User login
Immune Thrombocytopenia
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1). 
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2). 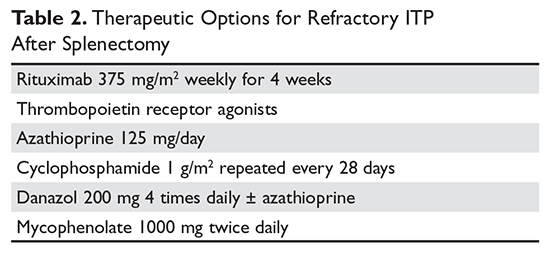
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79 
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1).
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2).
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
Introduction
Immune thrombocytopenia (ITP) is a common acquired autoimmune disease characterized by low platelet counts and an increased risk of bleeding. The incidence of ITP is approximately 3.3 per 100,000 adults.1 There is considerable controversy about all aspects of the disease, with little “hard” data on which to base decisions given the lack of randomized clinical trials to address most clinical questions. This article reviews the presentation and diagnosis of ITP and its treatment options and discusses management of ITP in specific clinical situations.
Pathogenesis and Epidemiology
ITP is caused by autoantibodies binding to platelet surface proteins, most often to the platelet receptor GP IIb/IIIa.2-4 These antibody-coated platelets then bind to Fc receptors in macrophages and are removed from circulation. The initiating event in ITP is unknown. It is speculated that the patient responds to a viral or bacterial infection by creating antibodies which cross-react with the platelet receptors. Continued exposure to platelets perpetuates the immune response. ITP that occurs in childhood appears to be an acute response to viral infection and usually resolves. ITP in adults may occur in any age group but is seen especially in young women.
Despite the increased platelet destruction that occurs in ITP, the production of new platelets often is not significantly increased. This is most likely due to lack of an increase in thrombopoietin, the predominant platelet growth factor.5
It had been thought that most adult patients who present with ITP go on to have a chronic course, but more recent studies have shown this is not the case. In modern series the percentage of patients who are “cured” with steroids ranges from 30% to 70%.6–9 In addition, it has been appreciated that even in patients with modest thrombocytopenia, no therapy is required if the platelet count remains higher than 30 × 103/µL. However, this leaves a considerable number of patients who will require chronic therapy.
Clinical Presentation
Presentation can range from a symptomatic patient with low platelets found on a routine blood count to a patient with massive bleeding. Typically, patients first present with petechiae (small bruises 1 mm in size) on the shins. True petechiae are seen only in severe thrombocytopenia. Patients will also report frequent bruising and bleeding from the gums. Patients with very low platelet counts will notice “wet purpura,” which is characterized by blood-filled bullae in the oral cavity. Life-threatening bleeding is a very unusual presenting sign unless other problems (trauma, ulcers) are present. The physical examination is only remarkable for stigmata of bleeding such as the petechiae. The presence of splenomegaly or lymphadenopathy weighs strongly against a diagnosis of ITP. Many patients with ITP will note fatigue when their platelets counts are lower.10
Diagnosis
Extremely low platelet counts with a normal blood smear and an otherwise healthy patient are diagnostic of ITP. The platelet count cutoff for considering ITP is 100 × 103/µL as the majority of patients with counts in the 100 to 150 × 103/µL range will not develop greater thrombocytopenia.11 Also, the platelet count decreases with age (9 × 103/µL per decade in one study), and this also needs to be factored into the evaluation.12 The finding of relatives with ITP should raise suspicion for congenital thrombocytopenia.13 One should question the patient carefully about drug exposure (see Drug-Induced Thrombocytopenia), especially about over-the-counter medicines, “natural” remedies, or recreational drugs.
There is no laboratory test that rules in ITP; rather, it is a diagnosis of exclusion. The blood smear should be carefully examined for evidence of microangiopathic hemolytic anemias (schistocytes), bone marrow disease (blasts, teardrop cells), or any other evidence of a primary bone marrow disease. In ITP, the platelets can be larger than normal, but finding some platelets the size of red cells should raise the issue of congenital thrombocytopenia.14 Pseudo-thrombocytopenia, which is the clumping of platelets due to a reaction to the EDTA anticoagulant in the tube, should be excluded. The diagnosis is established by drawing the blood in a citrated (blue-top) tube to perform the platelet count. There is no role for antiplatelet antibody assay because this test lacks sensitivity and specificity. In a patient without a history of autoimmune disease or symptoms, empiric testing for autoimmune disease is not recommended.
Patients who present with ITP should be tested for both HIV and hepatitis C infection.15,16 These are the most common viral causes of secondary ITP, and both have prognostic and treatment implications. Some authorities also recommend checking thyroid function as hypothyroidism can present or aggravate the thrombocytopenia.
The role of bone marrow examination is controversial.17 Patients with a classic presentation of ITP (young woman, normal blood smear) do not require a bone marrow exam before therapy is initiated, although patients who do not respond to initial therapy should have a bone marrow aspiration. The rare entity amegakaryocytic thrombocytopenia can present with a clinical picture similar to that of ITP, but amegakaryocytic thrombocytopenia will not respond to steroids. Bone marrow aspiration reveals the absence of megakaryocytes in this entity. It is rare, however, that another hematologic disease is diagnosed in patients with a classic clinical presentation of ITP.
In the future, measurement of thrombopoietin and reticulated platelets may provide clues to the diagnosis.4 Patients with ITP paradoxically have normal or only mildly elevated thrombopoietin levels. The finding of a significantly elevated thrombopoietin level should lead to questioning of the diagnosis. One can also measure “reticulated platelets,” which are analogous to red cell reticulocytes. Patients with ITP (or any platelet destructive disorders) will have high levels of reticulated platelets. These tests are not recommended for routine evaluation, but may be helpful in difficult cases.
Treatment
In general, therapy in ITP should be guided by the patient’s signs of bleeding and not by unquestioning adherence to measuring platelet levels,15 as patients tolerate thrombocytopenia well. It is unusual to have life-threatening bleeding with platelet counts greater than 5 × 103/µL in the absence of mechanical lesions. Despite the low platelet count in patients with ITP, the overall mortality is estimated to be only 0.3% to 1.3%.18 It is sobering that in one study the rate of death from infections was twice as high as that from bleeding.19 Rare patients will have antibodies that interfere with the function of the platelet, and these patients can have profound bleeding with only modestly lowered platelet counts.20 A suggested cut-off for treating newly diagnosed patients is 30 × 103/µL.21
Initial Therapy
The primary therapy of ITP is glucocorticoids, either prednisone or dexamethasone. In the past prednisone at a dose of 60 to 80 mg/day was started at the time of diagnosis (Table 1).
For rapid induction of a response, there are 2 options. A single dose of intravenous immune globulin (IVIG) at 1 g/kg or intravenous anti-D immunoglobulin (anti-D) at 50 to 75 µg/kg can induce a response in more than 80% of patients in 24 to 48 hours.21,24 IVIG has several drawbacks. It can cause aseptic meningitis, and in patients with vascular disease the increased viscosity can induce ischemia. There is also a considerable fluid load delivered with the IVIG, and it needs to be given over several hours.
The use of anti-D is limited to Rh-positive patients who have not had a splenectomy. It should not be used in patients who are Coombs positive due to the risk of provoking more hemolysis. Rarely anti-D has been reported to cause a severe hemolytic disseminated intravascular coagulation syndrome (1:20,000 patients), which has led to restrictions in its use.25 Although the drug can be rapidly given over 15 minutes, due to these concerns current recommendations are now to observe patients for 8 hours after their dose and to perform a urine dipstick test for blood at 2, 4, and 8 hours. Concerns about this rare but serious side effect have led to a dramatic decrease in the use of anti-D.
For patients who are severely thrombocytopenic and do not respond to initial therapy, there are 2 options for raising the platelet counts. One is to use a combination of IVIG, methylprednisolone, vincristine, and/or anti-D.26 The combination of IVIG and anti-D may be synergistic since these agents block different Fc receptors. A response of 71% has been reported for this 3- or 4-drug combination in a series of 35 patients.26 The other option is to treat with a continuous infusion of platelets (1 unit over 6 hours) and IVIG 1 g/kg for 24 hours. Response rates of 62.7% have been reported with this combination, and this rapid rise in platelets can allow time for other therapies to take effect.27,28
Patients with severe thrombocytopenia who relapse with reduction of steroids or who do not respond to steroids have several options for further management. Repeated doses of IVIG can transiently raise the platelet count, and some patients may only need several courses of therapy over the course of many months. One study showed that 60% of patients could delay or defer therapy by receiving multiple doses of anti-D. However, 30% of patients did eventually receive splenectomy and 20% of patients required ongoing therapy with anti-D.29 In a randomized trial comparing early use of anti-D to steroids to avoid splenectomy, there was no difference in splenectomy rate (38% versus 42%).30 Finally, an option as mentioned above is to try a 6-month course of pulse dexamethasone 40 mg/day for 4 days, repeated every 28 days.
Options for Refractory ITP
There are multiple options for patients who do not respond to initial ITP therapies. These can be divided into several broad groups: curative therapies (splenectomy and rituximab), thrombopoietin receptor agonists, and anecdotal therapies.
Splenectomy
In patients with severe thrombocytopenia who do not respond or who relapse with lower doses of prednisone, splenectomy should be strongly considered. Splenectomy will induce a good response in 60% to 70% of patients and is durable in most patients. In 2 recently published reviews of splenectomy, the complete response rate was 67% and the total response rate was 88% to 90%%.8,31 Between 15% and 28% of patients relapsed over 5 years, with most recurrences occurring in the first 2 years. Splenectomy carries a short-term surgical risk, and the life-long risk of increased susceptibility to overwhelming sepsis is discussed below. However, the absolute magnitude of these risks is low and is often lower than the risks of continued prednisone therapy or of continued cytotoxic therapy.
Timing of splenectomy depends on the patient’s presentation. Most patients should be given a 6-month trial of steroids or other therapies before proceeding to splenectomy.31 However, patients who persist with severe thrombocytopenia despite initial therapies or who are suffering intolerable side effects from therapy should be considered sooner for splenectomy.31 In the George review, multiple factors such as responding to IVIG were found not to be predictive of response to splenectomy.8
Method of splenectomy appears not to matter.21 Rates of finding accessory spleens are just as high or higher with laparoscopic splenectomy and the patient can recover faster. In patients who are severely thrombocytopenic, open splenectomy can allow for quicker control of the vascular access of the spleen.
Rates of splenectomy in recent years have decreased for many reasons,32 including the acceptance of lower platelet counts in asymptomatic patients and the availability of alternative therapies such as rituximab. In addition, despite abundant data for good outcomes, there is a concern that splenectomy responses are not durable. Although splenectomy will not cure every patient with ITP, splenectomy is the therapy with the most patients, the longest follow-up, and the most consistent rate of cure, and it should be discussed with every ITP patient who does not respond to initial therapy and needs further treatment.
The risk of overwhelming sepsis varies by indications for splenectomy but appears to be about 1%.33,34 The use of pneumococcal vaccine and recognition of this syndrome have helped reduce the risk. Asplenic patients need to be counseled about the risk of overwhelming infections, should be vaccinated for pneumococcus, meningococcus, and Haemophilus influenzae, and should wear an ID bracelet.35–37 Patients previously vaccinated for pneumococcus should be re-vaccinated every 3 to 5 years. The role of prophylactic antibiotics in adults is controversial, but patients under the age of 18 should be on penicillin VK 250 mg orally twice daily.
Rituximab
Rituximab has been shown to be very active in ITP. Most studies used the standard dose of 375 mg/m2 weekly for 4 weeks, but other studies have shown that 1000 mg twice 14 days apart (ie, on days 1 and 15) resulted in the same response rate and may be more convenient for patients.38,39 The response time can vary, with patients either showing a rapid response or requiring up to 8 weeks for their counts to go up. Although experience is limited, the response seems to be durable, especially in those patients whose counts rise higher than 150 × 103/µL; in patients who relapse, a response can be re-induced with a repeat course. Overall the response rate for rituximab is about 60%, but only approximately 20% to 40% of patients will remain in long-term remission.40–42 There is no evidence yet that “maintenance” therapy or monitoring CD19/CD20 cells can help further the duration of remission.
Whether to give rituximab pre- or post-splenectomy is also uncertain. An advantage of presplenectomy rituximab is that many patients will achieve remission, delaying the need for surgery. Also, rituximab is a good option for patients whose medical conditions put them at high risk for complications with splenectomy. However, it is unknown whether rituximab poses any long-term risks, while the long-term risks of splenectomy are well-defined. Rituximab is the only curative option left for patients who have failed splenectomy and is a reasonable option for these patients.
There is an intriguing trial in which patients were randomly assigned to dexamethasone alone versus dexamethasone plus rituximab upon presentation with ITP; those who were refractory to dexamethasone alone received salvage therapy with dexamethasone plus rituximab.43 The dexamethasone plus rituximab group had an overall higher rate of sustained remission at 6 months than the dexamethasone group, 63% versus 36%. Interestingly, patients who failed their first course of dexamethasone but then were “salvaged” with dexamethasone/rituximab had a similar overall response rate of 56%, suggesting that saving the addition of rituximab for steroid failures may be an effective option.
Although not “chemotherapy,” rituximab is not without risks. Patients can develop infusion reactions, which can be severe in 1% to 2% of patients. In a meta-analysis the fatal reaction rate was 2.9%.40 Patients with chronic hepatitis B infections can experience reactivation with rituximab, and thus all patients should be screened before treatment. Finally, the very rare but devastating complication of progressive multifocal leukoencephalopathy has been reported.
Thrombopoietin Receptor Agonists
Although patients with ITP have low platelet counts, studies starting with Dameshek have shown that these patients also have reduced production of platelets.44 Despite the very low circulating platelet count, levels of the platelet growth factor thrombopoietin (TPO) are not raised.45 Seminal studies with recombinant TPO in the 1990s showed that ITP patients responded to thrombopoietin-stimulating protein, but the formation of anti-TPO antibodies halted trials with the first generation of these agents. Two TPO receptor agonists (TPO-RA) are approved for use in patients with ITP.
Romiplostim. Romiplostim is a peptibody, a combination of a peptide that binds and stimulates the TPO receptor and an Fc domain to extend its half-life.46 It is administered in a weekly subcutaneous dose starting at 1 to 3 µg/kg. Use of romiplostim in ITP patients produces a response rate of 80% to 88%, with 87% of patients being able to wean off or decrease other anti-ITP medications.47 In a long-term extension study, the response was again high at 87%.48 These studies have also shown a reduced incidence of bleeding.
The major side effect of romiplostim seen in clinical trials was marrow reticulin formation, which occurred in up to 5.6% of patients.47,48 The clinical course in these patients is the development of anemia and a myelophthisic blood smear with teardrop cells and nucleated red cells. These changes appear to reverse with cessation of the drug. The bone marrow shows increased reticulin formation but rarely, if ever, shows the collagen deposition seen with primary myelofibrosis.
Thrombosis has also been seen, with a rate of 0.08 to 0.1 cases per 100 patient-weeks,49 but it remains unclear if this is due to the drug, part of the natural history of ITP, or expected complications in older patients undergoing any type of medical therapy. Surprisingly, despite the low platelet counts, patients with ITP in one study had double the risk of venous thrombosis, demonstrating that ITP itself can be a risk factor for thrombosis.50 These trials have shown no long-term concerns for other clinical problems such as liver disease.
Eltrombopag. The other available TPO-RA is eltrombopag,51 an oral agent that stimulates the TPO receptor by binding the transmembrane domain and activating it. The drug is given orally starting at 50 mg/day (25 mg for patients of Asian ancestry or with liver disease) and can be dose escalated to 75 mg/day. The drug needs to be taken on an empty stomach. Eltrombopag has been shown to be effective in chronic ITP, with response rates of 59% to 80% and reduction in use of rescue medications.47,51,52 As with romiplostim, the incidence of bleeding was also decreased with eltrombopag in these trials.47,51
Clinical trials demonstrated that eltrombopag shares with romiplostim the risk for marrow fibrosis. A side effect unique to eltrombopag observed in these trials was a 3% to 7% incidence of elevated liver function tests.21,52 These abnormal findings appeared to resolve in most patients, but liver function tests need to be monitored in patients receiving eltrombopag.
Clinical use. The clearest indication for the use of TPO-RAs is in patients who have failed several therapies and remain symptomatic or are on intolerable doses of other medications such as prednisone. The clear benefits are their relative safety and high rates of success. The main drawback of TPO-RAs is the need for continuing therapy as the platelet count will return to baseline shortly after these agents are stopped. Currently there is no clear indication for one medication over the other. The advantages of romiplostim are great flexibility in dosing (1–10 µg/kg week) and no concerns about drug interaction. The current drawback of romiplostim is the Food and Drug Administration’s requirement for patients to receive the drug from a clinic and not at home. Eltrombopag offers the advantage of oral use, but it has a limited dose range and potential for drug interactions. Both agents have been associated with marrow reticulin formation, although in clinical use this risk appears to be very low.53
Other Options
In the literature there are numerous options for the treatment of ITP.54,55 Most of these studies are anecdotal, enrolled small number of patients, and sometimes included patients with mild thrombocytopenia, but these therapeutic options can be tried in patients who are refractory to standard therapies and have bleeding. The agents with the greatest amount of supporting data are danazol, vincristine, azathioprine, cyclophosphamide, and fostamatinib.
Danazol 200 mg 4 times daily is thought to downregulate the macrophage Fc receptor. The onset of action may be delayed and a therapeutic trial of up to 4 to 6 months is advised. Danazol is very effective in patients with antiphospholipid antibody syndrome who develop ITP and may be more effective in premenopausal women.56 Once a response is seen, danazol should be continued for 6 months and then an attempt to wean the patient off the agent should be made. A partial response can be seen in 70% to 90% of patients, but a complete response is rare.54
Vincristine 1.4 mg/m2 weekly has a low response rate, but if a response is going to occur, it will occur rapidly within 2 weeks. Thus, a prolonged trial of vincristine is not needed; if no platelet rise is seen in several weeks, the drug should be stopped. Again, partial responses are more common than complete response—50% to 63% versus 0% to 6%.54Azathioprine 150 mg orally daily, like danazol, demonstrates a delayed response and requires several months to assess for response. However, 19% to 25% of patients may have a complete response.54 It has been reported that the related agent mycophenolate 1000 mg twice daily is also effective in ITP.57
Cyclophosphamide 1 g/m2 intravenously repeated every 28 days has been reported to have a response rate of up to 40%.58 Although considered more aggressive, this is a standard immunosuppressive dose and should be considered in patients with very low platelet counts. Patients who have not responded to single-agent cyclophosphamide may respond to multi-agent chemotherapy with agents such as etoposide and vincristine plus cyclophosphamide.59
Fostamatinib, a spleen tyrosine kinase (SYK) inhibitor, is currently under investigation for the treatment of ITP.60 This agent prevents phagocytosis of antibody-coated platelets by macrophages. In early studies fostamatinib has been well tolerated at a dose of 150 mg twice daily, with 75% of patients showing a response. Large phase 3 trials are underway, and if the earlier promising results hold up fostamatinib may be a novel option for refractory patients.
A Practical Approach to Refractory ITP
One approach is to divide patients into bleeders, or those with either very low platelet counts (< 5 × 103/µL) or who have had significant bleeding in the past, and nonbleeders, or those with platelet counts above 5 × 103/µL and no history of severe bleeding. Bleeders who do not respond adequately to splenectomy should first start with rituximab since it is not cytotoxic and is the only other “curative” therapy (Table 2).
Nonbleeders should be tried on danazol and other relatively safe agents. If this fails, rituximab or TPO-RAs can be considered. Before one considers cytotoxic therapy, the risk of the therapy must be weighed against the risk posed by the thrombocytopenia. The mortality from ITP is fairly low (5%) and is restricted to patients with severe disease. Patients with only moderate thrombocytopenia and no bleeding are better served with conservative management. There is little justification for the use of continuous steroid therapy in this group of patients given the long-term risks of this therapy.
Special Situations
Surgery
Patients with ITP who need surgery either for splenectomy or for other reasons should have their platelet counts raised to a level greater than 20 to 30 × 103/µL before surgery. Most patients with ITP have increased platelet function and will not have excessive bleeding with these platelet counts. For patients with platelet counts below this level, an infusion of immune globulin or anti-D may rapidly increase the platelet counts. If the surgery is elective, short-term use of TPO-RAs to raise the counts can also be considered.
Pregnancy
Up to 10% of pregnant women will develop low platelet counts during their pregnancy.61,62 The most common etiology is gestational thrombocytopenia, which is an exaggeration of the lowered platelet count seen in pregnancy. Counts may fall as low as 50 × 103/µL at the time of delivery. No therapy is required as the fetus is not affected and the mother does not have an increased risk of bleeding. Pregnancy complications such as HELLP syndrome and thrombotic microangiopathies also present with low platelet counts, but these can be diagnosed by history.61,63
Women with ITP can either develop the disease during pregnancy or have a worsening of the symptoms.64 Counts often drop dramatically during the first trimester. Early management should be conservative with low doses of prednisone to keep the count above 10 × 103/µL.21 Immunoglobulin is also effective,65 but there are rare reports of pulmonary edema. Rarely patients who are refractory will require splenectomy, which may be safely performed in the second trimester. For delivery the count should be greater than 30 × 103/µL and for an epidural greater than 50 × 103/µL.64 There are reports of the use of TPO-RAs in pregnancy, and this can be considered for refractory cases.66
Most controversy centers on management of the delivery. In the past it was feared that fetal thrombocytopenia could lead to intracranial hemorrhage, and Caesarean section was always recommended. It now appears that most cases of intracranial hemorrhage were due to alloimmune thrombocytopenia and not ITP. Furthermore, the nadir of the baby’s platelet count is not at birth but several days after. It appears the safest course is to proceed with a vaginal or C-section delivery determined by obstetrical indications and then immediately check the baby’s platelet count. If the platelet count is low in the neonate, immunoglobulin will raise the count. Since the neonatal thrombocytopenia is due to passive transfer of maternal antibody, the platelet destruction will abate in 4 to 6 weeks.
Pediatric Patients
The incidence of ITP in children is 2.2 to 5.3 per 100,000 children.1 There are several distinct differences in pediatric ITP. Most cases will resolve in weeks, with only a minority of patients transforming into chronic ITP (5%–10%). Also, the rates of serious bleeding are lower in children than in adults, with intracranial hemorrhage rates of 0.1% to 0.5% being seen.67 For most patients with no or mild bleeding, management now is observation alone regardless of platelet count because it is felt that the risks of therapies are higher than the risk of bleeding.21 For patients with bleeding, IVIG, anti-D, or a short course of steroids can be used. Given the risk of overwhelming sepsis, splenectomy is often deferred as long as possible. Rituximab is increasingly being used in children due to concerns about use of agents such a cyclophosphamide or azathioprine in children.68 Abundant data on use of TPO-RAs in children showing high response rates and safety support their use, and these should be considered in refractory ITP before any cytotoxic agent.69–71
Helicobacter Pylori Infection
There has been much interest in the relationship between H. pylori and ITP.16,72,73H. pylori infections have been associated with a variety of autoimmune diseases, and there is a confusing literature on this infection and ITP. Several meta-analyses have shown that eradication of H. pylori will result in an ITP response rate of 20% to 30%, but responses curiously appear to be limited to certain geographic areas such as Japan and Italy but not the United States. In patients with recalcitrant ITP, especially in geographic areas with high incidence, it may be worthwhile to check for H. pylori infection and treat accordingly if positive.
Drug-Induced Thrombocytopenia
Patients with drug-induced thrombocytopenia present with very low (< 10 × 103/µL) platelet counts 1 to 3 weeks after starting a new medication.74–76 In patients with a possible drug-induced thrombocytopenia, the primary therapy is to stop the suspect drug.77 If there are multiple new medications, the best approach is to stop any drug that has been strongly associated with thrombocytopenia (Table 3).74,78,79
Immune globulin, corticosteroids, or intravenous anti‑D have been suggested as useful in drug‑related thrombocytopenia. However, since most of these thrombocytopenic patients recover when the agent is cleared from the body, this therapy is probably not necessary and withholding treatment avoids exposing the patients to the adverse events associated with further therapy.
Evans Syndrome
Evans syndrome is defined as the combination of autoimmune hemolytic anemia (AIHA) and ITP.80,81 These cytopenias can present simultaneously or sequentially. Patients with Evans syndrome are thought to have a more severe disease process, to be more prone to bleeding, and to be more difficult to treat, but the rarity of this syndrome makes this hard to quantify.
The classic clinical presentation of Evans syndrome is severe anemia and thrombocytopenia. Children with Evans syndrome often have complex immunodeficiencies such as autoimmune lymphoproliferative syndrome.82,83 In adults, Evans syndrome most often complicates other autoimmune diseases such as lupus. There are increasing reports of Evans syndrome occurring as a complication of T-cell lymphomas. Often the autoimmune disease can predate the lymphoma diagnosis by months or even years.
In theory the diagnostic approach is straightforward by showing a Coombs-positive hemolytic anemia in the setting of a clinical diagnosis of immune thrombocytopenia. The blood smear will show spherocytes and a diminished platelet count. The presence of other abnormal red cell forms should raise the possibility of an alternative diagnosis. It is unclear how vigorously one should search for other underlying diseases. Many patients will already have the diagnosis of an underlying autoimmune disease. The presence of lymphadenopathy should raise concern for lymphoma.
Initial therapy is high-dose steroids (2 mg/kg/day). IVIG should be added if severe thrombocytopenia is present. Patients who cannot be weaned off prednisone or relapse after prednisone should be considered for splenectomy, although these patients are at higher risk of relapsing.80 Increasingly rituximab is being used with success.84,85 For patients who fail splenectomy and rituximab, aggressive immunosuppression should be considered. Increasing data support the benefits of sirolimus, and this should be considered for refractory patients.86 For patients with Evans syndrome due to underlying lymphoma, antineoplastic therapy often results in prompt resolution of the symptoms. Recurrence of the autoimmune cytopenias often heralds relapse.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
1. Terrell DR, Beebe LA, Vesely SK, et al. The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010;85:174–80.
2. McMillan R, Lopez-Dee J, Bowditch R. Clonal restriction of platelet-associated anti-GPIIb/IIIa autoantibodies in patients with chronic ITP. Thromb Haemost 2001;85:821–3.
3. Aster RH, George JN, McMillan R, Ganguly P. Workshop on autoimmune (idiopathic) thrombocytopenic purpura: Pathogenesis and new approaches to therapy. Am J Hematol 1998;58:231–4.
4. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol 2011;152:52–60.
5. Kuter DJ, Gernsheimer TB. Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1193–211.
6. Pamuk GE, Pamuk ON, Baslar Z, et al. Overview of 321 patients with idiopathic thrombocytopenic purpura. Retrospective analysis of the clinical features and response to therapy. Ann Hematol 2002;81:436–40.
7. Stasi R, Stipa E, Masi M, et al. Long-term observation of 208 adults with chronic idiopathic thrombocytopenic purpura. Am J Med 1995;98:436–42.
8. Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004;104:2623–34.
9. Matschke J, Muller-Beissenhirtz H, Novotny J, et al. A randomized trial of daily prednisone versus pulsed dexamethasone in treatment-naive adult patients with immune thrombocytopenia: EIS 2002 study. Acta Haematol 2016;136:101–7.
10. Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011;86:420–9.
11. Stasi R, Amadori S, Osborn J, et al. Long-term outcome of otherwise healthy individuals with incidentally discovered borderline thrombocytopenia. PLoS Med 2006;3:e24.
12. Biino G, Balduini CL, Casula L, et al. Analysis of 12,517 inhabitants of a Sardinian geographic isolate reveals that predispositions to thrombocytopenia and thrombocytosis are inherited traits. Haematologica 2011;96:96–101.
13. Drachman JG. Inherited thrombocytopenia: when a low platelet count does not mean ITP. Blood 2004;103:390–8.
14. Geddis AE, Balduini CL. Diagnosis of immune thrombocytopenic purpura in children. Curr Opin Hematol 2007;14:520–5.
15. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010;115:168–86.
16. Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009;23:1275–97.
17. Jubelirer SJ, Harpold R. The role of the bone marrow examination in the diagnosis of immune thrombocytopenic purpura: case series and literature review. Clin Appl Thromb Hemost 2002;8:73–6.
18. George JN. Management of patients with refractory immune thrombocytopenic purpura. J Thromb Haemost 2006;4:1664–72.
19. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001;97:2549–54.
20. McMillan R, Bowditch RD, Tani P, et al. A non-thrombocytopenic bleeding disorder due to an IgG4- kappa anti-GPIIb/IIIa autoantibody. Br J Haematol 1996;95:747–9.
21. Neunert C, Lim W, Crowther M, et al. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011;117:4190–207.22. Mazzucconi MG, Fazi P, Bernasconi S, et al. Therapy with high-dose dexamethasone (HD-DXM) in previously untreated patients affected by idiopathic thrombocytopenic purpura: a GIMEMA experience. Blood 2007;109:1401–7.
23. Wei Y, Ji XB, Wang YW, et al. High-dose dexamethasone vs prednisone for treatment of adult immune thrombocytopenia: a prospective multicenter randomized trial. Blood 2016;127:296–302.
24. Newman GC, Novoa MV, Fodero EM, et al. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001;112:1076–8.
25. Gaines AR. Acute onset hemoglobinemia and/or hemoglobinuria and sequelae following Rho(D) immune globulin intravenous administration in immune thrombocytopenic purpura patients. Blood 2000;95:2523–9.
26. Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007;110:3526–31.
27. Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008;83:122–5.
28. Olson SR, Chu C, Shatzel JJ, Deloughery TG. The “platelet boilermaker”: A treatment protocol to rapidly increase platelets in patients with immune-mediated thrombocytopenia. Am J Hematol 2016;91:E330–1.
29. Cooper N, Woloski BM, Fodero EM, et al. Does treatment with intermittent infusions of intravenous anti-D allow a proportion of adults with recently diagnosed immune thrombocytopenic purpura to avoid splenectomy? Blood 2002;99:1922–7.
30. George JN, Raskob GE, Vesely SK, et al. Initial management of immune thrombocytopenic purpura in adults: a randomized controlled trial comparing intermittent anti-D with routine care. Am J Hematol 2003;74:161–9.
31. Mikhael J, Northridge K, Lindquist K, et al. Short-term and long-term failure of laparoscopic splenectomy in adult immune thrombocytopenic purpura patients: a systematic review. Am J Hematol 2009;84:743–8.
32. Palandri F, Polverelli N, Sollazzo D, et al. Have splenectomy rate and main outcomes of ITP changed after the introduction of new treatments? A monocentric study in the outpatient setting during 35 years. Am J Hematol 2016;91:E267–72.
33. Landgren O, Bjorkholm M, Konradsen HB, et al. A prospective study on antibody response to repeated vaccinations with pneumococcal capsular polysaccharide in splenectomized individuals with special reference to Hodgkin’s lymphoma. J Intern Med 2004;255:664–73.
34. Bisharat N, Omari H, Lavi I, Raz R. Risk of infection and death among post-splenectomy patients. J Infect 2001;43:182–6.
35. Mileno MD, Bia FJ. The compromised traveler. Infect Dis Clin North Am 1998;12:369–412.
36. Guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force. BMJ 1996;312:430–4.
37. Ericsson CD. Travellers with pre-existing medical conditions. Int J Antimicrob Agents 2003;21:181–8.
38. Tran H, Brighton T, Grigg A, et al. A multi-centre, single-arm, open-label study evaluating the safety and efficacy of fixed dose rituximab in patients with refractory, relapsed or chronic idiopathic thrombocytopenic purpura (R-ITP1000 study). Br J Haematol 2014;167:243–51.
39. Mahevas M, Ebbo M, Audia S, et al. Efficacy and safety of rituximab given at 1,000 mg on days 1 and 15 compared to the standard regimen to treat adult immune thrombocytopenia. Am J Hematol 2013;88:858–61.
40. Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007;146:25–33.
41. Khellaf M, Charles-Nelson A, Fain O, et al. Safety and efficacy of rituximab in adult immune thrombocytopenia: results from a prospective registry including 248 patients. Blood 2014;124:3228–36.
42. Ghanima W, Khelif A, Waage A, et al. Rituximab as second-line treatment for adult immune thrombocytopenia (the RITP trial): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2015;385:1653–61.
43. Zaja F, Baccarani M, Mazza P, et al. Dexamethasone plus rituximab yields higher sustained response rates than dexamethasone monotherapy in adults with primary immune thrombocytopenia. Blood 2010;115:2755–62.
44. Dameshek W, Miller EB. The megakaryocytes in idiopathic thrombocytopenic purpura, a form of hypersplenism. Blood 1946;1:27–50.
45. Kuter DJ. Thrombopoietin and thrombopoietin mimetics in the treatment of thrombocytopenia. Annu Rev Med 2009;60:193–206.
46. Bussel JB, Kuter DJ, George JN, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med 2006;355:1672–81.
47. Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:641–8.
48. Bussel JB, Kuter DJ, Pullarkat V, et al. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009;113:2161–71.
49. Gernsheimer TB, George JN, Aledort LM, et al. Evaluation of bleeding and thrombotic events during long-term use of romiplostim in patients with chronic immune thrombocytopenia (ITP). J Thromb Haemost 2010;8:1372–82.
50. Severinsen MT, Engebjerg MC, Farkas DK, et al. Risk of venous thromboembolism in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol 2011;152:360–2.
51. Bussel JB, Cheng G, Saleh MN, et al. Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 2007;357:2237–47.
52. Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011;377:393–402.
53. Brynes RK, Orazi A, Theodore D, et al. Evaluation of bone marrow reticulin in patients with chronic immune thrombocytopenia treated with eltrombopag: Data from the EXTEND study. Am J Hematol 2015;90:598–601.
54. George JN, Kojouri K, Perdue JJ, Vesely SK. Management of patients with chronic, refractory idiopathic thrombocytopenic purpura. Semin Hematol 2000;37:290–8.
55. McMillan R. Therapy for adults with refractory chronic immune thrombocytopenic purpura. Ann Intern Med 1997;126:307–14.
56. Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, et al. Successful therapy with danazol in refractory autoimmune thrombocytopenia associated with rheumatic diseases. Br J Rheumatol 1997;36:1095–9.
57. Provan D, Moss AJ, Newland AC, Bussel JB. Efficacy of mycophenolate mofetil as single-agent therapy for refractory immune thrombocytopenic purpura. Am J Hematol 2006;81:19–25.
58. Reiner A, Gernsheimer T, Slichter SJ. Pulse cyclophosphamide therapy for refractory autoimmune thrombocytopenic purpura. Blood 1995;85:351–8.
59. Figueroa M, Gehlsen J, Hammond D, et al. Combination chemotherapy in refractory immune thrombocytopenic purpura. N Engl J Med 1993;328:1226–9.
60. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy 2 Oct 2017.
61. McCrae KR. Thrombocytopenia in pregnancy. Hematology Am Soc Hematol Educ Program 2010;2010:397–402.
62. Gernsheimer T, McCrae KR. Immune thrombocytopenic purpura in pregnancy. Curr Opin Hematol 2007;14:574–80.
63. DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin 2005;21:531–62.
64. Stavrou E, McCrae KR. Immune thrombocytopenia in pregnancy. Hematol Oncol Clin North Am 2009;23:1299–316.
65. Sun D, Shehata N, Ye XY, et al. Corticosteroids compared with intravenous immunoglobulin for the treatment of immune thrombocytopenia in pregnancy. Blood 2016;128:1329–35.
66. Kong Z, Qin P, Xiao S, et al. A novel recombinant human thrombopoietin therapy for the management of immune thrombocytopenia in pregnancy. Blood 2017;130:1097–103.
67. Psaila B, Petrovic A, Page LK, et al. Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009;114:4777–83.
68. Journeycake JM. Childhood immune thrombocytopenia: role of rituximab, recombinant thrombopoietin, and other new therapeutics. Hematology Am Soc Hematol Educ Program 2012;2012:444–9.
69. Zhang J, Liang Y, Ai Y, et al. Thrombopoietin-receptor agonists for children with immune thrombocytopenia: a systematic review. Expert Opin Pharmacother 2017;18:1543–51.
70. Tarantino MD, Bussel JB, Blanchette VS, et al. Romiplostim in children with immune thrombocytopenia: a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:45–54.71. Grainger JD, Locatelli F, Chotsampancharoen T, et al. Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015;386:1649–58.
72. Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009;113:1231–40.
73. Arnold DM, Bernotas A, Nazi I, et al. Platelet count response to H. pylori treatment in patients with immune thrombocytopenic purpura with and without H. pylori infection: a systematic review. Haematologica 2009;94:850–6.
74. Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med 2007;357:580–7.
75. Reese JA, Li X, Hauben M, et al. Identifying drugs that cause acute thrombocytopenia: an analysis using 3 distinct methods. Blood 2010;116:2127–33.
76. Aster RH, Curtis BR, McFarland JG, Bougie DW. Drug-induced immune thrombocytopenia: pathogenesis, diagnosis and management. J Thromb Haemost 2009;7:911–8.
77. Zondor SD, George JN, Medina PJ. Treatment of drug-induced thrombocytopenia. Expert Opin Drug Saf 2002;1:173–80.
78. George JN, Raskob GE, Shah SR, et al. Drug-induced thrombocytopenia: A systematic review of published case reports. Ann Intern Med 1998;129:886–90.
79. Green D, Hougie C, Kazmier FJ, et al. Report of the working party on acquired inhibitors of coagulation: studies of the “lupus” anticoagulant. Thromb Haemost 1983;49:144–6.
80. Michel M, Chanet V, Dechartres A, et al. The spectrum of Evans syndrome in adults: new insight into the disease based on the analysis of 68 cases. Blood 2009;114:3167–72.
81. Dhingra KK, Jain D, Mandal S, et al. Evans syndrome: a study of six cases with review of literature. Hematology 2008;13:356–60.
82. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematology Am Soc Hematol Educ Program 2009:139–43.
83. Martinez-Valdez L, Deya-Martinez A, Giner MT, et al. Evans syndrome as first manifestation of primary immunodeficiency in clinical practice. J Pediatr Hematol Oncol 2017;39:490–4.
84. Shanafelt TD, Madueme HL, Wolf RC, Tefferi A. Rituximab for immune cytopenia in adults: idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia, and Evans syndrome. Mayo Clin Proc 2003;78:1340–6.
85. Mantadakis E, Danilatou V, Stiakaki E, Kalmanti M. Rituximab for refractory Evans syndrome and other immune-mediated hematologic diseases. Am J Hematol 2004;77:303–10.
86. Jasinski S, Weinblatt ME, Glasser CL. Sirolimus as an effective agent in the treatment of immune thrombocytopenia (ITP) and Evans syndrome (ES): a single institution’s experience. J Pediatr Hematol Oncol 2017;39:420–4.
FDA grants priority review to NDA for avatrombopag
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for avatrombopag.
Avatrombopag is a second-generation thrombopoietin receptor agonist that is intended to address the limitations of existing treatments for thrombocytopenia.
With this NDA, Dova Pharmaceuticals, Inc., is seeking approval of avatrombopag for the treatment of thrombocytopenia in patients with chronic liver disease who are scheduled to undergo a procedure.
The FDA expects to make a decision on the NDA by May 21, 2018.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 3 trials
The NDA submission for avatrombopag is supported by 2 identically designed phase 3 trials, ADAPT 1 and ADAPT 2. Results from these trials were presented at the 2017 American Association for the Study of Liver Disease (AASLD) Meeting last month (abstract 217).
The studies randomized 435 patients with thrombocytopenia and chronic liver disease who were scheduled to undergo a procedure.
Patients with low baseline platelet counts (<40x 109/L) were randomized to receive 60 mg of avatrombopag or placebo daily for 5 days.
Patients with higher baseline platelet counts (40 to <50 x 109/L) were randomized to receive 40 mg of avatrombopag or placebo daily for 5 days.
Patients underwent their procedures 5 to 8 days after their last dose of avatrombopag.
In ADAPT-1, 85 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 78 controls completed the study.
In ADAPT-2, 68 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 68 controls completed the study.
Efficacy
The primary efficacy endpoint of these trials was the proportion of patients who did not require any bleeding rescue up to 7 days post-procedure. Bleeding rescue included platelet transfusion, fresh frozen plasma, cryoprecipitate, vitamin K (phytonadione), desmopressin, recombinant activated factor VII, aminocaproicacid, tranexamic acid, whole blood transfusion, packed red cell transfusion, surgical intervention, or interventional radiology.
In ADAPT-1, the primary endpoint was achieved by 66% of patients who received avatrombopag at 60 mg and 23% of those who received placebo in the low-platelet-count cohort (P<0.0001). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 38% of controls in the higher-platelet-count cohort (P<0.0001).
In ADAPT-2, the primary endpoint was achieved by 69% of patients who received avatrombopag at 60 mg and 35% of those who received placebo in the low-platelet-count cohort (P<0.0006). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 33% of controls in the higher- platelet-count cohort (P<0.0001).
A secondary efficacy endpoint was the proportion of patients achieving the target platelet count (≥50 x 109/L).
In ADAPT-1, this endpoint was met by 69% of patients who received avatrombopag at 60 mg and 4% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 88% of patients who received avatrombopag at 40 mg and 21% of controls in the higher-platelet-count cohort (P<0.0001)
In ADAPT-2, this endpoint was met by 67% of patients who received avatrombopag at 60 mg and 7% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 93% of patients who received avatrombopag at 40 mg and 39% of controls in the higher-platelet-count cohort (P<0.0001).
Safety
The researchers pooled safety data from the 2 trials.
Treatment-emergent adverse events (AEs) occurred in 58.2% of controls and 56% of avatrombopag-treated patients in the low-platelet-count cohort (60 mg). Treatment-emergent AEs also occurred in 50.8% of controls and 51.3% of avatrombopag-treated patients in the higher-platelet-count cohort (40 mg).
The most frequently reported treatment-emergent AEs were pyrexia, abdominal pain, nausea, headache, diarrhea, and fatigue.
One patient experienced partial portal vein thrombosis that was considered non-serious and potentially related to avatrombopag.
Treatment-related AEs occurred in 17.6% of controls and 11.3% of avatrombopag-treated patients in the low-platelet-count cohort. Treatment-related AEs also occurred in 6.2% of controls and 7% of avatrombopag-treated patients in the higher-platelet-count cohort.
Serious AEs occurred in 13.2%, 6.9%, 3.1%, and 7.8%, respectively.
There were 3 deaths—2 in the 40 mg avatrombopag arm in ADAPT-1 and 1 in the control group in ADAPT-2. None of the deaths was considered treatment-related.
Future directions
Dova Pharmaceuticals, Inc., is planning to explore the potential use of avatrombopag in a broader population of patients with thrombocytopenia. This includes patients undergoing surgical procedures associated with a high risk of bleeding and patients who develop thrombocytopenia after receiving chemotherapy.
In addition, the company is exploring a potential regulatory approval pathway for avatrombopag for the treatment of adults with chronic immune thrombocytopenic purpura based on results from a completed phase 3 trial in this patient population. ![]()
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for avatrombopag.
Avatrombopag is a second-generation thrombopoietin receptor agonist that is intended to address the limitations of existing treatments for thrombocytopenia.
With this NDA, Dova Pharmaceuticals, Inc., is seeking approval of avatrombopag for the treatment of thrombocytopenia in patients with chronic liver disease who are scheduled to undergo a procedure.
The FDA expects to make a decision on the NDA by May 21, 2018.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 3 trials
The NDA submission for avatrombopag is supported by 2 identically designed phase 3 trials, ADAPT 1 and ADAPT 2. Results from these trials were presented at the 2017 American Association for the Study of Liver Disease (AASLD) Meeting last month (abstract 217).
The studies randomized 435 patients with thrombocytopenia and chronic liver disease who were scheduled to undergo a procedure.
Patients with low baseline platelet counts (<40x 109/L) were randomized to receive 60 mg of avatrombopag or placebo daily for 5 days.
Patients with higher baseline platelet counts (40 to <50 x 109/L) were randomized to receive 40 mg of avatrombopag or placebo daily for 5 days.
Patients underwent their procedures 5 to 8 days after their last dose of avatrombopag.
In ADAPT-1, 85 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 78 controls completed the study.
In ADAPT-2, 68 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 68 controls completed the study.
Efficacy
The primary efficacy endpoint of these trials was the proportion of patients who did not require any bleeding rescue up to 7 days post-procedure. Bleeding rescue included platelet transfusion, fresh frozen plasma, cryoprecipitate, vitamin K (phytonadione), desmopressin, recombinant activated factor VII, aminocaproicacid, tranexamic acid, whole blood transfusion, packed red cell transfusion, surgical intervention, or interventional radiology.
In ADAPT-1, the primary endpoint was achieved by 66% of patients who received avatrombopag at 60 mg and 23% of those who received placebo in the low-platelet-count cohort (P<0.0001). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 38% of controls in the higher-platelet-count cohort (P<0.0001).
In ADAPT-2, the primary endpoint was achieved by 69% of patients who received avatrombopag at 60 mg and 35% of those who received placebo in the low-platelet-count cohort (P<0.0006). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 33% of controls in the higher- platelet-count cohort (P<0.0001).
A secondary efficacy endpoint was the proportion of patients achieving the target platelet count (≥50 x 109/L).
In ADAPT-1, this endpoint was met by 69% of patients who received avatrombopag at 60 mg and 4% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 88% of patients who received avatrombopag at 40 mg and 21% of controls in the higher-platelet-count cohort (P<0.0001)
In ADAPT-2, this endpoint was met by 67% of patients who received avatrombopag at 60 mg and 7% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 93% of patients who received avatrombopag at 40 mg and 39% of controls in the higher-platelet-count cohort (P<0.0001).
Safety
The researchers pooled safety data from the 2 trials.
Treatment-emergent adverse events (AEs) occurred in 58.2% of controls and 56% of avatrombopag-treated patients in the low-platelet-count cohort (60 mg). Treatment-emergent AEs also occurred in 50.8% of controls and 51.3% of avatrombopag-treated patients in the higher-platelet-count cohort (40 mg).
The most frequently reported treatment-emergent AEs were pyrexia, abdominal pain, nausea, headache, diarrhea, and fatigue.
One patient experienced partial portal vein thrombosis that was considered non-serious and potentially related to avatrombopag.
Treatment-related AEs occurred in 17.6% of controls and 11.3% of avatrombopag-treated patients in the low-platelet-count cohort. Treatment-related AEs also occurred in 6.2% of controls and 7% of avatrombopag-treated patients in the higher-platelet-count cohort.
Serious AEs occurred in 13.2%, 6.9%, 3.1%, and 7.8%, respectively.
There were 3 deaths—2 in the 40 mg avatrombopag arm in ADAPT-1 and 1 in the control group in ADAPT-2. None of the deaths was considered treatment-related.
Future directions
Dova Pharmaceuticals, Inc., is planning to explore the potential use of avatrombopag in a broader population of patients with thrombocytopenia. This includes patients undergoing surgical procedures associated with a high risk of bleeding and patients who develop thrombocytopenia after receiving chemotherapy.
In addition, the company is exploring a potential regulatory approval pathway for avatrombopag for the treatment of adults with chronic immune thrombocytopenic purpura based on results from a completed phase 3 trial in this patient population. ![]()
The US Food and Drug Administration (FDA) has granted priority review to the new drug application (NDA) for avatrombopag.
Avatrombopag is a second-generation thrombopoietin receptor agonist that is intended to address the limitations of existing treatments for thrombocytopenia.
With this NDA, Dova Pharmaceuticals, Inc., is seeking approval of avatrombopag for the treatment of thrombocytopenia in patients with chronic liver disease who are scheduled to undergo a procedure.
The FDA expects to make a decision on the NDA by May 21, 2018.
The FDA’s goal is to take action on a priority review application within 6 months of receiving it, rather than the standard 10 months.
The FDA grants priority review to applications for products that may provide significant improvements in the treatment, diagnosis, or prevention of serious conditions.
Phase 3 trials
The NDA submission for avatrombopag is supported by 2 identically designed phase 3 trials, ADAPT 1 and ADAPT 2. Results from these trials were presented at the 2017 American Association for the Study of Liver Disease (AASLD) Meeting last month (abstract 217).
The studies randomized 435 patients with thrombocytopenia and chronic liver disease who were scheduled to undergo a procedure.
Patients with low baseline platelet counts (<40x 109/L) were randomized to receive 60 mg of avatrombopag or placebo daily for 5 days.
Patients with higher baseline platelet counts (40 to <50 x 109/L) were randomized to receive 40 mg of avatrombopag or placebo daily for 5 days.
Patients underwent their procedures 5 to 8 days after their last dose of avatrombopag.
In ADAPT-1, 85 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 78 controls completed the study.
In ADAPT-2, 68 patients completed treatment with avatrombopag at 60 mg, 55 completed treatment with avatrombopag at 40 mg, and 68 controls completed the study.
Efficacy
The primary efficacy endpoint of these trials was the proportion of patients who did not require any bleeding rescue up to 7 days post-procedure. Bleeding rescue included platelet transfusion, fresh frozen plasma, cryoprecipitate, vitamin K (phytonadione), desmopressin, recombinant activated factor VII, aminocaproicacid, tranexamic acid, whole blood transfusion, packed red cell transfusion, surgical intervention, or interventional radiology.
In ADAPT-1, the primary endpoint was achieved by 66% of patients who received avatrombopag at 60 mg and 23% of those who received placebo in the low-platelet-count cohort (P<0.0001). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 38% of controls in the higher-platelet-count cohort (P<0.0001).
In ADAPT-2, the primary endpoint was achieved by 69% of patients who received avatrombopag at 60 mg and 35% of those who received placebo in the low-platelet-count cohort (P<0.0006). The endpoint was also achieved by 88% of patients who received avatrombopag at 40 mg and 33% of controls in the higher- platelet-count cohort (P<0.0001).
A secondary efficacy endpoint was the proportion of patients achieving the target platelet count (≥50 x 109/L).
In ADAPT-1, this endpoint was met by 69% of patients who received avatrombopag at 60 mg and 4% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 88% of patients who received avatrombopag at 40 mg and 21% of controls in the higher-platelet-count cohort (P<0.0001)
In ADAPT-2, this endpoint was met by 67% of patients who received avatrombopag at 60 mg and 7% of controls in the low-platelet-count cohort (P<0.0001). It was also met by 93% of patients who received avatrombopag at 40 mg and 39% of controls in the higher-platelet-count cohort (P<0.0001).
Safety
The researchers pooled safety data from the 2 trials.
Treatment-emergent adverse events (AEs) occurred in 58.2% of controls and 56% of avatrombopag-treated patients in the low-platelet-count cohort (60 mg). Treatment-emergent AEs also occurred in 50.8% of controls and 51.3% of avatrombopag-treated patients in the higher-platelet-count cohort (40 mg).
The most frequently reported treatment-emergent AEs were pyrexia, abdominal pain, nausea, headache, diarrhea, and fatigue.
One patient experienced partial portal vein thrombosis that was considered non-serious and potentially related to avatrombopag.
Treatment-related AEs occurred in 17.6% of controls and 11.3% of avatrombopag-treated patients in the low-platelet-count cohort. Treatment-related AEs also occurred in 6.2% of controls and 7% of avatrombopag-treated patients in the higher-platelet-count cohort.
Serious AEs occurred in 13.2%, 6.9%, 3.1%, and 7.8%, respectively.
There were 3 deaths—2 in the 40 mg avatrombopag arm in ADAPT-1 and 1 in the control group in ADAPT-2. None of the deaths was considered treatment-related.
Future directions
Dova Pharmaceuticals, Inc., is planning to explore the potential use of avatrombopag in a broader population of patients with thrombocytopenia. This includes patients undergoing surgical procedures associated with a high risk of bleeding and patients who develop thrombocytopenia after receiving chemotherapy.
In addition, the company is exploring a potential regulatory approval pathway for avatrombopag for the treatment of adults with chronic immune thrombocytopenic purpura based on results from a completed phase 3 trial in this patient population. ![]()
FDA grants orphan drug status to rofecoxib for hemophilic arthropathy
The Food and Drug Administration on Nov. 21 granted orphan drug designation to rofecoxib (TRM-201), a cyclooxygenase 2–selective nonsteroidal anti-inflammatory drug (NSAID) intended to treat patients with hemophilic arthropathy (HA).
HA, a joint disease caused by hemarthrosis, is the largest cause of morbidity for hemophilia patients. There are currently no approved treatments in the United States.
The attempt at a reintroduction of rofecoxib specifically for the treatment of HA is being developed by Tremeau Pharmaceuticals.
Patients with hemophilia look to avoid traditional NSAIDs, as those drugs risk gastrointestinal ulcers and impair platelet aggregation. The current standard of care for HA is opioid treatment.
Rofecoxib and other NSAIDs cause an increased risk of serious cardiovascular thrombotic events and gastrointestinal adverse events.
Orphan drug status is available to treatments for rare disorders and provides a 7-year marketing exclusivity period against competition, along with tax credits and a waiver of Prescription Drug User Fee Act filing fees.
The Food and Drug Administration on Nov. 21 granted orphan drug designation to rofecoxib (TRM-201), a cyclooxygenase 2–selective nonsteroidal anti-inflammatory drug (NSAID) intended to treat patients with hemophilic arthropathy (HA).
HA, a joint disease caused by hemarthrosis, is the largest cause of morbidity for hemophilia patients. There are currently no approved treatments in the United States.
The attempt at a reintroduction of rofecoxib specifically for the treatment of HA is being developed by Tremeau Pharmaceuticals.
Patients with hemophilia look to avoid traditional NSAIDs, as those drugs risk gastrointestinal ulcers and impair platelet aggregation. The current standard of care for HA is opioid treatment.
Rofecoxib and other NSAIDs cause an increased risk of serious cardiovascular thrombotic events and gastrointestinal adverse events.
Orphan drug status is available to treatments for rare disorders and provides a 7-year marketing exclusivity period against competition, along with tax credits and a waiver of Prescription Drug User Fee Act filing fees.
The Food and Drug Administration on Nov. 21 granted orphan drug designation to rofecoxib (TRM-201), a cyclooxygenase 2–selective nonsteroidal anti-inflammatory drug (NSAID) intended to treat patients with hemophilic arthropathy (HA).
HA, a joint disease caused by hemarthrosis, is the largest cause of morbidity for hemophilia patients. There are currently no approved treatments in the United States.
The attempt at a reintroduction of rofecoxib specifically for the treatment of HA is being developed by Tremeau Pharmaceuticals.
Patients with hemophilia look to avoid traditional NSAIDs, as those drugs risk gastrointestinal ulcers and impair platelet aggregation. The current standard of care for HA is opioid treatment.
Rofecoxib and other NSAIDs cause an increased risk of serious cardiovascular thrombotic events and gastrointestinal adverse events.
Orphan drug status is available to treatments for rare disorders and provides a 7-year marketing exclusivity period against competition, along with tax credits and a waiver of Prescription Drug User Fee Act filing fees.
FDA approves first-in-class drug for hemophilia A with Factor VIII
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.
The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.
The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.
The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
Product approved to treat patients with hemophilia A and inhibitors

The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
The US Food and Drug Administration (FDA) has approved use of emicizumab-kxwh (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
Emicizumab is approved as routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children who have hemophilia A and factor VIII (FVIII) inhibitors.
Emicizumab can be self-administered once-weekly via subcutaneous injection.
The labeling for emicizumab contains a boxed warning noting that patients who received emicizumab in conjunction with activated prothrombin complex concentrate developed thrombotic microangiopathy (TMA) and thromboembolic events (TEs).
Therefore, patients should discontinue prophylactic use of bypassing agents (BPAs) the day before starting prophylaxis with emicizumab.
The FDA granted the approval of emicizumab to Genentech, Inc. The agency granted the application for emicizumab priority review, and the product received breakthrough therapy and orphan drug designations.
Access to emicizumab
According to Genentech, emicizumab will be available shortly.
The company said it will be offering comprehensive services to help minimize barriers to access and reimbursement. Patients can call 866-436-5427 (866-HEMLIBRA) for more information.
For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is available at 866-422-2377 (866-4ACCESS) or http://www.Genentech-Access.com.
Emicizumab trials
The biologics license application for emicizumab was supported by results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July. Interim results from HAVEN 2 were presented at ISTH as well.
HAVEN 1
The study enrolled 109 patients (age 12 and older) with hemophilia A and FVIII inhibitors who were previously treated with BPAs on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced TEs, and 3 had TMA while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in children younger than 12 years of age who had hemophilia A with FVIII inhibitors.
The interim efficacy analysis, after at least 12 weeks of treatment, included 23 children.
After a median observation time of 38.1 weeks, 87% (95% CI: 66.4; 97.2) of children who received emicizumab experienced 0 treated bleeds. The percentage with 0 treated or non-treated bleeds was lower, at 34.8% (95% CI: 16.4; 57.3).
The most common adverse events (observed in at least 10% of patients) were mild injection site reactions and nasopharyngitis. No TEs or TMAs were observed.
HAVEN 3 and 4
Emicizumab is now being studied in 2 additional phase 3 trials.
In HAVEN 3, researchers are evaluating emicizumab prophylaxis dosed once weekly or once every other week in patients age 12 and older with hemophilia A without FVIII inhibitors.
In HAVEN 4, researchers are evaluating emicizumab prophylaxis dosed every 4 weeks in patients age 12 and older with hemophilia A, with or without inhibitors. ![]()
CHMP backs therapy for hemophilia A

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for rurioctocog alfa pegol (Adynovi).
Rurioctocog alfa pegol (formerly BAX 855) is a pegylated, full-length, recombinant factor VIII product built on a licensed recombinant factor VIII product (Advate).
The CHMP is recommending that rurioctocog alfa pegol be approved for the treatment and prophylaxis of bleeding in patients age 12 and older with hemophilia A.
The CHMP’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
If approved, rurioctocog alfa pegol would be available as a powder and solvent for solution for injection (250 IU, 500 IU, 1000 IU, and 2000 IU).
Phase 3 trials
Rurioctocog alfa pegol has been studied in 3 phase 3 trials.
One study (phase 2/3) included 137 patients, age 12 and older, with previously treated hemophilia A. Results from this trial were published in Blood in July 2015.
Another study included 15 patients with severe hemophilia A who were undergoing surgical procedures. Results were published in Haemophilia in June 2016.
A third study included 66 patients, age 12 and younger, who had previously treated hemophilia A. Results from this trial were presented at the World Federation of Hemophilia 2016 World Congress in July 2016. ![]()
CHMP recommends approval of romiplostim in kids

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for romiplostim (Nplate®) to include children.
The CHMP is recommending authorization of romiplostim to treat patients age 1 and older who have chronic immune thrombocytopenia (ITP) that is refractory to other treatments.
The committee’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, a centralized marketing authorization will be granted that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The recommendation for romiplostim was based on 5 studies of the drug in children with ITP. This includes 4 completed studies—a phase 1/2, a phase 3, and 2 long-term safety and efficacy studies—and 1 ongoing long-term study.
Results from the phase 1/2 trial were published in Blood in 2011. Phase 3 results were published in The Lancet in April of last year.
And results from 2 of the long-term trials were presented at 22nd Congress of the European Hematology Association in June (abstract P367 and abstract P727). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for romiplostim (Nplate®) to include children.
The CHMP is recommending authorization of romiplostim to treat patients age 1 and older who have chronic immune thrombocytopenia (ITP) that is refractory to other treatments.
The committee’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, a centralized marketing authorization will be granted that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The recommendation for romiplostim was based on 5 studies of the drug in children with ITP. This includes 4 completed studies—a phase 1/2, a phase 3, and 2 long-term safety and efficacy studies—and 1 ongoing long-term study.
Results from the phase 1/2 trial were published in Blood in 2011. Phase 3 results were published in The Lancet in April of last year.
And results from 2 of the long-term trials were presented at 22nd Congress of the European Hematology Association in June (abstract P367 and abstract P727). ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for romiplostim (Nplate®) to include children.
The CHMP is recommending authorization of romiplostim to treat patients age 1 and older who have chronic immune thrombocytopenia (ITP) that is refractory to other treatments.
The committee’s opinion will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, a centralized marketing authorization will be granted that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The recommendation for romiplostim was based on 5 studies of the drug in children with ITP. This includes 4 completed studies—a phase 1/2, a phase 3, and 2 long-term safety and efficacy studies—and 1 ongoing long-term study.
Results from the phase 1/2 trial were published in Blood in 2011. Phase 3 results were published in The Lancet in April of last year.
And results from 2 of the long-term trials were presented at 22nd Congress of the European Hematology Association in June (abstract P367 and abstract P727). ![]()
Drug receives orphan designation for treatment of PNH

The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to ACH-4471 for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
And the European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products has recommended the drug receive orphan status for the same indication in the European Economic Area.
ACH-4471 is a factor D inhibitor being developed by Achillion Pharmaceuticals, Inc.
In April, the company announced the initiation of a phase 2, three-month, dose-ranging trial with ACH-4471 for patients with untreated PNH (NCT03053102).
The primary objective of the trial is to assess the change from baseline in serum lactate dehydrogenase (LDH) levels. Secondary endpoints include changes in hemoglobin, PNH red blood cells, fatigue score (FACIT scale), changes in levels of complement pathway biomarkers such as Bb and factor D, pharmacokinetics, and safety.
The protocol allows for intra-patient dose-escalation. Patients initially receive 100 mg or 150 mg of ACH-4471 three times daily, and doses may be increased during the treatment period.
After patients complete 3 months of treatment and investigators have assessed safety and clinical benefit, patients may be enrolled in the long-term extension trial (NCT03181633).
To date, 200 mg three times daily has been the highest dose of ACH-4471 administered. And Achillion has collected data on 4 patients.
Two of the patients have completed the 3-month trial and entered the long-term extension trial. One patient continues to receive dosing in the 3-month trial, and the fourth patient voluntarily withdrew from the trial on day 41 for reasons unrelated to safety.
Thus far, ACH-4471 has produced clinically meaningful complement inhibition and demonstrated a favorable tolerability profile, with no reports of clinically meaningful increases in liver enzymes. ACH-4471 has improved LDH, hemoglobin, fatigue score, and other measures of response, including PNH clone size.
FDA’s orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
EMA’s orphan designation
The EMA’s orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. It also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure.
The EMA’s Committee for Orphan Medicinal Products adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision. The commission typically makes a decision within 30 days of the submission. ![]()
Sickle cell patients suffer discrimination, poor care – and shorter lives
For more than a year, NeDina Brocks-Capla avoided one room in her large, brightly colored San Francisco house – the bathroom on the second floor.
“It was really hard to bathe in here, and I found myself not wanting to touch the walls,” she explained. The bathroom is where Ms. Brocks-Capla’s son Kareem Jones died in 2013 at age 36, from sickle cell disease.
It’s not just the loss of her son that upsets Ms. Brocks-Capla; she believes that if Mr. Jones had gotten the proper medical care, he might still be alive today.
Sickle cell disease is an inherited disorder that causes some red blood cells to bend into a crescent shape. The misshapen, inflexible cells clog the blood vessels, preventing blood from circulating oxygen properly, which can cause chronic pain, multiorgan failure, and stroke. About 100,000 people in the United States have sickle cell disease, and most of them are African American.
Patients and experts alike say it’s no surprise then that while life expectancy for almost every major malady is improving, patients with sickle cell disease can expect to die younger than they did 20 years ago. In 1994, life expectancy for sickle cell patients was 42 for men and 48 for women. By 2005, life expectancy had dipped to 38 for men and 42 for women.
Sickle cell disease is “a microcosm of how issues of race, ethnicity and identity come into conflict with issues of health care,” said Keith Wailoo, PhD, a professor at Princeton University who writes about the history of the disease.
It is also an example of the broader discrimination experienced by African Americans in the medical system. Nearly a third report that they have experienced discrimination when going to the doctor, according to a poll by NPR, Robert Wood Johnson Foundation, and Harvard T.H. Chan School of Public Health.
“One of the national crises in health care is the care for adult sickle cell,” said leading researcher and physician Elliott Vichinsky, MD, who started the sickle cell center at UCSF Benioff Children’s Hospital Oakland in 1978. “This group of people can live much longer with the management we have, and they’re dying because we don’t have access to care.”
Indeed, with the proper care, Dr. Vichinsky’s center and the handful of other specialty clinics like it across the country have been able to increase life expectancy for sickle cell patients well into their 60s.
Dr. Vichinsky’s patient Derek Perkins, 45, knows he has already beaten the odds. He sits in an exam room decorated with cartoon characters at Children’s Hospital Oakland, but this is the adult sickle cell clinic. He’s been Dr. Vichinsky’s patient since childhood.
“Without the sickle cell clinic here in Oakland, I don’t know what I would do. I don’t know anywhere else I could go,” Mr. Perkins said.
When Mr. Perkins was 27, he once ended up at a different hospital where doctors misdiagnosed his crisis. He went into a coma and was near death before his mother insisted he be transferred.
“Dr. Vichinsky was able to get me here to Children’s Hospital, and he found out what was wrong and within 18 hours – all I needed was an emergency blood transfusion and I was awake,” Mr. Perkins recalled.
Kareem Jones lived just across the bay from Mr. Perkins, but he had a profoundly different experience.
Mr. Jones’ mother, Ms. Brocks-Capla, said her son received excellent medical care as a child, but once he turned 18 and aged out of his pediatric program, it felt like falling off a cliff. Mr. Jones was sent to a clinic at San Francisco General Hospital, but it was open only for a half-day, one day each week. If he was sick any other day, he had two options: leave a voicemail for a clinic nurse or go to the emergency room. “That’s not comprehensive care – that’s not consistent care for a disease of this type,” said Ms. Brocks-Capla.
Ms. Brocks-Capla is a retired supervisor at a worker’s compensation firm. She knew how to navigate the health care system, but she couldn’t get her son the care he needed. Like most sickle cell patients, Mr. Jones had frequent pain crises. Usually he ended up in the emergency department where, Ms. Brocks-Capla said, the doctors didn’t seem to know much about sickle cell disease.
When she tried to explain her son’s pain to the doctors and nurses, she recalled, “they say have a seat. ‘He can’t have a seat! Can’t you see him?’ ”
Studies have found that sickle cell patients have to wait up to 50% longer for help in the emergency department than do other pain patients. The opioid crisis has made things even worse, Dr. Vichinsky added, as patients in terrible pain are likely to be seen as drug seekers with addiction problems rather than patients in need.
Despite his illness, Mr. Jones fought to have a normal life. He lived with his girlfriend, had a daughter, and worked as much as he could between pain crises. He was an avid San Francisco Giants fan.
For years, he took hydroxyurea, but it had side effects, and after a while Mr. Jones had to stop taking it. “And that was it, because you know there isn’t any other medication out there,” said Ms. Brocks-Capla.
Indeed, hydroxyurea, which the Food and Drug Administration first approved in 1967 as a cancer drug, was the only drug on the market to treat sickle cell during Mr. Jones’ lifetime. In July, the FDA approved a second drug, Endari (L-glutamine oral powder), specifically to treat patients with sickle cell disease.
Funding by the federal government and private foundations for the disease pales in comparison to other disorders. Cystic fibrosis offers a good comparison. It is another inherited disorder that requires complex care and most often occurs in Caucasians. Cystic fibrosis gets 7-11 times more funding per patient than does sickle cell disease, according to a 2013 study in the journal Blood. From 2010 to 2013 alone, the FDA approved five new drugs for the treatment of cystic fibrosis.
“There’s no question in my mind that class and color are major factors in impairing their survival. Without question,” Dr. Vichinsky said of sickle cell patients. “The death rate is increasing. The quality of care is going down.”
Without a new medication, Mr. Jones got progressively worse. At 36, his kidneys began to fail, and he had to go on dialysis. He ended up in the hospital, with the worst pain of his life. The doctors stabilized him and gave him pain meds but did not diagnose the underlying cause of the crisis. He was released to his mother’s care, still in incredible pain.
At home, Ms. Brocks-Capla ran him a warm bath to try to soothe his pain and went downstairs to get him a change of clothes. As she came back up the stairs, she heard loud banging against the bathroom walls.
“So I run into the bathroom and he’s having a seizure. And I didn’t know what to do. I was like, ‘Oh come on, come on. Don’t do this. Don’t do this to me.’ ”
She called 911. The paramedics came but couldn’t revive him. “He died here with me,” she said.
It turned out Mr. Jones had a series of small strokes. His organs were in failure, something Ms. Brocks-Capla said the hospital missed. She believes his death could have been prevented with consistent care – the kind he got as a child. Dr. Vichinsky thinks she is probably right.
“I would say 40% or more of the deaths I’ve had recently have been preventable – I mean totally preventable,” he said, but he got to the cases too late. “It makes me so angry. I’ve spent my life trying to help these people, and the harder part is you can change this – this isn’t a knowledge issue. It’s an access issue.”
Dr. Vichinsky’s center and others like it have made major advances in screening patients for the early signs of organ failure and intervening to prevent premature death. Patients at these clinics live 2 decades longer than the average sickle cell patient.
Good care for sickle cell requires time and training for physicians, but it often doesn’t pay well, because many patients are on Medicaid or other government insurance programs. The result is that most adult sickle cell patients still struggle even to access treatments that have been around for decades, Dr. Vichinsky said.
The phenomenon is nothing new — the disease that used to be known as sickle cell anemia has had a long and sordid past. It was first identified in 1910 and helped launch the field of molecular biology. But most of the research was used to study science rather than improving care for sickle cell patients, Dr. Vichinsky said.
In the 1960s and 1970s, sickle cell became a lightning rod for the civil rights movement. At the time, the average patient died before age 20. The Black Panther Party took up the cause and began testing people at its “survival conferences” across the country.
“I’m sure we tested over four-and-a-half-thousand people for sickle cell anemia last night – and I think that the voter registration is running neck and neck with it,” Black Panther Party Chairman Bobby Seale told news crews at an event in Oakland in 1972.
The movement grew, and Washington listened. “It is a sad and shameful fact that the causes of this disease have been largely neglected throughout our history,” President Richard Nixon told Congress in 1971. “We cannot rewrite this record of neglect, but we can reverse it. To this end, this administration is increasing its budget for research and treatment of sickle cell disease.”
For a while, funding did increase, newborn screening took hold, and by the 1990s, life expectancy had doubled, with patients living into their 40s. But over time, funding waned, clinics closed, and life expectancy started dropping again.
Dr. Vichinsky pushes against that trend for patients like Derek Perkins. The father of four looks healthy and robust, but like most sickle cell patients, he has episodes of extreme pain and has problems with his kidneys, heart, hips, and breathing. Keeping him thriving requires regular checkups and constant monitoring for potential problems.
“The program Dr. Vichinsky is running here, I feel I owe my life to [it],” said Mr. Perkins. “If it wasn’t for him and the things that he did for me, my family wouldn’t have me.”
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation. KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
For more than a year, NeDina Brocks-Capla avoided one room in her large, brightly colored San Francisco house – the bathroom on the second floor.
“It was really hard to bathe in here, and I found myself not wanting to touch the walls,” she explained. The bathroom is where Ms. Brocks-Capla’s son Kareem Jones died in 2013 at age 36, from sickle cell disease.
It’s not just the loss of her son that upsets Ms. Brocks-Capla; she believes that if Mr. Jones had gotten the proper medical care, he might still be alive today.
Sickle cell disease is an inherited disorder that causes some red blood cells to bend into a crescent shape. The misshapen, inflexible cells clog the blood vessels, preventing blood from circulating oxygen properly, which can cause chronic pain, multiorgan failure, and stroke. About 100,000 people in the United States have sickle cell disease, and most of them are African American.
Patients and experts alike say it’s no surprise then that while life expectancy for almost every major malady is improving, patients with sickle cell disease can expect to die younger than they did 20 years ago. In 1994, life expectancy for sickle cell patients was 42 for men and 48 for women. By 2005, life expectancy had dipped to 38 for men and 42 for women.
Sickle cell disease is “a microcosm of how issues of race, ethnicity and identity come into conflict with issues of health care,” said Keith Wailoo, PhD, a professor at Princeton University who writes about the history of the disease.
It is also an example of the broader discrimination experienced by African Americans in the medical system. Nearly a third report that they have experienced discrimination when going to the doctor, according to a poll by NPR, Robert Wood Johnson Foundation, and Harvard T.H. Chan School of Public Health.
“One of the national crises in health care is the care for adult sickle cell,” said leading researcher and physician Elliott Vichinsky, MD, who started the sickle cell center at UCSF Benioff Children’s Hospital Oakland in 1978. “This group of people can live much longer with the management we have, and they’re dying because we don’t have access to care.”
Indeed, with the proper care, Dr. Vichinsky’s center and the handful of other specialty clinics like it across the country have been able to increase life expectancy for sickle cell patients well into their 60s.
Dr. Vichinsky’s patient Derek Perkins, 45, knows he has already beaten the odds. He sits in an exam room decorated with cartoon characters at Children’s Hospital Oakland, but this is the adult sickle cell clinic. He’s been Dr. Vichinsky’s patient since childhood.
“Without the sickle cell clinic here in Oakland, I don’t know what I would do. I don’t know anywhere else I could go,” Mr. Perkins said.
When Mr. Perkins was 27, he once ended up at a different hospital where doctors misdiagnosed his crisis. He went into a coma and was near death before his mother insisted he be transferred.
“Dr. Vichinsky was able to get me here to Children’s Hospital, and he found out what was wrong and within 18 hours – all I needed was an emergency blood transfusion and I was awake,” Mr. Perkins recalled.
Kareem Jones lived just across the bay from Mr. Perkins, but he had a profoundly different experience.
Mr. Jones’ mother, Ms. Brocks-Capla, said her son received excellent medical care as a child, but once he turned 18 and aged out of his pediatric program, it felt like falling off a cliff. Mr. Jones was sent to a clinic at San Francisco General Hospital, but it was open only for a half-day, one day each week. If he was sick any other day, he had two options: leave a voicemail for a clinic nurse or go to the emergency room. “That’s not comprehensive care – that’s not consistent care for a disease of this type,” said Ms. Brocks-Capla.
Ms. Brocks-Capla is a retired supervisor at a worker’s compensation firm. She knew how to navigate the health care system, but she couldn’t get her son the care he needed. Like most sickle cell patients, Mr. Jones had frequent pain crises. Usually he ended up in the emergency department where, Ms. Brocks-Capla said, the doctors didn’t seem to know much about sickle cell disease.
When she tried to explain her son’s pain to the doctors and nurses, she recalled, “they say have a seat. ‘He can’t have a seat! Can’t you see him?’ ”
Studies have found that sickle cell patients have to wait up to 50% longer for help in the emergency department than do other pain patients. The opioid crisis has made things even worse, Dr. Vichinsky added, as patients in terrible pain are likely to be seen as drug seekers with addiction problems rather than patients in need.
Despite his illness, Mr. Jones fought to have a normal life. He lived with his girlfriend, had a daughter, and worked as much as he could between pain crises. He was an avid San Francisco Giants fan.
For years, he took hydroxyurea, but it had side effects, and after a while Mr. Jones had to stop taking it. “And that was it, because you know there isn’t any other medication out there,” said Ms. Brocks-Capla.
Indeed, hydroxyurea, which the Food and Drug Administration first approved in 1967 as a cancer drug, was the only drug on the market to treat sickle cell during Mr. Jones’ lifetime. In July, the FDA approved a second drug, Endari (L-glutamine oral powder), specifically to treat patients with sickle cell disease.
Funding by the federal government and private foundations for the disease pales in comparison to other disorders. Cystic fibrosis offers a good comparison. It is another inherited disorder that requires complex care and most often occurs in Caucasians. Cystic fibrosis gets 7-11 times more funding per patient than does sickle cell disease, according to a 2013 study in the journal Blood. From 2010 to 2013 alone, the FDA approved five new drugs for the treatment of cystic fibrosis.
“There’s no question in my mind that class and color are major factors in impairing their survival. Without question,” Dr. Vichinsky said of sickle cell patients. “The death rate is increasing. The quality of care is going down.”
Without a new medication, Mr. Jones got progressively worse. At 36, his kidneys began to fail, and he had to go on dialysis. He ended up in the hospital, with the worst pain of his life. The doctors stabilized him and gave him pain meds but did not diagnose the underlying cause of the crisis. He was released to his mother’s care, still in incredible pain.
At home, Ms. Brocks-Capla ran him a warm bath to try to soothe his pain and went downstairs to get him a change of clothes. As she came back up the stairs, she heard loud banging against the bathroom walls.
“So I run into the bathroom and he’s having a seizure. And I didn’t know what to do. I was like, ‘Oh come on, come on. Don’t do this. Don’t do this to me.’ ”
She called 911. The paramedics came but couldn’t revive him. “He died here with me,” she said.
It turned out Mr. Jones had a series of small strokes. His organs were in failure, something Ms. Brocks-Capla said the hospital missed. She believes his death could have been prevented with consistent care – the kind he got as a child. Dr. Vichinsky thinks she is probably right.
“I would say 40% or more of the deaths I’ve had recently have been preventable – I mean totally preventable,” he said, but he got to the cases too late. “It makes me so angry. I’ve spent my life trying to help these people, and the harder part is you can change this – this isn’t a knowledge issue. It’s an access issue.”
Dr. Vichinsky’s center and others like it have made major advances in screening patients for the early signs of organ failure and intervening to prevent premature death. Patients at these clinics live 2 decades longer than the average sickle cell patient.
Good care for sickle cell requires time and training for physicians, but it often doesn’t pay well, because many patients are on Medicaid or other government insurance programs. The result is that most adult sickle cell patients still struggle even to access treatments that have been around for decades, Dr. Vichinsky said.
The phenomenon is nothing new — the disease that used to be known as sickle cell anemia has had a long and sordid past. It was first identified in 1910 and helped launch the field of molecular biology. But most of the research was used to study science rather than improving care for sickle cell patients, Dr. Vichinsky said.
In the 1960s and 1970s, sickle cell became a lightning rod for the civil rights movement. At the time, the average patient died before age 20. The Black Panther Party took up the cause and began testing people at its “survival conferences” across the country.
“I’m sure we tested over four-and-a-half-thousand people for sickle cell anemia last night – and I think that the voter registration is running neck and neck with it,” Black Panther Party Chairman Bobby Seale told news crews at an event in Oakland in 1972.
The movement grew, and Washington listened. “It is a sad and shameful fact that the causes of this disease have been largely neglected throughout our history,” President Richard Nixon told Congress in 1971. “We cannot rewrite this record of neglect, but we can reverse it. To this end, this administration is increasing its budget for research and treatment of sickle cell disease.”
For a while, funding did increase, newborn screening took hold, and by the 1990s, life expectancy had doubled, with patients living into their 40s. But over time, funding waned, clinics closed, and life expectancy started dropping again.
Dr. Vichinsky pushes against that trend for patients like Derek Perkins. The father of four looks healthy and robust, but like most sickle cell patients, he has episodes of extreme pain and has problems with his kidneys, heart, hips, and breathing. Keeping him thriving requires regular checkups and constant monitoring for potential problems.
“The program Dr. Vichinsky is running here, I feel I owe my life to [it],” said Mr. Perkins. “If it wasn’t for him and the things that he did for me, my family wouldn’t have me.”
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation. KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
For more than a year, NeDina Brocks-Capla avoided one room in her large, brightly colored San Francisco house – the bathroom on the second floor.
“It was really hard to bathe in here, and I found myself not wanting to touch the walls,” she explained. The bathroom is where Ms. Brocks-Capla’s son Kareem Jones died in 2013 at age 36, from sickle cell disease.
It’s not just the loss of her son that upsets Ms. Brocks-Capla; she believes that if Mr. Jones had gotten the proper medical care, he might still be alive today.
Sickle cell disease is an inherited disorder that causes some red blood cells to bend into a crescent shape. The misshapen, inflexible cells clog the blood vessels, preventing blood from circulating oxygen properly, which can cause chronic pain, multiorgan failure, and stroke. About 100,000 people in the United States have sickle cell disease, and most of them are African American.
Patients and experts alike say it’s no surprise then that while life expectancy for almost every major malady is improving, patients with sickle cell disease can expect to die younger than they did 20 years ago. In 1994, life expectancy for sickle cell patients was 42 for men and 48 for women. By 2005, life expectancy had dipped to 38 for men and 42 for women.
Sickle cell disease is “a microcosm of how issues of race, ethnicity and identity come into conflict with issues of health care,” said Keith Wailoo, PhD, a professor at Princeton University who writes about the history of the disease.
It is also an example of the broader discrimination experienced by African Americans in the medical system. Nearly a third report that they have experienced discrimination when going to the doctor, according to a poll by NPR, Robert Wood Johnson Foundation, and Harvard T.H. Chan School of Public Health.
“One of the national crises in health care is the care for adult sickle cell,” said leading researcher and physician Elliott Vichinsky, MD, who started the sickle cell center at UCSF Benioff Children’s Hospital Oakland in 1978. “This group of people can live much longer with the management we have, and they’re dying because we don’t have access to care.”
Indeed, with the proper care, Dr. Vichinsky’s center and the handful of other specialty clinics like it across the country have been able to increase life expectancy for sickle cell patients well into their 60s.
Dr. Vichinsky’s patient Derek Perkins, 45, knows he has already beaten the odds. He sits in an exam room decorated with cartoon characters at Children’s Hospital Oakland, but this is the adult sickle cell clinic. He’s been Dr. Vichinsky’s patient since childhood.
“Without the sickle cell clinic here in Oakland, I don’t know what I would do. I don’t know anywhere else I could go,” Mr. Perkins said.
When Mr. Perkins was 27, he once ended up at a different hospital where doctors misdiagnosed his crisis. He went into a coma and was near death before his mother insisted he be transferred.
“Dr. Vichinsky was able to get me here to Children’s Hospital, and he found out what was wrong and within 18 hours – all I needed was an emergency blood transfusion and I was awake,” Mr. Perkins recalled.
Kareem Jones lived just across the bay from Mr. Perkins, but he had a profoundly different experience.
Mr. Jones’ mother, Ms. Brocks-Capla, said her son received excellent medical care as a child, but once he turned 18 and aged out of his pediatric program, it felt like falling off a cliff. Mr. Jones was sent to a clinic at San Francisco General Hospital, but it was open only for a half-day, one day each week. If he was sick any other day, he had two options: leave a voicemail for a clinic nurse or go to the emergency room. “That’s not comprehensive care – that’s not consistent care for a disease of this type,” said Ms. Brocks-Capla.
Ms. Brocks-Capla is a retired supervisor at a worker’s compensation firm. She knew how to navigate the health care system, but she couldn’t get her son the care he needed. Like most sickle cell patients, Mr. Jones had frequent pain crises. Usually he ended up in the emergency department where, Ms. Brocks-Capla said, the doctors didn’t seem to know much about sickle cell disease.
When she tried to explain her son’s pain to the doctors and nurses, she recalled, “they say have a seat. ‘He can’t have a seat! Can’t you see him?’ ”
Studies have found that sickle cell patients have to wait up to 50% longer for help in the emergency department than do other pain patients. The opioid crisis has made things even worse, Dr. Vichinsky added, as patients in terrible pain are likely to be seen as drug seekers with addiction problems rather than patients in need.
Despite his illness, Mr. Jones fought to have a normal life. He lived with his girlfriend, had a daughter, and worked as much as he could between pain crises. He was an avid San Francisco Giants fan.
For years, he took hydroxyurea, but it had side effects, and after a while Mr. Jones had to stop taking it. “And that was it, because you know there isn’t any other medication out there,” said Ms. Brocks-Capla.
Indeed, hydroxyurea, which the Food and Drug Administration first approved in 1967 as a cancer drug, was the only drug on the market to treat sickle cell during Mr. Jones’ lifetime. In July, the FDA approved a second drug, Endari (L-glutamine oral powder), specifically to treat patients with sickle cell disease.
Funding by the federal government and private foundations for the disease pales in comparison to other disorders. Cystic fibrosis offers a good comparison. It is another inherited disorder that requires complex care and most often occurs in Caucasians. Cystic fibrosis gets 7-11 times more funding per patient than does sickle cell disease, according to a 2013 study in the journal Blood. From 2010 to 2013 alone, the FDA approved five new drugs for the treatment of cystic fibrosis.
“There’s no question in my mind that class and color are major factors in impairing their survival. Without question,” Dr. Vichinsky said of sickle cell patients. “The death rate is increasing. The quality of care is going down.”
Without a new medication, Mr. Jones got progressively worse. At 36, his kidneys began to fail, and he had to go on dialysis. He ended up in the hospital, with the worst pain of his life. The doctors stabilized him and gave him pain meds but did not diagnose the underlying cause of the crisis. He was released to his mother’s care, still in incredible pain.
At home, Ms. Brocks-Capla ran him a warm bath to try to soothe his pain and went downstairs to get him a change of clothes. As she came back up the stairs, she heard loud banging against the bathroom walls.
“So I run into the bathroom and he’s having a seizure. And I didn’t know what to do. I was like, ‘Oh come on, come on. Don’t do this. Don’t do this to me.’ ”
She called 911. The paramedics came but couldn’t revive him. “He died here with me,” she said.
It turned out Mr. Jones had a series of small strokes. His organs were in failure, something Ms. Brocks-Capla said the hospital missed. She believes his death could have been prevented with consistent care – the kind he got as a child. Dr. Vichinsky thinks she is probably right.
“I would say 40% or more of the deaths I’ve had recently have been preventable – I mean totally preventable,” he said, but he got to the cases too late. “It makes me so angry. I’ve spent my life trying to help these people, and the harder part is you can change this – this isn’t a knowledge issue. It’s an access issue.”
Dr. Vichinsky’s center and others like it have made major advances in screening patients for the early signs of organ failure and intervening to prevent premature death. Patients at these clinics live 2 decades longer than the average sickle cell patient.
Good care for sickle cell requires time and training for physicians, but it often doesn’t pay well, because many patients are on Medicaid or other government insurance programs. The result is that most adult sickle cell patients still struggle even to access treatments that have been around for decades, Dr. Vichinsky said.
The phenomenon is nothing new — the disease that used to be known as sickle cell anemia has had a long and sordid past. It was first identified in 1910 and helped launch the field of molecular biology. But most of the research was used to study science rather than improving care for sickle cell patients, Dr. Vichinsky said.
In the 1960s and 1970s, sickle cell became a lightning rod for the civil rights movement. At the time, the average patient died before age 20. The Black Panther Party took up the cause and began testing people at its “survival conferences” across the country.
“I’m sure we tested over four-and-a-half-thousand people for sickle cell anemia last night – and I think that the voter registration is running neck and neck with it,” Black Panther Party Chairman Bobby Seale told news crews at an event in Oakland in 1972.
The movement grew, and Washington listened. “It is a sad and shameful fact that the causes of this disease have been largely neglected throughout our history,” President Richard Nixon told Congress in 1971. “We cannot rewrite this record of neglect, but we can reverse it. To this end, this administration is increasing its budget for research and treatment of sickle cell disease.”
For a while, funding did increase, newborn screening took hold, and by the 1990s, life expectancy had doubled, with patients living into their 40s. But over time, funding waned, clinics closed, and life expectancy started dropping again.
Dr. Vichinsky pushes against that trend for patients like Derek Perkins. The father of four looks healthy and robust, but like most sickle cell patients, he has episodes of extreme pain and has problems with his kidneys, heart, hips, and breathing. Keeping him thriving requires regular checkups and constant monitoring for potential problems.
“The program Dr. Vichinsky is running here, I feel I owe my life to [it],” said Mr. Perkins. “If it wasn’t for him and the things that he did for me, my family wouldn’t have me.”
Kaiser Health News is a national health policy news service that is part of the nonpartisan Henry J. Kaiser Family Foundation. KHN’s coverage of children’s health care issues is supported in part by a grant from The Heising-Simons Foundation.
Product can improve joint health in hemophilia A
New research suggests prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc) can improve joint health over time in patients with hemophilia A.
Patients saw continuous improvement in joint health over a nearly 3-year period, regardless of prior treatment regimen, severity of joint damage, or target joints.
“Gradual joint destruction, which is the leading cause of morbidity for people with hemophilia, remains a significant challenge in the treatment of hemophilia A,” said Johannes Oldenburg, MD, of University Clinic Bonn in Germany.
“This is the first study to show that functional joint health can continue to improve using prophylactic treatment with an extended half-life factor therapy, even for those who have severe joint disease at the start of treatment.”
Dr Oldenburg and his colleagues reported these findings in Haemophilia. The research was sponsored by Biogen/Bioverativ and Sobi, the companies marketing rFVIIIFc (or efmoroctocog alfa) as Eloctate or Elocta.
This interim post hoc analysis was an evaluation of joint health in adults and adolescents who received rFVIIIFc prophylaxis in the A-LONG and ASPIRE studies.
In A-LONG, patients age 12 and older who had severe hemophilia A received rFVIIIFc at 25-65 IU/kg every 3 to 5 days (arm 1), at 65 IU/kg weekly (arm 2), or as episodic treatment (arm 3). Patients who completed A-LONG could then enroll in the ASPIRE extension study.
For the current analysis, Dr Oldenburg and his colleagues assessed joint health in ASPIRE enrollees using a modified version of the Hemophilia Joint Health Score (mHJHS). This tool grades joints by specific domains, including swelling, muscle atrophy, alignment, range of motion, joint pain, strength, and global gait.
The researchers examined mHJHS measurements (a decrease in score reflecting improvement) taken at A-LONG baseline, ASPIRE baseline, and annually thereafter for roughly 2.8 years of treatment.
There were 47 patients who had mHJHS data at both study baselines, ASPIRE year 1, and ASPIRE year 2.
These patients had a mean improvement in joint health score of -4.1 at ASPIRE year 2, compared with A-LONG baseline (P=0.001).
The mean improvement was -2.4 (P=0.09) for patients who received pre-study prophylaxis and -7.2 (P=0.003) for those who received pre-study episodic treatment.
The mean improvement was -5.6 (P=0.005) in patients with target joints and -8.8 (P=0.02) in those with severe joint destruction.
The mHJHS components with the greatest improvement at ASPIRE year 2 were swelling (-1.4, P=0.008), range of motion (-1.1, P=0.03), and strength (-0.8, P=0.04). ![]()
New research suggests prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc) can improve joint health over time in patients with hemophilia A.
Patients saw continuous improvement in joint health over a nearly 3-year period, regardless of prior treatment regimen, severity of joint damage, or target joints.
“Gradual joint destruction, which is the leading cause of morbidity for people with hemophilia, remains a significant challenge in the treatment of hemophilia A,” said Johannes Oldenburg, MD, of University Clinic Bonn in Germany.
“This is the first study to show that functional joint health can continue to improve using prophylactic treatment with an extended half-life factor therapy, even for those who have severe joint disease at the start of treatment.”
Dr Oldenburg and his colleagues reported these findings in Haemophilia. The research was sponsored by Biogen/Bioverativ and Sobi, the companies marketing rFVIIIFc (or efmoroctocog alfa) as Eloctate or Elocta.
This interim post hoc analysis was an evaluation of joint health in adults and adolescents who received rFVIIIFc prophylaxis in the A-LONG and ASPIRE studies.
In A-LONG, patients age 12 and older who had severe hemophilia A received rFVIIIFc at 25-65 IU/kg every 3 to 5 days (arm 1), at 65 IU/kg weekly (arm 2), or as episodic treatment (arm 3). Patients who completed A-LONG could then enroll in the ASPIRE extension study.
For the current analysis, Dr Oldenburg and his colleagues assessed joint health in ASPIRE enrollees using a modified version of the Hemophilia Joint Health Score (mHJHS). This tool grades joints by specific domains, including swelling, muscle atrophy, alignment, range of motion, joint pain, strength, and global gait.
The researchers examined mHJHS measurements (a decrease in score reflecting improvement) taken at A-LONG baseline, ASPIRE baseline, and annually thereafter for roughly 2.8 years of treatment.
There were 47 patients who had mHJHS data at both study baselines, ASPIRE year 1, and ASPIRE year 2.
These patients had a mean improvement in joint health score of -4.1 at ASPIRE year 2, compared with A-LONG baseline (P=0.001).
The mean improvement was -2.4 (P=0.09) for patients who received pre-study prophylaxis and -7.2 (P=0.003) for those who received pre-study episodic treatment.
The mean improvement was -5.6 (P=0.005) in patients with target joints and -8.8 (P=0.02) in those with severe joint destruction.
The mHJHS components with the greatest improvement at ASPIRE year 2 were swelling (-1.4, P=0.008), range of motion (-1.1, P=0.03), and strength (-0.8, P=0.04). ![]()
New research suggests prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc) can improve joint health over time in patients with hemophilia A.
Patients saw continuous improvement in joint health over a nearly 3-year period, regardless of prior treatment regimen, severity of joint damage, or target joints.
“Gradual joint destruction, which is the leading cause of morbidity for people with hemophilia, remains a significant challenge in the treatment of hemophilia A,” said Johannes Oldenburg, MD, of University Clinic Bonn in Germany.
“This is the first study to show that functional joint health can continue to improve using prophylactic treatment with an extended half-life factor therapy, even for those who have severe joint disease at the start of treatment.”
Dr Oldenburg and his colleagues reported these findings in Haemophilia. The research was sponsored by Biogen/Bioverativ and Sobi, the companies marketing rFVIIIFc (or efmoroctocog alfa) as Eloctate or Elocta.
This interim post hoc analysis was an evaluation of joint health in adults and adolescents who received rFVIIIFc prophylaxis in the A-LONG and ASPIRE studies.
In A-LONG, patients age 12 and older who had severe hemophilia A received rFVIIIFc at 25-65 IU/kg every 3 to 5 days (arm 1), at 65 IU/kg weekly (arm 2), or as episodic treatment (arm 3). Patients who completed A-LONG could then enroll in the ASPIRE extension study.
For the current analysis, Dr Oldenburg and his colleagues assessed joint health in ASPIRE enrollees using a modified version of the Hemophilia Joint Health Score (mHJHS). This tool grades joints by specific domains, including swelling, muscle atrophy, alignment, range of motion, joint pain, strength, and global gait.
The researchers examined mHJHS measurements (a decrease in score reflecting improvement) taken at A-LONG baseline, ASPIRE baseline, and annually thereafter for roughly 2.8 years of treatment.
There were 47 patients who had mHJHS data at both study baselines, ASPIRE year 1, and ASPIRE year 2.
These patients had a mean improvement in joint health score of -4.1 at ASPIRE year 2, compared with A-LONG baseline (P=0.001).
The mean improvement was -2.4 (P=0.09) for patients who received pre-study prophylaxis and -7.2 (P=0.003) for those who received pre-study episodic treatment.
The mean improvement was -5.6 (P=0.005) in patients with target joints and -8.8 (P=0.02) in those with severe joint destruction.
The mHJHS components with the greatest improvement at ASPIRE year 2 were swelling (-1.4, P=0.008), range of motion (-1.1, P=0.03), and strength (-0.8, P=0.04). ![]()