User login
Starting all CML patients on imatinib could cut costs, team says

Photo courtesy of the CDC
New research suggests that starting all patients with chronic myeloid leukemia (CML) on the generic form of Gleevec, imatinib, could result in substantial savings for patients and insurance companies in the US.
Researchers found that starting treatment with generic imatinib and then switching patients to the second-generation tyrosine kinase inhibitors (TKIs) dasatinib (Sprycel) and nilotinib (Tasigna) if necessary would be more cost-effective than the current standard of care, which allows physicians to start patients on any of the aforementioned TKIs.
In fact, the data suggest that starting all CML patients on generic imatinib could reduce the cost of treatment per patient over 5 years by nearly $90,000.
“If we start all patients on the generic form of Gleevec, and it works, then they are on a generic for the rest of their lives,” said study author William V. Padula, PhD, of Johns Hopkins University in Baltimore, Maryland.
“This amounts to a huge cost savings for them and their insurers.”
Dr Padula and his colleagues described these potential savings in the Journal of the National Cancer Institute.
The researchers compared the cost-effectiveness of the different TKIs by analyzing Truven Health Analytics MarketScan data from newly diagnosed CML patients between January 1, 2011, and December 31, 2012.
The team constructed Markov models to compare the 5-year cost-effectiveness of imatinib-first vs physician’s choice. The main outcome of the models was cost per quality-adjusted life-year (QALY).
The researchers interpreted outcomes based on a willingness-to-pay threshold of $100,000/QALY. They found that both imatinib-first and physician’s choice met that threshold.
However, imatinib-first cost less over 5 years and conferred only slightly fewer QALYs than physician’s choice—$277,401 and 3.87 QALYs for imatinib-first and $365,744 and 3.97 QALYs for physician’s choice.
So that’s a 0.10 decrement in QALYs and a savings of $88,343 over 5 years with imatinib-first. The imatinib-first incremental cost-effectiveness ratio was about $883,730/QALY.
“There is minimal risk to starting all patients on imatinib first,” Dr Padula said. “If the patient can’t tolerate the medication or it seems to be ineffective in that patient, then we can switch the patient to a more expensive drug.”
“Insurance companies have the ability to dictate which drugs physicians prescribe first, and they regularly do. Doing so here would mean very little risk to health and a lot of cost savings.” ![]()
Photo courtesy of the CDC
New research suggests that starting all patients with chronic myeloid leukemia (CML) on the generic form of Gleevec, imatinib, could result in substantial savings for patients and insurance companies in the US.
Researchers found that starting treatment with generic imatinib and then switching patients to the second-generation tyrosine kinase inhibitors (TKIs) dasatinib (Sprycel) and nilotinib (Tasigna) if necessary would be more cost-effective than the current standard of care, which allows physicians to start patients on any of the aforementioned TKIs.
In fact, the data suggest that starting all CML patients on generic imatinib could reduce the cost of treatment per patient over 5 years by nearly $90,000.
“If we start all patients on the generic form of Gleevec, and it works, then they are on a generic for the rest of their lives,” said study author William V. Padula, PhD, of Johns Hopkins University in Baltimore, Maryland.
“This amounts to a huge cost savings for them and their insurers.”
Dr Padula and his colleagues described these potential savings in the Journal of the National Cancer Institute.
The researchers compared the cost-effectiveness of the different TKIs by analyzing Truven Health Analytics MarketScan data from newly diagnosed CML patients between January 1, 2011, and December 31, 2012.
The team constructed Markov models to compare the 5-year cost-effectiveness of imatinib-first vs physician’s choice. The main outcome of the models was cost per quality-adjusted life-year (QALY).
The researchers interpreted outcomes based on a willingness-to-pay threshold of $100,000/QALY. They found that both imatinib-first and physician’s choice met that threshold.
However, imatinib-first cost less over 5 years and conferred only slightly fewer QALYs than physician’s choice—$277,401 and 3.87 QALYs for imatinib-first and $365,744 and 3.97 QALYs for physician’s choice.
So that’s a 0.10 decrement in QALYs and a savings of $88,343 over 5 years with imatinib-first. The imatinib-first incremental cost-effectiveness ratio was about $883,730/QALY.
“There is minimal risk to starting all patients on imatinib first,” Dr Padula said. “If the patient can’t tolerate the medication or it seems to be ineffective in that patient, then we can switch the patient to a more expensive drug.”
“Insurance companies have the ability to dictate which drugs physicians prescribe first, and they regularly do. Doing so here would mean very little risk to health and a lot of cost savings.” ![]()
Photo courtesy of the CDC
New research suggests that starting all patients with chronic myeloid leukemia (CML) on the generic form of Gleevec, imatinib, could result in substantial savings for patients and insurance companies in the US.
Researchers found that starting treatment with generic imatinib and then switching patients to the second-generation tyrosine kinase inhibitors (TKIs) dasatinib (Sprycel) and nilotinib (Tasigna) if necessary would be more cost-effective than the current standard of care, which allows physicians to start patients on any of the aforementioned TKIs.
In fact, the data suggest that starting all CML patients on generic imatinib could reduce the cost of treatment per patient over 5 years by nearly $90,000.
“If we start all patients on the generic form of Gleevec, and it works, then they are on a generic for the rest of their lives,” said study author William V. Padula, PhD, of Johns Hopkins University in Baltimore, Maryland.
“This amounts to a huge cost savings for them and their insurers.”
Dr Padula and his colleagues described these potential savings in the Journal of the National Cancer Institute.
The researchers compared the cost-effectiveness of the different TKIs by analyzing Truven Health Analytics MarketScan data from newly diagnosed CML patients between January 1, 2011, and December 31, 2012.
The team constructed Markov models to compare the 5-year cost-effectiveness of imatinib-first vs physician’s choice. The main outcome of the models was cost per quality-adjusted life-year (QALY).
The researchers interpreted outcomes based on a willingness-to-pay threshold of $100,000/QALY. They found that both imatinib-first and physician’s choice met that threshold.
However, imatinib-first cost less over 5 years and conferred only slightly fewer QALYs than physician’s choice—$277,401 and 3.87 QALYs for imatinib-first and $365,744 and 3.97 QALYs for physician’s choice.
So that’s a 0.10 decrement in QALYs and a savings of $88,343 over 5 years with imatinib-first. The imatinib-first incremental cost-effectiveness ratio was about $883,730/QALY.
“There is minimal risk to starting all patients on imatinib first,” Dr Padula said. “If the patient can’t tolerate the medication or it seems to be ineffective in that patient, then we can switch the patient to a more expensive drug.”
“Insurance companies have the ability to dictate which drugs physicians prescribe first, and they regularly do. Doing so here would mean very little risk to health and a lot of cost savings.” ![]()
Germline mutations linked to hematologic malignancies

A new study suggests mutations in the gene DDX41 occur in families where hematologic malignancies are common.
Previous research showed that both germline and acquired DDX41 mutations occur in families with multiple cases of late-onset myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).
The new study, published in Blood, has linked germline mutations in DDX41 to chronic myeloid leukemia and lymphomas as well.
“This is the first gene identified in families with lymphoma and represents a major breakthrough for the field,” said study author Hamish Scott, PhD, of the University of Adelaide in South Australia.
“Researchers are recognizing now that genetic predisposition to blood cancer is more common than previously thought, and our study shows the importance of taking a thorough family history at diagnosis.”
To conduct this study, Dr Scott and his colleagues screened 2 cohorts of families with a range of hematologic disorders (malignant and non-malignant). One cohort included 240 individuals from 93 families in Australia. The other included 246 individuals from 198 families in the US.
In all, 9 of the families (3%) had germline DDX41 mutations.
Three families carried the recurrent p.D140Gfs*2 mutation, which was linked to AML.
One family carried a germline mutation—p.R525H, c.1574G.A—that was previously described only as a somatic mutation at the time of progression to MDS or AML. In the current study, the mutation was again linked to MDS and AML.
Five families carried novel DDX41 mutations.
One of these mutations was a germline substitution—c.435-2_435-1delAGinsCA—that was linked to MDS in 1 family.
Two families had a missense start-loss substitution—c.3G.A, p.M1I—that was linked to MDS, AML, chronic myeloid leukemia, and non-Hodgkin lymphoma.
One family had a DDX41 missense variant—c.490C.T, p.R164W. This was linked to Hodgkin and non-Hodgkin lymphoma (including 3 cases of follicular lymphoma). There was a possible link to multiple myeloma as well, but the diagnosis could not be confirmed.
And 1 family had a missense mutation in the helicase domain—p.G530D—that was linked to AML.
“DDX41 is a new type of cancer predisposition gene, and we are still investigating its function,” Dr Scott noted.
“But it appears to have dual roles in regulating the correct expression of genes in the cell and also enabling the immune system to respond to threats such as bacteria and viruses, as well as the development of cancer cells. Immunotherapy is a promising approach for cancer treatment, and our research to understand the function of DDX41 will help design better therapies.” ![]()
A new study suggests mutations in the gene DDX41 occur in families where hematologic malignancies are common.
Previous research showed that both germline and acquired DDX41 mutations occur in families with multiple cases of late-onset myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).
The new study, published in Blood, has linked germline mutations in DDX41 to chronic myeloid leukemia and lymphomas as well.
“This is the first gene identified in families with lymphoma and represents a major breakthrough for the field,” said study author Hamish Scott, PhD, of the University of Adelaide in South Australia.
“Researchers are recognizing now that genetic predisposition to blood cancer is more common than previously thought, and our study shows the importance of taking a thorough family history at diagnosis.”
To conduct this study, Dr Scott and his colleagues screened 2 cohorts of families with a range of hematologic disorders (malignant and non-malignant). One cohort included 240 individuals from 93 families in Australia. The other included 246 individuals from 198 families in the US.
In all, 9 of the families (3%) had germline DDX41 mutations.
Three families carried the recurrent p.D140Gfs*2 mutation, which was linked to AML.
One family carried a germline mutation—p.R525H, c.1574G.A—that was previously described only as a somatic mutation at the time of progression to MDS or AML. In the current study, the mutation was again linked to MDS and AML.
Five families carried novel DDX41 mutations.
One of these mutations was a germline substitution—c.435-2_435-1delAGinsCA—that was linked to MDS in 1 family.
Two families had a missense start-loss substitution—c.3G.A, p.M1I—that was linked to MDS, AML, chronic myeloid leukemia, and non-Hodgkin lymphoma.
One family had a DDX41 missense variant—c.490C.T, p.R164W. This was linked to Hodgkin and non-Hodgkin lymphoma (including 3 cases of follicular lymphoma). There was a possible link to multiple myeloma as well, but the diagnosis could not be confirmed.
And 1 family had a missense mutation in the helicase domain—p.G530D—that was linked to AML.
“DDX41 is a new type of cancer predisposition gene, and we are still investigating its function,” Dr Scott noted.
“But it appears to have dual roles in regulating the correct expression of genes in the cell and also enabling the immune system to respond to threats such as bacteria and viruses, as well as the development of cancer cells. Immunotherapy is a promising approach for cancer treatment, and our research to understand the function of DDX41 will help design better therapies.” ![]()
A new study suggests mutations in the gene DDX41 occur in families where hematologic malignancies are common.
Previous research showed that both germline and acquired DDX41 mutations occur in families with multiple cases of late-onset myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).
The new study, published in Blood, has linked germline mutations in DDX41 to chronic myeloid leukemia and lymphomas as well.
“This is the first gene identified in families with lymphoma and represents a major breakthrough for the field,” said study author Hamish Scott, PhD, of the University of Adelaide in South Australia.
“Researchers are recognizing now that genetic predisposition to blood cancer is more common than previously thought, and our study shows the importance of taking a thorough family history at diagnosis.”
To conduct this study, Dr Scott and his colleagues screened 2 cohorts of families with a range of hematologic disorders (malignant and non-malignant). One cohort included 240 individuals from 93 families in Australia. The other included 246 individuals from 198 families in the US.
In all, 9 of the families (3%) had germline DDX41 mutations.
Three families carried the recurrent p.D140Gfs*2 mutation, which was linked to AML.
One family carried a germline mutation—p.R525H, c.1574G.A—that was previously described only as a somatic mutation at the time of progression to MDS or AML. In the current study, the mutation was again linked to MDS and AML.
Five families carried novel DDX41 mutations.
One of these mutations was a germline substitution—c.435-2_435-1delAGinsCA—that was linked to MDS in 1 family.
Two families had a missense start-loss substitution—c.3G.A, p.M1I—that was linked to MDS, AML, chronic myeloid leukemia, and non-Hodgkin lymphoma.
One family had a DDX41 missense variant—c.490C.T, p.R164W. This was linked to Hodgkin and non-Hodgkin lymphoma (including 3 cases of follicular lymphoma). There was a possible link to multiple myeloma as well, but the diagnosis could not be confirmed.
And 1 family had a missense mutation in the helicase domain—p.G530D—that was linked to AML.
“DDX41 is a new type of cancer predisposition gene, and we are still investigating its function,” Dr Scott noted.
“But it appears to have dual roles in regulating the correct expression of genes in the cell and also enabling the immune system to respond to threats such as bacteria and viruses, as well as the development of cancer cells. Immunotherapy is a promising approach for cancer treatment, and our research to understand the function of DDX41 will help design better therapies.” ![]()
Ponatinib effective in chronic phase CML regardless of baseline mutation status
In heavily pretreated patients with chronic phase chronic myeloid leukemia (CP-CML), response to the tyrosine kinase inhibitor ponatinib did not depend on baseline mutation status, and no single or compound mutation was a major driver of primary or secondary resistance to ponatinib, according to researchers.
Irrespective of baseline mutation status, responses to ponatinib were durable. As determined by next-generation sequencing (NGS), patients with zero, one, or two or more BCR-ABL1 mutations had rates of 50%-61% for major cytogenetic response (MCyR) by 1 year and 29%-45% for major molecular response (MMR) at any time. The rates were similar to those observed with mutation status determined by Sanger sequencing. Rates of sustained response at 2 years for MCyR and MMR were 87% and 65%, respectively (Blood. 2016 Feb 11. doi: 10.1182/blood-2015-08-660977).
Sanger sequencing typically is used to identify BCR-ABL1 mutations associated with tyrosine kinase inhibitor (TKI) resistance, but the method fails to detect low-level mutations that occur in less than 10%-20% of cells. Researchers used NGS to determine the impact of low-level mutations, as well as compound mutations, on the efficacy of the third generation TKI ponatinib.
Ponatinib is the most potent BCR-ABL1 TKI but is associated with considerable cardiovascular toxicity.
“The role of NGS in this setting may be to identify patients with (low level) T315I who are unlikely to derive lasting benefit from second-generation TKIs, but have a high likelihood of achieving durable cytogenetic and molecular responses to ponatinib, an important factor for balancing risks and benefits of salvage therapy selection,” wrote Dr. Michael W. Deininger, Chief of Hematology at the Huntsman Cancer Institute at the University of Utah, Salt Lake City, and his colleagues.
Patients with low-level mutations had similar response rates to those with no mutations: MCyR by one year and MMR at any time were 43% and 31%, respectively, compared with 50% and 29%. Response rates were higher in patients with compound mutations (64% and 52%) or one or more mutation (57% and 64%). The researchers speculated that the lower response rates in patients with low level or no mutations may reflect resistance mechanisms independent of BCR-ABL1.
Analysis of postbaseline samples from 127 patients (24 of whom had discontinued ponatinib for at least 1 month) determined the impact of acquired resistance. At a median follow-up of 30.1 months, emergence of previously undetected single and compound mutants during ponatinib therapy was observed in 8 patients.
The study analyzed patients from the PACE trial who had CP-CML with resistance or intolerance to dasatinib or nilotinib, or with a T315I mutation, and who were treated with ponatinib. All 267 patients had baseline mutation status determined by Sanger sequencing, with 161 mutations detected in 131 patients. NGS identified these and 105 additional mutations. Consistent with greater sensitivity of NGS, the proportion of patients with no baseline mutations by 39% by NGS vs. 51% by Sanger sequencing, and the proportion with multiple mutations was 23% vs. 10%.
The study was funded by ARIAD Pharmaceuticals. Dr. Deininger reported financial ties to ARIAD, Bristol Myers-Squibb, Novartis, Celgene, Genzyme, Gilead, Incyte, and Pfizer. Several of his coauthors reported ties to industry.
In heavily pretreated patients with chronic phase chronic myeloid leukemia (CP-CML), response to the tyrosine kinase inhibitor ponatinib did not depend on baseline mutation status, and no single or compound mutation was a major driver of primary or secondary resistance to ponatinib, according to researchers.
Irrespective of baseline mutation status, responses to ponatinib were durable. As determined by next-generation sequencing (NGS), patients with zero, one, or two or more BCR-ABL1 mutations had rates of 50%-61% for major cytogenetic response (MCyR) by 1 year and 29%-45% for major molecular response (MMR) at any time. The rates were similar to those observed with mutation status determined by Sanger sequencing. Rates of sustained response at 2 years for MCyR and MMR were 87% and 65%, respectively (Blood. 2016 Feb 11. doi: 10.1182/blood-2015-08-660977).
Sanger sequencing typically is used to identify BCR-ABL1 mutations associated with tyrosine kinase inhibitor (TKI) resistance, but the method fails to detect low-level mutations that occur in less than 10%-20% of cells. Researchers used NGS to determine the impact of low-level mutations, as well as compound mutations, on the efficacy of the third generation TKI ponatinib.
Ponatinib is the most potent BCR-ABL1 TKI but is associated with considerable cardiovascular toxicity.
“The role of NGS in this setting may be to identify patients with (low level) T315I who are unlikely to derive lasting benefit from second-generation TKIs, but have a high likelihood of achieving durable cytogenetic and molecular responses to ponatinib, an important factor for balancing risks and benefits of salvage therapy selection,” wrote Dr. Michael W. Deininger, Chief of Hematology at the Huntsman Cancer Institute at the University of Utah, Salt Lake City, and his colleagues.
Patients with low-level mutations had similar response rates to those with no mutations: MCyR by one year and MMR at any time were 43% and 31%, respectively, compared with 50% and 29%. Response rates were higher in patients with compound mutations (64% and 52%) or one or more mutation (57% and 64%). The researchers speculated that the lower response rates in patients with low level or no mutations may reflect resistance mechanisms independent of BCR-ABL1.
Analysis of postbaseline samples from 127 patients (24 of whom had discontinued ponatinib for at least 1 month) determined the impact of acquired resistance. At a median follow-up of 30.1 months, emergence of previously undetected single and compound mutants during ponatinib therapy was observed in 8 patients.
The study analyzed patients from the PACE trial who had CP-CML with resistance or intolerance to dasatinib or nilotinib, or with a T315I mutation, and who were treated with ponatinib. All 267 patients had baseline mutation status determined by Sanger sequencing, with 161 mutations detected in 131 patients. NGS identified these and 105 additional mutations. Consistent with greater sensitivity of NGS, the proportion of patients with no baseline mutations by 39% by NGS vs. 51% by Sanger sequencing, and the proportion with multiple mutations was 23% vs. 10%.
The study was funded by ARIAD Pharmaceuticals. Dr. Deininger reported financial ties to ARIAD, Bristol Myers-Squibb, Novartis, Celgene, Genzyme, Gilead, Incyte, and Pfizer. Several of his coauthors reported ties to industry.
In heavily pretreated patients with chronic phase chronic myeloid leukemia (CP-CML), response to the tyrosine kinase inhibitor ponatinib did not depend on baseline mutation status, and no single or compound mutation was a major driver of primary or secondary resistance to ponatinib, according to researchers.
Irrespective of baseline mutation status, responses to ponatinib were durable. As determined by next-generation sequencing (NGS), patients with zero, one, or two or more BCR-ABL1 mutations had rates of 50%-61% for major cytogenetic response (MCyR) by 1 year and 29%-45% for major molecular response (MMR) at any time. The rates were similar to those observed with mutation status determined by Sanger sequencing. Rates of sustained response at 2 years for MCyR and MMR were 87% and 65%, respectively (Blood. 2016 Feb 11. doi: 10.1182/blood-2015-08-660977).
Sanger sequencing typically is used to identify BCR-ABL1 mutations associated with tyrosine kinase inhibitor (TKI) resistance, but the method fails to detect low-level mutations that occur in less than 10%-20% of cells. Researchers used NGS to determine the impact of low-level mutations, as well as compound mutations, on the efficacy of the third generation TKI ponatinib.
Ponatinib is the most potent BCR-ABL1 TKI but is associated with considerable cardiovascular toxicity.
“The role of NGS in this setting may be to identify patients with (low level) T315I who are unlikely to derive lasting benefit from second-generation TKIs, but have a high likelihood of achieving durable cytogenetic and molecular responses to ponatinib, an important factor for balancing risks and benefits of salvage therapy selection,” wrote Dr. Michael W. Deininger, Chief of Hematology at the Huntsman Cancer Institute at the University of Utah, Salt Lake City, and his colleagues.
Patients with low-level mutations had similar response rates to those with no mutations: MCyR by one year and MMR at any time were 43% and 31%, respectively, compared with 50% and 29%. Response rates were higher in patients with compound mutations (64% and 52%) or one or more mutation (57% and 64%). The researchers speculated that the lower response rates in patients with low level or no mutations may reflect resistance mechanisms independent of BCR-ABL1.
Analysis of postbaseline samples from 127 patients (24 of whom had discontinued ponatinib for at least 1 month) determined the impact of acquired resistance. At a median follow-up of 30.1 months, emergence of previously undetected single and compound mutants during ponatinib therapy was observed in 8 patients.
The study analyzed patients from the PACE trial who had CP-CML with resistance or intolerance to dasatinib or nilotinib, or with a T315I mutation, and who were treated with ponatinib. All 267 patients had baseline mutation status determined by Sanger sequencing, with 161 mutations detected in 131 patients. NGS identified these and 105 additional mutations. Consistent with greater sensitivity of NGS, the proportion of patients with no baseline mutations by 39% by NGS vs. 51% by Sanger sequencing, and the proportion with multiple mutations was 23% vs. 10%.
The study was funded by ARIAD Pharmaceuticals. Dr. Deininger reported financial ties to ARIAD, Bristol Myers-Squibb, Novartis, Celgene, Genzyme, Gilead, Incyte, and Pfizer. Several of his coauthors reported ties to industry.
FROM BLOOD
Key clinical point: Baseline mutation status had little impact on ponatinib response, and no single or compound mutation was a major driver of primary or secondary resistance to ponatinib in patients with chronic phase chronic myeloid leukemia (CP-CML).
Major finding: In patients with zero, one, or two or more BCR-ABL1 mutations at baseline by next-generation sequencing were 50%-61% for major cytogenetic response (MCyR) by 1 year and 29%-45% for major molecular response (MMR) at any time; rates of sustained response at 2 years for MCyR and MMR were 87% and 65%, respectively.
Data source: From the PACE trial, 267 patients with CP-CML with resistance or intolerance to dasatinib or nilotinib, or with a T315I mutation, were treated with ponatinib.
Disclosures: The study was funded by ARIAD Pharmaceuticals. Dr. Deininger reported financial ties to ARIAD, Bristol Myers-Squibb, Novartis, Celgene, Genzyme, Gilead, Incyte, and Pfizer. Several of his coauthors reported ties to industry.
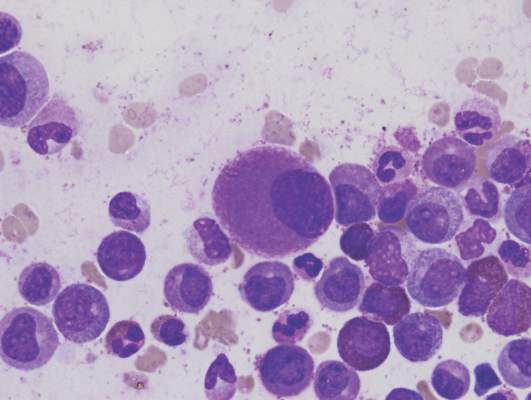
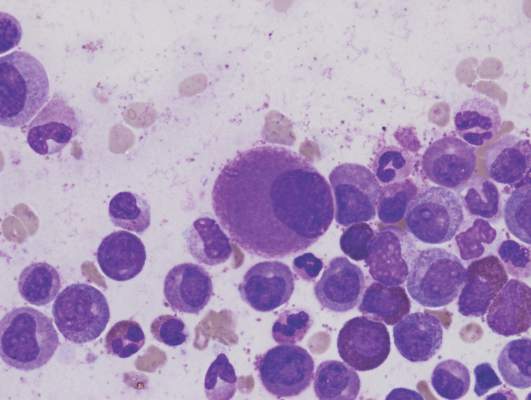
Pinpointing the cells that cause CML relapse

Image by Difu Wu
Preclinical research suggests chronic myeloid leukemia (CML) patients have a heterogeneous population of long-term hematopoietic stem cells (LTHSCs)—some that can initiate leukemia and some that cannot.
Researchers found they could identify the leukemia-initiating cells by measuring expression of the thrombopoietin receptor MPL. Cells with high MPL expression could initiate CML in mice.
The team said these results suggest the leukemic LTHSCs are the cells responsible for relapse in CML.
“This shows that not all leukemia stem cells are equal,” said study author Ravi Bhatia, MD, of the University of Alabama Birmingham.
“Some are more prone to causing leukemia and relapses, while some others may just hang around without potential for contributing to relapse.”
Dr Bhatia and his colleagues reported these findings in The Journal of Clinical Investigation.
In their experiments, the team used an inducible transgenic mouse model of CML, where the BCR-ABL gene fusion is under the control of a tetracycline-regulated enhancer. This model creates a chronic myeloproliferative disorder that resembles chronic phase CML.
Previous work had shown that only cells with an LTHSC phenotype were capable of long-term repopulation and leukemia-initiating capacity after transplantation to another mouse.
When the researchers transplanted LTHSCs from CML-model mice to other mice, 11 of 20 recipients developed CML, and 9 of 20 showed engraftment with CML cells but did not develop the leukocytosis characteristic of leukemia.
When the LTHSCs from the primary-recipient mice were transferred to secondary recipients, 7 of 17 mice receiving cells from leukemic mice developed CML, and none of the secondary-recipient mice receiving cells from the non-leukemic mice developed CML.
The researchers tested these 2 groups of LTHSCs for differences in gene expression. They found significant differences between the leukemic and non-leukemic LTHSCs for the genes Mpl, c-Myc, CD47, Pten, Sirt1, Ptch1, and Tie2.
The team then decided to focus on Mpl. They used flow cytometry to select LTHSCs with either high or low Mpl expression from CML-model BCR-ABL mice.
Seven of 16 mice receiving Mpl-Hi LTHSCs developed leukemia after transplantation, compared with 1 of 17 receiving Mpl-Lo LTHSCs. This suggested an increased leukemogenic capacity for the Mpl-Hi LTHSCs.
The researchers also investigated the impact of cell-cycle status. They found that CML Mpl-Hi LTHSCs that were in a resting stage of the cell cycle had enhanced long-term engraftment and leukemogenic capacity compared with cycling Mpl-Hi LTHSCs.
The team used virus vectors and shRNA to create Mpl knockdown BCR-ABL LTHSCs and showed that the knockdown cells had a greatly reduced ability to produce leukemia in recipient mice.
The Mpl knockdown cells, after stimulation by the Mpl ligand thrombopoietin, also had reduced expression of the activated transcription factors p-STAT3 and p-STAT5, compared with controls.
Finally, the researchers examined human CML cells for differences between MPL-Hi LTHSCs and MPL-Lo LTHSCs. The results were similar to those observed in mice.
The human MPL-Hi LTHSCs had a higher rate of engraftment than the human MPL-Lo LTHSCs, as tested in a xenograft model using immunodeficient mice.
Additionally, the human MPL-Hi LTHSCs had reduced sensitivity to nilotinib compared with MPL-Lo LTHSCs. However, a Jak/STAT inhibitor significantly reduced cell growth and increased apoptosis in human MPL-Hi LTHSCs.
The researchers concluded that MPL expression is a marker and key regulator of leukemogenic potential and drug sensitivity of CML LTHSCs. They said their findings support further investigation of approaches to antagonize MPL signaling as a potential therapeutic strategy to eliminate leukemia-initiating LTHSCs. ![]()
Image by Difu Wu
Preclinical research suggests chronic myeloid leukemia (CML) patients have a heterogeneous population of long-term hematopoietic stem cells (LTHSCs)—some that can initiate leukemia and some that cannot.
Researchers found they could identify the leukemia-initiating cells by measuring expression of the thrombopoietin receptor MPL. Cells with high MPL expression could initiate CML in mice.
The team said these results suggest the leukemic LTHSCs are the cells responsible for relapse in CML.
“This shows that not all leukemia stem cells are equal,” said study author Ravi Bhatia, MD, of the University of Alabama Birmingham.
“Some are more prone to causing leukemia and relapses, while some others may just hang around without potential for contributing to relapse.”
Dr Bhatia and his colleagues reported these findings in The Journal of Clinical Investigation.
In their experiments, the team used an inducible transgenic mouse model of CML, where the BCR-ABL gene fusion is under the control of a tetracycline-regulated enhancer. This model creates a chronic myeloproliferative disorder that resembles chronic phase CML.
Previous work had shown that only cells with an LTHSC phenotype were capable of long-term repopulation and leukemia-initiating capacity after transplantation to another mouse.
When the researchers transplanted LTHSCs from CML-model mice to other mice, 11 of 20 recipients developed CML, and 9 of 20 showed engraftment with CML cells but did not develop the leukocytosis characteristic of leukemia.
When the LTHSCs from the primary-recipient mice were transferred to secondary recipients, 7 of 17 mice receiving cells from leukemic mice developed CML, and none of the secondary-recipient mice receiving cells from the non-leukemic mice developed CML.
The researchers tested these 2 groups of LTHSCs for differences in gene expression. They found significant differences between the leukemic and non-leukemic LTHSCs for the genes Mpl, c-Myc, CD47, Pten, Sirt1, Ptch1, and Tie2.
The team then decided to focus on Mpl. They used flow cytometry to select LTHSCs with either high or low Mpl expression from CML-model BCR-ABL mice.
Seven of 16 mice receiving Mpl-Hi LTHSCs developed leukemia after transplantation, compared with 1 of 17 receiving Mpl-Lo LTHSCs. This suggested an increased leukemogenic capacity for the Mpl-Hi LTHSCs.
The researchers also investigated the impact of cell-cycle status. They found that CML Mpl-Hi LTHSCs that were in a resting stage of the cell cycle had enhanced long-term engraftment and leukemogenic capacity compared with cycling Mpl-Hi LTHSCs.
The team used virus vectors and shRNA to create Mpl knockdown BCR-ABL LTHSCs and showed that the knockdown cells had a greatly reduced ability to produce leukemia in recipient mice.
The Mpl knockdown cells, after stimulation by the Mpl ligand thrombopoietin, also had reduced expression of the activated transcription factors p-STAT3 and p-STAT5, compared with controls.
Finally, the researchers examined human CML cells for differences between MPL-Hi LTHSCs and MPL-Lo LTHSCs. The results were similar to those observed in mice.
The human MPL-Hi LTHSCs had a higher rate of engraftment than the human MPL-Lo LTHSCs, as tested in a xenograft model using immunodeficient mice.
Additionally, the human MPL-Hi LTHSCs had reduced sensitivity to nilotinib compared with MPL-Lo LTHSCs. However, a Jak/STAT inhibitor significantly reduced cell growth and increased apoptosis in human MPL-Hi LTHSCs.
The researchers concluded that MPL expression is a marker and key regulator of leukemogenic potential and drug sensitivity of CML LTHSCs. They said their findings support further investigation of approaches to antagonize MPL signaling as a potential therapeutic strategy to eliminate leukemia-initiating LTHSCs. ![]()
Image by Difu Wu
Preclinical research suggests chronic myeloid leukemia (CML) patients have a heterogeneous population of long-term hematopoietic stem cells (LTHSCs)—some that can initiate leukemia and some that cannot.
Researchers found they could identify the leukemia-initiating cells by measuring expression of the thrombopoietin receptor MPL. Cells with high MPL expression could initiate CML in mice.
The team said these results suggest the leukemic LTHSCs are the cells responsible for relapse in CML.
“This shows that not all leukemia stem cells are equal,” said study author Ravi Bhatia, MD, of the University of Alabama Birmingham.
“Some are more prone to causing leukemia and relapses, while some others may just hang around without potential for contributing to relapse.”
Dr Bhatia and his colleagues reported these findings in The Journal of Clinical Investigation.
In their experiments, the team used an inducible transgenic mouse model of CML, where the BCR-ABL gene fusion is under the control of a tetracycline-regulated enhancer. This model creates a chronic myeloproliferative disorder that resembles chronic phase CML.
Previous work had shown that only cells with an LTHSC phenotype were capable of long-term repopulation and leukemia-initiating capacity after transplantation to another mouse.
When the researchers transplanted LTHSCs from CML-model mice to other mice, 11 of 20 recipients developed CML, and 9 of 20 showed engraftment with CML cells but did not develop the leukocytosis characteristic of leukemia.
When the LTHSCs from the primary-recipient mice were transferred to secondary recipients, 7 of 17 mice receiving cells from leukemic mice developed CML, and none of the secondary-recipient mice receiving cells from the non-leukemic mice developed CML.
The researchers tested these 2 groups of LTHSCs for differences in gene expression. They found significant differences between the leukemic and non-leukemic LTHSCs for the genes Mpl, c-Myc, CD47, Pten, Sirt1, Ptch1, and Tie2.
The team then decided to focus on Mpl. They used flow cytometry to select LTHSCs with either high or low Mpl expression from CML-model BCR-ABL mice.
Seven of 16 mice receiving Mpl-Hi LTHSCs developed leukemia after transplantation, compared with 1 of 17 receiving Mpl-Lo LTHSCs. This suggested an increased leukemogenic capacity for the Mpl-Hi LTHSCs.
The researchers also investigated the impact of cell-cycle status. They found that CML Mpl-Hi LTHSCs that were in a resting stage of the cell cycle had enhanced long-term engraftment and leukemogenic capacity compared with cycling Mpl-Hi LTHSCs.
The team used virus vectors and shRNA to create Mpl knockdown BCR-ABL LTHSCs and showed that the knockdown cells had a greatly reduced ability to produce leukemia in recipient mice.
The Mpl knockdown cells, after stimulation by the Mpl ligand thrombopoietin, also had reduced expression of the activated transcription factors p-STAT3 and p-STAT5, compared with controls.
Finally, the researchers examined human CML cells for differences between MPL-Hi LTHSCs and MPL-Lo LTHSCs. The results were similar to those observed in mice.
The human MPL-Hi LTHSCs had a higher rate of engraftment than the human MPL-Lo LTHSCs, as tested in a xenograft model using immunodeficient mice.
Additionally, the human MPL-Hi LTHSCs had reduced sensitivity to nilotinib compared with MPL-Lo LTHSCs. However, a Jak/STAT inhibitor significantly reduced cell growth and increased apoptosis in human MPL-Hi LTHSCs.
The researchers concluded that MPL expression is a marker and key regulator of leukemogenic potential and drug sensitivity of CML LTHSCs. They said their findings support further investigation of approaches to antagonize MPL signaling as a potential therapeutic strategy to eliminate leukemia-initiating LTHSCs. ![]()
Novel test detects low levels of residual CML
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.

Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
FROM THE JOURNAL OF MOLECULAR DIAGNOSTICS
Key clinical point: A novel test may predict which CML patients in remission can safely stop TKI therapy.
Major finding: Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%).
Data source: DNA-based dPCR measures in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Disclosures: The researchers had no relevant disclosures.

New assay detects persistent CML better, team says
A new assay is more accurate than the current gold standard for detecting residual disease in patients with chronic myeloid leukemia (CML), according to a study published in The Journal of Molecular Diagnostics.
Investigators found this test, a DNA-based digital PCR (dPCR) assay, could detect persistent disease in 81% of samples taken from CML patients who were in remission according to reverse transcriptase-quantitative PCR (RT-qPCR).
RT-qPCR is currently the most widely used method for monitoring residual disease in CML patients.
“If validated in clinical trials of stopping TKIs [tyrosine kinase inhibitors], this technique [the dPCR assay] will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest chance of long-term remission,” said investigator Jane F. Apperley, MD, PhD, of Imperial College London in the UK.
For this study, Dr Apperley and her colleagues compared the sensitivity of the dPCR assay to 3 other quantitative PCR methods currently used to measure residual CML—RT-qPCR, quantitative PCR (qPCR), and reverse transcriptase-digital PCR (RT-dPCR).
Thirty-six samples were taken from 6 patients with early CML who were thought to be in deep molecular remission, as indicated by RT-qPCR results.
Repeat analysis using dPCR with preamplification detected persistent disease in 81% of the samples. In comparison, the detection rate was 25% using RT-dPCR and 19% for qPCR.
“We conclude that dPCR for BCR-ABL1 DNA is the most sensitive available method of residual disease detection in CML and may prove useful in the management of TKI withdrawal,” Dr Apperley said.
She and her colleagues believe the new assay has the potential to dramatically impact CML management. They foresee that, immediately after CML diagnosis, the patient’s genomic breakpoints would be identified, enabling the design of a patient-specific assay.
The patient’s response to therapy would be monitored using standard RT-qPCR until reaching molecular remission. At that point, routine monitoring would be augmented with dPCR, allowing better-informed treatment decisions and improved patient management.
According to Dr Apperley, the new method improves on previous methodologies in 2 key areas. First, the dPCR platform provides greater sensitivity.
And second, dPCR is a DNA-based method that allows identification of BCR-ABL1 fusion junctions by targeted next-generation sequencing. This enables the rapid generation of high-performing DNA-based hydrolysis probe assays that are specific to the individual molecular footprint of each patient’s CML clone, although the number and location of fusion junctions may vary among patients.
“The technique we describe, with which we successfully mapped a disease-specific junction in all patients tested, is relatively simple, cost-effective, and suited to a high-throughput laboratory,” Dr Apperley concluded. ![]()
A new assay is more accurate than the current gold standard for detecting residual disease in patients with chronic myeloid leukemia (CML), according to a study published in The Journal of Molecular Diagnostics.
Investigators found this test, a DNA-based digital PCR (dPCR) assay, could detect persistent disease in 81% of samples taken from CML patients who were in remission according to reverse transcriptase-quantitative PCR (RT-qPCR).
RT-qPCR is currently the most widely used method for monitoring residual disease in CML patients.
“If validated in clinical trials of stopping TKIs [tyrosine kinase inhibitors], this technique [the dPCR assay] will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest chance of long-term remission,” said investigator Jane F. Apperley, MD, PhD, of Imperial College London in the UK.
For this study, Dr Apperley and her colleagues compared the sensitivity of the dPCR assay to 3 other quantitative PCR methods currently used to measure residual CML—RT-qPCR, quantitative PCR (qPCR), and reverse transcriptase-digital PCR (RT-dPCR).
Thirty-six samples were taken from 6 patients with early CML who were thought to be in deep molecular remission, as indicated by RT-qPCR results.
Repeat analysis using dPCR with preamplification detected persistent disease in 81% of the samples. In comparison, the detection rate was 25% using RT-dPCR and 19% for qPCR.
“We conclude that dPCR for BCR-ABL1 DNA is the most sensitive available method of residual disease detection in CML and may prove useful in the management of TKI withdrawal,” Dr Apperley said.
She and her colleagues believe the new assay has the potential to dramatically impact CML management. They foresee that, immediately after CML diagnosis, the patient’s genomic breakpoints would be identified, enabling the design of a patient-specific assay.
The patient’s response to therapy would be monitored using standard RT-qPCR until reaching molecular remission. At that point, routine monitoring would be augmented with dPCR, allowing better-informed treatment decisions and improved patient management.
According to Dr Apperley, the new method improves on previous methodologies in 2 key areas. First, the dPCR platform provides greater sensitivity.
And second, dPCR is a DNA-based method that allows identification of BCR-ABL1 fusion junctions by targeted next-generation sequencing. This enables the rapid generation of high-performing DNA-based hydrolysis probe assays that are specific to the individual molecular footprint of each patient’s CML clone, although the number and location of fusion junctions may vary among patients.
“The technique we describe, with which we successfully mapped a disease-specific junction in all patients tested, is relatively simple, cost-effective, and suited to a high-throughput laboratory,” Dr Apperley concluded. ![]()
A new assay is more accurate than the current gold standard for detecting residual disease in patients with chronic myeloid leukemia (CML), according to a study published in The Journal of Molecular Diagnostics.
Investigators found this test, a DNA-based digital PCR (dPCR) assay, could detect persistent disease in 81% of samples taken from CML patients who were in remission according to reverse transcriptase-quantitative PCR (RT-qPCR).
RT-qPCR is currently the most widely used method for monitoring residual disease in CML patients.
“If validated in clinical trials of stopping TKIs [tyrosine kinase inhibitors], this technique [the dPCR assay] will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest chance of long-term remission,” said investigator Jane F. Apperley, MD, PhD, of Imperial College London in the UK.
For this study, Dr Apperley and her colleagues compared the sensitivity of the dPCR assay to 3 other quantitative PCR methods currently used to measure residual CML—RT-qPCR, quantitative PCR (qPCR), and reverse transcriptase-digital PCR (RT-dPCR).
Thirty-six samples were taken from 6 patients with early CML who were thought to be in deep molecular remission, as indicated by RT-qPCR results.
Repeat analysis using dPCR with preamplification detected persistent disease in 81% of the samples. In comparison, the detection rate was 25% using RT-dPCR and 19% for qPCR.
“We conclude that dPCR for BCR-ABL1 DNA is the most sensitive available method of residual disease detection in CML and may prove useful in the management of TKI withdrawal,” Dr Apperley said.
She and her colleagues believe the new assay has the potential to dramatically impact CML management. They foresee that, immediately after CML diagnosis, the patient’s genomic breakpoints would be identified, enabling the design of a patient-specific assay.
The patient’s response to therapy would be monitored using standard RT-qPCR until reaching molecular remission. At that point, routine monitoring would be augmented with dPCR, allowing better-informed treatment decisions and improved patient management.
According to Dr Apperley, the new method improves on previous methodologies in 2 key areas. First, the dPCR platform provides greater sensitivity.
And second, dPCR is a DNA-based method that allows identification of BCR-ABL1 fusion junctions by targeted next-generation sequencing. This enables the rapid generation of high-performing DNA-based hydrolysis probe assays that are specific to the individual molecular footprint of each patient’s CML clone, although the number and location of fusion junctions may vary among patients.
“The technique we describe, with which we successfully mapped a disease-specific junction in all patients tested, is relatively simple, cost-effective, and suited to a high-throughput laboratory,” Dr Apperley concluded. ![]()
Generic imatinib launched with savings program

Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Rhoda Baer
Sun Pharma has announced the US launch of imatinib mesylate tablets, which are a generic version of Novartis’s Gleevec, for indications approved by the US Food and Drug Administration (FDA).
As part of this launch, Sun Pharma has rolled out a savings card program. The goal is to provide greater access to imatinib mesylate tablets for patients who have commercial insurance, but their out-of-pocket cost may exceed an affordable amount.
Sun Pharma’s Imatinib Mesylate Savings Card will reduce patient’s co-payment to $10. The card will also offer patients an additional savings benefit of up to $700 for a 30-day fill to offset any additional out-of-pocket cost should they be required to meet their deductible or co-insurance.
Participating pharmacies across the US can use the patient’s card as part of this program.
Eligible patients can participate in Sun Pharma’s Imatinib Mesylate Savings Card program by registering at www.imatinibrx.com or by requesting a savings card from their oncologist. Sun Pharma will be supplying its Imatinib Mesylate Savings Cards to more than 4500 oncologists.
Sun Pharma has established a Hub service so patients can call and speak with a trained healthcare professional about imatinib mesylate. The number is 1-844-502-5950.
In addition, qualifying patients can receive Sun Pharma’s imatinib mesylate at no cost. Based on qualifications for applying and including a doctor’s prescription, the Hub service will determine if a patient is qualified to receive imatinib mesylate for free. Upon acceptance, the prescription will be processed and delivered to the qualifying patient at no cost.
Sun Pharma’s imatinib mesylate was approved by the FDA in December 2015 and was granted 180 days of marketing exclusivity from the time of its launch. The drug is available in 100 mg and 400 mg tablets.
It is approved to treat:
- Newly diagnosed adult and pediatric patients with Philadelphia-chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with Ph+ CML in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adults with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adults with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adults with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adults with hypereosinophilic syndrome and/or chronic eosinophilic leukemia, including those who have the FIP1L1-PDGFRα fusion kinase
- Adult patients with unresectable, recurrent, and/or metastatic dermatofibrosarcoma protuberans.
Sun Pharma’s imatinib mesylate is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
KIR2DL5B genotype predicts outcome in chronic phase–CML
The presence of KIR2DL5B was associated with lower rates of major molecular response (MMR), transformation-free survival, and event-free survival (but not overall survival) in patients with chronic phase–chronic myeloid leukemia (CP-CML) treated with sequential imatinib/nilotinib, according to researchers.
Univariate analysis demonstrated a significant association between KIR2DL5B and achievement of a major molecular response, with hazard ratio 0.423 (95% CI, 0.262-0.682; P less than .001). Other KIR genotypes, KIR2DL2pos and KIR2DS3pos, were also associated with inferior achievement of MMR, probably because of their association with KIR2DL5B due to linkage disequilibrium among KIR genes, according to the investigators.
“Our findings suggest that even with the potent second-generation TKI [tyrosine kinase inhibitor] nilotinib, KIR genotypes, a predetermined genetic host factor, may still be one of the most discriminatory prognostic markers available at baseline,” wrote Dr. David T. Yeung of the department of genetics and molecular pathology, Centre for Cancer Biology and the University of Adelaide, South Australia, and colleagues (Blood 2015 Dec 17. doi:10.1182/blood-2015-07-655589).
Killer immunoglobulin-like receptors (KIRs) contribute to natural killer (NK) cell–mediated killing of tumor cells, in both activating and inhibitory roles. Normal cells are spared through actions of inhibitory KIRs. Although the mechanism underlying the association between KIR2DL5B and CP-CML treatment outcomes is still unclear, the gene encodes an inhibitory KIR receptor, the absence of which may increase efficiency of NK-mediated killing of leukemic stem cells, researchers suggested.
The Therapeutic Intensification in De Novo Leukaemia (TIDEL-II) study included 210 patients with CP-CML who were treated with imatinib initially, and nilotinib subsequently if predetermined molecular targets were not met. The KIR substudy included 148 patients with samples available for genotyping.
KIR genotype frequencies observed in this study were similar to other white populations reported in the Allele Frequency Database.
Early molecular response was also significantly associated with treatment outcomes, independent of KIR prognostic significance, and may add additional prognostic information, available 3 months after treatment commences.
“In contrast, KIR2DL5B can identify, at baseline, the 20% of patients with a transformation risk of [about] 10% over 2 years versus the 80% of patients with a transformation risk of less than 3%,” the authors wrote. They suggest that KIR2DL5B, combined with other predictive markers, may enable targeted early interventions to improve outcomes.
The presence of KIR2DL5B was associated with lower rates of major molecular response (MMR), transformation-free survival, and event-free survival (but not overall survival) in patients with chronic phase–chronic myeloid leukemia (CP-CML) treated with sequential imatinib/nilotinib, according to researchers.
Univariate analysis demonstrated a significant association between KIR2DL5B and achievement of a major molecular response, with hazard ratio 0.423 (95% CI, 0.262-0.682; P less than .001). Other KIR genotypes, KIR2DL2pos and KIR2DS3pos, were also associated with inferior achievement of MMR, probably because of their association with KIR2DL5B due to linkage disequilibrium among KIR genes, according to the investigators.
“Our findings suggest that even with the potent second-generation TKI [tyrosine kinase inhibitor] nilotinib, KIR genotypes, a predetermined genetic host factor, may still be one of the most discriminatory prognostic markers available at baseline,” wrote Dr. David T. Yeung of the department of genetics and molecular pathology, Centre for Cancer Biology and the University of Adelaide, South Australia, and colleagues (Blood 2015 Dec 17. doi:10.1182/blood-2015-07-655589).
Killer immunoglobulin-like receptors (KIRs) contribute to natural killer (NK) cell–mediated killing of tumor cells, in both activating and inhibitory roles. Normal cells are spared through actions of inhibitory KIRs. Although the mechanism underlying the association between KIR2DL5B and CP-CML treatment outcomes is still unclear, the gene encodes an inhibitory KIR receptor, the absence of which may increase efficiency of NK-mediated killing of leukemic stem cells, researchers suggested.
The Therapeutic Intensification in De Novo Leukaemia (TIDEL-II) study included 210 patients with CP-CML who were treated with imatinib initially, and nilotinib subsequently if predetermined molecular targets were not met. The KIR substudy included 148 patients with samples available for genotyping.
KIR genotype frequencies observed in this study were similar to other white populations reported in the Allele Frequency Database.
Early molecular response was also significantly associated with treatment outcomes, independent of KIR prognostic significance, and may add additional prognostic information, available 3 months after treatment commences.
“In contrast, KIR2DL5B can identify, at baseline, the 20% of patients with a transformation risk of [about] 10% over 2 years versus the 80% of patients with a transformation risk of less than 3%,” the authors wrote. They suggest that KIR2DL5B, combined with other predictive markers, may enable targeted early interventions to improve outcomes.
The presence of KIR2DL5B was associated with lower rates of major molecular response (MMR), transformation-free survival, and event-free survival (but not overall survival) in patients with chronic phase–chronic myeloid leukemia (CP-CML) treated with sequential imatinib/nilotinib, according to researchers.
Univariate analysis demonstrated a significant association between KIR2DL5B and achievement of a major molecular response, with hazard ratio 0.423 (95% CI, 0.262-0.682; P less than .001). Other KIR genotypes, KIR2DL2pos and KIR2DS3pos, were also associated with inferior achievement of MMR, probably because of their association with KIR2DL5B due to linkage disequilibrium among KIR genes, according to the investigators.
“Our findings suggest that even with the potent second-generation TKI [tyrosine kinase inhibitor] nilotinib, KIR genotypes, a predetermined genetic host factor, may still be one of the most discriminatory prognostic markers available at baseline,” wrote Dr. David T. Yeung of the department of genetics and molecular pathology, Centre for Cancer Biology and the University of Adelaide, South Australia, and colleagues (Blood 2015 Dec 17. doi:10.1182/blood-2015-07-655589).
Killer immunoglobulin-like receptors (KIRs) contribute to natural killer (NK) cell–mediated killing of tumor cells, in both activating and inhibitory roles. Normal cells are spared through actions of inhibitory KIRs. Although the mechanism underlying the association between KIR2DL5B and CP-CML treatment outcomes is still unclear, the gene encodes an inhibitory KIR receptor, the absence of which may increase efficiency of NK-mediated killing of leukemic stem cells, researchers suggested.
The Therapeutic Intensification in De Novo Leukaemia (TIDEL-II) study included 210 patients with CP-CML who were treated with imatinib initially, and nilotinib subsequently if predetermined molecular targets were not met. The KIR substudy included 148 patients with samples available for genotyping.
KIR genotype frequencies observed in this study were similar to other white populations reported in the Allele Frequency Database.
Early molecular response was also significantly associated with treatment outcomes, independent of KIR prognostic significance, and may add additional prognostic information, available 3 months after treatment commences.
“In contrast, KIR2DL5B can identify, at baseline, the 20% of patients with a transformation risk of [about] 10% over 2 years versus the 80% of patients with a transformation risk of less than 3%,” the authors wrote. They suggest that KIR2DL5B, combined with other predictive markers, may enable targeted early interventions to improve outcomes.
FROM BLOOD
Key clinical point: The presence of KIR2DL5B was associated with worse outcomes in patients with chronic phase–chronic myeloid leukemia treated with sequential imatinib/nilotinib.
Major finding: Achievement of a major molecular response was associated with the KIR2DL5B genotype (HR, 0.423; 95% CI, 0.262-0.682; P less than .001).
Data source: A substudy of the Therapeutic Intensification in De Novo Leukaemia (TIDEL-II) study that included 148 patients with KIR genotype data available.
Disclosures: Support for the study was provided in part by Novartis. Dr. Yeung reported consulting or advisory roles with Novartis, BMS, and Ariad. Several coauthors reported ties to industry.
Cardiovascular risk assessment required with use of TKIs for CML
Treatment for chronic myeloid leukemia (CML) entails effective but mostly noncurative long-term use of tyrosine kinase inhibitors (TKIs) that require proactive, rational approaches to minimizing cardiovascular toxicities, according to a recent review.
Survival rates of patients with newly diagnosed CML are about 90%, and in those with a complete cytogenetic response, survival is comparable to that of age-matched controls. Although second-generation TKIs have increased efficacy, survival rates are similar to those of imatinib, possibly due in part to mortality from non-CML causes.
TKIs used in CML therapy target BCR-ABL1, but their potencies vary against other kinases, including receptors for vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF). The relationship between off-target activities and adverse events (AEs) remains unclear, and AE management is largely empirical, said Dr. Javid Moslehi of Vanderbilt University Medical Center, Nashville, Tenn., and Dr. Michael Deininger, professor at the University of Utah Huntsman Cancer Institute, Salt Lake City.
“Reports of cardiovascular AEs with nilotinib, pulmonary arterial hypertension (PAH) on dasatinib, and frequent cardiovascular AEs with ponatinib have caused a reassessment of the situation,” they noted.
“Given the high population frequency of cardiovascular disease and the increased frequency of vascular events with nilotinib and ponatinib, cardiovascular risk assessment and, if necessary, treatment need to be integrated into the management of patients with CML on TKIs,” they wrote (J Clin Onc. 2015 Dec 10. doi: 10.1200/JCO.2015.62.4718).
Retrospective studies have indicated that imatinib may have favorable metabolic and vascular effects, but prospective controlled trials are lacking. Defining the cardiovascular baseline risk of the specific CML population under study will be crucial in future studies.
Dasatinib was approved for front-line CML treatment based on superior cytogenic response rates, compared with imatinib, but in 2011 the Food and Drug Administration warned against cardiopulmonary risks and recommended that patients be evaluated for signs and symptoms of cardiopulmonary disease before and during dasatinib treatment. Results of DASISION (Dasatinib Versus Imatinib Study in Treatment-Naive CML Patients) showed that, at 36 months of follow-up, PAH was reported in 3% of patients on dasatinib and 0% on imatinib.
Nilotinib has shown superior efficacy to imatinib and was FDA approved for first-line therapy, with recommendations for arrhythmia monitoring and avoidance of QT interval–prolonging medications. There have been no subsequent reports of ventricular arrhythmias with nilotinib, but 36% of patients on nilotinib experienced hyperglycemia in the ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed Patients) study, compared with 20% on imatinib. Nilotinib also has been associated with hyperlipidemia and increased body mass. Recent results point to vascular toxicity with nilotinib. At the 6-year follow-up of the ENESTnd study, 10% of patients on nilotinib 300 mg twice per day and 16% on nilotinib 400 mg twice per day had cardiovascular events, compared with 2.5% of patients taking imatinib 400 mg once per day. The dose-dependent increased risk implicates a drug-dependent process.
Ponatinib is the only clinical TKI active against the BCR-ABL1T315I mutation. It is a potent inhibitor of numerous other kinases as well, including VEGF receptors. In the PACE (Ponatinib Ph-positive Acute Lymphoblastic Leukemia and CML Evaluation) study, 26% of patients on ponatinib developed hypertension, and traditional atherosclerosis risk factors (age, hypertension, and diabetes) predisposed patients to serious vascular AEs. Cardiovascular toxicity was shown to be dose dependent, and older patients with history of diabetes or ischemic events are the least tolerant of high dose intensity. A subset of patients will benefit from ponatinib, particularly those with BCR-ABL1T315I, but leukemia-related and cardiovascular risks must both be assessed.
Dr. Moslehi reported financial ties with Novartis, ARIAD, Takeda/Millennium, Bristol-Myers Squibb, and Acceleron Pharma. Dr. Deininger reported ties to Novartis, Bristol-Myers Squibb, Incyte, ARIAD, Pfizer, and Cellgene.
Treatment for chronic myeloid leukemia (CML) entails effective but mostly noncurative long-term use of tyrosine kinase inhibitors (TKIs) that require proactive, rational approaches to minimizing cardiovascular toxicities, according to a recent review.
Survival rates of patients with newly diagnosed CML are about 90%, and in those with a complete cytogenetic response, survival is comparable to that of age-matched controls. Although second-generation TKIs have increased efficacy, survival rates are similar to those of imatinib, possibly due in part to mortality from non-CML causes.
TKIs used in CML therapy target BCR-ABL1, but their potencies vary against other kinases, including receptors for vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF). The relationship between off-target activities and adverse events (AEs) remains unclear, and AE management is largely empirical, said Dr. Javid Moslehi of Vanderbilt University Medical Center, Nashville, Tenn., and Dr. Michael Deininger, professor at the University of Utah Huntsman Cancer Institute, Salt Lake City.
“Reports of cardiovascular AEs with nilotinib, pulmonary arterial hypertension (PAH) on dasatinib, and frequent cardiovascular AEs with ponatinib have caused a reassessment of the situation,” they noted.
“Given the high population frequency of cardiovascular disease and the increased frequency of vascular events with nilotinib and ponatinib, cardiovascular risk assessment and, if necessary, treatment need to be integrated into the management of patients with CML on TKIs,” they wrote (J Clin Onc. 2015 Dec 10. doi: 10.1200/JCO.2015.62.4718).
Retrospective studies have indicated that imatinib may have favorable metabolic and vascular effects, but prospective controlled trials are lacking. Defining the cardiovascular baseline risk of the specific CML population under study will be crucial in future studies.
Dasatinib was approved for front-line CML treatment based on superior cytogenic response rates, compared with imatinib, but in 2011 the Food and Drug Administration warned against cardiopulmonary risks and recommended that patients be evaluated for signs and symptoms of cardiopulmonary disease before and during dasatinib treatment. Results of DASISION (Dasatinib Versus Imatinib Study in Treatment-Naive CML Patients) showed that, at 36 months of follow-up, PAH was reported in 3% of patients on dasatinib and 0% on imatinib.
Nilotinib has shown superior efficacy to imatinib and was FDA approved for first-line therapy, with recommendations for arrhythmia monitoring and avoidance of QT interval–prolonging medications. There have been no subsequent reports of ventricular arrhythmias with nilotinib, but 36% of patients on nilotinib experienced hyperglycemia in the ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed Patients) study, compared with 20% on imatinib. Nilotinib also has been associated with hyperlipidemia and increased body mass. Recent results point to vascular toxicity with nilotinib. At the 6-year follow-up of the ENESTnd study, 10% of patients on nilotinib 300 mg twice per day and 16% on nilotinib 400 mg twice per day had cardiovascular events, compared with 2.5% of patients taking imatinib 400 mg once per day. The dose-dependent increased risk implicates a drug-dependent process.
Ponatinib is the only clinical TKI active against the BCR-ABL1T315I mutation. It is a potent inhibitor of numerous other kinases as well, including VEGF receptors. In the PACE (Ponatinib Ph-positive Acute Lymphoblastic Leukemia and CML Evaluation) study, 26% of patients on ponatinib developed hypertension, and traditional atherosclerosis risk factors (age, hypertension, and diabetes) predisposed patients to serious vascular AEs. Cardiovascular toxicity was shown to be dose dependent, and older patients with history of diabetes or ischemic events are the least tolerant of high dose intensity. A subset of patients will benefit from ponatinib, particularly those with BCR-ABL1T315I, but leukemia-related and cardiovascular risks must both be assessed.
Dr. Moslehi reported financial ties with Novartis, ARIAD, Takeda/Millennium, Bristol-Myers Squibb, and Acceleron Pharma. Dr. Deininger reported ties to Novartis, Bristol-Myers Squibb, Incyte, ARIAD, Pfizer, and Cellgene.
Treatment for chronic myeloid leukemia (CML) entails effective but mostly noncurative long-term use of tyrosine kinase inhibitors (TKIs) that require proactive, rational approaches to minimizing cardiovascular toxicities, according to a recent review.
Survival rates of patients with newly diagnosed CML are about 90%, and in those with a complete cytogenetic response, survival is comparable to that of age-matched controls. Although second-generation TKIs have increased efficacy, survival rates are similar to those of imatinib, possibly due in part to mortality from non-CML causes.
TKIs used in CML therapy target BCR-ABL1, but their potencies vary against other kinases, including receptors for vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and fibroblast growth factor (FGF). The relationship between off-target activities and adverse events (AEs) remains unclear, and AE management is largely empirical, said Dr. Javid Moslehi of Vanderbilt University Medical Center, Nashville, Tenn., and Dr. Michael Deininger, professor at the University of Utah Huntsman Cancer Institute, Salt Lake City.
“Reports of cardiovascular AEs with nilotinib, pulmonary arterial hypertension (PAH) on dasatinib, and frequent cardiovascular AEs with ponatinib have caused a reassessment of the situation,” they noted.
“Given the high population frequency of cardiovascular disease and the increased frequency of vascular events with nilotinib and ponatinib, cardiovascular risk assessment and, if necessary, treatment need to be integrated into the management of patients with CML on TKIs,” they wrote (J Clin Onc. 2015 Dec 10. doi: 10.1200/JCO.2015.62.4718).
Retrospective studies have indicated that imatinib may have favorable metabolic and vascular effects, but prospective controlled trials are lacking. Defining the cardiovascular baseline risk of the specific CML population under study will be crucial in future studies.
Dasatinib was approved for front-line CML treatment based on superior cytogenic response rates, compared with imatinib, but in 2011 the Food and Drug Administration warned against cardiopulmonary risks and recommended that patients be evaluated for signs and symptoms of cardiopulmonary disease before and during dasatinib treatment. Results of DASISION (Dasatinib Versus Imatinib Study in Treatment-Naive CML Patients) showed that, at 36 months of follow-up, PAH was reported in 3% of patients on dasatinib and 0% on imatinib.
Nilotinib has shown superior efficacy to imatinib and was FDA approved for first-line therapy, with recommendations for arrhythmia monitoring and avoidance of QT interval–prolonging medications. There have been no subsequent reports of ventricular arrhythmias with nilotinib, but 36% of patients on nilotinib experienced hyperglycemia in the ENESTnd (Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed Patients) study, compared with 20% on imatinib. Nilotinib also has been associated with hyperlipidemia and increased body mass. Recent results point to vascular toxicity with nilotinib. At the 6-year follow-up of the ENESTnd study, 10% of patients on nilotinib 300 mg twice per day and 16% on nilotinib 400 mg twice per day had cardiovascular events, compared with 2.5% of patients taking imatinib 400 mg once per day. The dose-dependent increased risk implicates a drug-dependent process.
Ponatinib is the only clinical TKI active against the BCR-ABL1T315I mutation. It is a potent inhibitor of numerous other kinases as well, including VEGF receptors. In the PACE (Ponatinib Ph-positive Acute Lymphoblastic Leukemia and CML Evaluation) study, 26% of patients on ponatinib developed hypertension, and traditional atherosclerosis risk factors (age, hypertension, and diabetes) predisposed patients to serious vascular AEs. Cardiovascular toxicity was shown to be dose dependent, and older patients with history of diabetes or ischemic events are the least tolerant of high dose intensity. A subset of patients will benefit from ponatinib, particularly those with BCR-ABL1T315I, but leukemia-related and cardiovascular risks must both be assessed.
Dr. Moslehi reported financial ties with Novartis, ARIAD, Takeda/Millennium, Bristol-Myers Squibb, and Acceleron Pharma. Dr. Deininger reported ties to Novartis, Bristol-Myers Squibb, Incyte, ARIAD, Pfizer, and Cellgene.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Most patients with chronic myeloid leukemia require long-term tyrosine kinase inhibitor (TKI) therapy, and cardiovascular effects are critical factors in treatment decisions.
Major finding: Second- and third-generation TKIs have been associated with more cardiovascular risk than first-generation imatinib.
Data source: Review of current literature on cardiovascular toxicity of BCR-ABL1 TKIs for treatment of chronic myeloid leukemia.
Disclosures: Dr. Moslehi reported financial ties with Novartis, ARIAD, Takeda/Millennium, Bristol-Myers Squibb, and Acceleron Pharma. Dr. Deininger reported ties to Novartis, Bristol-Myers Squibb, Incyte, ARIAD, Pfizer, and Cellgene.
Potential new alternative in CML when TKI therapy fails

ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.
ASH Annual Meeting
Photo courtesy of ASH
ORLANDO, FL—ABL001, an allosteric inhibitor of BCR-ABL1, has shown early evidence of single-agent activity in a multicenter, first-in-human, first-in-class trial of heavily treated patients with chronic myeloid leukemia (CML) that is resistant to or intolerant of prior tyrosine kinase inhibitors (TKIs), even at the lowest dose evaluated.
ABL001 and classical TKIs exhibit complementary mutation profiles, with ABL001 showing activity against TKI resistance mutations.
When combined with nilotinib in a mouse model of CML, ABL001 prevented the emergence of resistant disease even after treatment was discontinued.
“This produces a new therapeutic concept—that of allosteric inhibition,” said Oliver G. Ottmann, MD, of Cardiff University in the UK.
The ABL001 binding site is located in a region remote from the kinase domain and has the potential to combine with TKIs for greater pharmacologic control of BCR-ABL1.
“This obviously has the opportunity both for combining different treatments and for overcoming resistance to one or the other,” Dr Ottmann added.
Based on preliminary pharmacokinetic data and preclinical evidence, investigators proceeded to evaluate ABL001 in a phase 1 dose-escalation and dose-expansion study.
Their primary objective was to determine the maximum tolerated dose (MTD) in humans and the recommended dose for expansion (RDE). Secondary objectives were to evaluate the safety, tolerability, preliminary anti-CML activity, and pharmacokinetic and pharmacodynamic profile.
Dr Ottmann presented the findings during the 2015 ASH Annual Meeting as abstract 138.*
Study design
Patients received ABL001 orally as a single agent twice a day (BID) continuously until disease progression, unacceptable toxicity, consent withdrawal, or death.
The dose-escalation schema followed a Bayesian logistic regression model based on dose-limiting toxicities during cycle 1. Doses ranged from 10 mg to 200 mg BID.
A subsequent dose-expansion phase was planned to augment the data generated in the dose-escalation phase and to include patients with Ph-positive acute lymphoblastic leukemia resistant or intolerant to prior TKI therapy.
Dr Ottmann noted that there were 2 protocol amendments. The first amendment was made to include a once-daily (QD) dosing of ABL001 at 120 mg and 200 mg. The second amendment was made to evaluate the combination of 40 mg of ABL001 BID with nilotinib at 300 mg BID.
Inclusion/exclusion criteria
Patients had to be at least 18 years old with CML in chronic or accelerated phase. They had to have failed at least 2 prior TKIs or be intolerant of TKIs. Their performance status had to be 0–2.
Patients were excluded from the trial if they had an absolute neutrophil count less than 500/mm3, a platelet count less than 50,000/mm3, bilirubin level more than 1.5 x the upper limit of normal (ULN) or more than 3.0 x ULN in patients with Gilberts syndrome.
Their aspartate aminotransferase or alanine aminotransferase could not be above 3 x ULN, and creatinine could not be above 1.5 x ULN.
Patients were also excluded if they needed treatment with strong inhibitors or inducers of CYP3A4 or its substrates with narrow therapeutic index.
Patient demographics
Fifty-nine patients were enrolled on the trial at the time of the presentation and they had “typical” characteristics of patients at this stage, Dr Ottmann said.
Their median age was 56 (range, 23–78). Almost two-thirds (61%) were male and 39% female.
All but 1 patient had an ECOG performance status of 0, and patients had a median of 3.5 (range, 2–5) prior lines of therapy. Twenty-four patients (41%) had 2 prior TKIs, and 35 (59%) had 3 or more TKIs. Forty-five patients (76%) were resistant and 14 (24%) were intolerant to their prior TKI.
All but 1 patient had chronic phase CML, 18 (31%) were TKD nonmutated, 14 (24%) were mutated, and 17 (46%) were not evaluable.
Patient disposition
Of the 43 patients in the monotherapy BID cohort, 1 was treated at the 10 mg dose level, 5 at the 20 mg level, 12 at the 40 mg level, 12 at the 80 mg level, 8 at the 150 mg level, and 5 at the 200 mg level. They had a median duration of drug exposure ranging from 25 weeks to 67 weeks.
Of the 11 patients in the monotherapy QD group, 5 were treated at the 120 mg dose level and 6 at the 200 mg level. Their drug exposure was a median of 26 weeks for those receiving 120 mg and 9.8 weeks for those receiving the 200 mg dose.
And the 5 patients in the ABL001-plus-nilotinib group had a median of 6.3 weeks of drug exposure.
“We had a remarkably low rate of discontinuation to date,” Dr Ottmann pointed out.
Ten patients discontinued therapy, all in the monotherapy BID group, 1 at 10 mg, 2 at 40 mg, 2 at 80 mg, 3 at 150 mg, and 2 at 200 mg.
Seven patients discontinued for adverse events. Two patients withdrew consent, and 1 patient in the 40 mg group had disease progression, which is “quite remarkable in a phase 1,” Dr Ottmann said.
Pharmacokinetic profile
ABL001 is rapidly absorbed in a median of 2 to 3 hours, and there is a dose-proportional increase in exposure following single and repeated dosing.
The drug has an approximately 2-fold or lower accumulation on repeated dosing and a short elimination half-life of 5 to 6 hours.
Safety
“We have excellent tolerability,” Dr Ottmann said, with a small number of grade 3/4 adverse events (AEs).
Grade 3/4 AEs considered to be drug-related were mostly associated with hematologic suppression. Four patients (7%) had thrombocytopenia, 4 (7%) neutropenia, 3 (5%) anemia, 4 (7%) lipase increase, and 1 (2%) hypercholesterolemia.
AEs of all grades suspected of being related to the study drug and occurring in 5% or more of patients included thrombocytopenia (19%), neutropenia (15%), anemia (10%), nausea/vomiting/diarrhea (29%), arthralgia/myalgia (20%), rash (17%), fatigue (15%), lipase increase (14%), headache (14%), pruritus (10%), dry skin (7%), hypophosphatemia (7%), and acute pancreatitis (5%).
“The pancreatitis was reversible upon interruption or discontinuation of the drug,” Dr Ottmann explained.
There were 5 dose-limiting toxicities. Two patients had grade 3 lipase elevation in the 40 mg BID and 200 mg QD cohorts. One patient had grade 2 myalgia/arthralgia at 80 mg BID, 1 patient had a grade 3 acute coronary event at 150 mg BID, and 1 patient had a grade 3 bronchospasm at 200 mg BID.
No deaths occurred on the study, and the dose escalation is still ongoing.
Response
Twenty-nine patients with 3 months or more of follow-up were evaluable for response.
Twelve patients, who at baseline had hematologic relapse, achieved complete hematologic response within 2 months, and 8 who had cytogenetic relapse at baseline achieved a complete cytogenetic response within 3 to 6 months.
Of the 29 patients who had molecular relapse at baseline, 10 (34.5%) achieved a molecular response within 6 months, 7 (24.1%) had 1 log or more reduction in BCR-ABL1, 9 (31.0%) had less than a log reduction, and 3 (10.3%) had no reduction.
“The obvious question from the preclinical data,” Dr Ottmann said, “is do the mutations respond?”
And ABL001 has shown clinical activity across TKI-resistant mutations, such as V299L, F317L, and Y253H.
“So to conclude,” he said, “we have a new class, a new therapeutic category of drug, ABL001, which is quite well tolerated in extremely heavily treated patients with CML. We do consider this a promising approach.”
The trial was sponsored by Novartis. ![]()
*Data in the abstract differ from the presentation.