary endpoint of major bleed, stroke, transient ischemic attack, thromboembolic events and thrombosis (30 events vs. 39 events; RR, 0.86; P = .54), he said.
Specifically, hemorrhagic stroke occurred in 1 test patient and 2 controls (RR, 0.56) and ischemic stroke in 5 patients in each group (RR, 1.12), he said.
Valve-related mortality was also similar in the test and control groups (5 deaths vs. 4 deaths), as was total mortality (10 vs. 11), Dr. Puskas said.
Invited discussant Dr. A. Pieter Kappetein, a member of the RE-ALIGN trial steering committee and professor of thoracic surgery at Erasmus Medical Center in Rotterdam, the Netherlands, said the number of patients in the analysis was extremely low and questioned the validity of combining bleeding and thromboembolic events in the primary endpoint.
"In this study, you mix the efficacy endpoint with the safety endpoints," he said, observing that they move in opposite directions.
In light of such large-scale trials as ARISTOTLE and RELY, he asked whether PROACT should be considered a pilot trial and whether a new trial, designed with roughly 8,000 patients, should be performed that would also include newer anticoagulation agents to adequately evaluate reduced anticoagulation in mechanical valves.
"Is it not potentially dangerous if we do not know what the increase is for thrombosis and follow your conclusions?" he added.
Dr. Puskas said he shared Dr. Kappetein’s concern about the noninferiority design of the trial and that it was a topic of great discussion with the Food and Drug Administration (FDA). He also agreed that thrombotic events and bleeding events move in the opposite direction.
"What we are really looking for is to determine the sweet spot where those two curves intersect," he said. "While it is theoretically and intellectually correct to say that thrombosis is the efficacy issue and hemorrhage is the safety issue, we are obliged to combine those for two reasons.
"The first is practical; no company will sponsor an 8,000-patient trial, and second, this is, in fact, a trade-off in the minds of patients and clinicians. So, it is a relevant clinical endpoint – the unholy composite, if you will – of thrombosis and hemorrhage."
Finally, a member of the audience asked whether the results would hold up with standard management because universal point-of-care home testing is not the "real world" in the United States.
Dr. Puskas replied that it is in Scandinavia and other parts of the world, and admonished American clinicians, including himself, "to catch up to what should be standard of care." He noted that, based on the roughly 53,000 INR readings in PROACT, controlling INR within your range was more important in terms of adverse events, particularly hemorrhagic events, than what arm patients were assigned to.
"Home monitoring is available, it’s not high tech and it’s much easier for patients," he said. "To be perfectly blunt, there’s really no excuse for us not using it uniformly in America. Quite frankly, it is a conflict of interest between local caregivers and their patients’ well-being.
"There is a small revenue stream to cardiology offices and primary care doctors running Coumadin clinics, and that is keeping us in the system that we have now rather than home monitoring through bigger, centralized Coumadin clinics."
Dr. Puskas did not report data on PROACT’s low-risk arm managed with clopidogrel 75 mg/day plus aspirin 325 mg/day, or a third arm managed on warfarin at an INR of 2.0-2.5 plus aspirin 81 mg/day. The low-risk data will not be available for at least one more year, although the investigators are in discussion with the FDA about a possible interim analysis, he said in an interview.
The evaluable high-risk patients were 79% male, 93% were in sinus rhythm preoperatively, and concomitant procedures included coronary artery bypass grafting in 27%, aortic aneurysm repair in 14%, and other procedures in 25%. Their average age was 55 years.
Life Technologies sponsored the study. Dr. Puskas reported having no financial relationship with Life Technologies.
Correction, 6/18/2013: An earlier version of this article stated that the On-X aortic valve is investigational. This was a misstatement. The valve itself has been approved in the United States since 2001. The PROACT trial applications of lowered INR and an aspirin/Plavix regimen are not approved.
MINNEAPOLIS – High-risk patients receiving the On-X mechanical aortic valve can be safely managed with less aggressive anticoagulation than currently recommended, interim results of the PROACT trial suggest.*
The lower target international normalized ratio (INR) in the trial resulted in a decline of more than 50% in bleeding events and did not increase the risk of thromboembolism, reported Dr. John Puskas, international principal investigator for PROACT (Prospective Randomized On-X Anticoagulation Clinical Trial) and associate chief of cardiothoracic surgery at Emory University in Atlanta.
"This aortic bileaflet mechanical valve may be safely managed in these select patients at an INR of 1.5 to 2.0, with daily low-dose aspirin," he said at the annual meeting of the American Association for Thoracic Surgery.
Current American College of Cardiology and American Heart Association guidelines recommend that warfarin be dosed to achieve an INR of 2.0 to 3.0 after implantation of a bileaflet mechanical valve, and that once-daily aspirin 75-100 mg be added for all patients with mechanical heart valves.
Dr. Puskas and his associates analyzed data from 375 high-risk patients randomly assigned to lower-dose warfarin (INR 1.5-2.0) or to continue standard-dose warfarin (INR 2.0-3.0), 3 months after implantation with the On-X bileaflet mechanical heart valve. All patients received aspirin 81 mg daily. INR was adjusted by rigorous home self-monitoring, with an average of 9 days between readings and at least 96% compliance.
High-risk patients included those with chronic atrial fibrillation, left ventricular ejection fraction less than 30%, ventricular aneurysm, left atrium diameter greater than 50 mm, prior neurological events, on estrogen replacement therapy, hypercoagulability, or inadequate platelet response to aspirin or clopidogrel (Plavix). There were 185 patients in the experimental, test arm and 190 in the control arm. After randomization, 11 test patients had a neurological event (5 strokes, 6 transient ischemic attacks) and crossed over to the control group, per protocol.
After an average follow-up of 3.82 years, patients managed with a lower target INR had a significant benefit compared with controls with respect to number of major bleeding events (10 vs. 25, respectively), minor bleeds (8 vs. 25), total bleeds (18 vs. 50), and all bleeding and thrombus (38 vs. 64), Dr. Puskas said. The corresponding rate ratios (RRs) were 0.45, 0.36, 0.40, and 0.66.
There was no difference between groups in the composite primary endpoint of major bleed, stroke, transient ischemic attack, thromboembolic events and thrombosis (30 events vs. 39 events; RR, 0.86; P = .54), he said.
Specifically, hemorrhagic stroke occurred in 1 test patient and 2 controls (RR, 0.56) and ischemic stroke in 5 patients in each group (RR, 1.12), he said.
Valve-related mortality was also similar in the test and control groups (5 deaths vs. 4 deaths), as was total mortality (10 vs. 11), Dr. Puskas said.
Invited discussant Dr. A. Pieter Kappetein, a member of the RE-ALIGN trial steering committee and professor of thoracic surgery at Erasmus Medical Center in Rotterdam, the Netherlands, said the number of patients in the analysis was extremely low and questioned the validity of combining bleeding and thromboembolic events in the primary endpoint.
"In this study, you mix the efficacy endpoint with the safety endpoints," he said, observing that they move in opposite directions.
In light of such large-scale trials as ARISTOTLE and RELY, he asked whether PROACT should be considered a pilot trial and whether a new trial, designed with roughly 8,000 patients, should be performed that would also include newer anticoagulation agents to adequately evaluate reduced anticoagulation in mechanical valves.
"Is it not potentially dangerous if we do not know what the increase is for thrombosis and follow your conclusions?" he added.
Dr. Puskas said he shared Dr. Kappetein’s concern about the noninferiority design of the trial and that it was a topic of great discussion with the Food and Drug Administration (FDA). He also agreed that thrombotic events and bleeding events move in the opposite direction.
"What we are really looking for is to determine the sweet spot where those two curves intersect," he said. "While it is theoretically and intellectually correct to say that thrombosis is the efficacy issue and hemorrhage is the safety issue, we are obliged to combine those for two reasons.
"The first is practical; no company will sponsor an 8,000-patient trial, and second, this is, in fact, a trade-off in the minds of patients and clinicians. So, it is a relevant clinical endpoint – the unholy composite, if you will – of thrombosis and hemorrhage."
Finally, a member of the audience asked whether the results would hold up with standard management because universal point-of-care home testing is not the "real world" in the United States.
Dr. Puskas replied that it is in Scandinavia and other parts of the world, and admonished American clinicians, including himself, "to catch up to what should be standard of care." He noted that, based on the roughly 53,000 INR readings in PROACT, controlling INR within your range was more important in terms of adverse events, particularly hemorrhagic events, than what arm patients were assigned to.
"Home monitoring is available, it’s not high tech and it’s much easier for patients," he said. "To be perfectly blunt, there’s really no excuse for us not using it uniformly in America. Quite frankly, it is a conflict of interest between local caregivers and their patients’ well-being.
"There is a small revenue stream to cardiology offices and primary care doctors running Coumadin clinics, and that is keeping us in the system that we have now rather than home monitoring through bigger, centralized Coumadin clinics."
Dr. Puskas did not report data on PROACT’s low-risk arm managed with clopidogrel 75 mg/day plus aspirin 325 mg/day, or a third arm managed on warfarin at an INR of 2.0-2.5 plus aspirin 81 mg/day. The low-risk data will not be available for at least one more year, although the investigators are in discussion with the FDA about a possible interim analysis, he said in an interview.
The evaluable high-risk patients were 79% male, 93% were in sinus rhythm preoperatively, and concomitant procedures included coronary artery bypass grafting in 27%, aortic aneurysm repair in 14%, and other procedures in 25%. Their average age was 55 years.
Life Technologies sponsored the study. Dr. Puskas reported having no financial relationship with Life Technologies.
Correction, 6/18/2013: An earlier version of this article stated that the On-X aortic valve is investigational. This was a misstatement. The valve itself has been approved in the United States since 2001. The PROACT trial applications of lowered INR and an aspirin/Plavix regimen are not approved.
AT THE AATS ANNUAL MEETING
Major finding: The composite primary endpoint of major bleed, stroke, transient ischemic attack, thromboembolic events, and thrombosis occurred in 30 patients managed with less aggressive anticoagulation and in 39 managed with standard warfarin anticoagulation (rate ratio, 0.86; P = .54).
Data source: Interim analysis of 375 high-risk aortic valve replacement patients in the Prospective Randomized On-X Anticoagulation Trial.
Disclosures: Life Technologies sponsored the study. Dr. Puskas reported having no financial relationship with Life Technologies.

Progress, obstacles cited in building STEMI networks
SNOWMASS, COLO. – Competition among hospitals and between cardiology groups constitutes the greatest barrier to well-functioning regional networks for ST-elevation myocardial infarction therapy, according to Dr. Bernard J. Gersh.
"We’ve got the resources in this country, but we are competitive. That’s the name of the game. So this is a real challenge," said Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.

The 2013 ACC/American Heart Association STEMI (ST-elevation myocardial infarction) guidelines list as a class I recommendation that "each community should develop a STEMI system of care."
But a one-size-fits-all approach won’t work. Creating an efficient network to deliver reperfusion therapy to as many STEMI patients as quickly as possible in Los Angeles, where there is seemingly a percutaneous coronary intervention (PCI) center every few blocks, poses a very different set of challenges than in, say, Wyoming, with two cardiac catheterization laboratories to serve nearly a 100,000-quare-mile area, he said.
He recalled a recent conversation with a colleague from a midsize Eastern city with four PCI hospitals. All four run three call schedules per 24 hours so an interventional cardiologist is always available. But collectively the hospitals handle an average of only five or six STEMIs per week.
"Can you really justify that? Furthermore, if you look at all the epidemiology coming out of the U.S. and the Western World, STEMI is in decline. Only about 30% of MIs now are STEMIs, and it’s going to be less and less," Dr. Gersh continued.
Dr. Gersh noted that the American Heart Association Mission: Lifeline program, which was created to increase timely access to PCI for STEMI patients, recently published the first-ever national survey of regional STEMI systems. The purpose was to identify best practices, financing strategies, and barriers to system implementation. Responses were obtained from 381 STEMI networks with 899 PCI hospitals.
The single most commonly cited barrier to network implementation and optimal functioning was hospital competition, identified as a significant problem in 37% of the systems. Next came emergency medical services (EMS) transport and finances, cited by 26% of respondents. The third most common barrier was competition between cardiology groups, which was an issue in 21% of networks.
The predominant funding sources for STEMI systems were PCI hospitals and cardiology practices.
Based on his favorable personal experience with the Mayo Clinic STEMI network, which uses three helicopters, an airplane, and ground ambulances to serve 28 hospitals as far as 150 miles away, Dr. Gersh said it’s clear from the national survey results that most STEMI systems around the country are doing a lot of the important things right.
For example, 92% of systems activate the cath lab with a single phone call, 97% of PCI hospitals accept a STEMI patient 24/7 regardless of bed availability, and 84% of programs operate a data registry with continuous audit.
Two-thirds of STEMI systems have the capability to transmit ECGs from at least some of their ambulances (Circ. Cardiovasc. Qual. Outcomes 2012;5:423-8).
In 87% of the networks nationwide, an emergency department physician can activate the cath lab without cardiology consultation. However, the Mayo Clinic network takes a different approach: Transferred patients bypass the emergency department and are taken straight to the coronary care unit or cath lab.
In an editorial accompanying the report, Dr. Timothy D. Henry, who in 2002 helped organize the nation’s first regional STEMI system at the Minneapolis Heart Institute, said: "Public policy changes to provide financial incentives for more rational use of resources to support regional STEMI systems rather than building more catheterization laboratories would ... be helpful" (Circulation 2012;126:166-8).
Dr. Gersh reported that he serves as a consultant to a number of device and pharmaceutical companies.
SNOWMASS, COLO. – Competition among hospitals and between cardiology groups constitutes the greatest barrier to well-functioning regional networks for ST-elevation myocardial infarction therapy, according to Dr. Bernard J. Gersh.
"We’ve got the resources in this country, but we are competitive. That’s the name of the game. So this is a real challenge," said Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.
The 2013 ACC/American Heart Association STEMI (ST-elevation myocardial infarction) guidelines list as a class I recommendation that "each community should develop a STEMI system of care."
But a one-size-fits-all approach won’t work. Creating an efficient network to deliver reperfusion therapy to as many STEMI patients as quickly as possible in Los Angeles, where there is seemingly a percutaneous coronary intervention (PCI) center every few blocks, poses a very different set of challenges than in, say, Wyoming, with two cardiac catheterization laboratories to serve nearly a 100,000-quare-mile area, he said.
He recalled a recent conversation with a colleague from a midsize Eastern city with four PCI hospitals. All four run three call schedules per 24 hours so an interventional cardiologist is always available. But collectively the hospitals handle an average of only five or six STEMIs per week.
"Can you really justify that? Furthermore, if you look at all the epidemiology coming out of the U.S. and the Western World, STEMI is in decline. Only about 30% of MIs now are STEMIs, and it’s going to be less and less," Dr. Gersh continued.
Dr. Gersh noted that the American Heart Association Mission: Lifeline program, which was created to increase timely access to PCI for STEMI patients, recently published the first-ever national survey of regional STEMI systems. The purpose was to identify best practices, financing strategies, and barriers to system implementation. Responses were obtained from 381 STEMI networks with 899 PCI hospitals.
The single most commonly cited barrier to network implementation and optimal functioning was hospital competition, identified as a significant problem in 37% of the systems. Next came emergency medical services (EMS) transport and finances, cited by 26% of respondents. The third most common barrier was competition between cardiology groups, which was an issue in 21% of networks.
The predominant funding sources for STEMI systems were PCI hospitals and cardiology practices.
Based on his favorable personal experience with the Mayo Clinic STEMI network, which uses three helicopters, an airplane, and ground ambulances to serve 28 hospitals as far as 150 miles away, Dr. Gersh said it’s clear from the national survey results that most STEMI systems around the country are doing a lot of the important things right.
For example, 92% of systems activate the cath lab with a single phone call, 97% of PCI hospitals accept a STEMI patient 24/7 regardless of bed availability, and 84% of programs operate a data registry with continuous audit.
Two-thirds of STEMI systems have the capability to transmit ECGs from at least some of their ambulances (Circ. Cardiovasc. Qual. Outcomes 2012;5:423-8).
In 87% of the networks nationwide, an emergency department physician can activate the cath lab without cardiology consultation. However, the Mayo Clinic network takes a different approach: Transferred patients bypass the emergency department and are taken straight to the coronary care unit or cath lab.
In an editorial accompanying the report, Dr. Timothy D. Henry, who in 2002 helped organize the nation’s first regional STEMI system at the Minneapolis Heart Institute, said: "Public policy changes to provide financial incentives for more rational use of resources to support regional STEMI systems rather than building more catheterization laboratories would ... be helpful" (Circulation 2012;126:166-8).
Dr. Gersh reported that he serves as a consultant to a number of device and pharmaceutical companies.
SNOWMASS, COLO. – Competition among hospitals and between cardiology groups constitutes the greatest barrier to well-functioning regional networks for ST-elevation myocardial infarction therapy, according to Dr. Bernard J. Gersh.
"We’ve got the resources in this country, but we are competitive. That’s the name of the game. So this is a real challenge," said Dr. Gersh, professor of medicine at the Mayo Clinic, Rochester, Minn.
The 2013 ACC/American Heart Association STEMI (ST-elevation myocardial infarction) guidelines list as a class I recommendation that "each community should develop a STEMI system of care."
But a one-size-fits-all approach won’t work. Creating an efficient network to deliver reperfusion therapy to as many STEMI patients as quickly as possible in Los Angeles, where there is seemingly a percutaneous coronary intervention (PCI) center every few blocks, poses a very different set of challenges than in, say, Wyoming, with two cardiac catheterization laboratories to serve nearly a 100,000-quare-mile area, he said.
He recalled a recent conversation with a colleague from a midsize Eastern city with four PCI hospitals. All four run three call schedules per 24 hours so an interventional cardiologist is always available. But collectively the hospitals handle an average of only five or six STEMIs per week.
"Can you really justify that? Furthermore, if you look at all the epidemiology coming out of the U.S. and the Western World, STEMI is in decline. Only about 30% of MIs now are STEMIs, and it’s going to be less and less," Dr. Gersh continued.
Dr. Gersh noted that the American Heart Association Mission: Lifeline program, which was created to increase timely access to PCI for STEMI patients, recently published the first-ever national survey of regional STEMI systems. The purpose was to identify best practices, financing strategies, and barriers to system implementation. Responses were obtained from 381 STEMI networks with 899 PCI hospitals.
The single most commonly cited barrier to network implementation and optimal functioning was hospital competition, identified as a significant problem in 37% of the systems. Next came emergency medical services (EMS) transport and finances, cited by 26% of respondents. The third most common barrier was competition between cardiology groups, which was an issue in 21% of networks.
The predominant funding sources for STEMI systems were PCI hospitals and cardiology practices.
Based on his favorable personal experience with the Mayo Clinic STEMI network, which uses three helicopters, an airplane, and ground ambulances to serve 28 hospitals as far as 150 miles away, Dr. Gersh said it’s clear from the national survey results that most STEMI systems around the country are doing a lot of the important things right.
For example, 92% of systems activate the cath lab with a single phone call, 97% of PCI hospitals accept a STEMI patient 24/7 regardless of bed availability, and 84% of programs operate a data registry with continuous audit.
Two-thirds of STEMI systems have the capability to transmit ECGs from at least some of their ambulances (Circ. Cardiovasc. Qual. Outcomes 2012;5:423-8).
In 87% of the networks nationwide, an emergency department physician can activate the cath lab without cardiology consultation. However, the Mayo Clinic network takes a different approach: Transferred patients bypass the emergency department and are taken straight to the coronary care unit or cath lab.
In an editorial accompanying the report, Dr. Timothy D. Henry, who in 2002 helped organize the nation’s first regional STEMI system at the Minneapolis Heart Institute, said: "Public policy changes to provide financial incentives for more rational use of resources to support regional STEMI systems rather than building more catheterization laboratories would ... be helpful" (Circulation 2012;126:166-8).
Dr. Gersh reported that he serves as a consultant to a number of device and pharmaceutical companies.

Beta-blocker use fails as a CABG quality metric
LOS ANGELES – Single-institution reports regarding the benefits of beta-blocker use are conflicting, despite the fact that preoperative beta-blockade for coronary artery bypass grafting has become an accepted hospital quality metric, according to Dr. Damien J. LaPar and his colleagues.

Dr. LaPar of the University of Virginia, Charlottesville reported the results of research undertaken to assess this issue based on a study of patient records from a statewide, multi-institutional Society of Thoracic Surgeons (STS)–certified database for isolated coronary artery bypass grafting (CABG) operations (2001-2011). He and his colleagues found that there was no difference seen in mortality, length of stay, or readmission comparing patients with or without preoperative beta-blocker use.
Their prestigious Richard E. Clark Paper for Adult Cardiac Surgery, presented at the annual meeting of the Society of Thoracic Surgeons, utilized the STS Adult Cardiac Surgery Database to demonstrate that the perceived benefits of using beta-blockers before CABG do not stand up as statistically significant improvements in outcomes.
"Preoperative beta-blocker use is not associated with improved patient outcomes or hospital resource utilization following" CABG, said Dr. LaPar.
Patients were stratified by preoperative beta-blocker use and the influence of preoperative beta-blockers on risk-adjusted outcomes was assessed by hierarchical regression modeling with adjustment for preoperative risk using calculated STS predictive risk indices.
A total of 43,747 patients with a mean age of around 64 years were included in the study; 80.2% of these patients were treated with beta-blockers. The median STS-predicted risk of mortality scores for beta-blocker patients were incrementally lower, compared with non–beta blocker patients (1.2% vs. 1.4%, P less than .001). Non–beta blocker patients more frequently developed pneumonia (3.5% vs. 2.8%, P = .001), while beta-blocker patients had surprisingly greater intraoperative blood usage (16.0% vs. 11.2%, P less than .001).
There was, however, no difference in unadjusted mortality (beta-blocker, 1.9%, vs. non beta-blocker, 2.2%; P = 0.15). After risk adjustment, preoperative beta-blocker use was not associated with mortality (P = .63), morbidity, length of stay (P = .79), or hospital readmission (P = .97).
"These data suggest that the use of preoperative beta-blockers for [CABG] operations should not be used as a measure of surgical quality," Dr. LaPar said.
"In an era of increasing pressure on individual hospital and surgical outcomes, the identification of appropriate measures of surgical quality remains critical," Dr. LaPar added in an interview.
"More importantly, as public reporting of surgeon outcomes becomes more common, the cardiothoracic surgical community must play a central role in providing updated data from which to base health care policy, hospital and surgeon reimbursement strategies, and referral patterns for cardiac surgical patients," he stated.
"The results of our study provide an updated reexamination of an issue of increasing debate and provide current clinical estimates on the adjusted impact of preoperative beta-blocker use on outcomes following isolated CABG operations. Future randomized controlled trials are needed to more clearly define a cause-effect relationship between preoperative beta-blocker therapy and coronary artery bypass grafting outcomes before the routine use of beta-blockade should be adopted as a quality performance measure by the cardiothoracic surgical community," Dr. LaPar concluded.
Dr. LaPar reported having no relevant conflicts of interest with regard to this paper; two of his colleagues reported serving as speakers or receiving funding from a variety of drug and device companies.
LOS ANGELES – Single-institution reports regarding the benefits of beta-blocker use are conflicting, despite the fact that preoperative beta-blockade for coronary artery bypass grafting has become an accepted hospital quality metric, according to Dr. Damien J. LaPar and his colleagues.
Dr. LaPar of the University of Virginia, Charlottesville reported the results of research undertaken to assess this issue based on a study of patient records from a statewide, multi-institutional Society of Thoracic Surgeons (STS)–certified database for isolated coronary artery bypass grafting (CABG) operations (2001-2011). He and his colleagues found that there was no difference seen in mortality, length of stay, or readmission comparing patients with or without preoperative beta-blocker use.
Their prestigious Richard E. Clark Paper for Adult Cardiac Surgery, presented at the annual meeting of the Society of Thoracic Surgeons, utilized the STS Adult Cardiac Surgery Database to demonstrate that the perceived benefits of using beta-blockers before CABG do not stand up as statistically significant improvements in outcomes.
"Preoperative beta-blocker use is not associated with improved patient outcomes or hospital resource utilization following" CABG, said Dr. LaPar.
Patients were stratified by preoperative beta-blocker use and the influence of preoperative beta-blockers on risk-adjusted outcomes was assessed by hierarchical regression modeling with adjustment for preoperative risk using calculated STS predictive risk indices.
A total of 43,747 patients with a mean age of around 64 years were included in the study; 80.2% of these patients were treated with beta-blockers. The median STS-predicted risk of mortality scores for beta-blocker patients were incrementally lower, compared with non–beta blocker patients (1.2% vs. 1.4%, P less than .001). Non–beta blocker patients more frequently developed pneumonia (3.5% vs. 2.8%, P = .001), while beta-blocker patients had surprisingly greater intraoperative blood usage (16.0% vs. 11.2%, P less than .001).
There was, however, no difference in unadjusted mortality (beta-blocker, 1.9%, vs. non beta-blocker, 2.2%; P = 0.15). After risk adjustment, preoperative beta-blocker use was not associated with mortality (P = .63), morbidity, length of stay (P = .79), or hospital readmission (P = .97).
"These data suggest that the use of preoperative beta-blockers for [CABG] operations should not be used as a measure of surgical quality," Dr. LaPar said.
"In an era of increasing pressure on individual hospital and surgical outcomes, the identification of appropriate measures of surgical quality remains critical," Dr. LaPar added in an interview.
"More importantly, as public reporting of surgeon outcomes becomes more common, the cardiothoracic surgical community must play a central role in providing updated data from which to base health care policy, hospital and surgeon reimbursement strategies, and referral patterns for cardiac surgical patients," he stated.
"The results of our study provide an updated reexamination of an issue of increasing debate and provide current clinical estimates on the adjusted impact of preoperative beta-blocker use on outcomes following isolated CABG operations. Future randomized controlled trials are needed to more clearly define a cause-effect relationship between preoperative beta-blocker therapy and coronary artery bypass grafting outcomes before the routine use of beta-blockade should be adopted as a quality performance measure by the cardiothoracic surgical community," Dr. LaPar concluded.
Dr. LaPar reported having no relevant conflicts of interest with regard to this paper; two of his colleagues reported serving as speakers or receiving funding from a variety of drug and device companies.
LOS ANGELES – Single-institution reports regarding the benefits of beta-blocker use are conflicting, despite the fact that preoperative beta-blockade for coronary artery bypass grafting has become an accepted hospital quality metric, according to Dr. Damien J. LaPar and his colleagues.
Dr. LaPar of the University of Virginia, Charlottesville reported the results of research undertaken to assess this issue based on a study of patient records from a statewide, multi-institutional Society of Thoracic Surgeons (STS)–certified database for isolated coronary artery bypass grafting (CABG) operations (2001-2011). He and his colleagues found that there was no difference seen in mortality, length of stay, or readmission comparing patients with or without preoperative beta-blocker use.
Their prestigious Richard E. Clark Paper for Adult Cardiac Surgery, presented at the annual meeting of the Society of Thoracic Surgeons, utilized the STS Adult Cardiac Surgery Database to demonstrate that the perceived benefits of using beta-blockers before CABG do not stand up as statistically significant improvements in outcomes.
"Preoperative beta-blocker use is not associated with improved patient outcomes or hospital resource utilization following" CABG, said Dr. LaPar.
Patients were stratified by preoperative beta-blocker use and the influence of preoperative beta-blockers on risk-adjusted outcomes was assessed by hierarchical regression modeling with adjustment for preoperative risk using calculated STS predictive risk indices.
A total of 43,747 patients with a mean age of around 64 years were included in the study; 80.2% of these patients were treated with beta-blockers. The median STS-predicted risk of mortality scores for beta-blocker patients were incrementally lower, compared with non–beta blocker patients (1.2% vs. 1.4%, P less than .001). Non–beta blocker patients more frequently developed pneumonia (3.5% vs. 2.8%, P = .001), while beta-blocker patients had surprisingly greater intraoperative blood usage (16.0% vs. 11.2%, P less than .001).
There was, however, no difference in unadjusted mortality (beta-blocker, 1.9%, vs. non beta-blocker, 2.2%; P = 0.15). After risk adjustment, preoperative beta-blocker use was not associated with mortality (P = .63), morbidity, length of stay (P = .79), or hospital readmission (P = .97).
"These data suggest that the use of preoperative beta-blockers for [CABG] operations should not be used as a measure of surgical quality," Dr. LaPar said.
"In an era of increasing pressure on individual hospital and surgical outcomes, the identification of appropriate measures of surgical quality remains critical," Dr. LaPar added in an interview.
"More importantly, as public reporting of surgeon outcomes becomes more common, the cardiothoracic surgical community must play a central role in providing updated data from which to base health care policy, hospital and surgeon reimbursement strategies, and referral patterns for cardiac surgical patients," he stated.
"The results of our study provide an updated reexamination of an issue of increasing debate and provide current clinical estimates on the adjusted impact of preoperative beta-blocker use on outcomes following isolated CABG operations. Future randomized controlled trials are needed to more clearly define a cause-effect relationship between preoperative beta-blocker therapy and coronary artery bypass grafting outcomes before the routine use of beta-blockade should be adopted as a quality performance measure by the cardiothoracic surgical community," Dr. LaPar concluded.
Dr. LaPar reported having no relevant conflicts of interest with regard to this paper; two of his colleagues reported serving as speakers or receiving funding from a variety of drug and device companies.
AT THE STS ANNUAL MEETING
Major Finding: Preoperative beta-blocker use was not associated with mortality, morbidity, length of stay, or readmission.
Data Source: A retrospective database analysis of 43,747 CABG patients, 80% of whom were treated with beta-blockers.
Disclosures: Dr. LaPar reported having no relevant conflicts of interest; two of his colleagues reported serving as speakers or receiving funding from a variety of drug and device companies.

Stem cell treatment post PCI didn’t improve outcomes
Intracoronary delivery of bone marrow mononuclear cells to the infarct zone of patients who have undergone percutaneous coronary intervention following an ST-segment elevation myocardial infarction did not improve left ventricular function at 6 months in the TIME trial.
The trial was designed to examine the difference in effect of infusing the bone marrow mononuclear cells (BMCs) into the infarct-related artery at 7 days after PCI and at 3 days, Dr. Jay H. Traverse said at the American Heart Association meeting.
Unfortunately, neither of these approaches were any better than placebo infusion at improving the recovery of left ventricular function, improving left ventricular volume, or reducing infarct size at 6 months’ follow-up in this double-blind Timing in Myocardial Infarction Evaluation (TIME) clinical trial, reported Dr. Traverse, of the Minneapolis Heart Institute at Abbott Northwestern Hospital.
"However, long-term follow-up of these patients and the development of new composite end points may still reveal a role for this cell type after AMI [acute myocardial infarction]," Dr. Traverse and his associates said in an article published online simultaneously with his presentation (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.28726]).
Recent research indicates that the timing of BMC infusion after PCI due to ST-segment elevation myocardial infarction (STEMI) might be critical. In the days following STEMI, there are significant temporal changes in the release of cytokines and growth factors "that may support stem-cell homing and angiogenesis, leading to improved cell survival and engraftment," they noted.
In the TIME trial, a total of 132 high-risk patients were enrolled during a 3-year period after they had experienced STEMI, shown a left ventricular ejection fraction (LVEF) of 45% or less, and were slated for PCI with stenting. These subjects were randomly assigned to have stem cell therapy on either day 3 or day 7 after PCI. After some of these patients were excluded or withdrawn from the study, the remaining 120 patients underwent a second randomization to receive either the autologous BMCs (79 patients) or placebo infusions (41 patients).
The primary end points of TIME were changes in global and regional left ventricular function on MRI scanning at 6 months post PCI. There were no significant differences in these outcomes between subjects who received stem-cell therapy 3 days after PCI and those who received it 7 days after PCI, the investigators reported. There also were no significant differences among the study groups in secondary outcomes such as reduction in infarct volume or change in ventricular volumes.
However, there also were no significant differences in any outcomes between subjects who received active BMCs and those who received placebo infusions.
Dr. Traverse reported no conflicts.
The TIME trial, like two others in the series of studies from the Cardiovascular Cell Therapy Research Network, a consortium funded by the National Institutes of Health, is "well-intentioned, nicely designed, and impeccably executed, but difficult to interpret," said Dr. Eduardo Marban and Dr. Konstantinos Malliaras.
It may be that this treatment is ineffective in this patient population, "in which case the previous positive clinical studies were red herrings." Or it may be that, as Dr. Traverse suggests, the cell product used in TIME was somehow deficient, they said.
Dr. Marban and Dr. Malliaras are at Cedars-Sinai Heart Institute in Los Angeles. Dr. Marban reported being a founder of Capricor, a developer of cardiac stem-cell treatments; both he and Dr. Malliaras reported financial ties to Capricor. These remarks were taken from their editorial accompanying Dr. Traverse’s report (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.64751]).
The TIME trial, like two others in the series of studies from the Cardiovascular Cell Therapy Research Network, a consortium funded by the National Institutes of Health, is "well-intentioned, nicely designed, and impeccably executed, but difficult to interpret," said Dr. Eduardo Marban and Dr. Konstantinos Malliaras.
It may be that this treatment is ineffective in this patient population, "in which case the previous positive clinical studies were red herrings." Or it may be that, as Dr. Traverse suggests, the cell product used in TIME was somehow deficient, they said.
Dr. Marban and Dr. Malliaras are at Cedars-Sinai Heart Institute in Los Angeles. Dr. Marban reported being a founder of Capricor, a developer of cardiac stem-cell treatments; both he and Dr. Malliaras reported financial ties to Capricor. These remarks were taken from their editorial accompanying Dr. Traverse’s report (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.64751]).
The TIME trial, like two others in the series of studies from the Cardiovascular Cell Therapy Research Network, a consortium funded by the National Institutes of Health, is "well-intentioned, nicely designed, and impeccably executed, but difficult to interpret," said Dr. Eduardo Marban and Dr. Konstantinos Malliaras.
It may be that this treatment is ineffective in this patient population, "in which case the previous positive clinical studies were red herrings." Or it may be that, as Dr. Traverse suggests, the cell product used in TIME was somehow deficient, they said.
Dr. Marban and Dr. Malliaras are at Cedars-Sinai Heart Institute in Los Angeles. Dr. Marban reported being a founder of Capricor, a developer of cardiac stem-cell treatments; both he and Dr. Malliaras reported financial ties to Capricor. These remarks were taken from their editorial accompanying Dr. Traverse’s report (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.64751]).
Intracoronary delivery of bone marrow mononuclear cells to the infarct zone of patients who have undergone percutaneous coronary intervention following an ST-segment elevation myocardial infarction did not improve left ventricular function at 6 months in the TIME trial.
The trial was designed to examine the difference in effect of infusing the bone marrow mononuclear cells (BMCs) into the infarct-related artery at 7 days after PCI and at 3 days, Dr. Jay H. Traverse said at the American Heart Association meeting.
Unfortunately, neither of these approaches were any better than placebo infusion at improving the recovery of left ventricular function, improving left ventricular volume, or reducing infarct size at 6 months’ follow-up in this double-blind Timing in Myocardial Infarction Evaluation (TIME) clinical trial, reported Dr. Traverse, of the Minneapolis Heart Institute at Abbott Northwestern Hospital.
"However, long-term follow-up of these patients and the development of new composite end points may still reveal a role for this cell type after AMI [acute myocardial infarction]," Dr. Traverse and his associates said in an article published online simultaneously with his presentation (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.28726]).
Recent research indicates that the timing of BMC infusion after PCI due to ST-segment elevation myocardial infarction (STEMI) might be critical. In the days following STEMI, there are significant temporal changes in the release of cytokines and growth factors "that may support stem-cell homing and angiogenesis, leading to improved cell survival and engraftment," they noted.
In the TIME trial, a total of 132 high-risk patients were enrolled during a 3-year period after they had experienced STEMI, shown a left ventricular ejection fraction (LVEF) of 45% or less, and were slated for PCI with stenting. These subjects were randomly assigned to have stem cell therapy on either day 3 or day 7 after PCI. After some of these patients were excluded or withdrawn from the study, the remaining 120 patients underwent a second randomization to receive either the autologous BMCs (79 patients) or placebo infusions (41 patients).
The primary end points of TIME were changes in global and regional left ventricular function on MRI scanning at 6 months post PCI. There were no significant differences in these outcomes between subjects who received stem-cell therapy 3 days after PCI and those who received it 7 days after PCI, the investigators reported. There also were no significant differences among the study groups in secondary outcomes such as reduction in infarct volume or change in ventricular volumes.
However, there also were no significant differences in any outcomes between subjects who received active BMCs and those who received placebo infusions.
Dr. Traverse reported no conflicts.
Intracoronary delivery of bone marrow mononuclear cells to the infarct zone of patients who have undergone percutaneous coronary intervention following an ST-segment elevation myocardial infarction did not improve left ventricular function at 6 months in the TIME trial.
The trial was designed to examine the difference in effect of infusing the bone marrow mononuclear cells (BMCs) into the infarct-related artery at 7 days after PCI and at 3 days, Dr. Jay H. Traverse said at the American Heart Association meeting.
Unfortunately, neither of these approaches were any better than placebo infusion at improving the recovery of left ventricular function, improving left ventricular volume, or reducing infarct size at 6 months’ follow-up in this double-blind Timing in Myocardial Infarction Evaluation (TIME) clinical trial, reported Dr. Traverse, of the Minneapolis Heart Institute at Abbott Northwestern Hospital.
"However, long-term follow-up of these patients and the development of new composite end points may still reveal a role for this cell type after AMI [acute myocardial infarction]," Dr. Traverse and his associates said in an article published online simultaneously with his presentation (JAMA 2012 Nov. 6 [doi: 10.1001/jama.2012.28726]).
Recent research indicates that the timing of BMC infusion after PCI due to ST-segment elevation myocardial infarction (STEMI) might be critical. In the days following STEMI, there are significant temporal changes in the release of cytokines and growth factors "that may support stem-cell homing and angiogenesis, leading to improved cell survival and engraftment," they noted.
In the TIME trial, a total of 132 high-risk patients were enrolled during a 3-year period after they had experienced STEMI, shown a left ventricular ejection fraction (LVEF) of 45% or less, and were slated for PCI with stenting. These subjects were randomly assigned to have stem cell therapy on either day 3 or day 7 after PCI. After some of these patients were excluded or withdrawn from the study, the remaining 120 patients underwent a second randomization to receive either the autologous BMCs (79 patients) or placebo infusions (41 patients).
The primary end points of TIME were changes in global and regional left ventricular function on MRI scanning at 6 months post PCI. There were no significant differences in these outcomes between subjects who received stem-cell therapy 3 days after PCI and those who received it 7 days after PCI, the investigators reported. There also were no significant differences among the study groups in secondary outcomes such as reduction in infarct volume or change in ventricular volumes.
However, there also were no significant differences in any outcomes between subjects who received active BMCs and those who received placebo infusions.
Dr. Traverse reported no conflicts.
Major Finding: The recovery of left ventricular function and volume did not differ between patients who received stem-cell (BMC) therapy in an infarcted artery 3 days after PCI and those who received it 7 days after PCI.
Data Source: This was a randomized, double-blind, placebo-controlled clinical trial involving 120 STEMI patients who underwent PCI with stent placement, received BMC or placebo infusions either 3 or 7 days later, and were followed for 6 months.
Disclosures: This study was funded by the National Heart, Lung, and Blood Institute. Dr. Traverse reported no financial conflicts of interest, but his associates reported numerous ties to industry sources.