User login
Testosterone Trials’ cardiac, cognitive results disappoint
Testosterone treatment may have beneficial effects on unexplained anemia or bone density in men with age-related low testosterone, but at the cost of an increase in coronary artery plaque and with no benefit on cognitive function, new research suggests.
The results of four of the seven Testosterone Trials were published Feb. 21 in JAMA and JAMA Internal Medicine, adding to a growing body of research on the impact of testosterone supplementation but without finding clear evidence of an overall benefit.
In the double-blind, multicenter Anemia Trial, 788 men aged 65 years or older with average testosterone levels of less than 275 ng/dL were allocated to 12 months of testosterone gel or placebo. The group included 126 individuals with a hemoglobin level at or below 12.7 g/dL (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9540).
The study found that significantly more men who received testosterone treatment experienced increases in hemoglobin concentration of 1 g/dL or more above baseline, compared with those who received the placebo gel (54% vs. 15%; 95% CI 3.7-277.8; P = .002).
This effect was seen in men with known causes of anemia, such as myelodysplasia, iron deficiency, B12 deficiency, or chronic inflammation or disease; in men with anemia of unknown case; and in men who weren’t anemic.
After 12 months, more than half of the testosterone-treated men who started the study with unexplained anemia were no longer anemic, compared with around one-quarter of the placebo-treated men (58.3% vs. 22.2%). The men treated with placebo also had lower average hemoglobin level changes, compared with those treated with testosterone.
“Increases in hemoglobin levels were positively and significantly associated with participants’ global impression of change in overall health and energy,” wrote Cindy N. Roy, PhD, of Johns Hopkins University, Baltimore, and her coauthors.
Bone mineral density
A second trial examined the effect of 12 months of testosterone gel or placebo on bone mineral density in a group of 211 men with average testosterone concentrations less than 275 ng/L (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9539). The treatment increased median serum concentrations of total testosterone, free testosterone, and estradiol to within the normal ranges for young men.
The study showed significantly greater increases – measured by quantitative computed tomography – with testosterone treatment, compared with placebo, in spine trabecular, spine peripheral, hip trabecular, and peripheral volumetric bone mineral density, as well as in mean estimated strength of spine trabecular bone, spine peripheral bone, and hip trabecular and peripheral bone.
For the primary outcome of mean lumbar spine trabecular volumetric bone mineral density, testosterone treatment was associated with a mean increase of 7.5%, compared with a 0.8% increase with placebo.
Researchers also noted that the magnitude of the increase in spine trabecular bone mineral density from baseline was significantly associated with changes in total testosterone and estradiol.
However, there were no significant differences in fracture rate, with six fractures reported in each group during the year of treatment. In the observation year after treatment, three fractures were reported in the testosterone arm and four in the placebo arm.
“These results are unequivocal compared with prior studies of the effect of testosterone treatment on bone in older men, in spite of treatment limited to 1 year, perhaps because the mean pretreatment testosterone level was lower and the sample size larger than in prior studies, and because the primary outcome in this trial was vBMD by QCT,” wrote Peter J. Snyder, MD, of the University of Pennsylvania, Philadelphia, and his coauthors.
Coronary artery plaque
However, a third trial – this one in 170 men with low testosterone and symptoms suggestive of hypogonadism – found significantly greater increases in noncalcified plaque volume, median total plaque volume, and median coronary artery calcification score among the 88 men assigned to 12 months of testosterone gel, compared with those assigned to placebo.
The men treated with testosterone showed a mean increase in noncalcified coronary artery plaque volume of 40 mm3, compared with 4 mm3 in men given the placebo gel, and a mean increase in total plaque volume of 57 mm3 with testosterone and 21 mm3 with placebo (JAMA. 2017 Feb 21;317[7]:708-16).
There were no significant differences between the groups in change to coronary artery calcium score, and there were no adverse cardiovascular events reported in either group, despite the fact that around half the participants had severe atherosclerosis at baseline.
“The increase in coronary artery noncalcified and total plaque volumes in men treated with testosterone is concerning, because any limitation of the vascular lumen could be considered deleterious,” wrote Matthew J. Budoff, MD, of the Los Angeles Biomedical Research Institute, Torrance, Calif., and his coauthors. “The clinical significance of these increases could depend on the differential effects of testosterone on the individual components of noncalcified plaque.”
However, the investigators pointed out that the trial was neither large enough nor long enough to draw conclusions about the cardiovascular risks of testosterone treatment, and they called for larger studies to explore the association.
Cognitive function
The fourth study looked at mean change in cognitive function from baseline in 493 men with a serum testosterone level less than 275 ng/dL, impaired sexual function, physical function, or vitality, and who met the criteria for age-associated memory impairment. Half the participants were assigned to 12 months of testosterone gel, and half were assigned to placebo gel (JAMA. 2017 Feb 21;317[7]:717-27).
Researchers found no significant differences between the two groups from baseline to 6 months and 12 months in mean change in delayed paragraph recall score, visual memory, executive function, or spatial ability.
“The lack of association between testosterone treatment and cognition was apparent across all cognitive domains assessed among men with [age-associated memory impairment], in spite of an increase in circulating total and free testosterone concentrations in the testosterone group to levels typical of men aged 19-40 years,” wrote Susan M. Resnick, PhD, of the National Institute on Aging, and her coauthors.
The Testosterone Trials were supported by the National Institute on Aging, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, the National Institute of Child Health and Human Development, and AbbVie, which also provided the AndroGel, and placebo gel. Authors from the trials declared a range of funding, consultancies, and other support from the pharmaceutical industry, including AbbVie. One author declared a pending patent for a free testosterone calculator.
Today, 8 decades since the first clinical use of testosterone, the sole unequivocal indication for testosterone treatment is as replacement therapy for men with pathological hypogonadism (i.e., organic disorders of the reproductive system). Yet despite no proven new indications, global testosterone sales increased 100-fold over the last 3 decades, including increases of 40-fold in Canada and 10-fold in the United States from 2000 to 2011.
Overall, the findings from subtrials of the TTrials do not materially change the unfavorable balance of safety and efficacy to initiate testosterone treatment for age-related hypogonadism. With the results of the studies by Resnick et al. and by Budoff et al. in this issue of JAMA, the hopes for testosterone-led rejuvenation for older men are dimmed and disappointed if not yet finally dashed.
David J. Handelsman, MD, is from the ANZAC Research Institute, University of Sydney and Concord Hospital, Australia. These comments are taken from an editorial (JAMA 2017 Feb 21;317:699-701). Dr. Handelsman reported grants from Lawley Pharmaceuticals and Besins Healthcare and serving as a medical expert in testosterone litigation.
Today, 8 decades since the first clinical use of testosterone, the sole unequivocal indication for testosterone treatment is as replacement therapy for men with pathological hypogonadism (i.e., organic disorders of the reproductive system). Yet despite no proven new indications, global testosterone sales increased 100-fold over the last 3 decades, including increases of 40-fold in Canada and 10-fold in the United States from 2000 to 2011.
Overall, the findings from subtrials of the TTrials do not materially change the unfavorable balance of safety and efficacy to initiate testosterone treatment for age-related hypogonadism. With the results of the studies by Resnick et al. and by Budoff et al. in this issue of JAMA, the hopes for testosterone-led rejuvenation for older men are dimmed and disappointed if not yet finally dashed.
David J. Handelsman, MD, is from the ANZAC Research Institute, University of Sydney and Concord Hospital, Australia. These comments are taken from an editorial (JAMA 2017 Feb 21;317:699-701). Dr. Handelsman reported grants from Lawley Pharmaceuticals and Besins Healthcare and serving as a medical expert in testosterone litigation.
Today, 8 decades since the first clinical use of testosterone, the sole unequivocal indication for testosterone treatment is as replacement therapy for men with pathological hypogonadism (i.e., organic disorders of the reproductive system). Yet despite no proven new indications, global testosterone sales increased 100-fold over the last 3 decades, including increases of 40-fold in Canada and 10-fold in the United States from 2000 to 2011.
Overall, the findings from subtrials of the TTrials do not materially change the unfavorable balance of safety and efficacy to initiate testosterone treatment for age-related hypogonadism. With the results of the studies by Resnick et al. and by Budoff et al. in this issue of JAMA, the hopes for testosterone-led rejuvenation for older men are dimmed and disappointed if not yet finally dashed.
David J. Handelsman, MD, is from the ANZAC Research Institute, University of Sydney and Concord Hospital, Australia. These comments are taken from an editorial (JAMA 2017 Feb 21;317:699-701). Dr. Handelsman reported grants from Lawley Pharmaceuticals and Besins Healthcare and serving as a medical expert in testosterone litigation.
Testosterone treatment may have beneficial effects on unexplained anemia or bone density in men with age-related low testosterone, but at the cost of an increase in coronary artery plaque and with no benefit on cognitive function, new research suggests.
The results of four of the seven Testosterone Trials were published Feb. 21 in JAMA and JAMA Internal Medicine, adding to a growing body of research on the impact of testosterone supplementation but without finding clear evidence of an overall benefit.
In the double-blind, multicenter Anemia Trial, 788 men aged 65 years or older with average testosterone levels of less than 275 ng/dL were allocated to 12 months of testosterone gel or placebo. The group included 126 individuals with a hemoglobin level at or below 12.7 g/dL (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9540).
The study found that significantly more men who received testosterone treatment experienced increases in hemoglobin concentration of 1 g/dL or more above baseline, compared with those who received the placebo gel (54% vs. 15%; 95% CI 3.7-277.8; P = .002).
This effect was seen in men with known causes of anemia, such as myelodysplasia, iron deficiency, B12 deficiency, or chronic inflammation or disease; in men with anemia of unknown case; and in men who weren’t anemic.
After 12 months, more than half of the testosterone-treated men who started the study with unexplained anemia were no longer anemic, compared with around one-quarter of the placebo-treated men (58.3% vs. 22.2%). The men treated with placebo also had lower average hemoglobin level changes, compared with those treated with testosterone.
“Increases in hemoglobin levels were positively and significantly associated with participants’ global impression of change in overall health and energy,” wrote Cindy N. Roy, PhD, of Johns Hopkins University, Baltimore, and her coauthors.
Bone mineral density
A second trial examined the effect of 12 months of testosterone gel or placebo on bone mineral density in a group of 211 men with average testosterone concentrations less than 275 ng/L (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9539). The treatment increased median serum concentrations of total testosterone, free testosterone, and estradiol to within the normal ranges for young men.
The study showed significantly greater increases – measured by quantitative computed tomography – with testosterone treatment, compared with placebo, in spine trabecular, spine peripheral, hip trabecular, and peripheral volumetric bone mineral density, as well as in mean estimated strength of spine trabecular bone, spine peripheral bone, and hip trabecular and peripheral bone.
For the primary outcome of mean lumbar spine trabecular volumetric bone mineral density, testosterone treatment was associated with a mean increase of 7.5%, compared with a 0.8% increase with placebo.
Researchers also noted that the magnitude of the increase in spine trabecular bone mineral density from baseline was significantly associated with changes in total testosterone and estradiol.
However, there were no significant differences in fracture rate, with six fractures reported in each group during the year of treatment. In the observation year after treatment, three fractures were reported in the testosterone arm and four in the placebo arm.
“These results are unequivocal compared with prior studies of the effect of testosterone treatment on bone in older men, in spite of treatment limited to 1 year, perhaps because the mean pretreatment testosterone level was lower and the sample size larger than in prior studies, and because the primary outcome in this trial was vBMD by QCT,” wrote Peter J. Snyder, MD, of the University of Pennsylvania, Philadelphia, and his coauthors.
Coronary artery plaque
However, a third trial – this one in 170 men with low testosterone and symptoms suggestive of hypogonadism – found significantly greater increases in noncalcified plaque volume, median total plaque volume, and median coronary artery calcification score among the 88 men assigned to 12 months of testosterone gel, compared with those assigned to placebo.
The men treated with testosterone showed a mean increase in noncalcified coronary artery plaque volume of 40 mm3, compared with 4 mm3 in men given the placebo gel, and a mean increase in total plaque volume of 57 mm3 with testosterone and 21 mm3 with placebo (JAMA. 2017 Feb 21;317[7]:708-16).
There were no significant differences between the groups in change to coronary artery calcium score, and there were no adverse cardiovascular events reported in either group, despite the fact that around half the participants had severe atherosclerosis at baseline.
“The increase in coronary artery noncalcified and total plaque volumes in men treated with testosterone is concerning, because any limitation of the vascular lumen could be considered deleterious,” wrote Matthew J. Budoff, MD, of the Los Angeles Biomedical Research Institute, Torrance, Calif., and his coauthors. “The clinical significance of these increases could depend on the differential effects of testosterone on the individual components of noncalcified plaque.”
However, the investigators pointed out that the trial was neither large enough nor long enough to draw conclusions about the cardiovascular risks of testosterone treatment, and they called for larger studies to explore the association.
Cognitive function
The fourth study looked at mean change in cognitive function from baseline in 493 men with a serum testosterone level less than 275 ng/dL, impaired sexual function, physical function, or vitality, and who met the criteria for age-associated memory impairment. Half the participants were assigned to 12 months of testosterone gel, and half were assigned to placebo gel (JAMA. 2017 Feb 21;317[7]:717-27).
Researchers found no significant differences between the two groups from baseline to 6 months and 12 months in mean change in delayed paragraph recall score, visual memory, executive function, or spatial ability.
“The lack of association between testosterone treatment and cognition was apparent across all cognitive domains assessed among men with [age-associated memory impairment], in spite of an increase in circulating total and free testosterone concentrations in the testosterone group to levels typical of men aged 19-40 years,” wrote Susan M. Resnick, PhD, of the National Institute on Aging, and her coauthors.
The Testosterone Trials were supported by the National Institute on Aging, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, the National Institute of Child Health and Human Development, and AbbVie, which also provided the AndroGel, and placebo gel. Authors from the trials declared a range of funding, consultancies, and other support from the pharmaceutical industry, including AbbVie. One author declared a pending patent for a free testosterone calculator.
Testosterone treatment may have beneficial effects on unexplained anemia or bone density in men with age-related low testosterone, but at the cost of an increase in coronary artery plaque and with no benefit on cognitive function, new research suggests.
The results of four of the seven Testosterone Trials were published Feb. 21 in JAMA and JAMA Internal Medicine, adding to a growing body of research on the impact of testosterone supplementation but without finding clear evidence of an overall benefit.
In the double-blind, multicenter Anemia Trial, 788 men aged 65 years or older with average testosterone levels of less than 275 ng/dL were allocated to 12 months of testosterone gel or placebo. The group included 126 individuals with a hemoglobin level at or below 12.7 g/dL (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9540).
The study found that significantly more men who received testosterone treatment experienced increases in hemoglobin concentration of 1 g/dL or more above baseline, compared with those who received the placebo gel (54% vs. 15%; 95% CI 3.7-277.8; P = .002).
This effect was seen in men with known causes of anemia, such as myelodysplasia, iron deficiency, B12 deficiency, or chronic inflammation or disease; in men with anemia of unknown case; and in men who weren’t anemic.
After 12 months, more than half of the testosterone-treated men who started the study with unexplained anemia were no longer anemic, compared with around one-quarter of the placebo-treated men (58.3% vs. 22.2%). The men treated with placebo also had lower average hemoglobin level changes, compared with those treated with testosterone.
“Increases in hemoglobin levels were positively and significantly associated with participants’ global impression of change in overall health and energy,” wrote Cindy N. Roy, PhD, of Johns Hopkins University, Baltimore, and her coauthors.
Bone mineral density
A second trial examined the effect of 12 months of testosterone gel or placebo on bone mineral density in a group of 211 men with average testosterone concentrations less than 275 ng/L (JAMA Intern Med. 2017 Feb 21. doi: 10.1001/jamainternmed.2016.9539). The treatment increased median serum concentrations of total testosterone, free testosterone, and estradiol to within the normal ranges for young men.
The study showed significantly greater increases – measured by quantitative computed tomography – with testosterone treatment, compared with placebo, in spine trabecular, spine peripheral, hip trabecular, and peripheral volumetric bone mineral density, as well as in mean estimated strength of spine trabecular bone, spine peripheral bone, and hip trabecular and peripheral bone.
For the primary outcome of mean lumbar spine trabecular volumetric bone mineral density, testosterone treatment was associated with a mean increase of 7.5%, compared with a 0.8% increase with placebo.
Researchers also noted that the magnitude of the increase in spine trabecular bone mineral density from baseline was significantly associated with changes in total testosterone and estradiol.
However, there were no significant differences in fracture rate, with six fractures reported in each group during the year of treatment. In the observation year after treatment, three fractures were reported in the testosterone arm and four in the placebo arm.
“These results are unequivocal compared with prior studies of the effect of testosterone treatment on bone in older men, in spite of treatment limited to 1 year, perhaps because the mean pretreatment testosterone level was lower and the sample size larger than in prior studies, and because the primary outcome in this trial was vBMD by QCT,” wrote Peter J. Snyder, MD, of the University of Pennsylvania, Philadelphia, and his coauthors.
Coronary artery plaque
However, a third trial – this one in 170 men with low testosterone and symptoms suggestive of hypogonadism – found significantly greater increases in noncalcified plaque volume, median total plaque volume, and median coronary artery calcification score among the 88 men assigned to 12 months of testosterone gel, compared with those assigned to placebo.
The men treated with testosterone showed a mean increase in noncalcified coronary artery plaque volume of 40 mm3, compared with 4 mm3 in men given the placebo gel, and a mean increase in total plaque volume of 57 mm3 with testosterone and 21 mm3 with placebo (JAMA. 2017 Feb 21;317[7]:708-16).
There were no significant differences between the groups in change to coronary artery calcium score, and there were no adverse cardiovascular events reported in either group, despite the fact that around half the participants had severe atherosclerosis at baseline.
“The increase in coronary artery noncalcified and total plaque volumes in men treated with testosterone is concerning, because any limitation of the vascular lumen could be considered deleterious,” wrote Matthew J. Budoff, MD, of the Los Angeles Biomedical Research Institute, Torrance, Calif., and his coauthors. “The clinical significance of these increases could depend on the differential effects of testosterone on the individual components of noncalcified plaque.”
However, the investigators pointed out that the trial was neither large enough nor long enough to draw conclusions about the cardiovascular risks of testosterone treatment, and they called for larger studies to explore the association.
Cognitive function
The fourth study looked at mean change in cognitive function from baseline in 493 men with a serum testosterone level less than 275 ng/dL, impaired sexual function, physical function, or vitality, and who met the criteria for age-associated memory impairment. Half the participants were assigned to 12 months of testosterone gel, and half were assigned to placebo gel (JAMA. 2017 Feb 21;317[7]:717-27).
Researchers found no significant differences between the two groups from baseline to 6 months and 12 months in mean change in delayed paragraph recall score, visual memory, executive function, or spatial ability.
“The lack of association between testosterone treatment and cognition was apparent across all cognitive domains assessed among men with [age-associated memory impairment], in spite of an increase in circulating total and free testosterone concentrations in the testosterone group to levels typical of men aged 19-40 years,” wrote Susan M. Resnick, PhD, of the National Institute on Aging, and her coauthors.
The Testosterone Trials were supported by the National Institute on Aging, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, the National Institute of Child Health and Human Development, and AbbVie, which also provided the AndroGel, and placebo gel. Authors from the trials declared a range of funding, consultancies, and other support from the pharmaceutical industry, including AbbVie. One author declared a pending patent for a free testosterone calculator.
FROM JAMA AND JAMA INTERNAL MEDICINE
Key clinical point: Testosterone treatment may have beneficial effects on unexplained anemia or bone density in men with age-related low testosterone, but at the cost of an increase in coronary artery plaque and with no benefit for cognitive function.
Major finding: Testosterone treatment was associated with significantly greater increases in bone mineral density, hemoglobin, and noncalcified coronary artery plaque, compared with placebo, but no significant effects on cognitive function.
Data source: The Testosterone Trials in men aged 65 years or older with age-related testosterone decline.
Disclosures: The Testosterone Trials were supported by the National Institute on Aging, the National Heart, Lung, and Blood Institute, the National Institute of Neurological Diseases and Stroke, the National Institute of Child Health and Human Development, and AbbVie, which provided the AndroGel and placebo gel. Authors from the trials declared a range of funding, consultancies, and other support from the pharmaceutical industry, including AbbVie. One author declared a pending patent for a free testosterone calculator.
Model illustrates progression to MDS, AML
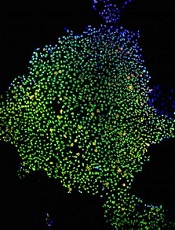
Researchers say they have created a model that shows the step-by-step progression from normal blood cells to acute myeloid leukemia (AML).
The team generated induced pluripotent stem cell (iPSC) lines capturing disease stages that included preleukemia, low-risk myelodysplastic syndrome (MDS), high-risk MDS, and AML.
The researchers then used CRISPR/Cas9 genome editing to induce disease progression and reversal.
And they used the iPSCs to uncover disease-stage-specific effects of 2 drugs.
Eirini P. Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York, and her colleagues described this work in Cell Stem Cell.
The researchers first explained how they generated patient-derived iPSCs that represented familial predisposition to myeloid malignancy, low-risk and high-risk MDS, and AML.
By studying these iPSC lines, the team uncovered “a phenotypic road map of disease progression” that led to a “serially transplantable leukemia.”
“We are encouraged by the discovery that it was possible to generate potent, engraftable leukemia derived from AML induced pluripotent stem cells,” said study author Michael G. Kharas, PhD, of the Icahn School of Medicine at Mount Sinai.
The researchers also showed that they could revert a high-risk MDS iPSC line to a premalignant state by correcting a chromosome 7q deletion.
And they could force progression in a preleukemic iPSC line. The team induced progression to low-risk MDS by inactivating the second GATA2 allele and progression to high-risk MDS by deleting chromosome 7q.
“This work shows that integrated patient cell reprogramming and cancer genetics is a powerful way to dissect cancer progression,” Dr Kharas said.
The researchers reported that, ultimately, they were able to model the stepwise progression of normal cells to preleukemia and MDS by sequentially introducing genetic lesions associated with earlier and later disease stages (ASXL1 truncation and chromosome 7q deletion, respectively).
“The new model will empower investigation into the cellular and molecular events underlying the development of leukemia in ways that were not possible before,” Dr Papapetrou said.
She added that the group’s findings provide a framework to aid investigation into disease mechanisms, events driving progression, and drug responses.
In fact, the researchers did use hematopoietic progenitor cells (HPCs) derived from their iPSCs to analyze the disease-stage-specific effects of 2 drugs—5-azacytidine and rigosertib.
The team said they found evidence to suggest that 5-azacytidine may work in low-risk MDS by affecting differentiation, and the drug’s main therapeutic action in high-risk MDS might be mediated through selective inhibition of the MDS clone.
The researchers tested rigosertib in HPCs derived from 2 AML lines (from the same patient) that captured 2 different disease stages. One line was derived from the dominant clone (del 7q), and the other was derived from a KRAS-mutated subclone.
The team found that HPCs derived from the KRAS-mutated line demonstrated “marked sensitivity” to rigosertib, but the other HPCs were “marginally affected.” ![]()
Researchers say they have created a model that shows the step-by-step progression from normal blood cells to acute myeloid leukemia (AML).
The team generated induced pluripotent stem cell (iPSC) lines capturing disease stages that included preleukemia, low-risk myelodysplastic syndrome (MDS), high-risk MDS, and AML.
The researchers then used CRISPR/Cas9 genome editing to induce disease progression and reversal.
And they used the iPSCs to uncover disease-stage-specific effects of 2 drugs.
Eirini P. Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York, and her colleagues described this work in Cell Stem Cell.
The researchers first explained how they generated patient-derived iPSCs that represented familial predisposition to myeloid malignancy, low-risk and high-risk MDS, and AML.
By studying these iPSC lines, the team uncovered “a phenotypic road map of disease progression” that led to a “serially transplantable leukemia.”
“We are encouraged by the discovery that it was possible to generate potent, engraftable leukemia derived from AML induced pluripotent stem cells,” said study author Michael G. Kharas, PhD, of the Icahn School of Medicine at Mount Sinai.
The researchers also showed that they could revert a high-risk MDS iPSC line to a premalignant state by correcting a chromosome 7q deletion.
And they could force progression in a preleukemic iPSC line. The team induced progression to low-risk MDS by inactivating the second GATA2 allele and progression to high-risk MDS by deleting chromosome 7q.
“This work shows that integrated patient cell reprogramming and cancer genetics is a powerful way to dissect cancer progression,” Dr Kharas said.
The researchers reported that, ultimately, they were able to model the stepwise progression of normal cells to preleukemia and MDS by sequentially introducing genetic lesions associated with earlier and later disease stages (ASXL1 truncation and chromosome 7q deletion, respectively).
“The new model will empower investigation into the cellular and molecular events underlying the development of leukemia in ways that were not possible before,” Dr Papapetrou said.
She added that the group’s findings provide a framework to aid investigation into disease mechanisms, events driving progression, and drug responses.
In fact, the researchers did use hematopoietic progenitor cells (HPCs) derived from their iPSCs to analyze the disease-stage-specific effects of 2 drugs—5-azacytidine and rigosertib.
The team said they found evidence to suggest that 5-azacytidine may work in low-risk MDS by affecting differentiation, and the drug’s main therapeutic action in high-risk MDS might be mediated through selective inhibition of the MDS clone.
The researchers tested rigosertib in HPCs derived from 2 AML lines (from the same patient) that captured 2 different disease stages. One line was derived from the dominant clone (del 7q), and the other was derived from a KRAS-mutated subclone.
The team found that HPCs derived from the KRAS-mutated line demonstrated “marked sensitivity” to rigosertib, but the other HPCs were “marginally affected.” ![]()
Researchers say they have created a model that shows the step-by-step progression from normal blood cells to acute myeloid leukemia (AML).
The team generated induced pluripotent stem cell (iPSC) lines capturing disease stages that included preleukemia, low-risk myelodysplastic syndrome (MDS), high-risk MDS, and AML.
The researchers then used CRISPR/Cas9 genome editing to induce disease progression and reversal.
And they used the iPSCs to uncover disease-stage-specific effects of 2 drugs.
Eirini P. Papapetrou, MD, PhD, of the Icahn School of Medicine at Mount Sinai in New York, New York, and her colleagues described this work in Cell Stem Cell.
The researchers first explained how they generated patient-derived iPSCs that represented familial predisposition to myeloid malignancy, low-risk and high-risk MDS, and AML.
By studying these iPSC lines, the team uncovered “a phenotypic road map of disease progression” that led to a “serially transplantable leukemia.”
“We are encouraged by the discovery that it was possible to generate potent, engraftable leukemia derived from AML induced pluripotent stem cells,” said study author Michael G. Kharas, PhD, of the Icahn School of Medicine at Mount Sinai.
The researchers also showed that they could revert a high-risk MDS iPSC line to a premalignant state by correcting a chromosome 7q deletion.
And they could force progression in a preleukemic iPSC line. The team induced progression to low-risk MDS by inactivating the second GATA2 allele and progression to high-risk MDS by deleting chromosome 7q.
“This work shows that integrated patient cell reprogramming and cancer genetics is a powerful way to dissect cancer progression,” Dr Kharas said.
The researchers reported that, ultimately, they were able to model the stepwise progression of normal cells to preleukemia and MDS by sequentially introducing genetic lesions associated with earlier and later disease stages (ASXL1 truncation and chromosome 7q deletion, respectively).
“The new model will empower investigation into the cellular and molecular events underlying the development of leukemia in ways that were not possible before,” Dr Papapetrou said.
She added that the group’s findings provide a framework to aid investigation into disease mechanisms, events driving progression, and drug responses.
In fact, the researchers did use hematopoietic progenitor cells (HPCs) derived from their iPSCs to analyze the disease-stage-specific effects of 2 drugs—5-azacytidine and rigosertib.
The team said they found evidence to suggest that 5-azacytidine may work in low-risk MDS by affecting differentiation, and the drug’s main therapeutic action in high-risk MDS might be mediated through selective inhibition of the MDS clone.
The researchers tested rigosertib in HPCs derived from 2 AML lines (from the same patient) that captured 2 different disease stages. One line was derived from the dominant clone (del 7q), and the other was derived from a KRAS-mutated subclone.
The team found that HPCs derived from the KRAS-mutated line demonstrated “marked sensitivity” to rigosertib, but the other HPCs were “marginally affected.” ![]()
Understanding a rare hemoglobin mutation

Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Smoking can prevent anemia in individuals with a rare hemoglobin mutation, according to research published in the Journal of Biological Chemistry.
The so-called Kirklareli mutation was found to be the cause of mild anemia in a young woman in Germany.
But a smoking habit protected the young woman’s father, who also carried the mutation, from developing anemia.
The Kirklareli mutation is one of more than 1000 discovered so far in adult human hemoglobin.
Most of these mutations appear to have no effect on people, but when medical problems occur, the disease is called a hemoglobinopathy and often named after the city or hospital where it was discovered. In this case, the family was living in Mannheim, Germany, but the father was born in the Turkish city of Kirklareli.
The Kirklareli mutation did not affect the iron content of the father’s blood, but it did appear to be the root cause of the young woman’s chronic anemia, according to researchers.
Further investigation revealed that absorbing carbon monoxide from cigarette smoke is therapeutic for individuals with this rare genetic disorder.
The Kirklareli mutation is in the alpha subunit of human hemoglobin (H58L) and causes it to rapidly auto-oxidize, which causes the protein to fall apart, lose heme, and precipitate. As a result, the protein loses its ability to carry oxygen. Eventually, red cells become deformed and are destroyed.
This mutation also gives the protein an 80,000-fold higher affinity for carbon monoxide than for oxygen. Carbon monoxide from a cigarette will be selectively taken up by the mutant hemoglobin and prevent it from oxidizing and denaturing.
This high affinity for carbon monoxide explained why the father showed no signs of anemia, the researchers said.
“He may never be an athlete because his blood can’t carry as much oxygen, but smoking has prevented him from being anemic,” said study author John Olson, PhD, of Rice University in Houston, Texas.
“And there’s a side benefit. People with this trait are more resistant to carbon monoxide poisoning.”
Dr Olson said he doesn’t know how or if doctors treated the young woman, but he suspects her iron-deficiency anemia was more an annoyance than a threat to her life and would not recommend she start smoking to relieve it.
“She shouldn’t smoke,” Dr Olson said. “But she could take antioxidants, such as a lot of vitamin C, which would help prevent oxidation of her mutant hemoglobin. Her anemia is not that severe. At the same time, she shouldn’t worry too much about secondhand smoke, which might have a positive effect.”
After ruling out common causes of anemia—such as blood loss, gastritis, or congenital defects—the woman’s doctors were curious enough about her ailment to call upon Emmanuel Bissé, MD, PhD, a researcher at Universitätsklinikum Freiburg in Freiburg, Germany, who discovered the Kirklareli mutation after sequencing the woman’s DNA.
Dr Bissé, in turn, recruited Dr Olson and his team to help determine why the histidine-to-leucine change caused anemia in the daughter but not the father.
Coincidentally, Ivan Birukou, a graduate student in Dr Olson’s lab, had already generated the Kirklareli mutation in human hemoglobin to study how the protein rapidly and selectively binds oxygen.
“Emmanuel wrote to me and said, ‘I know you’ve been making all these mutants in hemoglobin, and you’ve probably done the H58L mutation in [alpha] chains. Does this phenotype make sense?’” Dr Olson recalled.
“I said, ‘We can do a really neat study here, because we’ve already made the mutant hemoglobin in a recombinant system.’ We actually had a crystal structure [matching Kirklareli] that Ivan and [staff scientist] Jayashree Soman never published but had deposited in the Protein Data Bank. We had made this mutation to try to understand what the distal histidine was doing in alpha subunits.”
The researchers found in a 2010 study that replacing the histidine, which forms a strong hydrogen bond to oxygen, with leucine caused a dramatic decrease in oxygen affinity and an increase in carbon monoxide binding.
Dr Olson and Birukou realized back then that histidine played a key role in discriminating between oxygen and carbon monoxide in hemoglobin.
“When Emmanuel wrote to me about his discovery, I already ‘knew’ what was happening with respect to carbon monoxide binding,” Dr Olson said.
He said the normal hydrogen bond causes bound oxygen to stick more tightly to hemoglobin in the same way hydrogen bonds cause spilled soda to feel sticky.
“When you touch it, the sugar oxygens and hydrogens make hydrogen bonds with the polysaccharides on your finger,” Dr Olson said. “That stickiness helps hold onto oxygen. But leucine is more like an oil, like butane or hexane, and oxygen does not stick well inside hemoglobin. In contrast, bound carbon monoxide is more like methane or ethane and can’t form hydrogen bonds.”
Andres Benitez Cardenas, PhD, a researcher in Dr Olson’s lab, did the experiment in which he put carbon monoxide on the mutant alpha subunit of hemoglobin Kirklareli. The bound carbon monoxide slowed down oxidation of the protein and prevented loss of heme and precipitation.
“In effect, Andres did the ‘smoking experiment’ to show why the father’s hemoglobin didn’t denature and cause anemia,” Dr Olson said.
He noted that the effect caused by Kirklareli, though unusual, is not unique. Patients with hemoglobin Zurich also have an abnormal form of hemoglobin that more readily binds to carbon monoxide. ![]()
Study reveals patterns of ED use in SCD patients

Photo courtesy of St. Jude
Children’s Research Hospital
Population-based surveillance data has revealed patterns of emergency department (ED) visits among Californians with sickle cell disease (SCD).
Previous research suggested that between one-half and two-thirds of SCD patients’ ED visits end in a discharge from the ED, called a treat-and-release visit.
The remainder result in admission to a hospital or other treatment facility.
The purpose of the current study was to use data from the Sickle Cell Data Collection program to describe patterns of ED use for treat-and-release visits by California’s SCD population and compare these new findings with results of previous studies.
The current study was published in Pediatric Blood and Cancer.
Researchers looked at ED and hospital discharge data in California from 2005 to 2014. This included 4636 patients with SCD.
The data showed that 88% of patients had 1 or more treat-and-release ED visits during the 10-year study period.
This group of 4100 patients had 90,904 treat-and-release ED visits. The average number of visits each year was 2.1 (rage, 0-185).
In a single year (2005):
- 53% of patients had no treat-and-release ED visits (no ED use)
- 35% had between 1 and 3 visits (low ED use)
- 9% had between 4 and 10 visits (medium ED use)
- 3% had 11 or more visits (high ED use).
The youngest patients (age 0 to 9.9) and the oldest patients (80 and older) were the least likely to have at least 1 treat-and-release ED visit.
The proportion of patients with at least 1 ED visit over the study period was:
- 68% among patients age 0 to 9.9 at the close of the study
- 80% among patients age 10 to 19.9
- 92% among patients age 20 to 29.9
- 94% among patients age 30 to 39.9
- 93% among patients age 40 to 49.9
- 92% among patients age 50 to 59.9
- 92% among patients age 60 to 69.9
- 85% among patients age 70 to 79.9
- 73% among patients age 80 and older.
The researchers said this study highlights the utility of a multisource, longitudinal data collection effort for SCD. And further study of patients with the highest ED utilization may highlight areas where changes could improve and extend the lives of patients with SCD. ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Population-based surveillance data has revealed patterns of emergency department (ED) visits among Californians with sickle cell disease (SCD).
Previous research suggested that between one-half and two-thirds of SCD patients’ ED visits end in a discharge from the ED, called a treat-and-release visit.
The remainder result in admission to a hospital or other treatment facility.
The purpose of the current study was to use data from the Sickle Cell Data Collection program to describe patterns of ED use for treat-and-release visits by California’s SCD population and compare these new findings with results of previous studies.
The current study was published in Pediatric Blood and Cancer.
Researchers looked at ED and hospital discharge data in California from 2005 to 2014. This included 4636 patients with SCD.
The data showed that 88% of patients had 1 or more treat-and-release ED visits during the 10-year study period.
This group of 4100 patients had 90,904 treat-and-release ED visits. The average number of visits each year was 2.1 (rage, 0-185).
In a single year (2005):
- 53% of patients had no treat-and-release ED visits (no ED use)
- 35% had between 1 and 3 visits (low ED use)
- 9% had between 4 and 10 visits (medium ED use)
- 3% had 11 or more visits (high ED use).
The youngest patients (age 0 to 9.9) and the oldest patients (80 and older) were the least likely to have at least 1 treat-and-release ED visit.
The proportion of patients with at least 1 ED visit over the study period was:
- 68% among patients age 0 to 9.9 at the close of the study
- 80% among patients age 10 to 19.9
- 92% among patients age 20 to 29.9
- 94% among patients age 30 to 39.9
- 93% among patients age 40 to 49.9
- 92% among patients age 50 to 59.9
- 92% among patients age 60 to 69.9
- 85% among patients age 70 to 79.9
- 73% among patients age 80 and older.
The researchers said this study highlights the utility of a multisource, longitudinal data collection effort for SCD. And further study of patients with the highest ED utilization may highlight areas where changes could improve and extend the lives of patients with SCD. ![]()
Photo courtesy of St. Jude
Children’s Research Hospital
Population-based surveillance data has revealed patterns of emergency department (ED) visits among Californians with sickle cell disease (SCD).
Previous research suggested that between one-half and two-thirds of SCD patients’ ED visits end in a discharge from the ED, called a treat-and-release visit.
The remainder result in admission to a hospital or other treatment facility.
The purpose of the current study was to use data from the Sickle Cell Data Collection program to describe patterns of ED use for treat-and-release visits by California’s SCD population and compare these new findings with results of previous studies.
The current study was published in Pediatric Blood and Cancer.
Researchers looked at ED and hospital discharge data in California from 2005 to 2014. This included 4636 patients with SCD.
The data showed that 88% of patients had 1 or more treat-and-release ED visits during the 10-year study period.
This group of 4100 patients had 90,904 treat-and-release ED visits. The average number of visits each year was 2.1 (rage, 0-185).
In a single year (2005):
- 53% of patients had no treat-and-release ED visits (no ED use)
- 35% had between 1 and 3 visits (low ED use)
- 9% had between 4 and 10 visits (medium ED use)
- 3% had 11 or more visits (high ED use).
The youngest patients (age 0 to 9.9) and the oldest patients (80 and older) were the least likely to have at least 1 treat-and-release ED visit.
The proportion of patients with at least 1 ED visit over the study period was:
- 68% among patients age 0 to 9.9 at the close of the study
- 80% among patients age 10 to 19.9
- 92% among patients age 20 to 29.9
- 94% among patients age 30 to 39.9
- 93% among patients age 40 to 49.9
- 92% among patients age 50 to 59.9
- 92% among patients age 60 to 69.9
- 85% among patients age 70 to 79.9
- 73% among patients age 80 and older.
The researchers said this study highlights the utility of a multisource, longitudinal data collection effort for SCD. And further study of patients with the highest ED utilization may highlight areas where changes could improve and extend the lives of patients with SCD. ![]()
Genetic profiling can guide HSCT in MDS, team says

Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
Genetic profiling can be used to determine which patients with myelodysplastic syndrome (MDS) are likely to benefit from allogeneic hematopoietic stem cell transplant (HSCT), according to research published in NEJM.
Targeted sequencing of 129 genes revealed mutations that, after adjustment for clinical variables, were associated with shorter survival and/or relapse after HSCT.
Patients with mutations in TP53, JAK2, and the RAS pathway tended to have worse outcomes after HSCT than patients without such mutations.
“Although donor stem cell transplantation is the only curative therapy for MDS, many patients die after transplantation, largely due to relapse of the disease or complications relating to the transplant itself,” said study author R. Coleman Lindsley, MD, PhD, of Dana-Farber Cancer Institute in Boston, Massachusetts.
“As physicians, one of our major challenges is to be able to predict which patients are most likely to benefit from a transplant. Improving our ability to identify patients who are most likely to have a relapse or to experience life-threatening complications from a transplant could lead to better pre-transplant therapies and strategies for preventing relapse.”
Researchers have long known that specific genetic mutations are closely related to the course MDS takes. With this study, Dr Lindsley and his colleagues sought to discover whether mutations can be used to predict how patients will fare following allogeneic HSCT.
The team analyzed blood samples from 1514 MDS patients, performing targeted sequencing of 129 genes. The genes were selected based on their known or suspected involvement in the pathogenesis of myeloid cancers or bone marrow failure syndromes.
Dr Lindsley and his colleagues then evaluated the association between mutations and HSCT outcomes, including overall survival, relapse, and death without relapse.
After adjusting for significant clinical variables, the researchers found that having mutated TP53 was significantly associated with shorter survival and shorter time to relapse after HSCT (P<0.001 for both comparisons). This was true whether patients received standard conditioning or reduced-intensity conditioning.
In patients age 40 and older who did not have TP53 mutations, mutations in RAS pathway genes (P=0.004) or JAK2 (P=0.001) were significantly associated with shorter survival.
The shorter survival in patients with mutated RAS pathway genes was due to a higher risk of relapse, while the shorter survival in patients with JAK2 mutations was due to a higher risk of death without relapse.
In contrast to TP53 mutations, the adverse effect of RAS mutations on survival and risk of relapse was evident only in patients who received reduced-intensity conditioning (P<0.001). This suggests these patients may benefit from higher intensity conditioning regimens, the researchers said.
This study also yielded insights about the biology of MDS in specific groups of patients.
For example, the researchers found that 4% of MDS patients between the ages of 18 and 40 had mutations associated with Shwachman-Diamond syndrome (in the SBDS gene), but most of them had not previously been diagnosed with the syndrome.
In each case, the patients had acquired a TP53 mutation, suggesting not only how MDS develops in patients with Schwachman-Diamond syndrome but also what underlies their poor prognosis after HSCT.
The researchers also analyzed patients with therapy-related MDS. The team found that TP53 mutations and mutations in PPM1D, a gene that regulates TP53 function, were far more common in these patients than in those with primary MDS (15% and 3%, respectively, P<0.001).
“In deciding whether a stem cell transplant is appropriate for a patient with MDS, it’s always necessary to balance the potential benefit with the risk of complications,” Dr Lindsley noted.
“Our findings offer physicians a guide—based on the genetic profile of the disease and certain clinical factors—to identifying patients for whom a transplant is appropriate, and the intensity of treatment most likely to be effective.” ![]()
iPSCs used to identify potential treatment for DBA
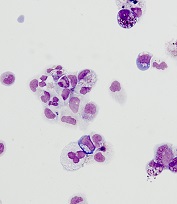
from cells from a DBA patient
Image courtesy of
Boston Children’s Hospital
Researchers have used induced pluripotent stem cells (iPSCs) to identify a compound that could treat Diamond Blackfan anemia (DBA).
The team used iPSCs to generate expandable hematopoietic progenitor cells (HPCs) that recapitulate the defects in erythroid differentiation observed in patients with DBA.
The researchers then used the HPCs to screen chemical compounds that might be used to treat DBA.
One of these compounds, SMER28, enhanced erythropoiesis in models of DBA.
“It is very satisfying as physician-scientists to find new potential treatments for rare blood diseases such as Diamond Blackfan anemia,” said study author Leonard Zon, MD, of Boston Children’s Hospital and Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Zon and his colleagues described this work in Science Translational Medicine.
The researchers first took fibroblasts from 2 patients with DBA and reprogrammed them into iPSCs. From the iPSCs, the team generated HPCs, which they loaded into a high-throughput drug screening system.
Testing a library of 1440 chemicals, the researchers found several that showed promise for treating DBA in vitro. One compound, SMER28, was able to induce red blood cell (RBC) production in mice and zebrafish.
The researchers believe this study marks an important advance in the stem cell field.
“[iPSCs] have been hard to instruct when it comes to making blood,” said study author Sergei Doulatov, PhD, of the University of Washington in Seattle.
“This is the first time [iPSCs] have been used to identify a drug to treat a blood disorder.”
Making RBCs
As in DBA itself, the patient-derived HPCs, studied in vitro, failed to generate erythroid cells. The same was true when the cells were transplanted into mice.
However, the chemical screen got several “hits.” In wells loaded with certain chemicals, erythroid cells began appearing. Because of its especially strong effect, SMER28 was put through additional testing.
When SMER28 was used to treat the marrow in zebrafish and mouse models of DBA, the animals made erythroid progenitor cells that, in turn, made RBCs, reversing or stabilizing anemia. The same was true in cells from DBA patients transplanted into mice.
The higher the dose of SMER28, the more RBCs were produced, and no ill effects were found. However, formal toxicity studies have not been conducted.
SMER28 has been tested preclinically for some neurodegenerative diseases. It activates an autophagy pathway that recycles damaged cellular components.
In DBA, SMER28 appears to turn on autophagy in erythroid progenitors. Dr Doulatov plans to further explore how this interferes with RBC production. ![]()
from cells from a DBA patient
Image courtesy of
Boston Children’s Hospital
Researchers have used induced pluripotent stem cells (iPSCs) to identify a compound that could treat Diamond Blackfan anemia (DBA).
The team used iPSCs to generate expandable hematopoietic progenitor cells (HPCs) that recapitulate the defects in erythroid differentiation observed in patients with DBA.
The researchers then used the HPCs to screen chemical compounds that might be used to treat DBA.
One of these compounds, SMER28, enhanced erythropoiesis in models of DBA.
“It is very satisfying as physician-scientists to find new potential treatments for rare blood diseases such as Diamond Blackfan anemia,” said study author Leonard Zon, MD, of Boston Children’s Hospital and Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Zon and his colleagues described this work in Science Translational Medicine.
The researchers first took fibroblasts from 2 patients with DBA and reprogrammed them into iPSCs. From the iPSCs, the team generated HPCs, which they loaded into a high-throughput drug screening system.
Testing a library of 1440 chemicals, the researchers found several that showed promise for treating DBA in vitro. One compound, SMER28, was able to induce red blood cell (RBC) production in mice and zebrafish.
The researchers believe this study marks an important advance in the stem cell field.
“[iPSCs] have been hard to instruct when it comes to making blood,” said study author Sergei Doulatov, PhD, of the University of Washington in Seattle.
“This is the first time [iPSCs] have been used to identify a drug to treat a blood disorder.”
Making RBCs
As in DBA itself, the patient-derived HPCs, studied in vitro, failed to generate erythroid cells. The same was true when the cells were transplanted into mice.
However, the chemical screen got several “hits.” In wells loaded with certain chemicals, erythroid cells began appearing. Because of its especially strong effect, SMER28 was put through additional testing.
When SMER28 was used to treat the marrow in zebrafish and mouse models of DBA, the animals made erythroid progenitor cells that, in turn, made RBCs, reversing or stabilizing anemia. The same was true in cells from DBA patients transplanted into mice.
The higher the dose of SMER28, the more RBCs were produced, and no ill effects were found. However, formal toxicity studies have not been conducted.
SMER28 has been tested preclinically for some neurodegenerative diseases. It activates an autophagy pathway that recycles damaged cellular components.
In DBA, SMER28 appears to turn on autophagy in erythroid progenitors. Dr Doulatov plans to further explore how this interferes with RBC production. ![]()
from cells from a DBA patient
Image courtesy of
Boston Children’s Hospital
Researchers have used induced pluripotent stem cells (iPSCs) to identify a compound that could treat Diamond Blackfan anemia (DBA).
The team used iPSCs to generate expandable hematopoietic progenitor cells (HPCs) that recapitulate the defects in erythroid differentiation observed in patients with DBA.
The researchers then used the HPCs to screen chemical compounds that might be used to treat DBA.
One of these compounds, SMER28, enhanced erythropoiesis in models of DBA.
“It is very satisfying as physician-scientists to find new potential treatments for rare blood diseases such as Diamond Blackfan anemia,” said study author Leonard Zon, MD, of Boston Children’s Hospital and Dana-Farber Cancer Institute in Boston, Massachusetts.
Dr Zon and his colleagues described this work in Science Translational Medicine.
The researchers first took fibroblasts from 2 patients with DBA and reprogrammed them into iPSCs. From the iPSCs, the team generated HPCs, which they loaded into a high-throughput drug screening system.
Testing a library of 1440 chemicals, the researchers found several that showed promise for treating DBA in vitro. One compound, SMER28, was able to induce red blood cell (RBC) production in mice and zebrafish.
The researchers believe this study marks an important advance in the stem cell field.
“[iPSCs] have been hard to instruct when it comes to making blood,” said study author Sergei Doulatov, PhD, of the University of Washington in Seattle.
“This is the first time [iPSCs] have been used to identify a drug to treat a blood disorder.”
Making RBCs
As in DBA itself, the patient-derived HPCs, studied in vitro, failed to generate erythroid cells. The same was true when the cells were transplanted into mice.
However, the chemical screen got several “hits.” In wells loaded with certain chemicals, erythroid cells began appearing. Because of its especially strong effect, SMER28 was put through additional testing.
When SMER28 was used to treat the marrow in zebrafish and mouse models of DBA, the animals made erythroid progenitor cells that, in turn, made RBCs, reversing or stabilizing anemia. The same was true in cells from DBA patients transplanted into mice.
The higher the dose of SMER28, the more RBCs were produced, and no ill effects were found. However, formal toxicity studies have not been conducted.
SMER28 has been tested preclinically for some neurodegenerative diseases. It activates an autophagy pathway that recycles damaged cellular components.
In DBA, SMER28 appears to turn on autophagy in erythroid progenitors. Dr Doulatov plans to further explore how this interferes with RBC production. ![]()
FDA approves IVIG product for PI and chronic ITP

Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin (IVIG) product (Gammaplex® 10%) for the treatment of primary immunodeficiency (PI) and chronic immune thrombocytopenia (ITP) in adults.
PI includes, but is not limited to,
the humoral immune defect in common variable immunodeficiency, X-linked and congenital
agammaglobulinemia, Wiskott-Aldrich
syndrome, and severe combined immunodeficiencies.
Gammaplex 10% is manufactured by Bio Products Laboratory Limited (BPL).
Gammaplex 10% is made with the same process as BPL’s previously approved IVIG treatment, Gammaplex® 5% (immune globulin intravenous [human], 5% liquid).
Gammaplex 10% is more concentrated than Gammaplex 5%, with an immune globulin G (IgG) concentration of 100 g/L, and is stabilized with glycine.
The FDA’s approval of Gammaplex 10% was based on a 2-phase, crossover bioequivalence study comparing Gammaplex 10% and Gammaplex 5% in 33 adult patients with PI. This study is the first direct comparison of 10% and 5% IVIG products in the treatment of PI.
The primary endpoint of bioequivalence between the products was achieved, and trough levels of IgG were well maintained throughout the study.
Both Gammaplex 10% and Gammaplex 5% infusion rates were increased incrementally at 15-minute intervals if tolerated by the subject. The Gammaplex 10% infusion rate was increased to the maximum in 96% of infusions.
The mean infusion time for Gammaplex 10% was 1 hour and 51 minutes, which was 57 minutes faster than Gammaplex 5%.
There were no notable differences in the safety and tolerability of the 2 products.
The most common adverse events in patients receiving Gammaplex 10% were headache (12.5%), migraine (6.3%), and pyrexia (6.3%). There were no serious product-related adverse events.
The safety of Gammaplex 10% has not been established in adults with chronic ITP. The safety profile for Gammaplex 5% has been studied in a phase 3 trial of adults with chronic ITP, and it is anticipated that the safety profile for both formulations are comparable for ITP patients.
The most common adverse events in adults with chronic ITP receiving Gammaplex 5% were headache, vomiting, nausea, pyrexia, arthralgia, and dehydration. Serious adverse events were headache, vomiting, and dehydration.
The full prescribing information for Gammaplex 10%, which includes trial data, is available at http://www.gammaplex.com. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin (IVIG) product (Gammaplex® 10%) for the treatment of primary immunodeficiency (PI) and chronic immune thrombocytopenia (ITP) in adults.
PI includes, but is not limited to,
the humoral immune defect in common variable immunodeficiency, X-linked and congenital
agammaglobulinemia, Wiskott-Aldrich
syndrome, and severe combined immunodeficiencies.
Gammaplex 10% is manufactured by Bio Products Laboratory Limited (BPL).
Gammaplex 10% is made with the same process as BPL’s previously approved IVIG treatment, Gammaplex® 5% (immune globulin intravenous [human], 5% liquid).
Gammaplex 10% is more concentrated than Gammaplex 5%, with an immune globulin G (IgG) concentration of 100 g/L, and is stabilized with glycine.
The FDA’s approval of Gammaplex 10% was based on a 2-phase, crossover bioequivalence study comparing Gammaplex 10% and Gammaplex 5% in 33 adult patients with PI. This study is the first direct comparison of 10% and 5% IVIG products in the treatment of PI.
The primary endpoint of bioequivalence between the products was achieved, and trough levels of IgG were well maintained throughout the study.
Both Gammaplex 10% and Gammaplex 5% infusion rates were increased incrementally at 15-minute intervals if tolerated by the subject. The Gammaplex 10% infusion rate was increased to the maximum in 96% of infusions.
The mean infusion time for Gammaplex 10% was 1 hour and 51 minutes, which was 57 minutes faster than Gammaplex 5%.
There were no notable differences in the safety and tolerability of the 2 products.
The most common adverse events in patients receiving Gammaplex 10% were headache (12.5%), migraine (6.3%), and pyrexia (6.3%). There were no serious product-related adverse events.
The safety of Gammaplex 10% has not been established in adults with chronic ITP. The safety profile for Gammaplex 5% has been studied in a phase 3 trial of adults with chronic ITP, and it is anticipated that the safety profile for both formulations are comparable for ITP patients.
The most common adverse events in adults with chronic ITP receiving Gammaplex 5% were headache, vomiting, nausea, pyrexia, arthralgia, and dehydration. Serious adverse events were headache, vomiting, and dehydration.
The full prescribing information for Gammaplex 10%, which includes trial data, is available at http://www.gammaplex.com. ![]()
Photo by Bill Branson
The US Food and Drug Administration (FDA) has approved an intravenous immunoglobulin (IVIG) product (Gammaplex® 10%) for the treatment of primary immunodeficiency (PI) and chronic immune thrombocytopenia (ITP) in adults.
PI includes, but is not limited to,
the humoral immune defect in common variable immunodeficiency, X-linked and congenital
agammaglobulinemia, Wiskott-Aldrich
syndrome, and severe combined immunodeficiencies.
Gammaplex 10% is manufactured by Bio Products Laboratory Limited (BPL).
Gammaplex 10% is made with the same process as BPL’s previously approved IVIG treatment, Gammaplex® 5% (immune globulin intravenous [human], 5% liquid).
Gammaplex 10% is more concentrated than Gammaplex 5%, with an immune globulin G (IgG) concentration of 100 g/L, and is stabilized with glycine.
The FDA’s approval of Gammaplex 10% was based on a 2-phase, crossover bioequivalence study comparing Gammaplex 10% and Gammaplex 5% in 33 adult patients with PI. This study is the first direct comparison of 10% and 5% IVIG products in the treatment of PI.
The primary endpoint of bioequivalence between the products was achieved, and trough levels of IgG were well maintained throughout the study.
Both Gammaplex 10% and Gammaplex 5% infusion rates were increased incrementally at 15-minute intervals if tolerated by the subject. The Gammaplex 10% infusion rate was increased to the maximum in 96% of infusions.
The mean infusion time for Gammaplex 10% was 1 hour and 51 minutes, which was 57 minutes faster than Gammaplex 5%.
There were no notable differences in the safety and tolerability of the 2 products.
The most common adverse events in patients receiving Gammaplex 10% were headache (12.5%), migraine (6.3%), and pyrexia (6.3%). There were no serious product-related adverse events.
The safety of Gammaplex 10% has not been established in adults with chronic ITP. The safety profile for Gammaplex 5% has been studied in a phase 3 trial of adults with chronic ITP, and it is anticipated that the safety profile for both formulations are comparable for ITP patients.
The most common adverse events in adults with chronic ITP receiving Gammaplex 5% were headache, vomiting, nausea, pyrexia, arthralgia, and dehydration. Serious adverse events were headache, vomiting, and dehydration.
The full prescribing information for Gammaplex 10%, which includes trial data, is available at http://www.gammaplex.com. ![]()
Sickle cell trait may confound blood sugar readings

Photo by Juan D. Alfonso
A new study suggests that hemoglobin A1c (HbA1c), a biomarker used to measure blood sugar over time, may not perform as accurately among African-Americans with sickle cell trait (SCT) and could be leading to a systemic underestimation of blood sugar control in that population.
Researchers analyzed data from more than 4600 people and found that HbA1c readings were significantly lower in individuals with SCT than in those without it, even after accounting for several possible confounding factors.
The team reported these findings in JAMA.
“We found that HbA1c was systematically lower in African-Americans with sickle cell trait than those without sickle cell trait, despite similar blood sugar measurements using other tests,” said study author Mary Lacy, a doctoral candidate at the Brown University School of Public Health in Providence, Rhode Island.
“We might be missing an opportunity for diagnosis and treatment of a serious disease.”
Lacy and her co-authors found that using standard clinical HbA1c cutoffs resulted in identifying 40% fewer potential cases of prediabetes and 48% fewer potential cases of diabetes in people with SCT than in people without SCT.
However, when the researchers used other blood glucose measures as the diagnostic criteria, they found no significant difference in the likelihood of diabetes and prediabetes among patients with or without SCT.
The questions the study raises about using HbA1c among SCT carriers matter for treatment as well as diagnosis, said study author Wen-Chih Wu, MD, of Brown University.
“The clinical implications of these results are highly relevant,” Dr Wu said. “For patients with diabetes, HbA1c is often used as a marker of how well they are managing their diabetes, so having an underestimation of their blood sugars is problematic because they might have a false sense of security, thinking they are doing okay when they are not.”
Study details
For this study, the researchers analyzed data from 2 major public health studies—the Coronary Artery Risk Development in Young Adults (CARDIA) study and the Jackson Heart Study (JHS). Of the 4620 participants included in the analysis, 367 had SCT.
Among all the patients included in the analysis, HbA1c readings came from either of 2 widely used, clinically accepted assays made by Tosoh Bioscience Inc. that rely on a process called high-performance liquid chromatography.
In addition to those measures, the researchers also compared fasting and 2-hour blood glucose and statistically controlled for demographic and medical factors, such as gender, age, body-mass index, whether diabetes had already been diagnosed, and whether it was being treated.
Study author Gregory Wellenius, ScD, of Brown University, said the study’s scale and breadth allowed for the most definitive comparison to date of HbA1c readings in patients with and without SCT. Two previous, smaller studies had not detected a similar discrepancy.
“The strengths of the study are that it’s the largest sample size ever used, it’s across 2 different studies with somewhat different populations, and it’s a more thorough evaluation than prior studies,” Dr Wellenius said.
While the study showed that HbA1c readings were significantly different between people with and without SCT, it also showed that blood glucose readings were not, suggesting that glucose metabolism is not necessarily different between the 2 groups, as the HbA1c readings alone would suggest.
Implications for practice
The study does not explain why the HbA1c readings differ. While it could be related to the assay method, which is approved by the NGSP (formerly National Glycohemoglobin Standardization Program) for use in patients with SCT, it could also be a consequence of the underlying biology of SCT.
Hypothetically, according to the researchers, if the hemoglobin variant of the trait endows red blood cells with a shorter lifespan, the cells’ hemoglobin would carry less accumulated blood glucose, leading to falsely low HbA1c readings.
“Irrespective of the reason of the underestimation, the underestimation is very real, and clinicians should consider screening for sickle cell trait and account for the difference in HbA1c,” Dr Wu said.
Yet not many people with SCT in the US know they carry the variant, especially those born before routine screenings at birth began, Lacy said.
The researchers therefore recommend that practitioners following African-American patients whose HbA1c levels are within 0.3 percentage points of a diagnostic cutoff also consider using additional blood glucose measures. Current diagnostic thresholds for A1C are ≥5.7% for prediabetes and ≥6.5% for diabetes. ![]()
Photo by Juan D. Alfonso
A new study suggests that hemoglobin A1c (HbA1c), a biomarker used to measure blood sugar over time, may not perform as accurately among African-Americans with sickle cell trait (SCT) and could be leading to a systemic underestimation of blood sugar control in that population.
Researchers analyzed data from more than 4600 people and found that HbA1c readings were significantly lower in individuals with SCT than in those without it, even after accounting for several possible confounding factors.
The team reported these findings in JAMA.
“We found that HbA1c was systematically lower in African-Americans with sickle cell trait than those without sickle cell trait, despite similar blood sugar measurements using other tests,” said study author Mary Lacy, a doctoral candidate at the Brown University School of Public Health in Providence, Rhode Island.
“We might be missing an opportunity for diagnosis and treatment of a serious disease.”
Lacy and her co-authors found that using standard clinical HbA1c cutoffs resulted in identifying 40% fewer potential cases of prediabetes and 48% fewer potential cases of diabetes in people with SCT than in people without SCT.
However, when the researchers used other blood glucose measures as the diagnostic criteria, they found no significant difference in the likelihood of diabetes and prediabetes among patients with or without SCT.
The questions the study raises about using HbA1c among SCT carriers matter for treatment as well as diagnosis, said study author Wen-Chih Wu, MD, of Brown University.
“The clinical implications of these results are highly relevant,” Dr Wu said. “For patients with diabetes, HbA1c is often used as a marker of how well they are managing their diabetes, so having an underestimation of their blood sugars is problematic because they might have a false sense of security, thinking they are doing okay when they are not.”
Study details
For this study, the researchers analyzed data from 2 major public health studies—the Coronary Artery Risk Development in Young Adults (CARDIA) study and the Jackson Heart Study (JHS). Of the 4620 participants included in the analysis, 367 had SCT.
Among all the patients included in the analysis, HbA1c readings came from either of 2 widely used, clinically accepted assays made by Tosoh Bioscience Inc. that rely on a process called high-performance liquid chromatography.
In addition to those measures, the researchers also compared fasting and 2-hour blood glucose and statistically controlled for demographic and medical factors, such as gender, age, body-mass index, whether diabetes had already been diagnosed, and whether it was being treated.
Study author Gregory Wellenius, ScD, of Brown University, said the study’s scale and breadth allowed for the most definitive comparison to date of HbA1c readings in patients with and without SCT. Two previous, smaller studies had not detected a similar discrepancy.
“The strengths of the study are that it’s the largest sample size ever used, it’s across 2 different studies with somewhat different populations, and it’s a more thorough evaluation than prior studies,” Dr Wellenius said.
While the study showed that HbA1c readings were significantly different between people with and without SCT, it also showed that blood glucose readings were not, suggesting that glucose metabolism is not necessarily different between the 2 groups, as the HbA1c readings alone would suggest.
Implications for practice
The study does not explain why the HbA1c readings differ. While it could be related to the assay method, which is approved by the NGSP (formerly National Glycohemoglobin Standardization Program) for use in patients with SCT, it could also be a consequence of the underlying biology of SCT.
Hypothetically, according to the researchers, if the hemoglobin variant of the trait endows red blood cells with a shorter lifespan, the cells’ hemoglobin would carry less accumulated blood glucose, leading to falsely low HbA1c readings.
“Irrespective of the reason of the underestimation, the underestimation is very real, and clinicians should consider screening for sickle cell trait and account for the difference in HbA1c,” Dr Wu said.
Yet not many people with SCT in the US know they carry the variant, especially those born before routine screenings at birth began, Lacy said.
The researchers therefore recommend that practitioners following African-American patients whose HbA1c levels are within 0.3 percentage points of a diagnostic cutoff also consider using additional blood glucose measures. Current diagnostic thresholds for A1C are ≥5.7% for prediabetes and ≥6.5% for diabetes. ![]()
Photo by Juan D. Alfonso
A new study suggests that hemoglobin A1c (HbA1c), a biomarker used to measure blood sugar over time, may not perform as accurately among African-Americans with sickle cell trait (SCT) and could be leading to a systemic underestimation of blood sugar control in that population.
Researchers analyzed data from more than 4600 people and found that HbA1c readings were significantly lower in individuals with SCT than in those without it, even after accounting for several possible confounding factors.
The team reported these findings in JAMA.
“We found that HbA1c was systematically lower in African-Americans with sickle cell trait than those without sickle cell trait, despite similar blood sugar measurements using other tests,” said study author Mary Lacy, a doctoral candidate at the Brown University School of Public Health in Providence, Rhode Island.
“We might be missing an opportunity for diagnosis and treatment of a serious disease.”
Lacy and her co-authors found that using standard clinical HbA1c cutoffs resulted in identifying 40% fewer potential cases of prediabetes and 48% fewer potential cases of diabetes in people with SCT than in people without SCT.
However, when the researchers used other blood glucose measures as the diagnostic criteria, they found no significant difference in the likelihood of diabetes and prediabetes among patients with or without SCT.
The questions the study raises about using HbA1c among SCT carriers matter for treatment as well as diagnosis, said study author Wen-Chih Wu, MD, of Brown University.
“The clinical implications of these results are highly relevant,” Dr Wu said. “For patients with diabetes, HbA1c is often used as a marker of how well they are managing their diabetes, so having an underestimation of their blood sugars is problematic because they might have a false sense of security, thinking they are doing okay when they are not.”
Study details
For this study, the researchers analyzed data from 2 major public health studies—the Coronary Artery Risk Development in Young Adults (CARDIA) study and the Jackson Heart Study (JHS). Of the 4620 participants included in the analysis, 367 had SCT.
Among all the patients included in the analysis, HbA1c readings came from either of 2 widely used, clinically accepted assays made by Tosoh Bioscience Inc. that rely on a process called high-performance liquid chromatography.
In addition to those measures, the researchers also compared fasting and 2-hour blood glucose and statistically controlled for demographic and medical factors, such as gender, age, body-mass index, whether diabetes had already been diagnosed, and whether it was being treated.
Study author Gregory Wellenius, ScD, of Brown University, said the study’s scale and breadth allowed for the most definitive comparison to date of HbA1c readings in patients with and without SCT. Two previous, smaller studies had not detected a similar discrepancy.
“The strengths of the study are that it’s the largest sample size ever used, it’s across 2 different studies with somewhat different populations, and it’s a more thorough evaluation than prior studies,” Dr Wellenius said.
While the study showed that HbA1c readings were significantly different between people with and without SCT, it also showed that blood glucose readings were not, suggesting that glucose metabolism is not necessarily different between the 2 groups, as the HbA1c readings alone would suggest.
Implications for practice
The study does not explain why the HbA1c readings differ. While it could be related to the assay method, which is approved by the NGSP (formerly National Glycohemoglobin Standardization Program) for use in patients with SCT, it could also be a consequence of the underlying biology of SCT.
Hypothetically, according to the researchers, if the hemoglobin variant of the trait endows red blood cells with a shorter lifespan, the cells’ hemoglobin would carry less accumulated blood glucose, leading to falsely low HbA1c readings.
“Irrespective of the reason of the underestimation, the underestimation is very real, and clinicians should consider screening for sickle cell trait and account for the difference in HbA1c,” Dr Wu said.
Yet not many people with SCT in the US know they carry the variant, especially those born before routine screenings at birth began, Lacy said.
The researchers therefore recommend that practitioners following African-American patients whose HbA1c levels are within 0.3 percentage points of a diagnostic cutoff also consider using additional blood glucose measures. Current diagnostic thresholds for A1C are ≥5.7% for prediabetes and ≥6.5% for diabetes.
‘Alternative’ BMT deemed ‘promising’ for SAA

Photo by Chad McNeeley
Researchers have reported a “promising” treatment approach for
refractory, severe aplastic anemia (SAA).
The
regimen consists of nonmyeloablative conditioning, bone marrow
transplants (BMTs) from “alternative” donors, and graft-vs-host disease
(GVHD) prophylaxis.
All 16 SAA patients who received this treatment
achieved engraftment and were completely cleared of disease.
There were 2 cases of acute and chronic GVHD, but they resolved.
All patients were ultimately able to stop immunosuppressive therapy.
Robert Brodsky, MD, of Sidney Kimmel Cancer Center in Baltimore, Maryland, and his colleagues reported these findings in Biology of Blood and Marrow Transplantation.
“Our findings have the potential to greatly widen treatment options for the vast majority of severe aplastic anemia patients,” Dr Brodsky said.
He and his colleagues tested their approach in 16 SAA patients between 11 and 69 years of age. Each of the patients had failed to respond to immunosuppressive therapy and other treatments.
The patients received conditioning with antithymocyte globulin, fludarabine, low-dose cyclophosphamide, and total body irradiation.
They then received BMTs. Thirteen of the donors were haploidentical related, 2 were fully matched unrelated, and 1 was mismatched unrelated.
Three and 4 days after BMT, the patients received cyclophosphamide at 50 mg/kg/day as GVHD prophylaxis. They then received mycophenolate mofetil on days 5 through 35 and tacrolimus from day 5 through 1 year.
The median time to neutrophil recovery (over 1000 × 103/mm3 for 3 consecutive days) was 19 days (range, 16 to 27). The median time to red cell engraftment was 25 days (range, 2 to 58). And the median time to the last platelet transfusion (to keep platelet counts over 50 × 103/mm3) was 27.5 days (range, 22 to 108).
At a median follow-up of 21 months (range, 3 to 64), all 16 patients were still alive, disease-free, and no longer required transfusions.
Two patients did develop grade 1/2 acute skin GVHD. They also had mild chronic GVHD of the skin/mouth, which required systemic steroids.
One of these patients was able to come off all immunosuppressive therapy by 15 months, and the other was able to do so by 17 months. All of the other patients stopped immunosuppressive therapy at 1 year.
Ending all therapy related to their disease has been life-changing for these patients, said study author Amy DeZern, MD, also of the Sidney Kimmel Cancer Center.
“It’s like night and day,” she said. “They go from not knowing if they have a future to hoping for what they’d hoped for before they got sick. It’s that transformative.”
Successful BMTs using partially matched donors open up the transplant option to nearly all patients with SAA, especially minority patients, added Dr Brodsky.
“Now, a therapy that used to be available to 25% to 30% of patients with severe aplastic anemia is potentially available to more than 95%,” he said.
Photo by Chad McNeeley
Researchers have reported a “promising” treatment approach for
refractory, severe aplastic anemia (SAA).
The
regimen consists of nonmyeloablative conditioning, bone marrow
transplants (BMTs) from “alternative” donors, and graft-vs-host disease
(GVHD) prophylaxis.
All 16 SAA patients who received this treatment
achieved engraftment and were completely cleared of disease.
There were 2 cases of acute and chronic GVHD, but they resolved.
All patients were ultimately able to stop immunosuppressive therapy.
Robert Brodsky, MD, of Sidney Kimmel Cancer Center in Baltimore, Maryland, and his colleagues reported these findings in Biology of Blood and Marrow Transplantation.
“Our findings have the potential to greatly widen treatment options for the vast majority of severe aplastic anemia patients,” Dr Brodsky said.
He and his colleagues tested their approach in 16 SAA patients between 11 and 69 years of age. Each of the patients had failed to respond to immunosuppressive therapy and other treatments.
The patients received conditioning with antithymocyte globulin, fludarabine, low-dose cyclophosphamide, and total body irradiation.
They then received BMTs. Thirteen of the donors were haploidentical related, 2 were fully matched unrelated, and 1 was mismatched unrelated.
Three and 4 days after BMT, the patients received cyclophosphamide at 50 mg/kg/day as GVHD prophylaxis. They then received mycophenolate mofetil on days 5 through 35 and tacrolimus from day 5 through 1 year.
The median time to neutrophil recovery (over 1000 × 103/mm3 for 3 consecutive days) was 19 days (range, 16 to 27). The median time to red cell engraftment was 25 days (range, 2 to 58). And the median time to the last platelet transfusion (to keep platelet counts over 50 × 103/mm3) was 27.5 days (range, 22 to 108).
At a median follow-up of 21 months (range, 3 to 64), all 16 patients were still alive, disease-free, and no longer required transfusions.
Two patients did develop grade 1/2 acute skin GVHD. They also had mild chronic GVHD of the skin/mouth, which required systemic steroids.
One of these patients was able to come off all immunosuppressive therapy by 15 months, and the other was able to do so by 17 months. All of the other patients stopped immunosuppressive therapy at 1 year.
Ending all therapy related to their disease has been life-changing for these patients, said study author Amy DeZern, MD, also of the Sidney Kimmel Cancer Center.
“It’s like night and day,” she said. “They go from not knowing if they have a future to hoping for what they’d hoped for before they got sick. It’s that transformative.”
Successful BMTs using partially matched donors open up the transplant option to nearly all patients with SAA, especially minority patients, added Dr Brodsky.
“Now, a therapy that used to be available to 25% to 30% of patients with severe aplastic anemia is potentially available to more than 95%,” he said.
Photo by Chad McNeeley
Researchers have reported a “promising” treatment approach for
refractory, severe aplastic anemia (SAA).
The
regimen consists of nonmyeloablative conditioning, bone marrow
transplants (BMTs) from “alternative” donors, and graft-vs-host disease
(GVHD) prophylaxis.
All 16 SAA patients who received this treatment
achieved engraftment and were completely cleared of disease.
There were 2 cases of acute and chronic GVHD, but they resolved.
All patients were ultimately able to stop immunosuppressive therapy.
Robert Brodsky, MD, of Sidney Kimmel Cancer Center in Baltimore, Maryland, and his colleagues reported these findings in Biology of Blood and Marrow Transplantation.
“Our findings have the potential to greatly widen treatment options for the vast majority of severe aplastic anemia patients,” Dr Brodsky said.
He and his colleagues tested their approach in 16 SAA patients between 11 and 69 years of age. Each of the patients had failed to respond to immunosuppressive therapy and other treatments.
The patients received conditioning with antithymocyte globulin, fludarabine, low-dose cyclophosphamide, and total body irradiation.
They then received BMTs. Thirteen of the donors were haploidentical related, 2 were fully matched unrelated, and 1 was mismatched unrelated.
Three and 4 days after BMT, the patients received cyclophosphamide at 50 mg/kg/day as GVHD prophylaxis. They then received mycophenolate mofetil on days 5 through 35 and tacrolimus from day 5 through 1 year.
The median time to neutrophil recovery (over 1000 × 103/mm3 for 3 consecutive days) was 19 days (range, 16 to 27). The median time to red cell engraftment was 25 days (range, 2 to 58). And the median time to the last platelet transfusion (to keep platelet counts over 50 × 103/mm3) was 27.5 days (range, 22 to 108).
At a median follow-up of 21 months (range, 3 to 64), all 16 patients were still alive, disease-free, and no longer required transfusions.
Two patients did develop grade 1/2 acute skin GVHD. They also had mild chronic GVHD of the skin/mouth, which required systemic steroids.
One of these patients was able to come off all immunosuppressive therapy by 15 months, and the other was able to do so by 17 months. All of the other patients stopped immunosuppressive therapy at 1 year.
Ending all therapy related to their disease has been life-changing for these patients, said study author Amy DeZern, MD, also of the Sidney Kimmel Cancer Center.
“It’s like night and day,” she said. “They go from not knowing if they have a future to hoping for what they’d hoped for before they got sick. It’s that transformative.”
Successful BMTs using partially matched donors open up the transplant option to nearly all patients with SAA, especially minority patients, added Dr Brodsky.
“Now, a therapy that used to be available to 25% to 30% of patients with severe aplastic anemia is potentially available to more than 95%,” he said.
Azathioprine may increase risk of MDS, AML

Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research.
Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research.
Results of a large, retrospective study suggest that taking azathioprine, a drug commonly used to treat autoimmune disease, may increase a person’s risk of developing myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML).
Researchers analyzed data on more than 40,000 patients with 27 common autoimmune diseases and found that azathioprine use was significantly associated with an increased risk of MDS and AML.
“Similar associations were already documented in case reports and case series but have never been evaluated in a broad spectrum of autoimmune diseases in that many patients and in context of individual medications,” said study author Raoul Tibes, MD, PhD, of the Mayo Clinic in Phoenix, Arizona.
“Interestingly, there was no association with length of time on therapy and resulting myeloid neoplasm.”
Dr Tibes and his colleagues reported these findings in JAMA Oncology.
The researchers reviewed data on 40,011 patients with primary autoimmune disorders, such as lupus and rheumatoid arthritis, who were seen at 2 centers from January 1, 2004, to December 31, 2014.
There were 311 patients with MDS or AML, but only 86 met strict inclusion criteria. Fifty-five patients had MDS, 21 had de novo AML, and 10 had AML and a history of MDS.
The researchers collected detailed data on each patient’s drug exposures, treatment duration, and disease characteristics and compared this information to data from patients with autoimmune disorders who did not have MDS or AML.
This revealed that use of azathioprine sodium was more frequent in cases than controls, and azathioprine was significantly associated with an increased risk of MDS and AML. The odds ratio was 7.05 (P<0.001).
Other agents used showed a similar trend, but the results were not statistically significant. The odds ratios were 3.58 for cyclophosphamide and 2.73 for mitoxantrone hydrochloride.
The researchers said that, while these results are intriguing, they should not change or replace the clinical judgments, monitoring, and current standard treatments for patients with autoimmune diseases.
Despite its large size, this study had limitations, including its retrospective nature, the fact that many different autoimmune diseases were analyzed, and that the researchers only looked at cases of MDS and AML.
No definitive causal association was made between taking a particular drug and MDS or AML. The number of patients with autoimmune disease developing MDS or AML is still low overall, and no prediction for individual patients can be concluded from the study.
The researchers plan to perform molecular investigations into the genetic susceptibility for therapy-related myeloid neoplasms as the next phase of this research.