User login
CML outcomes in the age of TKIs
CHICAGO – Major molecular response rates at 12 months were better among chronic myeloid leukemia patients treated with dasatinib/nilotinib vs. imatinib for first-line therapy, and among patients enrolled vs. not enrolled in clinical trials, in a retrospective review of patients treated with first-line tyrosine kinase inhibitors between 2002 and 2014.
The overall rates of major molecular response (MMR) at 12 and 24 months in the 51 patients in the study were 23.5% (12 patients) and 44.9% (22 patients), which was comparable to historical data, Dr. Isabelle Phuong Le reported in a poster at the American Society of Hematology Meeting on Hematologic Malignancies.
The MMR rate among those treated with dasatinib/nilotinib first line was 53%, compared with 10% for those treated with imatinib, said Dr. Le of the University of Texas Health Science Center, San Antonio.
Further the MMR rates at 12 and 24 months among those enrolled vs. not enrolled in clinical trials were 83% vs 15% and 100% vs. 45%, respectively, she noted.
Patients in the analysis were adults with a mean age of 44.6 years who had chronic phase chronic myeloid leukemia (CML). Almost half (49%) were Hispanic, and 56.9% were women. Two-thirds (66.7%) received imatinib first line, and 33.3% received dasatinib/nilotinib first line. More than a third (37.2%) were younger than age 40 years, 15% had a language barrier, and 11.7% were enrolled in clinical trials.
No differences in the MMR rates at 12 and 24 months were seen between Hispanics and non-Hispanics, those with and without language barrier, or men and women, nor were any differences seen between those with more than three lines of therapy and those with three or fewer lines of therapy, or between patients older vs. younger than 40 years, although there was a trend toward improved response rates in older patients, she noted.
“Our patients have similar MMR rates at 12 months and 24 months compared to historical data,” Dr. Le wrote, noting that the findings regarding improved MMR rates with dasatinib/nilotinib were expected.
The findings regarding improved MMR rates among those enrolled in clinical trials could be due to “selective patients as clinical trial patients, more controlled and monitored treatment, frequent follow-ups, and better education regarding disease and treatment,” she said, concluding that “ we can improve treatment response rates in our patients with better disease education.”
Dr. Le reported having no disclosures.
CHICAGO – Major molecular response rates at 12 months were better among chronic myeloid leukemia patients treated with dasatinib/nilotinib vs. imatinib for first-line therapy, and among patients enrolled vs. not enrolled in clinical trials, in a retrospective review of patients treated with first-line tyrosine kinase inhibitors between 2002 and 2014.
The overall rates of major molecular response (MMR) at 12 and 24 months in the 51 patients in the study were 23.5% (12 patients) and 44.9% (22 patients), which was comparable to historical data, Dr. Isabelle Phuong Le reported in a poster at the American Society of Hematology Meeting on Hematologic Malignancies.
The MMR rate among those treated with dasatinib/nilotinib first line was 53%, compared with 10% for those treated with imatinib, said Dr. Le of the University of Texas Health Science Center, San Antonio.
Further the MMR rates at 12 and 24 months among those enrolled vs. not enrolled in clinical trials were 83% vs 15% and 100% vs. 45%, respectively, she noted.
Patients in the analysis were adults with a mean age of 44.6 years who had chronic phase chronic myeloid leukemia (CML). Almost half (49%) were Hispanic, and 56.9% were women. Two-thirds (66.7%) received imatinib first line, and 33.3% received dasatinib/nilotinib first line. More than a third (37.2%) were younger than age 40 years, 15% had a language barrier, and 11.7% were enrolled in clinical trials.
No differences in the MMR rates at 12 and 24 months were seen between Hispanics and non-Hispanics, those with and without language barrier, or men and women, nor were any differences seen between those with more than three lines of therapy and those with three or fewer lines of therapy, or between patients older vs. younger than 40 years, although there was a trend toward improved response rates in older patients, she noted.
“Our patients have similar MMR rates at 12 months and 24 months compared to historical data,” Dr. Le wrote, noting that the findings regarding improved MMR rates with dasatinib/nilotinib were expected.
The findings regarding improved MMR rates among those enrolled in clinical trials could be due to “selective patients as clinical trial patients, more controlled and monitored treatment, frequent follow-ups, and better education regarding disease and treatment,” she said, concluding that “ we can improve treatment response rates in our patients with better disease education.”
Dr. Le reported having no disclosures.
CHICAGO – Major molecular response rates at 12 months were better among chronic myeloid leukemia patients treated with dasatinib/nilotinib vs. imatinib for first-line therapy, and among patients enrolled vs. not enrolled in clinical trials, in a retrospective review of patients treated with first-line tyrosine kinase inhibitors between 2002 and 2014.
The overall rates of major molecular response (MMR) at 12 and 24 months in the 51 patients in the study were 23.5% (12 patients) and 44.9% (22 patients), which was comparable to historical data, Dr. Isabelle Phuong Le reported in a poster at the American Society of Hematology Meeting on Hematologic Malignancies.
The MMR rate among those treated with dasatinib/nilotinib first line was 53%, compared with 10% for those treated with imatinib, said Dr. Le of the University of Texas Health Science Center, San Antonio.
Further the MMR rates at 12 and 24 months among those enrolled vs. not enrolled in clinical trials were 83% vs 15% and 100% vs. 45%, respectively, she noted.
Patients in the analysis were adults with a mean age of 44.6 years who had chronic phase chronic myeloid leukemia (CML). Almost half (49%) were Hispanic, and 56.9% were women. Two-thirds (66.7%) received imatinib first line, and 33.3% received dasatinib/nilotinib first line. More than a third (37.2%) were younger than age 40 years, 15% had a language barrier, and 11.7% were enrolled in clinical trials.
No differences in the MMR rates at 12 and 24 months were seen between Hispanics and non-Hispanics, those with and without language barrier, or men and women, nor were any differences seen between those with more than three lines of therapy and those with three or fewer lines of therapy, or between patients older vs. younger than 40 years, although there was a trend toward improved response rates in older patients, she noted.
“Our patients have similar MMR rates at 12 months and 24 months compared to historical data,” Dr. Le wrote, noting that the findings regarding improved MMR rates with dasatinib/nilotinib were expected.
The findings regarding improved MMR rates among those enrolled in clinical trials could be due to “selective patients as clinical trial patients, more controlled and monitored treatment, frequent follow-ups, and better education regarding disease and treatment,” she said, concluding that “ we can improve treatment response rates in our patients with better disease education.”
Dr. Le reported having no disclosures.
AT MHM 2015
Key clinical point: Major molecular response rates at 12 months were better among CML patients treated with dasatinib/nilotinib vs. imatinib for first-line therapy, and among patients enrolled vs. not enrolled in clinical trials.
Major finding: The major molecular response rates at 12 and 24 months were 23.5% and 44.9%, which is comparable to historical data.
Data source: A retrospective review of 51 cases.
Disclosures: Dr. Le reported having no disclosures.
CML: Select TKI based on comorbidities, monitor toxicity and adherence
CHICAGO – Chronic myeloid leukemia is highly treatable, and a “functional cure” appears to be within reach, according to Dr. Michael J. Mauro.
In fact, an “embarrassment of riches” exists when it comes to initial therapy for CML: In the United States there are five approved tyrosine kinase inhibitors (TKIs), and three are approved for front-line therapy, Dr. Mauro of Memorial Sloan Kettering Cancer Center, New York, said at the American Society of Hematology Meeting on Hematologic Malignancies.

Success, however, is contingent on managing reversible early toxicity, adherence to therapy, achieving landmarks of response, and remaining vigilant for late effects of therapy, he said.
Given the multitude of treatment options and the breadth of available data, Dr. Mauro said he counsels newly diagnosed patients that various treatment options are valid, and that “there may not be a right or wrong answer” for initial therapy. He also counsels patients that tolerability is manageable – and finding the right fit is an important process, and that response milestones are crucial and should be optimized.
It is important, he said, to discuss late toxicity concerns, to review comorbid conditions to help predict potential problems and identify risk, to consider the ramifications of potential toxicity, and to consider adherence.
“We need to portray therapy as really being medium- to long-term,” he said, noting that the urgency to think about treatment-free remission should be tempered by the reality that years of treatment are required first.
Risks and benefits of treatment should be discussed, and the acceptable balance determined in conjunction with the patient, he said, explaining that toxicities vary for the different TKIs.
Imatinib, for example, can be associated with edema/fluid retention, myalgias, hypophosphatemia, and gastrointestinal effects. Dasatinib can be associated with pleural/pericardial effusion, pulmonary arterial hypertension, and bleeding risk. Other toxicities associated with certain TKIs include pancreatic enzyme elevation, rash, and vascular adverse events.
Whether newly diagnosed patients should be directed away from certain agents remains unclear, as available data are open to interpretation, and the mechanism of action for some crucial late effects is unknown. Vascular disease should, however, be considered when making the decision, he said.
Given the available data on late toxicity with various therapies, a cardiovascular evaluation is advisable when initiating TKI therapy, he added.
Consider partnering with primary care, cardiology, or cardio-oncology specialists, and manage risk factors and findings of the evaluation as appropriate, irrespective of the CML, he said. Monitor for progression of cardiovascular risks or adverse events carefully, he added.
His approach for following recently diagnosed CML patients involves:
• A cardiovascular evaluation, at least including age- and comorbidity-appropriate studies, and an up-to-date cardiovascular risk profile. If nilotinib is used, he screens for peripheral, cerebral, and cardiovascular disease – an approach increasingly supported by data. If dasatinib is used, echocardiography is warranted to look for changes that suggest pulmonary hypertension. “And of course we should monitor blood pressure, lipid, and glycemic control,” he added.
• Initial studies, including bone marrow and quantitative polymerase chain reaction – international scale (qPCR IS).
• Lab studies every 1-2 weeks for at least 6 weeks, with titration thereafter as indicated, including for change in therapy.
• A 3-month assessment using qPCR IS. This is very important for following patient response, he said, noting that if the response surpasses compete cytogenetic remission and blood count is acceptable and stable, a repeat bone marrow study may be unnecessary.
• Sequential molecular analyses at least every 3 months.
• Repeat cardiovascular evaluation if/when indicated.
The 3-month response is an opportunity to critically appraise therapy choice and response trajectory; a therapy change is possible based on this assessment, he said. Responses at 6 and 12 months are also important, and changes in therapy for missed milestones at these time points are warranted as deeper remissions are sought.
At 18 months, the focus is on major molecular response, he added, noting that as patients get into deeper molecular remissions, plateaus and fluctuations are common; the nuances of determining who is well enough to consider for treatment-free remission remain to be sorted out in clinical trials.
In general, it appears that 3 years of therapy with about 2 years of optimal minimal residual disease is required prior to consideration for treatment-free remission, he said.
Dr. Mauro has consulted for and/or received research funding from Ariad, Bristol-Myers Squibb, Novartis, and Oregon Health & Science University.
CHICAGO – Chronic myeloid leukemia is highly treatable, and a “functional cure” appears to be within reach, according to Dr. Michael J. Mauro.
In fact, an “embarrassment of riches” exists when it comes to initial therapy for CML: In the United States there are five approved tyrosine kinase inhibitors (TKIs), and three are approved for front-line therapy, Dr. Mauro of Memorial Sloan Kettering Cancer Center, New York, said at the American Society of Hematology Meeting on Hematologic Malignancies.
Success, however, is contingent on managing reversible early toxicity, adherence to therapy, achieving landmarks of response, and remaining vigilant for late effects of therapy, he said.
Given the multitude of treatment options and the breadth of available data, Dr. Mauro said he counsels newly diagnosed patients that various treatment options are valid, and that “there may not be a right or wrong answer” for initial therapy. He also counsels patients that tolerability is manageable – and finding the right fit is an important process, and that response milestones are crucial and should be optimized.
It is important, he said, to discuss late toxicity concerns, to review comorbid conditions to help predict potential problems and identify risk, to consider the ramifications of potential toxicity, and to consider adherence.
“We need to portray therapy as really being medium- to long-term,” he said, noting that the urgency to think about treatment-free remission should be tempered by the reality that years of treatment are required first.
Risks and benefits of treatment should be discussed, and the acceptable balance determined in conjunction with the patient, he said, explaining that toxicities vary for the different TKIs.
Imatinib, for example, can be associated with edema/fluid retention, myalgias, hypophosphatemia, and gastrointestinal effects. Dasatinib can be associated with pleural/pericardial effusion, pulmonary arterial hypertension, and bleeding risk. Other toxicities associated with certain TKIs include pancreatic enzyme elevation, rash, and vascular adverse events.
Whether newly diagnosed patients should be directed away from certain agents remains unclear, as available data are open to interpretation, and the mechanism of action for some crucial late effects is unknown. Vascular disease should, however, be considered when making the decision, he said.
Given the available data on late toxicity with various therapies, a cardiovascular evaluation is advisable when initiating TKI therapy, he added.
Consider partnering with primary care, cardiology, or cardio-oncology specialists, and manage risk factors and findings of the evaluation as appropriate, irrespective of the CML, he said. Monitor for progression of cardiovascular risks or adverse events carefully, he added.
His approach for following recently diagnosed CML patients involves:
• A cardiovascular evaluation, at least including age- and comorbidity-appropriate studies, and an up-to-date cardiovascular risk profile. If nilotinib is used, he screens for peripheral, cerebral, and cardiovascular disease – an approach increasingly supported by data. If dasatinib is used, echocardiography is warranted to look for changes that suggest pulmonary hypertension. “And of course we should monitor blood pressure, lipid, and glycemic control,” he added.
• Initial studies, including bone marrow and quantitative polymerase chain reaction – international scale (qPCR IS).
• Lab studies every 1-2 weeks for at least 6 weeks, with titration thereafter as indicated, including for change in therapy.
• A 3-month assessment using qPCR IS. This is very important for following patient response, he said, noting that if the response surpasses compete cytogenetic remission and blood count is acceptable and stable, a repeat bone marrow study may be unnecessary.
• Sequential molecular analyses at least every 3 months.
• Repeat cardiovascular evaluation if/when indicated.
The 3-month response is an opportunity to critically appraise therapy choice and response trajectory; a therapy change is possible based on this assessment, he said. Responses at 6 and 12 months are also important, and changes in therapy for missed milestones at these time points are warranted as deeper remissions are sought.
At 18 months, the focus is on major molecular response, he added, noting that as patients get into deeper molecular remissions, plateaus and fluctuations are common; the nuances of determining who is well enough to consider for treatment-free remission remain to be sorted out in clinical trials.
In general, it appears that 3 years of therapy with about 2 years of optimal minimal residual disease is required prior to consideration for treatment-free remission, he said.
Dr. Mauro has consulted for and/or received research funding from Ariad, Bristol-Myers Squibb, Novartis, and Oregon Health & Science University.
CHICAGO – Chronic myeloid leukemia is highly treatable, and a “functional cure” appears to be within reach, according to Dr. Michael J. Mauro.
In fact, an “embarrassment of riches” exists when it comes to initial therapy for CML: In the United States there are five approved tyrosine kinase inhibitors (TKIs), and three are approved for front-line therapy, Dr. Mauro of Memorial Sloan Kettering Cancer Center, New York, said at the American Society of Hematology Meeting on Hematologic Malignancies.
Success, however, is contingent on managing reversible early toxicity, adherence to therapy, achieving landmarks of response, and remaining vigilant for late effects of therapy, he said.
Given the multitude of treatment options and the breadth of available data, Dr. Mauro said he counsels newly diagnosed patients that various treatment options are valid, and that “there may not be a right or wrong answer” for initial therapy. He also counsels patients that tolerability is manageable – and finding the right fit is an important process, and that response milestones are crucial and should be optimized.
It is important, he said, to discuss late toxicity concerns, to review comorbid conditions to help predict potential problems and identify risk, to consider the ramifications of potential toxicity, and to consider adherence.
“We need to portray therapy as really being medium- to long-term,” he said, noting that the urgency to think about treatment-free remission should be tempered by the reality that years of treatment are required first.
Risks and benefits of treatment should be discussed, and the acceptable balance determined in conjunction with the patient, he said, explaining that toxicities vary for the different TKIs.
Imatinib, for example, can be associated with edema/fluid retention, myalgias, hypophosphatemia, and gastrointestinal effects. Dasatinib can be associated with pleural/pericardial effusion, pulmonary arterial hypertension, and bleeding risk. Other toxicities associated with certain TKIs include pancreatic enzyme elevation, rash, and vascular adverse events.
Whether newly diagnosed patients should be directed away from certain agents remains unclear, as available data are open to interpretation, and the mechanism of action for some crucial late effects is unknown. Vascular disease should, however, be considered when making the decision, he said.
Given the available data on late toxicity with various therapies, a cardiovascular evaluation is advisable when initiating TKI therapy, he added.
Consider partnering with primary care, cardiology, or cardio-oncology specialists, and manage risk factors and findings of the evaluation as appropriate, irrespective of the CML, he said. Monitor for progression of cardiovascular risks or adverse events carefully, he added.
His approach for following recently diagnosed CML patients involves:
• A cardiovascular evaluation, at least including age- and comorbidity-appropriate studies, and an up-to-date cardiovascular risk profile. If nilotinib is used, he screens for peripheral, cerebral, and cardiovascular disease – an approach increasingly supported by data. If dasatinib is used, echocardiography is warranted to look for changes that suggest pulmonary hypertension. “And of course we should monitor blood pressure, lipid, and glycemic control,” he added.
• Initial studies, including bone marrow and quantitative polymerase chain reaction – international scale (qPCR IS).
• Lab studies every 1-2 weeks for at least 6 weeks, with titration thereafter as indicated, including for change in therapy.
• A 3-month assessment using qPCR IS. This is very important for following patient response, he said, noting that if the response surpasses compete cytogenetic remission and blood count is acceptable and stable, a repeat bone marrow study may be unnecessary.
• Sequential molecular analyses at least every 3 months.
• Repeat cardiovascular evaluation if/when indicated.
The 3-month response is an opportunity to critically appraise therapy choice and response trajectory; a therapy change is possible based on this assessment, he said. Responses at 6 and 12 months are also important, and changes in therapy for missed milestones at these time points are warranted as deeper remissions are sought.
At 18 months, the focus is on major molecular response, he added, noting that as patients get into deeper molecular remissions, plateaus and fluctuations are common; the nuances of determining who is well enough to consider for treatment-free remission remain to be sorted out in clinical trials.
In general, it appears that 3 years of therapy with about 2 years of optimal minimal residual disease is required prior to consideration for treatment-free remission, he said.
Dr. Mauro has consulted for and/or received research funding from Ariad, Bristol-Myers Squibb, Novartis, and Oregon Health & Science University.
EXPERT ANALYSIS FROM MHM 2015

Ponatinib’s toxicity limits use for upfront chronic phase CML
The tyrosine kinase inhibitor ponatinib produced a high degree of complete cytogenetic responses when used in first-line treatment of patients with chronic myeloid leukemia in chronic phase. But the drug’s toxicities, especially its propensity for causing thromboembolic events, mitigate against its upfront use, investigators say.
In a small phase II study, 90% of evaluable patients with chronic-phase chronic myeloid leukemia (CML) treated with the tyrosine kinase inhibitor (TKI) had a complete cytogenetic response (CCyR) after 3 months, and 94% had a CCyR after 6 months, but that efficacy came at the cost of significant adverse events requiring dose reductions, treatment interruptions, and early termination of the trial for safety reasons at the recommendation of the Food and Drug Administration, reported Dr. Preetesh Jain of MD Anderson Cancer Center in Houston, Tex., and colleagues.
“[O]ur study shows that ponatinib is a very potent TKI with high clinical activity in the first-line treatment of patients with chronic-phase CML. However, at the doses currently used in other settings, the safety profile might not be appropriate for treatment of this patient population who have other treatment options with high efficacy,” the investigators wrote (Lancet Haematol. 2015;2[9]:e376-e383).
Ponatinib (Iclusig) is a third-generation TKI with action against malignancies with mutations in the ABL kinase domain that make the cancers resistant to first- and second-generation TKIs such as imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna).
Ponatinib is approved in the United States for treatment of adult patients with CML positive for the T3151 mutation in chronic phase, accelerated phase, or blast phase, or T3151-positive Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ALL). It is also approved for treatment of adults with chronic phase, accelerated phase, or blast phase CML or Ph+ALL for whom no other TKI is indicated.
The drug labeling carries a boxed warning about increased risk for vascular occlusion, heart failure, and hepatotoxicity. The warning notes that “[a]rterial and venous thrombosis and occlusions have occurred in at least 27% of Iclusig-treated patients, including fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures.”
In a single-arm, open label trial, the investigators enrolled 51 patients with recently diagnosed CML in chronic phase, starting them on doses of oral ponatinib 45 mg daily. The protocol was later amended to a starting dose of 30 mg daily because of the high frequency of dose reductions required among patients started at 45 mg. Following a warning from the FDA about vascular complications with the drug, patients were started on daily low-dose aspirin (81 mg), and all drug doses were reduced to either 30 mg or 15 mg daily.
One of the patients discontinued therapy prior to assessment because of the FDA warning, leaving 50 for assessment at 6 months.
Of 46 patients evaluable for the primary outcome of CCyR by 6 months, 43 (94%) achieved it. Major molecular responses, defined as a BCR-ABL to ABL transcript ratio of 0.1% or lower, occurred in 40 of 50 patients (80%) evaluable for this secondary endpoint, and deep molecular response (MR4.5), defined as a ratio less than 0.0032% or lower, occurred in 28 of 50 (55%).
Cardiovascular events, primarily hypertension, occurred in 25 patients (49%), 15 (29%) had grade 3-4 myelosuppression, and 5 patients (10%) developed cerebrovascular or vaso-occlusive disease.
In all, 43 patients (85%) needed treatment interruptions at some time and 45 (88%) needed dose reductions. The study was terminated in June 2014, a little more than a year after recruitment began.
The study was sponsored by MD Anderson Cancer Center with partial support from Ariad Pharmaceuticals. Coauthors Hagop Kantarjian, Elias Jabbour, and Jorge Cortes report receiving grants, research support, or serving as consultants to Ariad.
The tyrosine kinase inhibitor ponatinib produced a high degree of complete cytogenetic responses when used in first-line treatment of patients with chronic myeloid leukemia in chronic phase. But the drug’s toxicities, especially its propensity for causing thromboembolic events, mitigate against its upfront use, investigators say.
In a small phase II study, 90% of evaluable patients with chronic-phase chronic myeloid leukemia (CML) treated with the tyrosine kinase inhibitor (TKI) had a complete cytogenetic response (CCyR) after 3 months, and 94% had a CCyR after 6 months, but that efficacy came at the cost of significant adverse events requiring dose reductions, treatment interruptions, and early termination of the trial for safety reasons at the recommendation of the Food and Drug Administration, reported Dr. Preetesh Jain of MD Anderson Cancer Center in Houston, Tex., and colleagues.
“[O]ur study shows that ponatinib is a very potent TKI with high clinical activity in the first-line treatment of patients with chronic-phase CML. However, at the doses currently used in other settings, the safety profile might not be appropriate for treatment of this patient population who have other treatment options with high efficacy,” the investigators wrote (Lancet Haematol. 2015;2[9]:e376-e383).
Ponatinib (Iclusig) is a third-generation TKI with action against malignancies with mutations in the ABL kinase domain that make the cancers resistant to first- and second-generation TKIs such as imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna).
Ponatinib is approved in the United States for treatment of adult patients with CML positive for the T3151 mutation in chronic phase, accelerated phase, or blast phase, or T3151-positive Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ALL). It is also approved for treatment of adults with chronic phase, accelerated phase, or blast phase CML or Ph+ALL for whom no other TKI is indicated.
The drug labeling carries a boxed warning about increased risk for vascular occlusion, heart failure, and hepatotoxicity. The warning notes that “[a]rterial and venous thrombosis and occlusions have occurred in at least 27% of Iclusig-treated patients, including fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures.”
In a single-arm, open label trial, the investigators enrolled 51 patients with recently diagnosed CML in chronic phase, starting them on doses of oral ponatinib 45 mg daily. The protocol was later amended to a starting dose of 30 mg daily because of the high frequency of dose reductions required among patients started at 45 mg. Following a warning from the FDA about vascular complications with the drug, patients were started on daily low-dose aspirin (81 mg), and all drug doses were reduced to either 30 mg or 15 mg daily.
One of the patients discontinued therapy prior to assessment because of the FDA warning, leaving 50 for assessment at 6 months.
Of 46 patients evaluable for the primary outcome of CCyR by 6 months, 43 (94%) achieved it. Major molecular responses, defined as a BCR-ABL to ABL transcript ratio of 0.1% or lower, occurred in 40 of 50 patients (80%) evaluable for this secondary endpoint, and deep molecular response (MR4.5), defined as a ratio less than 0.0032% or lower, occurred in 28 of 50 (55%).
Cardiovascular events, primarily hypertension, occurred in 25 patients (49%), 15 (29%) had grade 3-4 myelosuppression, and 5 patients (10%) developed cerebrovascular or vaso-occlusive disease.
In all, 43 patients (85%) needed treatment interruptions at some time and 45 (88%) needed dose reductions. The study was terminated in June 2014, a little more than a year after recruitment began.
The study was sponsored by MD Anderson Cancer Center with partial support from Ariad Pharmaceuticals. Coauthors Hagop Kantarjian, Elias Jabbour, and Jorge Cortes report receiving grants, research support, or serving as consultants to Ariad.
The tyrosine kinase inhibitor ponatinib produced a high degree of complete cytogenetic responses when used in first-line treatment of patients with chronic myeloid leukemia in chronic phase. But the drug’s toxicities, especially its propensity for causing thromboembolic events, mitigate against its upfront use, investigators say.
In a small phase II study, 90% of evaluable patients with chronic-phase chronic myeloid leukemia (CML) treated with the tyrosine kinase inhibitor (TKI) had a complete cytogenetic response (CCyR) after 3 months, and 94% had a CCyR after 6 months, but that efficacy came at the cost of significant adverse events requiring dose reductions, treatment interruptions, and early termination of the trial for safety reasons at the recommendation of the Food and Drug Administration, reported Dr. Preetesh Jain of MD Anderson Cancer Center in Houston, Tex., and colleagues.
“[O]ur study shows that ponatinib is a very potent TKI with high clinical activity in the first-line treatment of patients with chronic-phase CML. However, at the doses currently used in other settings, the safety profile might not be appropriate for treatment of this patient population who have other treatment options with high efficacy,” the investigators wrote (Lancet Haematol. 2015;2[9]:e376-e383).
Ponatinib (Iclusig) is a third-generation TKI with action against malignancies with mutations in the ABL kinase domain that make the cancers resistant to first- and second-generation TKIs such as imatinib (Gleevec), dasatinib (Sprycel), and nilotinib (Tasigna).
Ponatinib is approved in the United States for treatment of adult patients with CML positive for the T3151 mutation in chronic phase, accelerated phase, or blast phase, or T3151-positive Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ALL). It is also approved for treatment of adults with chronic phase, accelerated phase, or blast phase CML or Ph+ALL for whom no other TKI is indicated.
The drug labeling carries a boxed warning about increased risk for vascular occlusion, heart failure, and hepatotoxicity. The warning notes that “[a]rterial and venous thrombosis and occlusions have occurred in at least 27% of Iclusig-treated patients, including fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures.”
In a single-arm, open label trial, the investigators enrolled 51 patients with recently diagnosed CML in chronic phase, starting them on doses of oral ponatinib 45 mg daily. The protocol was later amended to a starting dose of 30 mg daily because of the high frequency of dose reductions required among patients started at 45 mg. Following a warning from the FDA about vascular complications with the drug, patients were started on daily low-dose aspirin (81 mg), and all drug doses were reduced to either 30 mg or 15 mg daily.
One of the patients discontinued therapy prior to assessment because of the FDA warning, leaving 50 for assessment at 6 months.
Of 46 patients evaluable for the primary outcome of CCyR by 6 months, 43 (94%) achieved it. Major molecular responses, defined as a BCR-ABL to ABL transcript ratio of 0.1% or lower, occurred in 40 of 50 patients (80%) evaluable for this secondary endpoint, and deep molecular response (MR4.5), defined as a ratio less than 0.0032% or lower, occurred in 28 of 50 (55%).
Cardiovascular events, primarily hypertension, occurred in 25 patients (49%), 15 (29%) had grade 3-4 myelosuppression, and 5 patients (10%) developed cerebrovascular or vaso-occlusive disease.
In all, 43 patients (85%) needed treatment interruptions at some time and 45 (88%) needed dose reductions. The study was terminated in June 2014, a little more than a year after recruitment began.
The study was sponsored by MD Anderson Cancer Center with partial support from Ariad Pharmaceuticals. Coauthors Hagop Kantarjian, Elias Jabbour, and Jorge Cortes report receiving grants, research support, or serving as consultants to Ariad.
FROM LANCET HAEMATOLOGY
Key clinical point: The multi-tyrosine kinase inhibitor ponatinib (Iclusig) may not be suitable as first-line therapy for CML in chronic phase.
Major finding: Among evaluable patients, 94% had a complete cytogenic response by 6 months, but most also required treatment interruptions or dose reductions because of toxicities.
Data source: Single-arm phase II trial of 51 patients with recently diagnosed chronic myeloid leukemia in chronic phase.
Disclosures: The study was sponsored by MD Anderson Cancer Center with partial support from Ariad Pharmaceuticals. Coauthors Hagop Kantarjian, Elias Jabbour, and Jorge Cortes report receiving grants, research support, or serving as consultants to Ariad.
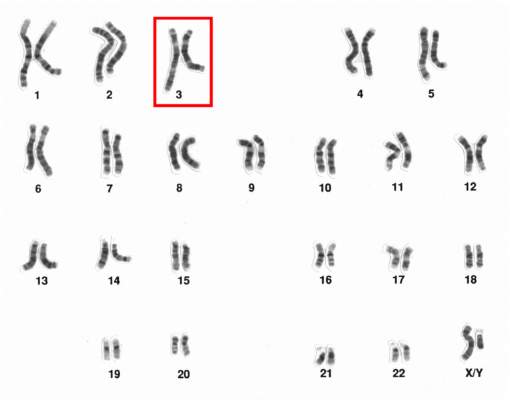
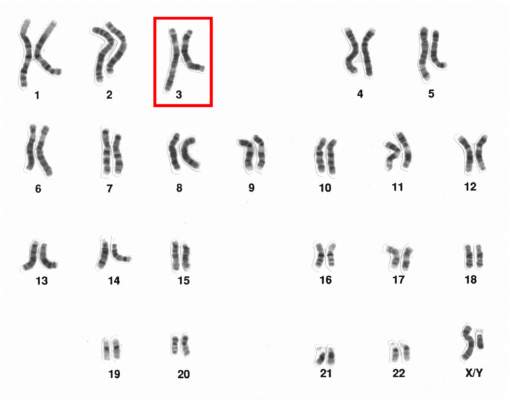
Chromosome 3 abnormalities linked to poor CML outcomes
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
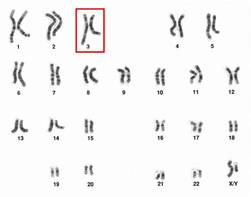
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities linked to poor CML outcomes
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Chromosome 3 abnormalities, specifically 3q26.2 rearrangements, were associated with treatment resistance and poor prognosis in patients with chronic myelogenous leukemia (CML), report Dr. Wei Wang and coauthors of the department of hematopathology at the University of Texas MD Anderson Cancer Center in Houston.
A study of 2,013 CML patients found that just 6% of those with 3q26.2 abnormalities achieved complete cytogenetic response during the course of tyrosine kinase inhibitor (TKI) treatment. Patients with other chromosome 3 abnormalities had a significantly better response rate of 42% the investigators found.
Additionally, patients with 3q26.2 chromosome rearrangements had significantly worse survival rates than those with abnormalities involving other chromosomes, with 2-year overall survival rates of 22% and 60%, respectively.
The lack of response to TKI treatment “raises the issue of how to manage these patients,” Dr. Wang and associates said in the report.
“TKIs themselves are not sufficient to control the disease with 3q26.2 abnormalities,” they added. “Intensive therapy, stem cell transplantation, or investigational therapy targeted to EVI1 should be considered,” concluded the authors, who declared that they had no competing financial interests.
Read the full article in Blood.
Variations in blood cancer survival across Europe

chemotherapy
Photo by Rhoda Baer
VIENNA—Results of the EUROCARE-5 study have revealed regional differences in survival for European patients with hematologic malignancies.
The data showed regional variations in 5-year relative survival rates for a number of cancers.
But the differences were particularly pronounced for leukemias, non-Hodgkin lymphomas (NHLs), and plasma cell neoplasms (PCNs).
Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy, presented these results at the 2015 European Cancer Congress (LBA 1).
Data from this study have also been published in several articles in the October 2015 issue of the European Journal of Cancer.
EUROCARE-5 includes records from 22 million cancer patients diagnosed between 1978 and 2007. The latest data encompass more than 10 million patients (ages 15 and older) diagnosed from 1995 to 2007 and followed up to 2008.
The data came from 107 cancer registries in 29 countries. The researchers estimated 5-year relative survival and trends from 1999 to 2007 according to region—Ireland/UK, Northern Europe, Central Europe, Southern Europe, and Eastern Europe.
“In general, 5-year relative survival—survival that is adjusted for causes of death other than cancer—increased steadily over time in Europe, particularly in Eastern Europe, for most cancers,” Dr Sant said.
“However, the most dramatic geographical variations were observed for cancers of the blood where there have been recent advances in treatment, such as chronic myeloid and lymphocytic leukemias, non-Hodgkin lymphoma and 2 of its subtypes (follicular and diffuse large B-cell lymphoma), and multiple myeloma. Hodgkin lymphoma was the exception, with smaller regional variations and a fairly good prognosis in most countries.”
Hodgkin lymphoma and NHL
Of all the hematologic malignancies, 5-year relative survival was highest for Hodgkin lymphoma, at 80.8% (40,625 cases). Five-year survival was 79.4% in Ireland and the UK, 85% in Northern countries, and 74.3% in Eastern Europe, which was significantly below the European average (P<0.0001).
For NHL, the 5-year relative survival was 59.4% (329,204 cases). Survival rates for NHL patients ranged from 49.7% in Eastern Europe to 63.3% in Northern Europe.
CLL/SLL
For chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), the 5-year relative survival was 70.4% (81,914 cases). CLL/SLL survival ranged from 58% in Eastern Europe to about 74% in Central and Northern Europe.
The researchers noted that between-country variations in CLL/SLL survival were high in all regions. Outliers that were significantly below the regional average were Austria (67%), Croatia (52%), and Bulgaria (45.5%).
PCNs
PCNs included multiple myeloma, plasmacytoma, and plasma cell leukemias. The 5-year relative survival for all PCNs was 39.2% (94,024 cases).
PCN survival rates were lowest in Eastern Europe (31.7%), slightly higher in the UK/Ireland (35.9%), and between 39.1% and 42% in the rest of Europe.
Myeloid leukemias
Of all the hematologic malignancies, 5-year relative survival was poorest for patients with acute myeloid leukemia (AML), at 17.1% (57,026 cases).
AML survival rates in Ireland/UK (15.0%) and Eastern Europe (13.0%) were significantly below the European average. But AML survival in Sweden, Belgium, France, and Germany was significantly higher than the average (P<0.005).
Five-year relative survival for chronic myeloid leukemia (CML) was 52.9% (17,713 cases).
Of all the hematologic malignancies, the survival gap between Eastern Europe and the rest of Europe was highest for CML. Five-year survival for CML patients was 33% in Eastern Europe and ranged from 51% to 58% in the rest of Europe.
The researchers also said there were striking survival variations by country in all areas. They found significant deviations from the regional average in Sweden (69.7%), Scotland (64.6%), France (71.7%), Austria (48.2%), Croatia (37.8%), Estonia (48.9%), Czech Republic (45.2%), and Latvia (22.1%).
“Results from EUROCARE can help to identify regions of low survival where action is needed to improve patients’ outcomes,” Dr Sant noted.
“Population-based survival information is essential for physicians, policy-makers, administrators, researchers, and patient organizations who deal with the needs of cancer patients, as well as with the issue of the growing expenditure on healthcare.” ![]()
chemotherapy
Photo by Rhoda Baer
VIENNA—Results of the EUROCARE-5 study have revealed regional differences in survival for European patients with hematologic malignancies.
The data showed regional variations in 5-year relative survival rates for a number of cancers.
But the differences were particularly pronounced for leukemias, non-Hodgkin lymphomas (NHLs), and plasma cell neoplasms (PCNs).
Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy, presented these results at the 2015 European Cancer Congress (LBA 1).
Data from this study have also been published in several articles in the October 2015 issue of the European Journal of Cancer.
EUROCARE-5 includes records from 22 million cancer patients diagnosed between 1978 and 2007. The latest data encompass more than 10 million patients (ages 15 and older) diagnosed from 1995 to 2007 and followed up to 2008.
The data came from 107 cancer registries in 29 countries. The researchers estimated 5-year relative survival and trends from 1999 to 2007 according to region—Ireland/UK, Northern Europe, Central Europe, Southern Europe, and Eastern Europe.
“In general, 5-year relative survival—survival that is adjusted for causes of death other than cancer—increased steadily over time in Europe, particularly in Eastern Europe, for most cancers,” Dr Sant said.
“However, the most dramatic geographical variations were observed for cancers of the blood where there have been recent advances in treatment, such as chronic myeloid and lymphocytic leukemias, non-Hodgkin lymphoma and 2 of its subtypes (follicular and diffuse large B-cell lymphoma), and multiple myeloma. Hodgkin lymphoma was the exception, with smaller regional variations and a fairly good prognosis in most countries.”
Hodgkin lymphoma and NHL
Of all the hematologic malignancies, 5-year relative survival was highest for Hodgkin lymphoma, at 80.8% (40,625 cases). Five-year survival was 79.4% in Ireland and the UK, 85% in Northern countries, and 74.3% in Eastern Europe, which was significantly below the European average (P<0.0001).
For NHL, the 5-year relative survival was 59.4% (329,204 cases). Survival rates for NHL patients ranged from 49.7% in Eastern Europe to 63.3% in Northern Europe.
CLL/SLL
For chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), the 5-year relative survival was 70.4% (81,914 cases). CLL/SLL survival ranged from 58% in Eastern Europe to about 74% in Central and Northern Europe.
The researchers noted that between-country variations in CLL/SLL survival were high in all regions. Outliers that were significantly below the regional average were Austria (67%), Croatia (52%), and Bulgaria (45.5%).
PCNs
PCNs included multiple myeloma, plasmacytoma, and plasma cell leukemias. The 5-year relative survival for all PCNs was 39.2% (94,024 cases).
PCN survival rates were lowest in Eastern Europe (31.7%), slightly higher in the UK/Ireland (35.9%), and between 39.1% and 42% in the rest of Europe.
Myeloid leukemias
Of all the hematologic malignancies, 5-year relative survival was poorest for patients with acute myeloid leukemia (AML), at 17.1% (57,026 cases).
AML survival rates in Ireland/UK (15.0%) and Eastern Europe (13.0%) were significantly below the European average. But AML survival in Sweden, Belgium, France, and Germany was significantly higher than the average (P<0.005).
Five-year relative survival for chronic myeloid leukemia (CML) was 52.9% (17,713 cases).
Of all the hematologic malignancies, the survival gap between Eastern Europe and the rest of Europe was highest for CML. Five-year survival for CML patients was 33% in Eastern Europe and ranged from 51% to 58% in the rest of Europe.
The researchers also said there were striking survival variations by country in all areas. They found significant deviations from the regional average in Sweden (69.7%), Scotland (64.6%), France (71.7%), Austria (48.2%), Croatia (37.8%), Estonia (48.9%), Czech Republic (45.2%), and Latvia (22.1%).
“Results from EUROCARE can help to identify regions of low survival where action is needed to improve patients’ outcomes,” Dr Sant noted.
“Population-based survival information is essential for physicians, policy-makers, administrators, researchers, and patient organizations who deal with the needs of cancer patients, as well as with the issue of the growing expenditure on healthcare.” ![]()
chemotherapy
Photo by Rhoda Baer
VIENNA—Results of the EUROCARE-5 study have revealed regional differences in survival for European patients with hematologic malignancies.
The data showed regional variations in 5-year relative survival rates for a number of cancers.
But the differences were particularly pronounced for leukemias, non-Hodgkin lymphomas (NHLs), and plasma cell neoplasms (PCNs).
Milena Sant, MD, of the Fondazione IRCCS Istituto Nazionale dei Tumori in Milan, Italy, presented these results at the 2015 European Cancer Congress (LBA 1).
Data from this study have also been published in several articles in the October 2015 issue of the European Journal of Cancer.
EUROCARE-5 includes records from 22 million cancer patients diagnosed between 1978 and 2007. The latest data encompass more than 10 million patients (ages 15 and older) diagnosed from 1995 to 2007 and followed up to 2008.
The data came from 107 cancer registries in 29 countries. The researchers estimated 5-year relative survival and trends from 1999 to 2007 according to region—Ireland/UK, Northern Europe, Central Europe, Southern Europe, and Eastern Europe.
“In general, 5-year relative survival—survival that is adjusted for causes of death other than cancer—increased steadily over time in Europe, particularly in Eastern Europe, for most cancers,” Dr Sant said.
“However, the most dramatic geographical variations were observed for cancers of the blood where there have been recent advances in treatment, such as chronic myeloid and lymphocytic leukemias, non-Hodgkin lymphoma and 2 of its subtypes (follicular and diffuse large B-cell lymphoma), and multiple myeloma. Hodgkin lymphoma was the exception, with smaller regional variations and a fairly good prognosis in most countries.”
Hodgkin lymphoma and NHL
Of all the hematologic malignancies, 5-year relative survival was highest for Hodgkin lymphoma, at 80.8% (40,625 cases). Five-year survival was 79.4% in Ireland and the UK, 85% in Northern countries, and 74.3% in Eastern Europe, which was significantly below the European average (P<0.0001).
For NHL, the 5-year relative survival was 59.4% (329,204 cases). Survival rates for NHL patients ranged from 49.7% in Eastern Europe to 63.3% in Northern Europe.
CLL/SLL
For chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), the 5-year relative survival was 70.4% (81,914 cases). CLL/SLL survival ranged from 58% in Eastern Europe to about 74% in Central and Northern Europe.
The researchers noted that between-country variations in CLL/SLL survival were high in all regions. Outliers that were significantly below the regional average were Austria (67%), Croatia (52%), and Bulgaria (45.5%).
PCNs
PCNs included multiple myeloma, plasmacytoma, and plasma cell leukemias. The 5-year relative survival for all PCNs was 39.2% (94,024 cases).
PCN survival rates were lowest in Eastern Europe (31.7%), slightly higher in the UK/Ireland (35.9%), and between 39.1% and 42% in the rest of Europe.
Myeloid leukemias
Of all the hematologic malignancies, 5-year relative survival was poorest for patients with acute myeloid leukemia (AML), at 17.1% (57,026 cases).
AML survival rates in Ireland/UK (15.0%) and Eastern Europe (13.0%) were significantly below the European average. But AML survival in Sweden, Belgium, France, and Germany was significantly higher than the average (P<0.005).
Five-year relative survival for chronic myeloid leukemia (CML) was 52.9% (17,713 cases).
Of all the hematologic malignancies, the survival gap between Eastern Europe and the rest of Europe was highest for CML. Five-year survival for CML patients was 33% in Eastern Europe and ranged from 51% to 58% in the rest of Europe.
The researchers also said there were striking survival variations by country in all areas. They found significant deviations from the regional average in Sweden (69.7%), Scotland (64.6%), France (71.7%), Austria (48.2%), Croatia (37.8%), Estonia (48.9%), Czech Republic (45.2%), and Latvia (22.1%).
“Results from EUROCARE can help to identify regions of low survival where action is needed to improve patients’ outcomes,” Dr Sant noted.
“Population-based survival information is essential for physicians, policy-makers, administrators, researchers, and patient organizations who deal with the needs of cancer patients, as well as with the issue of the growing expenditure on healthcare.” ![]()
Mutation testing aids CML treatment decisions
Patients with Ph+ CML-CP (Philadelphia chromosome–positive chronic myeloid leukemia, chronic phase) who fail to achieve and maintain treatment response at key milestones should be considered for mutation screening, based on data from the DASISION trial.
Patients with mutations had poor outcomes and high rates of treatment discontinuation in an extended 4-year minimum follow-up of patients in the trial; 14 of 17 dasatinib-treated patients and 14 of 18 imatinib-treated patients with mutations discontinued treatment. The primary reason for treatment discontinuation was protocol-defined disease progression (dasatinib, n = 11; imatinib, n = 8); patients with mutations accounted for 61% of discontinuations on dasatinib (n = 11/18) and 42% on imatinib (n = 8/19).
“With the introduction of generic imatinib into the market in 2016, choosing the most appropriate second-line tyrosine-kinase inhibitor for patients, based on factors such as mutation status, will become increasingly important,” Dr. Tim Hughes of the South Australian Health and Medical Research Institute in Adelaide and his colleagues wrote. Having the option to choose the most suitable second-line therapy may ensure improved outcomes and decreased health care costs.
In the DASISION (Dasatinib vs. Imatinib Study in Treatment-Naive CML-CP) trial, all participants had newly diagnosed Ph+ CML-CP; they were treated with dasatinib (n = 259) or imatinib (n = 260) and followed for a minimum of 3 years (Leukemia. 2015 Sep;29[9]:1832-8). Dr. Hughes and his colleagues conducted a retrospective study of the patients who were potentially at a higher risk for developing mutations. This included patients on treatment who had at least one clinically relevant event – no confirmed complete cytogenetic response (cCCyR) within 12 months, no major molecular response (MMR) within 12 months; a fivefold increase in BCR-ABL1 transcript levels with loss of MMR; loss of CCyR – and/or who discontinued treatment for any reason.
Screening identified only a small number of patients with mutations (dasatinib, n = 17; imatinib, n = 18). Those on dasatinib had a narrower spectrum of mutations (4 sites for dasatinib vs. 12 sites for imatinib), fewer phosphate-binding loop mutations (1 mutation for dasatinib vs, 9 mutations for imatinib), and fewer multiple mutations (1 patient on dasatinib vs. 6 patients on imatinib).
However, patients on dasatinib had a greater occurrence of T315I mutations (11 patients on dasatinib vs. 0 patients on imatinib). The researchers hypothesized that this finding resulted from differences in competitive advantage between mutant clones. For example, P-loop mutations Y253F, E255K were found to have higher transformation potency and proliferation rates, compared with T315I, even in the absence of BCR-ABL1 inhibitors. If one assumes that imatinib has lower activity than dasatinib against these mutations, then mutant clones with select P-loop mutations might expand more rapidly than clones with the T315I mutation when exposed to imatinib.
Consistent with this idea, T315I is less common than all P-loop mutations in CML-CP patients with imatinib resistance. In addition, dasatinib suppresses P-loop mutations to a greater extent than does T315I; therefore, T315I may be able to develop during dasatinib treatment with relatively little competition from rapidly proliferating clones.
“Dasatinib, nilotinib, bosutinib, and ponatinib have enabled many patients, including those with mutations, to overcome imatinib resistance; however, each lack[s] efficacy against a small number of different leukemic clones, and all except ponatinib lack efficacy against T315I,” the researchers wrote.
The study was sponsored by Bristol-Myers Squibb. Dr. Hughes reported receiving honoraria and research funding from ARIAD, the maker of ponatinib; Bristol-Myers Squibb, the maker of dasatinib; and Novartis, the maker of imatinib.
Patients with Ph+ CML-CP (Philadelphia chromosome–positive chronic myeloid leukemia, chronic phase) who fail to achieve and maintain treatment response at key milestones should be considered for mutation screening, based on data from the DASISION trial.
Patients with mutations had poor outcomes and high rates of treatment discontinuation in an extended 4-year minimum follow-up of patients in the trial; 14 of 17 dasatinib-treated patients and 14 of 18 imatinib-treated patients with mutations discontinued treatment. The primary reason for treatment discontinuation was protocol-defined disease progression (dasatinib, n = 11; imatinib, n = 8); patients with mutations accounted for 61% of discontinuations on dasatinib (n = 11/18) and 42% on imatinib (n = 8/19).
“With the introduction of generic imatinib into the market in 2016, choosing the most appropriate second-line tyrosine-kinase inhibitor for patients, based on factors such as mutation status, will become increasingly important,” Dr. Tim Hughes of the South Australian Health and Medical Research Institute in Adelaide and his colleagues wrote. Having the option to choose the most suitable second-line therapy may ensure improved outcomes and decreased health care costs.
In the DASISION (Dasatinib vs. Imatinib Study in Treatment-Naive CML-CP) trial, all participants had newly diagnosed Ph+ CML-CP; they were treated with dasatinib (n = 259) or imatinib (n = 260) and followed for a minimum of 3 years (Leukemia. 2015 Sep;29[9]:1832-8). Dr. Hughes and his colleagues conducted a retrospective study of the patients who were potentially at a higher risk for developing mutations. This included patients on treatment who had at least one clinically relevant event – no confirmed complete cytogenetic response (cCCyR) within 12 months, no major molecular response (MMR) within 12 months; a fivefold increase in BCR-ABL1 transcript levels with loss of MMR; loss of CCyR – and/or who discontinued treatment for any reason.
Screening identified only a small number of patients with mutations (dasatinib, n = 17; imatinib, n = 18). Those on dasatinib had a narrower spectrum of mutations (4 sites for dasatinib vs. 12 sites for imatinib), fewer phosphate-binding loop mutations (1 mutation for dasatinib vs, 9 mutations for imatinib), and fewer multiple mutations (1 patient on dasatinib vs. 6 patients on imatinib).
However, patients on dasatinib had a greater occurrence of T315I mutations (11 patients on dasatinib vs. 0 patients on imatinib). The researchers hypothesized that this finding resulted from differences in competitive advantage between mutant clones. For example, P-loop mutations Y253F, E255K were found to have higher transformation potency and proliferation rates, compared with T315I, even in the absence of BCR-ABL1 inhibitors. If one assumes that imatinib has lower activity than dasatinib against these mutations, then mutant clones with select P-loop mutations might expand more rapidly than clones with the T315I mutation when exposed to imatinib.
Consistent with this idea, T315I is less common than all P-loop mutations in CML-CP patients with imatinib resistance. In addition, dasatinib suppresses P-loop mutations to a greater extent than does T315I; therefore, T315I may be able to develop during dasatinib treatment with relatively little competition from rapidly proliferating clones.
“Dasatinib, nilotinib, bosutinib, and ponatinib have enabled many patients, including those with mutations, to overcome imatinib resistance; however, each lack[s] efficacy against a small number of different leukemic clones, and all except ponatinib lack efficacy against T315I,” the researchers wrote.
The study was sponsored by Bristol-Myers Squibb. Dr. Hughes reported receiving honoraria and research funding from ARIAD, the maker of ponatinib; Bristol-Myers Squibb, the maker of dasatinib; and Novartis, the maker of imatinib.
Patients with Ph+ CML-CP (Philadelphia chromosome–positive chronic myeloid leukemia, chronic phase) who fail to achieve and maintain treatment response at key milestones should be considered for mutation screening, based on data from the DASISION trial.
Patients with mutations had poor outcomes and high rates of treatment discontinuation in an extended 4-year minimum follow-up of patients in the trial; 14 of 17 dasatinib-treated patients and 14 of 18 imatinib-treated patients with mutations discontinued treatment. The primary reason for treatment discontinuation was protocol-defined disease progression (dasatinib, n = 11; imatinib, n = 8); patients with mutations accounted for 61% of discontinuations on dasatinib (n = 11/18) and 42% on imatinib (n = 8/19).
“With the introduction of generic imatinib into the market in 2016, choosing the most appropriate second-line tyrosine-kinase inhibitor for patients, based on factors such as mutation status, will become increasingly important,” Dr. Tim Hughes of the South Australian Health and Medical Research Institute in Adelaide and his colleagues wrote. Having the option to choose the most suitable second-line therapy may ensure improved outcomes and decreased health care costs.
In the DASISION (Dasatinib vs. Imatinib Study in Treatment-Naive CML-CP) trial, all participants had newly diagnosed Ph+ CML-CP; they were treated with dasatinib (n = 259) or imatinib (n = 260) and followed for a minimum of 3 years (Leukemia. 2015 Sep;29[9]:1832-8). Dr. Hughes and his colleagues conducted a retrospective study of the patients who were potentially at a higher risk for developing mutations. This included patients on treatment who had at least one clinically relevant event – no confirmed complete cytogenetic response (cCCyR) within 12 months, no major molecular response (MMR) within 12 months; a fivefold increase in BCR-ABL1 transcript levels with loss of MMR; loss of CCyR – and/or who discontinued treatment for any reason.
Screening identified only a small number of patients with mutations (dasatinib, n = 17; imatinib, n = 18). Those on dasatinib had a narrower spectrum of mutations (4 sites for dasatinib vs. 12 sites for imatinib), fewer phosphate-binding loop mutations (1 mutation for dasatinib vs, 9 mutations for imatinib), and fewer multiple mutations (1 patient on dasatinib vs. 6 patients on imatinib).
However, patients on dasatinib had a greater occurrence of T315I mutations (11 patients on dasatinib vs. 0 patients on imatinib). The researchers hypothesized that this finding resulted from differences in competitive advantage between mutant clones. For example, P-loop mutations Y253F, E255K were found to have higher transformation potency and proliferation rates, compared with T315I, even in the absence of BCR-ABL1 inhibitors. If one assumes that imatinib has lower activity than dasatinib against these mutations, then mutant clones with select P-loop mutations might expand more rapidly than clones with the T315I mutation when exposed to imatinib.
Consistent with this idea, T315I is less common than all P-loop mutations in CML-CP patients with imatinib resistance. In addition, dasatinib suppresses P-loop mutations to a greater extent than does T315I; therefore, T315I may be able to develop during dasatinib treatment with relatively little competition from rapidly proliferating clones.
“Dasatinib, nilotinib, bosutinib, and ponatinib have enabled many patients, including those with mutations, to overcome imatinib resistance; however, each lack[s] efficacy against a small number of different leukemic clones, and all except ponatinib lack efficacy against T315I,” the researchers wrote.
The study was sponsored by Bristol-Myers Squibb. Dr. Hughes reported receiving honoraria and research funding from ARIAD, the maker of ponatinib; Bristol-Myers Squibb, the maker of dasatinib; and Novartis, the maker of imatinib.
FROM LEUKEMIA
Key clinical point:Mutation testing may aid treatment selection in patients with chronic myeloid leukemia (CML) when selecting an alternative therapy because of treatment failure.
Major finding: Patients with mutations accounted for 61% of discontinuations on dasatinib (n = 11/18) and 42% on imatinib (n = 8/19).
Data source: A retrospective analysis of the DASISION trial results of 259 patients treated with dasatinib and 260 treated with imatinib.
Disclosures: The study was sponsored by Bristol-Myers Squibb. Dr. Hughes reported receiving honoraria and research funding from ARIAD, the maker of ponatinib; Bristol-Myers Squibb, the maker of dasatinib; and Novartis, the maker of imatinib.
Blood cancer drugs set to be removed from CDF

Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Medical Roundtable: Practical Management of Chronic Myelogenous Leukemia
Moderator: Matt Kalaycio, MD1
Discussants: Michael Mauro, MD2; Michael Deininger, MD, PhD3
From Cleveland Clinic, Cleveland, OH1; Memorial Sloan Kettering Cancer Center, New York, NY2; Huntsman Cancer Institute, Salt Lake City, UT3
Address for correspondence: Matt Kalaycio, MD, Cleveland Clinic Main Campus, Mail Code R32, 9500 Euclid Avenue, Cleveland, OH 44195
E-mail: [email protected]
Biographical sketch:

Dr. Kalaycio has been published in numerous scientific publications including Bone Marrow Transplantation, Journal of Clinical Oncology, and Leukemia. He also is the editor of a book on leukemia and co-editor of a book on clinical malignant hematology. His research interests focus on testing new treatments for leukemia.
Dr. Kalaycio received his degree from West Virginia University School of Medicine in Morgantown. He completed his residency in internal medicine at Mercy Hospital of Pittsburgh and fellowships in hematology and medical oncology and bone marrow transplantation at Cleveland Clinic.

Michael Deininger, MD, PhD, is Professor and Chief of Hematology and Hematologic Malignancies for the Department of Internal Medicine and for the Huntsman Cancer Institute (HCI) at the University of Utah. He is an HCI investigator and member of the Experimental Therapeutics program. He has extensive experience treating patients with blood cancers, including chronic myeloid leukemia (CML) and myeloproliferative neoplasms, a group of blood cancers related to leukemia.
Dr
Dr. Deininger’s scientific focus is leukemia, specifically myeloproliferative neoplasms including chronic myeloid leukemia (CML). As a clinician-scientist with a translational research focus Dr. Deininger is heading an extramurally funded research laboratory that is dedicated to the study of signaling pathways, drug resistance and new molecular therapies in leukemia. Dr. Deininger’s work describing the selective effects of imatinib on CML cells provided the rationale for clinical trials that led to the approval of Gleevec as the first molecularly-based therapy for leukemia. Current work in his lab is focused on understanding the role of the bone marrow microenvironment in leukemia drug resistance, discovering novel therapeutic targets and developing more specific signal transduction inhibitors. Dr. Deininger’s work encompasses more than 170 articles in the peer-reviewed literature, including journals like Blood, Journal of Clinical Investigation and the New England Journal of Medicine. He has co-authored more than 10 book chapters, with contributions in leading textbooks such as deVita’s Principles of Oncology. He is a regular speaker at major international scientific meetings, such as the American Society of Hematology and the European Hematology Association and a peer reviewer for journals like Nature Genetics and Cancer Cell. His honors include the Alexandra Kefalides Prize for Leukemia Research and membership on the Editorial Board of Blood, the leading journal in Hematology. Dr. Deininger was named among the world's Highly Cited Researchers by Thomson Reuters in 2014.
Dr. Kalaycio: My name is Matt Kalaycio and I'm the Chairman of the Department of Hematology and Medical Oncology at the Cleveland Clinic. Today I’m joined by Drs. Mike Deininger, Division Chief of Hematology and Hematologic Malignancies at the University of Utah Huntsman Cancer Institute and Michael Mauro, Myeloproliferative Neoplasm Program Director at Memorial Sloan Kettering Cancer Center, New York, New York – together we will discuss practical issues surrounding CML diagnosis, management and treatment options including TKIs and investigational therapies.
Initial Presentation: Assessment & Treatment Options
Dr. Kalaycio: Often, the patient with chronic myelogenous leukemia (CML) gets admitted to the hospital, or an emergency consult is called, because the white count is up for a concern of leukemia. The treatment team sees the differential circulating blasts, and they worry about acute leukemia. Then the hematologist comes and needs to make a decision about whether or not to do a bone marrow biopsy.
When the patient presents in such a manner, I often see that bone marrow biopsies are not performed. I would like to start by asking where you both stand with regard to the necessity of a bone marrow biopsy at the time of diagnosis. I'll start with Dr. Deininger.
Dr. Deininger: I would strongly be in favor of a diagnostic biopsy as well as a smear. The reason is that I think this is the one and only chance to get a clear disease classification into chronic phase, accelerated phase, and blast crisis.
The third may be rare. There are occasional patients who have sheets of blasts who would not be seen on just a differential. For these patients, of course, the treatment decisions are going to be very different compared to a patient in chronic phase.
I think this is an opportunity that shouldn't be missed and I would always recommend that.
Dr. Kalaycio: Dr. Mauro.
Dr. Mauro: I couldn't concur more. With so much focus on the change in disease status from presentation to early response, I think understanding the scope of the disease at diagnosis, including the bone marrow, is essential and either revealing or reassuring. Although we focus mostly on early molecular response, when expectations go awry, not having as full a picture of the disease prior to treatment leaves you less informed about best treatment.
Making sense of accelerated phase features is a good example; the difference in outcomes between chronic phase patients and those who have morphologic features of accelerated phase—with or without cytogenetic features (clonal evolution)—compared with those with clonal evolution can only drive initial and long term treatment decisions. Without cytogenetics and morphology together, such key pieces of the puzzle are missing; it is worth it for the practitioner and patient to have all the data in hand at all times.
Dr. Kalaycio: Once the diagnosis has been made and the biopsy was not done and now you're seeing the patient having either been on hydroxyurea for a month or having had tyrosine kinase inhibitor (TKI) for a month, do you bother with the bone marrow biopsy at that point?
Dr. Deininger: Well, that is a very good question. I think we would probably still do it most of the time. I think it's very clear that the information you can gather from that is less valuable. I think one should try to make up for the omissions as much as possible. We would still go for it.
Dr. Kalaycio: Interesting. The other thing that happens a lot is a patient will be started on hydrea while you're waiting for BCR-ABL to return either by fluorescence in situ hybridization or quantitative polymerase chain reaction (PCR).
I wonder if you have your own set of internal guidelines for when hydroxyurea should be employed or not.
Dr. Mauro: If you look back at some of the original imatinib trials—the phase I trials—patients initiated TKI with higher blood counts, a median around 25,000 and up to 200,000, and responded with excellent tolerance.1 I think there's a bit of overapplication of hydroxyurea early in CML, prior to initiation of TKI, which often confounds and complicates the early myelosuppressive toxicity of TKIs. With use of more potent TKIs, there may be greater amounts of myelosuppression as the leukemic clone may clear faster.
I think we may get ourselves into a bit of a bind by overusing hydroxyurea and then needing to hold and lower TKI dosages quickly when that may not have been necessary had we simply deployed the TKI sooner.
My general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. – Michael Mauro, MDMy general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. The other question that often comes up is about tumor lysis and hydration, and how closely these need to be managed. The likelihood of tumor lysis is low in CML treated with TKIs, but more frequent early labs and good hydration are always the right thing to do; better safe than sorry!
Dr. Deininger: I second what Michael just said. To your question about whether we have an algorithm, I have to admit we don't. I think it's up to the discretion of the treating physician to initiate hydroxyurea or not.
Dr. Kalaycio: Sure. When you start hydroxyurea, do you routinely add allopurinol?
Dr. Deininger: We tend to do that. I know that some people think it's unnecessary. It's such a low-risk and low-cost intervention that I think that it's hard to get anything wrong here.
Dr. Mauro: Right, we tend to do the same. Whether we need to or not is a different question, I suppose.
Dr. Kalaycio: One more thing about the initial presentation and assessment of a patient with what you think might be a myeloproliferative disorder, CML, how important do you find it, Dr. Mauro, to measure splenomegaly and calculate a risk score?
Dr. Mauro: I think it's very useful information. The spleen size factors heavily into the risk score and the risk score does forecast response to a degree. We've looked at calculation of risk score in recent large observational studies and it is under-reported and underutilized. It winds up being useful for two reasons. One, it does set expectations for response, and US treatment guidelines (National Comprehensive Cancer Network [NCCN] guidelines) note that treatment choice may be different for low versus high risk Sokal score.2
I think the second and most intriguing reason to assess Sokal risk seems now to be the impact that risk score has on the ability to proceed to a treatment-free remission (TFR). There appear to be differences in outcomes in patients with high-risk versus low-risk disease despite both having what is required to proceed to TFR in trials, namely consistent and deep molecular remission over a number of years.
Given these implications, we really ought to be gathering the initial risk stratification and quantifying spleen size. It's an important part of our initial assessment.
Dr. Kalaycio: Dr. Deininger?
Dr. Deininger: I think there's a lot of agreement today. I absolutely think that measuring the spleen size ascertains that you've got all these diagnostic parameters available. I think that should be part of the initial evaluation and work-up and will allow some prognostication.
Testing, Risk Factors & Considerations of Treatment: Nilotinib & Dasatinib
Dr. Kalaycio: I'm starting with the less controversial questions to begin with. I think the next set have the potential for some more controversy. Before we leave the initial assessment of CML, do either of you have observations regarding referrals that you get about which you would like to either dispel myths or remind practitioners about best practices in patients newly diagnosed with CML?
I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. – Michael Deininger, MD, PhDDr. Mauro: I can mention one thing. I think when thinking about initial molecular diagnostic results, it is important to point out that testing should screen broadly for different fusions, namely P190 and P210, or variant (p230) transcripts. There are rare patients in chronic phase with non-p210 fusion who need to be followed with specific PCR. On this same topic, the measurement of transcripts at presentation (ie, before treatment) has become quite important and whereas formerly had not been emphasized, presently all patients should have ”baseline” transcript levels.
Dr. Deininger: I think one issue that comes up once in a while is that spleen measurements are done by ultrasound. Technically it's more accurate, but all the clinical risk scores and the prognostication is based on the old fashioned—but probably highly inaccurate—technology of palpation. This is what counts, this is the value to document. I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. Getting a good handle of those risk factors at diagnosis is also important.
Dr. Kalaycio: I think that's a very important point. That's where I was going to go next with this conversation. I was going to avoid the conversation about which TKI to choose as initial treatment. I think that's a debate unto itself.
I would like to ask you how you might assess cardiovascular risk before placing a patient on nilotinib. You got to that a little bit, Dr. Deininger. Could you expand on what else you might review as far as whether or not you feel a patient is a good candidate to start on nilotinib?
Dr. Deininger: Specifically, with regard to nilotinib, we would always get a baseline electrocardiogram. We would do a clinical exam. We would not do an echocardiogram just routinely in the absence of a cardiovascular history or any clinical evidence for heart failure or other cardiovascular issues.
We've adopted the practice of doing a lipid panel. Of course we would include fasting glucose as well. Some of these recommendations are probably somewhat on the soft side, because it's not yet clear what to do with the information.
On the other hand, I think for a patient who is being considered for nilotinib one wants to make sure that one really does the best to minimize the cardiovascular risk factors. Of course that would include smoking history and taking blood pressure and making sure that these risk factors are controlled.
If people have a presentation that is really out of whack in terms of their risk factor management, I would send them to an internist or even a cardiologist to help me optimize the cardiovascular prevention strategy.
Dr. Kalaycio: Great. Similarly, Dr. Mauro, how do you assess pulmonary risk before placing a patient on dasatinib?
Dr. Mauro: I think here we are focused on the less frequent and also less well understood potential toxicity of pulmonary hypertension, coupled with the more common risk of pleural and pericardial effusions.
I'm not sure how much we've learned in clinical studies looking at baseline chest X-rays or timing of X-rays during treatment. I think our best tool in the prevention and management of pleural and pericardial effusions is full discussion with patients about risk and what to look for, attention to any and all symptoms, and appropriate deployment of diagnostics as indicated.
It's interesting to consider whether baseline echocardiography for measurement of pulmonary pressures is warranted. I would say now we're on probably somewhat softer ground, first because on routine echocardiogram pulmonary pressure can't be measured readily unless there is some valvular regurgitation. As well, it is stated that pulmonary hypertension is only properly diagnosed by right heart catheterization. While I'm tempted to do routine echocardiogram studies, I think that such a recommendation still may be perhaps the realm of a clinical study. We need to explore that further. I think with dasatinib there may be certain patients at higher risk, although the data are somewhat limited. There seem to be certain conditions potentially associated with more pleural and pericardial toxicity, including cardiovascular disease and autoimmune disease. There may be circumstances during treatment—lymphocytosis, for example—that may be associated with greater risk. I think expectant management may still be the right approach and echocardiography and more aggressive diagnostics be reserved for patients in whom there might be much more clinical consequence.
Dr. Kalaycio: I'd like to pursue that a little bit further because sometimes the patients will come to us having already had an echocardiogram that may actually show some mild pulmonary hypertension and maybe they've got significant cardiovascular risk factors where you would otherwise be thinking about using dasatinib. Here's someone with pulmonary hypertension, at least by echocardiographic criteria, would that be enough to dissuade you from the use of dasatinib?
Dr. Mauro: I think it would certainly require significant consideration, understanding what the basis of the pulmonary hypertension is for that patient, and risk with adding dasatinib. I think the good news is the low incidence and the reversibility for the most part of dasatinib-associated pulmonary hypertension.
Again, I think the mechanism of action and the pathophysiology isn't completely understood, although there is the intriguing notion that imatinib has been reported to potentially mitigate pulmonary hypertension whereas dasatinib triggers it—a ”closed loop” if you will and an area ripe for research.
I would probably think that a patient with preexisting pulmonary hypertension in the new diagnosis setting might be the kind of patient for whom you really might weigh the pluses versus the minuses of a second generation TKI versus imatinib.
After Evaluation & Diagnosis: Following the Patient
Dr. Kalaycio: I agree. We have fully evaluated our patient and we've made the diagnosis. Now, we start therapy with a TKI based on patient risk profile and side effect potentials. It's time to follow the patient and determine next steps.
Current guidelines3,4 suggest monitoring quantitative PCR after 3 months of therapy and to gauge response. Dr. Deininger, how do you interpret and act on those results following the first 3 months of therapy?
Dr. Deininger: I think what you're getting at is the 10% mark that is a highly predictive value in terms of subsequent achievement of major molecular response and also overall progression free and overall survival.
We'll certainly get this data point. Then we'll put it in a clinical context. I think this context needs to take into account the initial BCR-ABL transcript level. I think Dr. Mauro mentioned that they always determine that. I think that is really good advice, so you can make a comparison with the diagnostic value.
One scenario, of course, is the patient is well below 10% and then things are just in the green range and you would wave people through and reassess in 3 months. If people are very high, I think then you have to ask yourself why that's the case.
Some people have a lot of toxicity issues. For example, they may not have been able to take the required amount of medication. It's not always easy to clearly distinguish between resistance and intolerance.
No matter what, I think it's going to be critical to consider a potential change in treatment if people are in the 70%—80% range. In my mind, there's a gray area.
These are the people who are maybe between 10%—20%. Here, I think this initial value can really help. If there's a significant reduction compared to baseline, I would not necessarily change at that point.
If there is literally no change compared to baseline, I would strongly consider a change unless I am concerned about other issues like noncompliance or drug interactions. What I'm trying to say in a rather long-winded way is that the 10% value shouldn't be seen as a dogma.
It still needs to be placed in a clinical context. One should not rush to any conclusions. Ten percent, 11%, and 9% are identical values in the world of PCR testing. One should not over-interpret that.
Dr. Kalaycio: I think that you're making an important point about absolutes in the interpretation of these tests. Dr. Mauro, how do you interpret the 3-month data that comes back?
Dr. Mauro: I agree with my colleague on the approach that it has to be put in a clinical context. I think what we've learned is the importance of the starting value and relative reduction and to not consider response milestones as black and white guides.
I think there are certain scenarios that require some caution. The patient who is on imatinib who is close but has not reached the landmark may be different that a similar patient on dasatinib or nilotinib who has not had significant reduction—that's probably a more pressing situation.
It's ironic that guidelines—maybe because of lack of options—don't encourage us to think about changing therapy early in someone who hasn't met milestones in 3 months when they've been put on a more potent agent.
I also think that careful consideration is needed regarding treatment intensity and the impact of interruptions, in conjunction with all the other facets—the rate of change, the absolute change from the patient's baseline, the starting response level and the timing of the PCR. We need to look at the actual day it's performed. If it's really not 3 months of therapy, we can misjudge.
Dr. Kalaycio: Right. Now, as you're monitoring patients, Dr. Mauro, at what point would you consider testing for kinase domain mutations?
Dr. Mauro: I think we have pretty good guidance from studies to date regarding when mutation testing is indicated and of higher yield. It's generally earlier on into treatment when we have clinical scenarios that are of greater risk, namely failure to achieve cytogenetic responses. In such scenarios if mutation testing informs treatment decision making it is very helpful.
Patients who have defined themselves as not achieving early molecular response—which we discussed earlier—especially someone who's had a second-generation TKI, warrants mutation testing in my view. Patients who don't achieve classical cytogenetic response landmarks at 6 and 12 (or 18) months, who thus have a higher residual volume of disease and perhaps as a function more clonal instability, I think also warrant attention.
I think we run into trouble when we start to, in an overly critical manner, assess patients’ longer term and deeper molecular response trajectories. Mutation testing becomes difficult or impossible for patients who are at or near major molecular remission (MMR). Mutation yield is generally very low for patients who have not lost MMR and also likely those with small volume of change around the MMR threshold. Looking ahead, I think further investigation into early time points and the setting of minimal residual disease may yield data to be able to predict potential resistance earlier than observing it clinically.
Dr. Kalaycio: Right. Dr. Deininger, if you're monitoring a patient and he’s missing milestones and you do obtain a mutation analysis and find a T315I mutation, do you offer that patient an opportunity to see how well they're going to do with ponatinib, or do you refer that patient to your transplant team for consideration of transplant as soon as a donor is available?As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated. – Matt Kalaycio, MD
Dr. Deininger: I think we do the first and half of the second. We would certainly offer a trial of ponatinib or, if possible, a clinical trial unless there are insurmountable contraindications.
I don't really think there are any such circumstances with optimized cardiovascular management at the same time, and we may involve a cardiologist at that point to help us if there are cardiovascular risk factors.
We would also do a referral to a transplant center unless the patient is as per performance status, just not a transplant candidate or is too old. Otherwise, we would always make a referral, but we would not pursue a transplant as the first modality, provided that the patient is in chronic phase. Progression to accelerated phase or blastic phase would be looked at totally differently. Here we would certainly put people on a transplant course.
Dr. Kalaycio: I agree. As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated.
As the two of you know, we're doing fewer and fewer transplants for CML these days. The results we're getting in folks who are failing all of these agents are not as good as they used to be when we were transplanting in the first 3 months of a new diagnosis. Bone marrow transplant is something to defer for as long as possible, at least in my mind.
TKI Dosage Considerations
Dr. Kalaycio: Now we touched a little bit on the side effects of nilotinib and dasatinib. We've talked about progressive disease, perhaps with or without mutations. Dr. Mauro, could you comment on where you think bosutinib might play a role in patients with relapsed or progressive CML?
Dr. Mauro: I think bosutinib remains an underutilized TKI. It is potent and thus offers good salvage activity. I think it offers a fairly distinct side effect profile making it a good choice for cases of intolerance. I think we've learned more about how to initiate bosutinib and manage its side effects; one element of this is that current trials start at slightly lower doses of bosutinib to avoid some of the early gastrointestinal toxicity.
Bosutinib continues to seek a place as a first-line therapy. I think this may be worthwhile as we are looking for the right balance of safety and side effect risk to maximize early response. Regarding the salvage setting—intolerance and resistance to other agents—I find myself speaking to patients about bosutinib as a potent and viable alternative. Bosutinib offers a similar spectrum of activity as dasatinib but can allow us to avoid cardiovascular toxicity and/or allow clearance of pleural and pericardial toxicity occurring with dasatinib.
The one place I would say bosutinib may not hold up as strongly to its competition would be as a third-line therapy after failure of a second-generation TKI. I think while there is reasonable activity with bosutinib in this setting of somewhat fairly drug-resistant CML, the performance of ponatinib is better. There is likely often a struggle with the long-term safety question of ponatinib, making some feel that a trial of bosutinib is often a worthwhile, logical, or even necessary step before ponatinib. My concerns about this approach are related to treatment decision-making based on safety and theoretical risk of adverse effects rather than efficacy and risk of progression; these have to be weighed carefully and appropriately against each other.
Dr. Kalaycio: That's an interesting perspective. You mentioned starting with a lower dose. Dr. Deininger, I'd like to ask you your thoughts about dose modifications of the TKIs in general. It used to be anathema to either start with a low dose or to maintain patients on a lower dose.
I'm aware of at least some data suggesting that those patients who tolerate these TKIs poorly particularly with regard to myelosuppression can be treated long term at lower than typically prescribed doses without adverse effect. What are your thoughts surrounding dose modification of the TKIs?
Dr. Deininger: I would stratify according to TKIs. I think imatinib at 400 mg is probably just rightly dosed, maybe not even at the optimal dose. It could be 600 mg. If you go to 300 mg, that is still acceptable.
If you go to 200 mg, I think I would make double sure that I monitor patients very frequently. People should be, in my mind, in a major molecular response to treat them with 200 mg long term. With the second-generation TKIs, it's a little different.
I cannot really speak to bosutinib because I'm not aware of a lot of data on those modifications. What seems to be clear from dasatinib is that the initial recommended dose of 100 mg is quite high, especially in older people.
I believe that's been corroborated by a study that hasn't been published yet. Apparently the drug excretion in the older individuals is quite a bit slower than in younger people. I think in our practice and in other people's practices as well, about 50% of those people end at doses substantially lower than 100 mg, maybe 50 mg, maybe 40 mg, some even 20 mg per day.
I think in the case of dasatinib you have a lot of maneuvering space. You can adjust the dose according to tolerability and molecular response. With nilotinib I think it's kind of similar. We have a few patients who are on maybe 150 mg twice a day because they have issues in terms of side effects at the higher doses.
I should again thoroughly qualify that by saying if you give up through dose reductions the goal of a major molecular response, then I think you really have to think about the strategy in general because at that point, I think you basically say "this patient is not ever going to be reaching a safe haven and is not tolerating inhibitors well enough to get him there."
That's quite a significant statement to make. Myelosuppression is frequently a situation where you cannot deliver enough dose but you also don't get a good response, whereas other side effects like pleural effusions or excessive fluid retention may well be cause for dose reduction and yet responses may be acceptable. Maybe Dr. Mauro could chime in here and share his experience.
Dr. Mauro: I agree. I think some patients, based on their disease status, may not tolerate the dose that is going to—at least in theory—get them to a deep molecular remission. The biggest problem we often face is myelosuppression. Some patients may have indolent disease and still have a good outcome, at least in the short to medium term, with lower doses of drug. In general I try to work through myelosuppression, especially early, as the combination of myelosuppression and suboptimal or response failure is dangerous.
I think treatment shouldn't be too chaotic and change too much, if possible. That was a very nice summary of the way we view the doses. There's still a fair bit of flexibility amongst the TKI doses, although I don't advocate for starting low and titration up.
In general I think, with regard to optimal TKI dose, we might have overshot; for example, with dasatinib. I think we might have overshot with bosutinib. Trials ongoing now initiate bosutinib at 400 mg rather than 500 mg. That's what I was alluding to.
Treatment Free Remission: Approaches
Dr. Kalaycio: Very interesting discussions. As we wind down the conversation, I want to get to the idea of stopping therapy. We're all aware of data that suggest it's at least possible for a proportion of patients to stop treatment.5
However, the guidelines3,4 such as they are, suggest that stopping should not be done outside the context of a clinical trial. I think most of us would agree with that. I wonder in a practical sense, Dr. Mauro, in your practice, do you have your own set of internal criteria for stopping a TKI and observing patients in the absence of any therapy?
Dr. Mauro: I think I try to incorporate all the experience we have to date when considering this question. That being said I'm a little hesitant to agree entirely with the current thinking regarding retreatment during a Treatment Free Remission trial, which is waiting until patients lose MMR in order to resume treatment but agree it might be hard to retreat based on lesser degrees of molecular relapse. I just worry a little bit about that amount of proliferation without treatment.
On the other hand I think we're coming to the realization that TFR may be feasible in patients who are simply nonproliferative and have low volume of disease based on newer data that patients may not need to have consistent "complete molecular response" or disease reduction below 4.5 logs in order to consider TFR. This will mean more patients may be eligible for such a strategy.
With that being said, I strongly advocate for discontinuation occurring in trials still, especially in the United States. I think we still have to ensure regular monitoring and not run the risk of loss of CML remission as a result of this endeavor. I think we have to have a clear message that this is still an investigational approach.
As we apply TFR strategies more broadly and explore it in patients with different circumstances, rather than those already studied in clinical trials, we may see slightly different results. We may need to exercise more caution.
Outside of a trial I don't discontinue TKI therapy unless there's a clear medical indication and there's no way around it, such as pregnancy or other illness that precludes TKI treatment. I would still pursue TFR only in a clinical trial.
Dr. Kalaycio: Dr. Deininger, I'll give you the last word for your thoughts regarding both treatment discontinuation and the future of CML therapies.
Dr. Deininger: As far as TFR is concerned, I agree with Dr. Mauro. Unfortunately, in the United States we're not quite as far advanced as the Europeans at monitoring diligence. There's a bit of a concern that if we elevate that into daily practice, we may see that patients are not monitored frequently enough and then relapse, but that doesn't get caught. You could imagine a scenario where we're actually seeing a decline in outcomes because people are discontinued and then not restarted if they have a recurrence at the molecular level.
All in all, where this is going, I think it will be really interesting to see more confirmatory data for second-generation TKIs. Let me put that differently: whether a similar proportion of patients in deep molecular response can maintain responses and have TFR if they needed a second-generation TKI to get there rather than just imatinib.
If so, this would be very promising. That would mean that second-generation TKIs actually somewhat impact the natural history of the disease and its biology. Then TFR may become a reality for a substantial proportion of our patients.
I don't think we have the data yet, but at least there are some suggestions that this may be the case. If you do the math, you will still see that many patients with CML will never reach a state where they can consider TFR.
They'll never get into a deep molecular response or they will have been diagnosed with accelerated phase. For all these patients who are not candidates for TFR, we still have to think about treatment optimization in order to make them catch up with the rest.
Now I have a couple of interesting developments. One is clearly to see whether ponatinib administered at a lower dose will have a more practical, therapeutic window in terms of toxicity and yet maintain the excellent efficacy that it has in the setting.
These data need to be produced. Another scenario would be that people would get started on intense induction treatment with higher-risk drugs such as ponatinib. Then if they are a good responder, they can switch to something that is lower-risk and that may be better tolerated.
Actually some of these trials are underway in Europe. Of course there's the question, are there still other drugs that will enter the CML space. There's one promising molecule called ABL001, a TKI with a different mode of action, which exploits an allosteric site rather than the catalytic center of the kinase.
It’s really conceptually very interesting. The expectation would be that this molecule has fewer side effects but still may be quite potent. This could be a very interesting development and add something to the armamentarium that we currently don't have.
I also think there will be patients whose diseases are just beyond the reach of a TKI alone for many reasons, maybe additional mutations or things that have to do with the host and metabolism.
In these cases, we'll still have to think about combination treatments and non-TKI treatments. Here, I think an honest answer is the labs have pulled out a lot of interesting leads, but so far, nothing has really made it into a serious clinical context, either because of side effects or because the target may not be as good in humans as it seemed from mouse models.
I think a lot more work is required here to find the best combination therapies and to define those pathways that need to be inhibited together with BCR-ABL. I think there's a field that we'll develop further and that will be the cutting edge.
The Wrap-Up
Dr. Kalaycio: Although an uncommon diagnosis, CML in chronic phase can be treated with an expectation for nearly 100% 5-year survival. For that reason, clinicians need to manage their patients expertly. Steps taken at diagnosis are critical to subsequent decision-making. Once treatment begins, close monitoring is required to screen for side effects as well as ensuring treatment success. With so many effective agents available, patients can be selected for the agent least likely to cause them long-term harm. Perhaps in the future we will learn which patients can stop treatment altogether.
Gentleman, thank you so much for your time. Every time I talk to you I learn something.
1. Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942.
2. National Comprehensive Cancer Network guidelines for treatment of cancer by site. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed August 20, 2015.
3. O'Brien S, Radich JP, Abboud CN, et al. Chronic myelogenous leukemia, version 1.2015. J Natl Compr Canc Netw. 2014;12(11):1590–1610.
4. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884.
5. Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035.
Moderator: Matt Kalaycio, MD1
Discussants: Michael Mauro, MD2; Michael Deininger, MD, PhD3
From Cleveland Clinic, Cleveland, OH1; Memorial Sloan Kettering Cancer Center, New York, NY2; Huntsman Cancer Institute, Salt Lake City, UT3
Address for correspondence: Matt Kalaycio, MD, Cleveland Clinic Main Campus, Mail Code R32, 9500 Euclid Avenue, Cleveland, OH 44195
E-mail: [email protected]
Biographical sketch:
Dr. Kalaycio has been published in numerous scientific publications including Bone Marrow Transplantation, Journal of Clinical Oncology, and Leukemia. He also is the editor of a book on leukemia and co-editor of a book on clinical malignant hematology. His research interests focus on testing new treatments for leukemia.
Dr. Kalaycio received his degree from West Virginia University School of Medicine in Morgantown. He completed his residency in internal medicine at Mercy Hospital of Pittsburgh and fellowships in hematology and medical oncology and bone marrow transplantation at Cleveland Clinic.
Michael Deininger, MD, PhD, is Professor and Chief of Hematology and Hematologic Malignancies for the Department of Internal Medicine and for the Huntsman Cancer Institute (HCI) at the University of Utah. He is an HCI investigator and member of the Experimental Therapeutics program. He has extensive experience treating patients with blood cancers, including chronic myeloid leukemia (CML) and myeloproliferative neoplasms, a group of blood cancers related to leukemia.
Dr
Dr. Deininger’s scientific focus is leukemia, specifically myeloproliferative neoplasms including chronic myeloid leukemia (CML). As a clinician-scientist with a translational research focus Dr. Deininger is heading an extramurally funded research laboratory that is dedicated to the study of signaling pathways, drug resistance and new molecular therapies in leukemia. Dr. Deininger’s work describing the selective effects of imatinib on CML cells provided the rationale for clinical trials that led to the approval of Gleevec as the first molecularly-based therapy for leukemia. Current work in his lab is focused on understanding the role of the bone marrow microenvironment in leukemia drug resistance, discovering novel therapeutic targets and developing more specific signal transduction inhibitors. Dr. Deininger’s work encompasses more than 170 articles in the peer-reviewed literature, including journals like Blood, Journal of Clinical Investigation and the New England Journal of Medicine. He has co-authored more than 10 book chapters, with contributions in leading textbooks such as deVita’s Principles of Oncology. He is a regular speaker at major international scientific meetings, such as the American Society of Hematology and the European Hematology Association and a peer reviewer for journals like Nature Genetics and Cancer Cell. His honors include the Alexandra Kefalides Prize for Leukemia Research and membership on the Editorial Board of Blood, the leading journal in Hematology. Dr. Deininger was named among the world's Highly Cited Researchers by Thomson Reuters in 2014.
Dr. Kalaycio: My name is Matt Kalaycio and I'm the Chairman of the Department of Hematology and Medical Oncology at the Cleveland Clinic. Today I’m joined by Drs. Mike Deininger, Division Chief of Hematology and Hematologic Malignancies at the University of Utah Huntsman Cancer Institute and Michael Mauro, Myeloproliferative Neoplasm Program Director at Memorial Sloan Kettering Cancer Center, New York, New York – together we will discuss practical issues surrounding CML diagnosis, management and treatment options including TKIs and investigational therapies.
Initial Presentation: Assessment & Treatment Options
Dr. Kalaycio: Often, the patient with chronic myelogenous leukemia (CML) gets admitted to the hospital, or an emergency consult is called, because the white count is up for a concern of leukemia. The treatment team sees the differential circulating blasts, and they worry about acute leukemia. Then the hematologist comes and needs to make a decision about whether or not to do a bone marrow biopsy.
When the patient presents in such a manner, I often see that bone marrow biopsies are not performed. I would like to start by asking where you both stand with regard to the necessity of a bone marrow biopsy at the time of diagnosis. I'll start with Dr. Deininger.
Dr. Deininger: I would strongly be in favor of a diagnostic biopsy as well as a smear. The reason is that I think this is the one and only chance to get a clear disease classification into chronic phase, accelerated phase, and blast crisis.
The third may be rare. There are occasional patients who have sheets of blasts who would not be seen on just a differential. For these patients, of course, the treatment decisions are going to be very different compared to a patient in chronic phase.
I think this is an opportunity that shouldn't be missed and I would always recommend that.
Dr. Kalaycio: Dr. Mauro.
Dr. Mauro: I couldn't concur more. With so much focus on the change in disease status from presentation to early response, I think understanding the scope of the disease at diagnosis, including the bone marrow, is essential and either revealing or reassuring. Although we focus mostly on early molecular response, when expectations go awry, not having as full a picture of the disease prior to treatment leaves you less informed about best treatment.
Making sense of accelerated phase features is a good example; the difference in outcomes between chronic phase patients and those who have morphologic features of accelerated phase—with or without cytogenetic features (clonal evolution)—compared with those with clonal evolution can only drive initial and long term treatment decisions. Without cytogenetics and morphology together, such key pieces of the puzzle are missing; it is worth it for the practitioner and patient to have all the data in hand at all times.
Dr. Kalaycio: Once the diagnosis has been made and the biopsy was not done and now you're seeing the patient having either been on hydroxyurea for a month or having had tyrosine kinase inhibitor (TKI) for a month, do you bother with the bone marrow biopsy at that point?
Dr. Deininger: Well, that is a very good question. I think we would probably still do it most of the time. I think it's very clear that the information you can gather from that is less valuable. I think one should try to make up for the omissions as much as possible. We would still go for it.
Dr. Kalaycio: Interesting. The other thing that happens a lot is a patient will be started on hydrea while you're waiting for BCR-ABL to return either by fluorescence in situ hybridization or quantitative polymerase chain reaction (PCR).
I wonder if you have your own set of internal guidelines for when hydroxyurea should be employed or not.
Dr. Mauro: If you look back at some of the original imatinib trials—the phase I trials—patients initiated TKI with higher blood counts, a median around 25,000 and up to 200,000, and responded with excellent tolerance.1 I think there's a bit of overapplication of hydroxyurea early in CML, prior to initiation of TKI, which often confounds and complicates the early myelosuppressive toxicity of TKIs. With use of more potent TKIs, there may be greater amounts of myelosuppression as the leukemic clone may clear faster.
I think we may get ourselves into a bit of a bind by overusing hydroxyurea and then needing to hold and lower TKI dosages quickly when that may not have been necessary had we simply deployed the TKI sooner.
My general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. – Michael Mauro, MDMy general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. The other question that often comes up is about tumor lysis and hydration, and how closely these need to be managed. The likelihood of tumor lysis is low in CML treated with TKIs, but more frequent early labs and good hydration are always the right thing to do; better safe than sorry!
Dr. Deininger: I second what Michael just said. To your question about whether we have an algorithm, I have to admit we don't. I think it's up to the discretion of the treating physician to initiate hydroxyurea or not.
Dr. Kalaycio: Sure. When you start hydroxyurea, do you routinely add allopurinol?
Dr. Deininger: We tend to do that. I know that some people think it's unnecessary. It's such a low-risk and low-cost intervention that I think that it's hard to get anything wrong here.
Dr. Mauro: Right, we tend to do the same. Whether we need to or not is a different question, I suppose.
Dr. Kalaycio: One more thing about the initial presentation and assessment of a patient with what you think might be a myeloproliferative disorder, CML, how important do you find it, Dr. Mauro, to measure splenomegaly and calculate a risk score?
Dr. Mauro: I think it's very useful information. The spleen size factors heavily into the risk score and the risk score does forecast response to a degree. We've looked at calculation of risk score in recent large observational studies and it is under-reported and underutilized. It winds up being useful for two reasons. One, it does set expectations for response, and US treatment guidelines (National Comprehensive Cancer Network [NCCN] guidelines) note that treatment choice may be different for low versus high risk Sokal score.2
I think the second and most intriguing reason to assess Sokal risk seems now to be the impact that risk score has on the ability to proceed to a treatment-free remission (TFR). There appear to be differences in outcomes in patients with high-risk versus low-risk disease despite both having what is required to proceed to TFR in trials, namely consistent and deep molecular remission over a number of years.
Given these implications, we really ought to be gathering the initial risk stratification and quantifying spleen size. It's an important part of our initial assessment.
Dr. Kalaycio: Dr. Deininger?
Dr. Deininger: I think there's a lot of agreement today. I absolutely think that measuring the spleen size ascertains that you've got all these diagnostic parameters available. I think that should be part of the initial evaluation and work-up and will allow some prognostication.
Testing, Risk Factors & Considerations of Treatment: Nilotinib & Dasatinib
Dr. Kalaycio: I'm starting with the less controversial questions to begin with. I think the next set have the potential for some more controversy. Before we leave the initial assessment of CML, do either of you have observations regarding referrals that you get about which you would like to either dispel myths or remind practitioners about best practices in patients newly diagnosed with CML?
I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. – Michael Deininger, MD, PhDDr. Mauro: I can mention one thing. I think when thinking about initial molecular diagnostic results, it is important to point out that testing should screen broadly for different fusions, namely P190 and P210, or variant (p230) transcripts. There are rare patients in chronic phase with non-p210 fusion who need to be followed with specific PCR. On this same topic, the measurement of transcripts at presentation (ie, before treatment) has become quite important and whereas formerly had not been emphasized, presently all patients should have ”baseline” transcript levels.
Dr. Deininger: I think one issue that comes up once in a while is that spleen measurements are done by ultrasound. Technically it's more accurate, but all the clinical risk scores and the prognostication is based on the old fashioned—but probably highly inaccurate—technology of palpation. This is what counts, this is the value to document. I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. Getting a good handle of those risk factors at diagnosis is also important.
Dr. Kalaycio: I think that's a very important point. That's where I was going to go next with this conversation. I was going to avoid the conversation about which TKI to choose as initial treatment. I think that's a debate unto itself.
I would like to ask you how you might assess cardiovascular risk before placing a patient on nilotinib. You got to that a little bit, Dr. Deininger. Could you expand on what else you might review as far as whether or not you feel a patient is a good candidate to start on nilotinib?
Dr. Deininger: Specifically, with regard to nilotinib, we would always get a baseline electrocardiogram. We would do a clinical exam. We would not do an echocardiogram just routinely in the absence of a cardiovascular history or any clinical evidence for heart failure or other cardiovascular issues.
We've adopted the practice of doing a lipid panel. Of course we would include fasting glucose as well. Some of these recommendations are probably somewhat on the soft side, because it's not yet clear what to do with the information.
On the other hand, I think for a patient who is being considered for nilotinib one wants to make sure that one really does the best to minimize the cardiovascular risk factors. Of course that would include smoking history and taking blood pressure and making sure that these risk factors are controlled.
If people have a presentation that is really out of whack in terms of their risk factor management, I would send them to an internist or even a cardiologist to help me optimize the cardiovascular prevention strategy.
Dr. Kalaycio: Great. Similarly, Dr. Mauro, how do you assess pulmonary risk before placing a patient on dasatinib?
Dr. Mauro: I think here we are focused on the less frequent and also less well understood potential toxicity of pulmonary hypertension, coupled with the more common risk of pleural and pericardial effusions.
I'm not sure how much we've learned in clinical studies looking at baseline chest X-rays or timing of X-rays during treatment. I think our best tool in the prevention and management of pleural and pericardial effusions is full discussion with patients about risk and what to look for, attention to any and all symptoms, and appropriate deployment of diagnostics as indicated.
It's interesting to consider whether baseline echocardiography for measurement of pulmonary pressures is warranted. I would say now we're on probably somewhat softer ground, first because on routine echocardiogram pulmonary pressure can't be measured readily unless there is some valvular regurgitation. As well, it is stated that pulmonary hypertension is only properly diagnosed by right heart catheterization. While I'm tempted to do routine echocardiogram studies, I think that such a recommendation still may be perhaps the realm of a clinical study. We need to explore that further. I think with dasatinib there may be certain patients at higher risk, although the data are somewhat limited. There seem to be certain conditions potentially associated with more pleural and pericardial toxicity, including cardiovascular disease and autoimmune disease. There may be circumstances during treatment—lymphocytosis, for example—that may be associated with greater risk. I think expectant management may still be the right approach and echocardiography and more aggressive diagnostics be reserved for patients in whom there might be much more clinical consequence.
Dr. Kalaycio: I'd like to pursue that a little bit further because sometimes the patients will come to us having already had an echocardiogram that may actually show some mild pulmonary hypertension and maybe they've got significant cardiovascular risk factors where you would otherwise be thinking about using dasatinib. Here's someone with pulmonary hypertension, at least by echocardiographic criteria, would that be enough to dissuade you from the use of dasatinib?
Dr. Mauro: I think it would certainly require significant consideration, understanding what the basis of the pulmonary hypertension is for that patient, and risk with adding dasatinib. I think the good news is the low incidence and the reversibility for the most part of dasatinib-associated pulmonary hypertension.
Again, I think the mechanism of action and the pathophysiology isn't completely understood, although there is the intriguing notion that imatinib has been reported to potentially mitigate pulmonary hypertension whereas dasatinib triggers it—a ”closed loop” if you will and an area ripe for research.
I would probably think that a patient with preexisting pulmonary hypertension in the new diagnosis setting might be the kind of patient for whom you really might weigh the pluses versus the minuses of a second generation TKI versus imatinib.
After Evaluation & Diagnosis: Following the Patient
Dr. Kalaycio: I agree. We have fully evaluated our patient and we've made the diagnosis. Now, we start therapy with a TKI based on patient risk profile and side effect potentials. It's time to follow the patient and determine next steps.
Current guidelines3,4 suggest monitoring quantitative PCR after 3 months of therapy and to gauge response. Dr. Deininger, how do you interpret and act on those results following the first 3 months of therapy?
Dr. Deininger: I think what you're getting at is the 10% mark that is a highly predictive value in terms of subsequent achievement of major molecular response and also overall progression free and overall survival.
We'll certainly get this data point. Then we'll put it in a clinical context. I think this context needs to take into account the initial BCR-ABL transcript level. I think Dr. Mauro mentioned that they always determine that. I think that is really good advice, so you can make a comparison with the diagnostic value.
One scenario, of course, is the patient is well below 10% and then things are just in the green range and you would wave people through and reassess in 3 months. If people are very high, I think then you have to ask yourself why that's the case.
Some people have a lot of toxicity issues. For example, they may not have been able to take the required amount of medication. It's not always easy to clearly distinguish between resistance and intolerance.
No matter what, I think it's going to be critical to consider a potential change in treatment if people are in the 70%—80% range. In my mind, there's a gray area.
These are the people who are maybe between 10%—20%. Here, I think this initial value can really help. If there's a significant reduction compared to baseline, I would not necessarily change at that point.
If there is literally no change compared to baseline, I would strongly consider a change unless I am concerned about other issues like noncompliance or drug interactions. What I'm trying to say in a rather long-winded way is that the 10% value shouldn't be seen as a dogma.
It still needs to be placed in a clinical context. One should not rush to any conclusions. Ten percent, 11%, and 9% are identical values in the world of PCR testing. One should not over-interpret that.
Dr. Kalaycio: I think that you're making an important point about absolutes in the interpretation of these tests. Dr. Mauro, how do you interpret the 3-month data that comes back?
Dr. Mauro: I agree with my colleague on the approach that it has to be put in a clinical context. I think what we've learned is the importance of the starting value and relative reduction and to not consider response milestones as black and white guides.
I think there are certain scenarios that require some caution. The patient who is on imatinib who is close but has not reached the landmark may be different that a similar patient on dasatinib or nilotinib who has not had significant reduction—that's probably a more pressing situation.
It's ironic that guidelines—maybe because of lack of options—don't encourage us to think about changing therapy early in someone who hasn't met milestones in 3 months when they've been put on a more potent agent.
I also think that careful consideration is needed regarding treatment intensity and the impact of interruptions, in conjunction with all the other facets—the rate of change, the absolute change from the patient's baseline, the starting response level and the timing of the PCR. We need to look at the actual day it's performed. If it's really not 3 months of therapy, we can misjudge.
Dr. Kalaycio: Right. Now, as you're monitoring patients, Dr. Mauro, at what point would you consider testing for kinase domain mutations?
Dr. Mauro: I think we have pretty good guidance from studies to date regarding when mutation testing is indicated and of higher yield. It's generally earlier on into treatment when we have clinical scenarios that are of greater risk, namely failure to achieve cytogenetic responses. In such scenarios if mutation testing informs treatment decision making it is very helpful.
Patients who have defined themselves as not achieving early molecular response—which we discussed earlier—especially someone who's had a second-generation TKI, warrants mutation testing in my view. Patients who don't achieve classical cytogenetic response landmarks at 6 and 12 (or 18) months, who thus have a higher residual volume of disease and perhaps as a function more clonal instability, I think also warrant attention.
I think we run into trouble when we start to, in an overly critical manner, assess patients’ longer term and deeper molecular response trajectories. Mutation testing becomes difficult or impossible for patients who are at or near major molecular remission (MMR). Mutation yield is generally very low for patients who have not lost MMR and also likely those with small volume of change around the MMR threshold. Looking ahead, I think further investigation into early time points and the setting of minimal residual disease may yield data to be able to predict potential resistance earlier than observing it clinically.
Dr. Kalaycio: Right. Dr. Deininger, if you're monitoring a patient and he’s missing milestones and you do obtain a mutation analysis and find a T315I mutation, do you offer that patient an opportunity to see how well they're going to do with ponatinib, or do you refer that patient to your transplant team for consideration of transplant as soon as a donor is available?As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated. – Matt Kalaycio, MD
Dr. Deininger: I think we do the first and half of the second. We would certainly offer a trial of ponatinib or, if possible, a clinical trial unless there are insurmountable contraindications.
I don't really think there are any such circumstances with optimized cardiovascular management at the same time, and we may involve a cardiologist at that point to help us if there are cardiovascular risk factors.
We would also do a referral to a transplant center unless the patient is as per performance status, just not a transplant candidate or is too old. Otherwise, we would always make a referral, but we would not pursue a transplant as the first modality, provided that the patient is in chronic phase. Progression to accelerated phase or blastic phase would be looked at totally differently. Here we would certainly put people on a transplant course.
Dr. Kalaycio: I agree. As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated.
As the two of you know, we're doing fewer and fewer transplants for CML these days. The results we're getting in folks who are failing all of these agents are not as good as they used to be when we were transplanting in the first 3 months of a new diagnosis. Bone marrow transplant is something to defer for as long as possible, at least in my mind.
TKI Dosage Considerations
Dr. Kalaycio: Now we touched a little bit on the side effects of nilotinib and dasatinib. We've talked about progressive disease, perhaps with or without mutations. Dr. Mauro, could you comment on where you think bosutinib might play a role in patients with relapsed or progressive CML?
Dr. Mauro: I think bosutinib remains an underutilized TKI. It is potent and thus offers good salvage activity. I think it offers a fairly distinct side effect profile making it a good choice for cases of intolerance. I think we've learned more about how to initiate bosutinib and manage its side effects; one element of this is that current trials start at slightly lower doses of bosutinib to avoid some of the early gastrointestinal toxicity.
Bosutinib continues to seek a place as a first-line therapy. I think this may be worthwhile as we are looking for the right balance of safety and side effect risk to maximize early response. Regarding the salvage setting—intolerance and resistance to other agents—I find myself speaking to patients about bosutinib as a potent and viable alternative. Bosutinib offers a similar spectrum of activity as dasatinib but can allow us to avoid cardiovascular toxicity and/or allow clearance of pleural and pericardial toxicity occurring with dasatinib.
The one place I would say bosutinib may not hold up as strongly to its competition would be as a third-line therapy after failure of a second-generation TKI. I think while there is reasonable activity with bosutinib in this setting of somewhat fairly drug-resistant CML, the performance of ponatinib is better. There is likely often a struggle with the long-term safety question of ponatinib, making some feel that a trial of bosutinib is often a worthwhile, logical, or even necessary step before ponatinib. My concerns about this approach are related to treatment decision-making based on safety and theoretical risk of adverse effects rather than efficacy and risk of progression; these have to be weighed carefully and appropriately against each other.
Dr. Kalaycio: That's an interesting perspective. You mentioned starting with a lower dose. Dr. Deininger, I'd like to ask you your thoughts about dose modifications of the TKIs in general. It used to be anathema to either start with a low dose or to maintain patients on a lower dose.
I'm aware of at least some data suggesting that those patients who tolerate these TKIs poorly particularly with regard to myelosuppression can be treated long term at lower than typically prescribed doses without adverse effect. What are your thoughts surrounding dose modification of the TKIs?
Dr. Deininger: I would stratify according to TKIs. I think imatinib at 400 mg is probably just rightly dosed, maybe not even at the optimal dose. It could be 600 mg. If you go to 300 mg, that is still acceptable.
If you go to 200 mg, I think I would make double sure that I monitor patients very frequently. People should be, in my mind, in a major molecular response to treat them with 200 mg long term. With the second-generation TKIs, it's a little different.
I cannot really speak to bosutinib because I'm not aware of a lot of data on those modifications. What seems to be clear from dasatinib is that the initial recommended dose of 100 mg is quite high, especially in older people.
I believe that's been corroborated by a study that hasn't been published yet. Apparently the drug excretion in the older individuals is quite a bit slower than in younger people. I think in our practice and in other people's practices as well, about 50% of those people end at doses substantially lower than 100 mg, maybe 50 mg, maybe 40 mg, some even 20 mg per day.
I think in the case of dasatinib you have a lot of maneuvering space. You can adjust the dose according to tolerability and molecular response. With nilotinib I think it's kind of similar. We have a few patients who are on maybe 150 mg twice a day because they have issues in terms of side effects at the higher doses.
I should again thoroughly qualify that by saying if you give up through dose reductions the goal of a major molecular response, then I think you really have to think about the strategy in general because at that point, I think you basically say "this patient is not ever going to be reaching a safe haven and is not tolerating inhibitors well enough to get him there."
That's quite a significant statement to make. Myelosuppression is frequently a situation where you cannot deliver enough dose but you also don't get a good response, whereas other side effects like pleural effusions or excessive fluid retention may well be cause for dose reduction and yet responses may be acceptable. Maybe Dr. Mauro could chime in here and share his experience.
Dr. Mauro: I agree. I think some patients, based on their disease status, may not tolerate the dose that is going to—at least in theory—get them to a deep molecular remission. The biggest problem we often face is myelosuppression. Some patients may have indolent disease and still have a good outcome, at least in the short to medium term, with lower doses of drug. In general I try to work through myelosuppression, especially early, as the combination of myelosuppression and suboptimal or response failure is dangerous.
I think treatment shouldn't be too chaotic and change too much, if possible. That was a very nice summary of the way we view the doses. There's still a fair bit of flexibility amongst the TKI doses, although I don't advocate for starting low and titration up.
In general I think, with regard to optimal TKI dose, we might have overshot; for example, with dasatinib. I think we might have overshot with bosutinib. Trials ongoing now initiate bosutinib at 400 mg rather than 500 mg. That's what I was alluding to.
Treatment Free Remission: Approaches
Dr. Kalaycio: Very interesting discussions. As we wind down the conversation, I want to get to the idea of stopping therapy. We're all aware of data that suggest it's at least possible for a proportion of patients to stop treatment.5
However, the guidelines3,4 such as they are, suggest that stopping should not be done outside the context of a clinical trial. I think most of us would agree with that. I wonder in a practical sense, Dr. Mauro, in your practice, do you have your own set of internal criteria for stopping a TKI and observing patients in the absence of any therapy?
Dr. Mauro: I think I try to incorporate all the experience we have to date when considering this question. That being said I'm a little hesitant to agree entirely with the current thinking regarding retreatment during a Treatment Free Remission trial, which is waiting until patients lose MMR in order to resume treatment but agree it might be hard to retreat based on lesser degrees of molecular relapse. I just worry a little bit about that amount of proliferation without treatment.
On the other hand I think we're coming to the realization that TFR may be feasible in patients who are simply nonproliferative and have low volume of disease based on newer data that patients may not need to have consistent "complete molecular response" or disease reduction below 4.5 logs in order to consider TFR. This will mean more patients may be eligible for such a strategy.
With that being said, I strongly advocate for discontinuation occurring in trials still, especially in the United States. I think we still have to ensure regular monitoring and not run the risk of loss of CML remission as a result of this endeavor. I think we have to have a clear message that this is still an investigational approach.
As we apply TFR strategies more broadly and explore it in patients with different circumstances, rather than those already studied in clinical trials, we may see slightly different results. We may need to exercise more caution.
Outside of a trial I don't discontinue TKI therapy unless there's a clear medical indication and there's no way around it, such as pregnancy or other illness that precludes TKI treatment. I would still pursue TFR only in a clinical trial.
Dr. Kalaycio: Dr. Deininger, I'll give you the last word for your thoughts regarding both treatment discontinuation and the future of CML therapies.
Dr. Deininger: As far as TFR is concerned, I agree with Dr. Mauro. Unfortunately, in the United States we're not quite as far advanced as the Europeans at monitoring diligence. There's a bit of a concern that if we elevate that into daily practice, we may see that patients are not monitored frequently enough and then relapse, but that doesn't get caught. You could imagine a scenario where we're actually seeing a decline in outcomes because people are discontinued and then not restarted if they have a recurrence at the molecular level.
All in all, where this is going, I think it will be really interesting to see more confirmatory data for second-generation TKIs. Let me put that differently: whether a similar proportion of patients in deep molecular response can maintain responses and have TFR if they needed a second-generation TKI to get there rather than just imatinib.
If so, this would be very promising. That would mean that second-generation TKIs actually somewhat impact the natural history of the disease and its biology. Then TFR may become a reality for a substantial proportion of our patients.
I don't think we have the data yet, but at least there are some suggestions that this may be the case. If you do the math, you will still see that many patients with CML will never reach a state where they can consider TFR.
They'll never get into a deep molecular response or they will have been diagnosed with accelerated phase. For all these patients who are not candidates for TFR, we still have to think about treatment optimization in order to make them catch up with the rest.
Now I have a couple of interesting developments. One is clearly to see whether ponatinib administered at a lower dose will have a more practical, therapeutic window in terms of toxicity and yet maintain the excellent efficacy that it has in the setting.
These data need to be produced. Another scenario would be that people would get started on intense induction treatment with higher-risk drugs such as ponatinib. Then if they are a good responder, they can switch to something that is lower-risk and that may be better tolerated.
Actually some of these trials are underway in Europe. Of course there's the question, are there still other drugs that will enter the CML space. There's one promising molecule called ABL001, a TKI with a different mode of action, which exploits an allosteric site rather than the catalytic center of the kinase.
It’s really conceptually very interesting. The expectation would be that this molecule has fewer side effects but still may be quite potent. This could be a very interesting development and add something to the armamentarium that we currently don't have.
I also think there will be patients whose diseases are just beyond the reach of a TKI alone for many reasons, maybe additional mutations or things that have to do with the host and metabolism.
In these cases, we'll still have to think about combination treatments and non-TKI treatments. Here, I think an honest answer is the labs have pulled out a lot of interesting leads, but so far, nothing has really made it into a serious clinical context, either because of side effects or because the target may not be as good in humans as it seemed from mouse models.
I think a lot more work is required here to find the best combination therapies and to define those pathways that need to be inhibited together with BCR-ABL. I think there's a field that we'll develop further and that will be the cutting edge.
The Wrap-Up
Dr. Kalaycio: Although an uncommon diagnosis, CML in chronic phase can be treated with an expectation for nearly 100% 5-year survival. For that reason, clinicians need to manage their patients expertly. Steps taken at diagnosis are critical to subsequent decision-making. Once treatment begins, close monitoring is required to screen for side effects as well as ensuring treatment success. With so many effective agents available, patients can be selected for the agent least likely to cause them long-term harm. Perhaps in the future we will learn which patients can stop treatment altogether.
Gentleman, thank you so much for your time. Every time I talk to you I learn something.
Moderator: Matt Kalaycio, MD1
Discussants: Michael Mauro, MD2; Michael Deininger, MD, PhD3
From Cleveland Clinic, Cleveland, OH1; Memorial Sloan Kettering Cancer Center, New York, NY2; Huntsman Cancer Institute, Salt Lake City, UT3
Address for correspondence: Matt Kalaycio, MD, Cleveland Clinic Main Campus, Mail Code R32, 9500 Euclid Avenue, Cleveland, OH 44195
E-mail: [email protected]
Biographical sketch:
Dr. Kalaycio has been published in numerous scientific publications including Bone Marrow Transplantation, Journal of Clinical Oncology, and Leukemia. He also is the editor of a book on leukemia and co-editor of a book on clinical malignant hematology. His research interests focus on testing new treatments for leukemia.
Dr. Kalaycio received his degree from West Virginia University School of Medicine in Morgantown. He completed his residency in internal medicine at Mercy Hospital of Pittsburgh and fellowships in hematology and medical oncology and bone marrow transplantation at Cleveland Clinic.
Michael Deininger, MD, PhD, is Professor and Chief of Hematology and Hematologic Malignancies for the Department of Internal Medicine and for the Huntsman Cancer Institute (HCI) at the University of Utah. He is an HCI investigator and member of the Experimental Therapeutics program. He has extensive experience treating patients with blood cancers, including chronic myeloid leukemia (CML) and myeloproliferative neoplasms, a group of blood cancers related to leukemia.
Dr
Dr. Deininger’s scientific focus is leukemia, specifically myeloproliferative neoplasms including chronic myeloid leukemia (CML). As a clinician-scientist with a translational research focus Dr. Deininger is heading an extramurally funded research laboratory that is dedicated to the study of signaling pathways, drug resistance and new molecular therapies in leukemia. Dr. Deininger’s work describing the selective effects of imatinib on CML cells provided the rationale for clinical trials that led to the approval of Gleevec as the first molecularly-based therapy for leukemia. Current work in his lab is focused on understanding the role of the bone marrow microenvironment in leukemia drug resistance, discovering novel therapeutic targets and developing more specific signal transduction inhibitors. Dr. Deininger’s work encompasses more than 170 articles in the peer-reviewed literature, including journals like Blood, Journal of Clinical Investigation and the New England Journal of Medicine. He has co-authored more than 10 book chapters, with contributions in leading textbooks such as deVita’s Principles of Oncology. He is a regular speaker at major international scientific meetings, such as the American Society of Hematology and the European Hematology Association and a peer reviewer for journals like Nature Genetics and Cancer Cell. His honors include the Alexandra Kefalides Prize for Leukemia Research and membership on the Editorial Board of Blood, the leading journal in Hematology. Dr. Deininger was named among the world's Highly Cited Researchers by Thomson Reuters in 2014.
Dr. Kalaycio: My name is Matt Kalaycio and I'm the Chairman of the Department of Hematology and Medical Oncology at the Cleveland Clinic. Today I’m joined by Drs. Mike Deininger, Division Chief of Hematology and Hematologic Malignancies at the University of Utah Huntsman Cancer Institute and Michael Mauro, Myeloproliferative Neoplasm Program Director at Memorial Sloan Kettering Cancer Center, New York, New York – together we will discuss practical issues surrounding CML diagnosis, management and treatment options including TKIs and investigational therapies.
Initial Presentation: Assessment & Treatment Options
Dr. Kalaycio: Often, the patient with chronic myelogenous leukemia (CML) gets admitted to the hospital, or an emergency consult is called, because the white count is up for a concern of leukemia. The treatment team sees the differential circulating blasts, and they worry about acute leukemia. Then the hematologist comes and needs to make a decision about whether or not to do a bone marrow biopsy.
When the patient presents in such a manner, I often see that bone marrow biopsies are not performed. I would like to start by asking where you both stand with regard to the necessity of a bone marrow biopsy at the time of diagnosis. I'll start with Dr. Deininger.
Dr. Deininger: I would strongly be in favor of a diagnostic biopsy as well as a smear. The reason is that I think this is the one and only chance to get a clear disease classification into chronic phase, accelerated phase, and blast crisis.
The third may be rare. There are occasional patients who have sheets of blasts who would not be seen on just a differential. For these patients, of course, the treatment decisions are going to be very different compared to a patient in chronic phase.
I think this is an opportunity that shouldn't be missed and I would always recommend that.
Dr. Kalaycio: Dr. Mauro.
Dr. Mauro: I couldn't concur more. With so much focus on the change in disease status from presentation to early response, I think understanding the scope of the disease at diagnosis, including the bone marrow, is essential and either revealing or reassuring. Although we focus mostly on early molecular response, when expectations go awry, not having as full a picture of the disease prior to treatment leaves you less informed about best treatment.
Making sense of accelerated phase features is a good example; the difference in outcomes between chronic phase patients and those who have morphologic features of accelerated phase—with or without cytogenetic features (clonal evolution)—compared with those with clonal evolution can only drive initial and long term treatment decisions. Without cytogenetics and morphology together, such key pieces of the puzzle are missing; it is worth it for the practitioner and patient to have all the data in hand at all times.
Dr. Kalaycio: Once the diagnosis has been made and the biopsy was not done and now you're seeing the patient having either been on hydroxyurea for a month or having had tyrosine kinase inhibitor (TKI) for a month, do you bother with the bone marrow biopsy at that point?
Dr. Deininger: Well, that is a very good question. I think we would probably still do it most of the time. I think it's very clear that the information you can gather from that is less valuable. I think one should try to make up for the omissions as much as possible. We would still go for it.
Dr. Kalaycio: Interesting. The other thing that happens a lot is a patient will be started on hydrea while you're waiting for BCR-ABL to return either by fluorescence in situ hybridization or quantitative polymerase chain reaction (PCR).
I wonder if you have your own set of internal guidelines for when hydroxyurea should be employed or not.
Dr. Mauro: If you look back at some of the original imatinib trials—the phase I trials—patients initiated TKI with higher blood counts, a median around 25,000 and up to 200,000, and responded with excellent tolerance.1 I think there's a bit of overapplication of hydroxyurea early in CML, prior to initiation of TKI, which often confounds and complicates the early myelosuppressive toxicity of TKIs. With use of more potent TKIs, there may be greater amounts of myelosuppression as the leukemic clone may clear faster.
I think we may get ourselves into a bit of a bind by overusing hydroxyurea and then needing to hold and lower TKI dosages quickly when that may not have been necessary had we simply deployed the TKI sooner.
My general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. – Michael Mauro, MDMy general rule would be to use hydroxyurea for symptoms if necessary or for more extreme counts where leukostasis is a concern. The other question that often comes up is about tumor lysis and hydration, and how closely these need to be managed. The likelihood of tumor lysis is low in CML treated with TKIs, but more frequent early labs and good hydration are always the right thing to do; better safe than sorry!
Dr. Deininger: I second what Michael just said. To your question about whether we have an algorithm, I have to admit we don't. I think it's up to the discretion of the treating physician to initiate hydroxyurea or not.
Dr. Kalaycio: Sure. When you start hydroxyurea, do you routinely add allopurinol?
Dr. Deininger: We tend to do that. I know that some people think it's unnecessary. It's such a low-risk and low-cost intervention that I think that it's hard to get anything wrong here.
Dr. Mauro: Right, we tend to do the same. Whether we need to or not is a different question, I suppose.
Dr. Kalaycio: One more thing about the initial presentation and assessment of a patient with what you think might be a myeloproliferative disorder, CML, how important do you find it, Dr. Mauro, to measure splenomegaly and calculate a risk score?
Dr. Mauro: I think it's very useful information. The spleen size factors heavily into the risk score and the risk score does forecast response to a degree. We've looked at calculation of risk score in recent large observational studies and it is under-reported and underutilized. It winds up being useful for two reasons. One, it does set expectations for response, and US treatment guidelines (National Comprehensive Cancer Network [NCCN] guidelines) note that treatment choice may be different for low versus high risk Sokal score.2
I think the second and most intriguing reason to assess Sokal risk seems now to be the impact that risk score has on the ability to proceed to a treatment-free remission (TFR). There appear to be differences in outcomes in patients with high-risk versus low-risk disease despite both having what is required to proceed to TFR in trials, namely consistent and deep molecular remission over a number of years.
Given these implications, we really ought to be gathering the initial risk stratification and quantifying spleen size. It's an important part of our initial assessment.
Dr. Kalaycio: Dr. Deininger?
Dr. Deininger: I think there's a lot of agreement today. I absolutely think that measuring the spleen size ascertains that you've got all these diagnostic parameters available. I think that should be part of the initial evaluation and work-up and will allow some prognostication.
Testing, Risk Factors & Considerations of Treatment: Nilotinib & Dasatinib
Dr. Kalaycio: I'm starting with the less controversial questions to begin with. I think the next set have the potential for some more controversy. Before we leave the initial assessment of CML, do either of you have observations regarding referrals that you get about which you would like to either dispel myths or remind practitioners about best practices in patients newly diagnosed with CML?
I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. – Michael Deininger, MD, PhDDr. Mauro: I can mention one thing. I think when thinking about initial molecular diagnostic results, it is important to point out that testing should screen broadly for different fusions, namely P190 and P210, or variant (p230) transcripts. There are rare patients in chronic phase with non-p210 fusion who need to be followed with specific PCR. On this same topic, the measurement of transcripts at presentation (ie, before treatment) has become quite important and whereas formerly had not been emphasized, presently all patients should have ”baseline” transcript levels.
Dr. Deininger: I think one issue that comes up once in a while is that spleen measurements are done by ultrasound. Technically it's more accurate, but all the clinical risk scores and the prognostication is based on the old fashioned—but probably highly inaccurate—technology of palpation. This is what counts, this is the value to document. I think, moving forward it would be really important to have documentation of concomitant risk factors such as smoking and so on that are essentially driving outcomes more than the TKI treatment or the CML itself. Getting a good handle of those risk factors at diagnosis is also important.
Dr. Kalaycio: I think that's a very important point. That's where I was going to go next with this conversation. I was going to avoid the conversation about which TKI to choose as initial treatment. I think that's a debate unto itself.
I would like to ask you how you might assess cardiovascular risk before placing a patient on nilotinib. You got to that a little bit, Dr. Deininger. Could you expand on what else you might review as far as whether or not you feel a patient is a good candidate to start on nilotinib?
Dr. Deininger: Specifically, with regard to nilotinib, we would always get a baseline electrocardiogram. We would do a clinical exam. We would not do an echocardiogram just routinely in the absence of a cardiovascular history or any clinical evidence for heart failure or other cardiovascular issues.
We've adopted the practice of doing a lipid panel. Of course we would include fasting glucose as well. Some of these recommendations are probably somewhat on the soft side, because it's not yet clear what to do with the information.
On the other hand, I think for a patient who is being considered for nilotinib one wants to make sure that one really does the best to minimize the cardiovascular risk factors. Of course that would include smoking history and taking blood pressure and making sure that these risk factors are controlled.
If people have a presentation that is really out of whack in terms of their risk factor management, I would send them to an internist or even a cardiologist to help me optimize the cardiovascular prevention strategy.
Dr. Kalaycio: Great. Similarly, Dr. Mauro, how do you assess pulmonary risk before placing a patient on dasatinib?
Dr. Mauro: I think here we are focused on the less frequent and also less well understood potential toxicity of pulmonary hypertension, coupled with the more common risk of pleural and pericardial effusions.
I'm not sure how much we've learned in clinical studies looking at baseline chest X-rays or timing of X-rays during treatment. I think our best tool in the prevention and management of pleural and pericardial effusions is full discussion with patients about risk and what to look for, attention to any and all symptoms, and appropriate deployment of diagnostics as indicated.
It's interesting to consider whether baseline echocardiography for measurement of pulmonary pressures is warranted. I would say now we're on probably somewhat softer ground, first because on routine echocardiogram pulmonary pressure can't be measured readily unless there is some valvular regurgitation. As well, it is stated that pulmonary hypertension is only properly diagnosed by right heart catheterization. While I'm tempted to do routine echocardiogram studies, I think that such a recommendation still may be perhaps the realm of a clinical study. We need to explore that further. I think with dasatinib there may be certain patients at higher risk, although the data are somewhat limited. There seem to be certain conditions potentially associated with more pleural and pericardial toxicity, including cardiovascular disease and autoimmune disease. There may be circumstances during treatment—lymphocytosis, for example—that may be associated with greater risk. I think expectant management may still be the right approach and echocardiography and more aggressive diagnostics be reserved for patients in whom there might be much more clinical consequence.
Dr. Kalaycio: I'd like to pursue that a little bit further because sometimes the patients will come to us having already had an echocardiogram that may actually show some mild pulmonary hypertension and maybe they've got significant cardiovascular risk factors where you would otherwise be thinking about using dasatinib. Here's someone with pulmonary hypertension, at least by echocardiographic criteria, would that be enough to dissuade you from the use of dasatinib?
Dr. Mauro: I think it would certainly require significant consideration, understanding what the basis of the pulmonary hypertension is for that patient, and risk with adding dasatinib. I think the good news is the low incidence and the reversibility for the most part of dasatinib-associated pulmonary hypertension.
Again, I think the mechanism of action and the pathophysiology isn't completely understood, although there is the intriguing notion that imatinib has been reported to potentially mitigate pulmonary hypertension whereas dasatinib triggers it—a ”closed loop” if you will and an area ripe for research.
I would probably think that a patient with preexisting pulmonary hypertension in the new diagnosis setting might be the kind of patient for whom you really might weigh the pluses versus the minuses of a second generation TKI versus imatinib.
After Evaluation & Diagnosis: Following the Patient
Dr. Kalaycio: I agree. We have fully evaluated our patient and we've made the diagnosis. Now, we start therapy with a TKI based on patient risk profile and side effect potentials. It's time to follow the patient and determine next steps.
Current guidelines3,4 suggest monitoring quantitative PCR after 3 months of therapy and to gauge response. Dr. Deininger, how do you interpret and act on those results following the first 3 months of therapy?
Dr. Deininger: I think what you're getting at is the 10% mark that is a highly predictive value in terms of subsequent achievement of major molecular response and also overall progression free and overall survival.
We'll certainly get this data point. Then we'll put it in a clinical context. I think this context needs to take into account the initial BCR-ABL transcript level. I think Dr. Mauro mentioned that they always determine that. I think that is really good advice, so you can make a comparison with the diagnostic value.
One scenario, of course, is the patient is well below 10% and then things are just in the green range and you would wave people through and reassess in 3 months. If people are very high, I think then you have to ask yourself why that's the case.
Some people have a lot of toxicity issues. For example, they may not have been able to take the required amount of medication. It's not always easy to clearly distinguish between resistance and intolerance.
No matter what, I think it's going to be critical to consider a potential change in treatment if people are in the 70%—80% range. In my mind, there's a gray area.
These are the people who are maybe between 10%—20%. Here, I think this initial value can really help. If there's a significant reduction compared to baseline, I would not necessarily change at that point.
If there is literally no change compared to baseline, I would strongly consider a change unless I am concerned about other issues like noncompliance or drug interactions. What I'm trying to say in a rather long-winded way is that the 10% value shouldn't be seen as a dogma.
It still needs to be placed in a clinical context. One should not rush to any conclusions. Ten percent, 11%, and 9% are identical values in the world of PCR testing. One should not over-interpret that.
Dr. Kalaycio: I think that you're making an important point about absolutes in the interpretation of these tests. Dr. Mauro, how do you interpret the 3-month data that comes back?
Dr. Mauro: I agree with my colleague on the approach that it has to be put in a clinical context. I think what we've learned is the importance of the starting value and relative reduction and to not consider response milestones as black and white guides.
I think there are certain scenarios that require some caution. The patient who is on imatinib who is close but has not reached the landmark may be different that a similar patient on dasatinib or nilotinib who has not had significant reduction—that's probably a more pressing situation.
It's ironic that guidelines—maybe because of lack of options—don't encourage us to think about changing therapy early in someone who hasn't met milestones in 3 months when they've been put on a more potent agent.
I also think that careful consideration is needed regarding treatment intensity and the impact of interruptions, in conjunction with all the other facets—the rate of change, the absolute change from the patient's baseline, the starting response level and the timing of the PCR. We need to look at the actual day it's performed. If it's really not 3 months of therapy, we can misjudge.
Dr. Kalaycio: Right. Now, as you're monitoring patients, Dr. Mauro, at what point would you consider testing for kinase domain mutations?
Dr. Mauro: I think we have pretty good guidance from studies to date regarding when mutation testing is indicated and of higher yield. It's generally earlier on into treatment when we have clinical scenarios that are of greater risk, namely failure to achieve cytogenetic responses. In such scenarios if mutation testing informs treatment decision making it is very helpful.
Patients who have defined themselves as not achieving early molecular response—which we discussed earlier—especially someone who's had a second-generation TKI, warrants mutation testing in my view. Patients who don't achieve classical cytogenetic response landmarks at 6 and 12 (or 18) months, who thus have a higher residual volume of disease and perhaps as a function more clonal instability, I think also warrant attention.
I think we run into trouble when we start to, in an overly critical manner, assess patients’ longer term and deeper molecular response trajectories. Mutation testing becomes difficult or impossible for patients who are at or near major molecular remission (MMR). Mutation yield is generally very low for patients who have not lost MMR and also likely those with small volume of change around the MMR threshold. Looking ahead, I think further investigation into early time points and the setting of minimal residual disease may yield data to be able to predict potential resistance earlier than observing it clinically.
Dr. Kalaycio: Right. Dr. Deininger, if you're monitoring a patient and he’s missing milestones and you do obtain a mutation analysis and find a T315I mutation, do you offer that patient an opportunity to see how well they're going to do with ponatinib, or do you refer that patient to your transplant team for consideration of transplant as soon as a donor is available?As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated. – Matt Kalaycio, MD
Dr. Deininger: I think we do the first and half of the second. We would certainly offer a trial of ponatinib or, if possible, a clinical trial unless there are insurmountable contraindications.
I don't really think there are any such circumstances with optimized cardiovascular management at the same time, and we may involve a cardiologist at that point to help us if there are cardiovascular risk factors.
We would also do a referral to a transplant center unless the patient is as per performance status, just not a transplant candidate or is too old. Otherwise, we would always make a referral, but we would not pursue a transplant as the first modality, provided that the patient is in chronic phase. Progression to accelerated phase or blastic phase would be looked at totally differently. Here we would certainly put people on a transplant course.
Dr. Kalaycio: I agree. As a transplanter, even I would not suggest transplant upon the recognition of the T315I. I would wait until all available TKIs have been tried and have either not worked or weren't tolerated.
As the two of you know, we're doing fewer and fewer transplants for CML these days. The results we're getting in folks who are failing all of these agents are not as good as they used to be when we were transplanting in the first 3 months of a new diagnosis. Bone marrow transplant is something to defer for as long as possible, at least in my mind.
TKI Dosage Considerations
Dr. Kalaycio: Now we touched a little bit on the side effects of nilotinib and dasatinib. We've talked about progressive disease, perhaps with or without mutations. Dr. Mauro, could you comment on where you think bosutinib might play a role in patients with relapsed or progressive CML?
Dr. Mauro: I think bosutinib remains an underutilized TKI. It is potent and thus offers good salvage activity. I think it offers a fairly distinct side effect profile making it a good choice for cases of intolerance. I think we've learned more about how to initiate bosutinib and manage its side effects; one element of this is that current trials start at slightly lower doses of bosutinib to avoid some of the early gastrointestinal toxicity.
Bosutinib continues to seek a place as a first-line therapy. I think this may be worthwhile as we are looking for the right balance of safety and side effect risk to maximize early response. Regarding the salvage setting—intolerance and resistance to other agents—I find myself speaking to patients about bosutinib as a potent and viable alternative. Bosutinib offers a similar spectrum of activity as dasatinib but can allow us to avoid cardiovascular toxicity and/or allow clearance of pleural and pericardial toxicity occurring with dasatinib.
The one place I would say bosutinib may not hold up as strongly to its competition would be as a third-line therapy after failure of a second-generation TKI. I think while there is reasonable activity with bosutinib in this setting of somewhat fairly drug-resistant CML, the performance of ponatinib is better. There is likely often a struggle with the long-term safety question of ponatinib, making some feel that a trial of bosutinib is often a worthwhile, logical, or even necessary step before ponatinib. My concerns about this approach are related to treatment decision-making based on safety and theoretical risk of adverse effects rather than efficacy and risk of progression; these have to be weighed carefully and appropriately against each other.
Dr. Kalaycio: That's an interesting perspective. You mentioned starting with a lower dose. Dr. Deininger, I'd like to ask you your thoughts about dose modifications of the TKIs in general. It used to be anathema to either start with a low dose or to maintain patients on a lower dose.
I'm aware of at least some data suggesting that those patients who tolerate these TKIs poorly particularly with regard to myelosuppression can be treated long term at lower than typically prescribed doses without adverse effect. What are your thoughts surrounding dose modification of the TKIs?
Dr. Deininger: I would stratify according to TKIs. I think imatinib at 400 mg is probably just rightly dosed, maybe not even at the optimal dose. It could be 600 mg. If you go to 300 mg, that is still acceptable.
If you go to 200 mg, I think I would make double sure that I monitor patients very frequently. People should be, in my mind, in a major molecular response to treat them with 200 mg long term. With the second-generation TKIs, it's a little different.
I cannot really speak to bosutinib because I'm not aware of a lot of data on those modifications. What seems to be clear from dasatinib is that the initial recommended dose of 100 mg is quite high, especially in older people.
I believe that's been corroborated by a study that hasn't been published yet. Apparently the drug excretion in the older individuals is quite a bit slower than in younger people. I think in our practice and in other people's practices as well, about 50% of those people end at doses substantially lower than 100 mg, maybe 50 mg, maybe 40 mg, some even 20 mg per day.
I think in the case of dasatinib you have a lot of maneuvering space. You can adjust the dose according to tolerability and molecular response. With nilotinib I think it's kind of similar. We have a few patients who are on maybe 150 mg twice a day because they have issues in terms of side effects at the higher doses.
I should again thoroughly qualify that by saying if you give up through dose reductions the goal of a major molecular response, then I think you really have to think about the strategy in general because at that point, I think you basically say "this patient is not ever going to be reaching a safe haven and is not tolerating inhibitors well enough to get him there."
That's quite a significant statement to make. Myelosuppression is frequently a situation where you cannot deliver enough dose but you also don't get a good response, whereas other side effects like pleural effusions or excessive fluid retention may well be cause for dose reduction and yet responses may be acceptable. Maybe Dr. Mauro could chime in here and share his experience.
Dr. Mauro: I agree. I think some patients, based on their disease status, may not tolerate the dose that is going to—at least in theory—get them to a deep molecular remission. The biggest problem we often face is myelosuppression. Some patients may have indolent disease and still have a good outcome, at least in the short to medium term, with lower doses of drug. In general I try to work through myelosuppression, especially early, as the combination of myelosuppression and suboptimal or response failure is dangerous.
I think treatment shouldn't be too chaotic and change too much, if possible. That was a very nice summary of the way we view the doses. There's still a fair bit of flexibility amongst the TKI doses, although I don't advocate for starting low and titration up.
In general I think, with regard to optimal TKI dose, we might have overshot; for example, with dasatinib. I think we might have overshot with bosutinib. Trials ongoing now initiate bosutinib at 400 mg rather than 500 mg. That's what I was alluding to.
Treatment Free Remission: Approaches
Dr. Kalaycio: Very interesting discussions. As we wind down the conversation, I want to get to the idea of stopping therapy. We're all aware of data that suggest it's at least possible for a proportion of patients to stop treatment.5
However, the guidelines3,4 such as they are, suggest that stopping should not be done outside the context of a clinical trial. I think most of us would agree with that. I wonder in a practical sense, Dr. Mauro, in your practice, do you have your own set of internal criteria for stopping a TKI and observing patients in the absence of any therapy?
Dr. Mauro: I think I try to incorporate all the experience we have to date when considering this question. That being said I'm a little hesitant to agree entirely with the current thinking regarding retreatment during a Treatment Free Remission trial, which is waiting until patients lose MMR in order to resume treatment but agree it might be hard to retreat based on lesser degrees of molecular relapse. I just worry a little bit about that amount of proliferation without treatment.
On the other hand I think we're coming to the realization that TFR may be feasible in patients who are simply nonproliferative and have low volume of disease based on newer data that patients may not need to have consistent "complete molecular response" or disease reduction below 4.5 logs in order to consider TFR. This will mean more patients may be eligible for such a strategy.
With that being said, I strongly advocate for discontinuation occurring in trials still, especially in the United States. I think we still have to ensure regular monitoring and not run the risk of loss of CML remission as a result of this endeavor. I think we have to have a clear message that this is still an investigational approach.
As we apply TFR strategies more broadly and explore it in patients with different circumstances, rather than those already studied in clinical trials, we may see slightly different results. We may need to exercise more caution.
Outside of a trial I don't discontinue TKI therapy unless there's a clear medical indication and there's no way around it, such as pregnancy or other illness that precludes TKI treatment. I would still pursue TFR only in a clinical trial.
Dr. Kalaycio: Dr. Deininger, I'll give you the last word for your thoughts regarding both treatment discontinuation and the future of CML therapies.
Dr. Deininger: As far as TFR is concerned, I agree with Dr. Mauro. Unfortunately, in the United States we're not quite as far advanced as the Europeans at monitoring diligence. There's a bit of a concern that if we elevate that into daily practice, we may see that patients are not monitored frequently enough and then relapse, but that doesn't get caught. You could imagine a scenario where we're actually seeing a decline in outcomes because people are discontinued and then not restarted if they have a recurrence at the molecular level.
All in all, where this is going, I think it will be really interesting to see more confirmatory data for second-generation TKIs. Let me put that differently: whether a similar proportion of patients in deep molecular response can maintain responses and have TFR if they needed a second-generation TKI to get there rather than just imatinib.
If so, this would be very promising. That would mean that second-generation TKIs actually somewhat impact the natural history of the disease and its biology. Then TFR may become a reality for a substantial proportion of our patients.
I don't think we have the data yet, but at least there are some suggestions that this may be the case. If you do the math, you will still see that many patients with CML will never reach a state where they can consider TFR.
They'll never get into a deep molecular response or they will have been diagnosed with accelerated phase. For all these patients who are not candidates for TFR, we still have to think about treatment optimization in order to make them catch up with the rest.
Now I have a couple of interesting developments. One is clearly to see whether ponatinib administered at a lower dose will have a more practical, therapeutic window in terms of toxicity and yet maintain the excellent efficacy that it has in the setting.
These data need to be produced. Another scenario would be that people would get started on intense induction treatment with higher-risk drugs such as ponatinib. Then if they are a good responder, they can switch to something that is lower-risk and that may be better tolerated.
Actually some of these trials are underway in Europe. Of course there's the question, are there still other drugs that will enter the CML space. There's one promising molecule called ABL001, a TKI with a different mode of action, which exploits an allosteric site rather than the catalytic center of the kinase.
It’s really conceptually very interesting. The expectation would be that this molecule has fewer side effects but still may be quite potent. This could be a very interesting development and add something to the armamentarium that we currently don't have.
I also think there will be patients whose diseases are just beyond the reach of a TKI alone for many reasons, maybe additional mutations or things that have to do with the host and metabolism.
In these cases, we'll still have to think about combination treatments and non-TKI treatments. Here, I think an honest answer is the labs have pulled out a lot of interesting leads, but so far, nothing has really made it into a serious clinical context, either because of side effects or because the target may not be as good in humans as it seemed from mouse models.
I think a lot more work is required here to find the best combination therapies and to define those pathways that need to be inhibited together with BCR-ABL. I think there's a field that we'll develop further and that will be the cutting edge.
The Wrap-Up
Dr. Kalaycio: Although an uncommon diagnosis, CML in chronic phase can be treated with an expectation for nearly 100% 5-year survival. For that reason, clinicians need to manage their patients expertly. Steps taken at diagnosis are critical to subsequent decision-making. Once treatment begins, close monitoring is required to screen for side effects as well as ensuring treatment success. With so many effective agents available, patients can be selected for the agent least likely to cause them long-term harm. Perhaps in the future we will learn which patients can stop treatment altogether.
Gentleman, thank you so much for your time. Every time I talk to you I learn something.
1. Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942.
2. National Comprehensive Cancer Network guidelines for treatment of cancer by site. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed August 20, 2015.
3. O'Brien S, Radich JP, Abboud CN, et al. Chronic myelogenous leukemia, version 1.2015. J Natl Compr Canc Netw. 2014;12(11):1590–1610.
4. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884.
5. Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035.
1. Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942.
2. National Comprehensive Cancer Network guidelines for treatment of cancer by site. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed August 20, 2015.
3. O'Brien S, Radich JP, Abboud CN, et al. Chronic myelogenous leukemia, version 1.2015. J Natl Compr Canc Netw. 2014;12(11):1590–1610.
4. Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884.
5. Mahon FX, Réa D, Guilhot J, et al. Discontinuation of imatinib in patients with chronic myeloid leukemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010;11(11):1029–1035.

Inhibitors can target CML stem cells
Preclinical research has revealed nutrients that support the activity of chronic myelogenous leukemia (CML) stem cells and suggests these nutrients may be promising targets for CML therapy.
Investigators discovered that CML stem cells accumulate high levels of certain dipeptide species, and these dipeptides act as nutrients for the cells.
When the team inhibited dipeptide uptake, they observed decreased CML stem cell activity.
Combining agents that inhibit dipeptide uptake with tyrosine kinase inhibitors (TKIs) proved more effective against CML than TKI treatment alone, both in vitro and in vivo.
Kazuhito Naka, PhD, of Hiroshima University in Japan, and his colleagues conducted this research and disclosed the results in Nature Communications.
The team began by analyzing CML stem cells isolated from a mouse model of the disease. They found that CML stem cells accumulate significantly higher levels of certain dipeptide species—such as Ala-Leu, Asp-Leu, Ser-Tyr, and Thr-Val—than normal hematopoietic stem cells.
Additional investigation revealed that CML stem cells take up dipeptides via the Slc15A2 transporter. And once internalized, the dipeptides act as nutrients and play a role in CML stem cell maintenance.
The dipeptides activate amino-acid signaling via a pathway involving p38MAPK and Smad3, and this promotes CML stem cell maintenance.
To build upon these findings, the investigators assessed the effects of cefadroxil, which inhibits Slc15A2-mediated nutrient signaling, in combination with imatinib as CML treatment.
In murine CML stem cell cultures, the 2 drugs in combination reduced colony formation more effectively than imatinib alone.
In vivo, mice with CML responded to imatinib alone but eventually experienced disease recurrence. And cefadroxil alone promoted disease development. But when cefadroxil was given in combination with imatinib, disease recurrence was significantly lower than in mice that received imatinib alone.
The investigators then showed that cefadroxil decreases the number of CML stem cells in CML-affected mice. And cefadroxil combined with imatinib reduces CML stem cell numbers more effectively than imatinib alone.
In serial transplantation experiments, CML stem cells isolated from cefadroxil-treated mice completely lost their ability to drive BCR-ABL1+ disease in new recipients. These animals survived for more than 90 days, whereas mice that received CML stem cells from vehicle-treated mice developed BCR-ABL1+ disease and died before 80 days.
The investigators also tested cefadroxil in stem cells derived from humans with chronic CML. Cefadroxil suppressed the colony-forming capacity of all 3 samples tested. And combining cefadroxil with imatinib or dasatinib reduced colony formation more effectively than either TKI alone.
Lastly, the team decided to test 3 clinical-grade p38MAPK inhibitors that are already approved for use in the US—Ly2228820 (ralimetinib), VX-702, and BIRB796 (doramapimod). When cultured with CML stem cells, each of these drugs significantly decreased colony formation.
In addition, Ly2228820 combined with dasatinib delayed CML onset in mice and improved their survival when compared with dasatinib alone.
These results suggest p38MAPK inhibitors and cefadroxil may be useful additions to TKI therapy in CML, the investigators said.
“Our proposed approach of using inhibitors to shut down a key nutrient uptake process specific to CML stem cells, in combination with TKI therapy, may thus provide concrete therapeutic benefits to patients with CML,” Dr Naka said. “It will open up a novel avenue for curative CML therapy.” ![]()
Preclinical research has revealed nutrients that support the activity of chronic myelogenous leukemia (CML) stem cells and suggests these nutrients may be promising targets for CML therapy.
Investigators discovered that CML stem cells accumulate high levels of certain dipeptide species, and these dipeptides act as nutrients for the cells.
When the team inhibited dipeptide uptake, they observed decreased CML stem cell activity.
Combining agents that inhibit dipeptide uptake with tyrosine kinase inhibitors (TKIs) proved more effective against CML than TKI treatment alone, both in vitro and in vivo.
Kazuhito Naka, PhD, of Hiroshima University in Japan, and his colleagues conducted this research and disclosed the results in Nature Communications.
The team began by analyzing CML stem cells isolated from a mouse model of the disease. They found that CML stem cells accumulate significantly higher levels of certain dipeptide species—such as Ala-Leu, Asp-Leu, Ser-Tyr, and Thr-Val—than normal hematopoietic stem cells.
Additional investigation revealed that CML stem cells take up dipeptides via the Slc15A2 transporter. And once internalized, the dipeptides act as nutrients and play a role in CML stem cell maintenance.
The dipeptides activate amino-acid signaling via a pathway involving p38MAPK and Smad3, and this promotes CML stem cell maintenance.
To build upon these findings, the investigators assessed the effects of cefadroxil, which inhibits Slc15A2-mediated nutrient signaling, in combination with imatinib as CML treatment.
In murine CML stem cell cultures, the 2 drugs in combination reduced colony formation more effectively than imatinib alone.
In vivo, mice with CML responded to imatinib alone but eventually experienced disease recurrence. And cefadroxil alone promoted disease development. But when cefadroxil was given in combination with imatinib, disease recurrence was significantly lower than in mice that received imatinib alone.
The investigators then showed that cefadroxil decreases the number of CML stem cells in CML-affected mice. And cefadroxil combined with imatinib reduces CML stem cell numbers more effectively than imatinib alone.
In serial transplantation experiments, CML stem cells isolated from cefadroxil-treated mice completely lost their ability to drive BCR-ABL1+ disease in new recipients. These animals survived for more than 90 days, whereas mice that received CML stem cells from vehicle-treated mice developed BCR-ABL1+ disease and died before 80 days.
The investigators also tested cefadroxil in stem cells derived from humans with chronic CML. Cefadroxil suppressed the colony-forming capacity of all 3 samples tested. And combining cefadroxil with imatinib or dasatinib reduced colony formation more effectively than either TKI alone.
Lastly, the team decided to test 3 clinical-grade p38MAPK inhibitors that are already approved for use in the US—Ly2228820 (ralimetinib), VX-702, and BIRB796 (doramapimod). When cultured with CML stem cells, each of these drugs significantly decreased colony formation.
In addition, Ly2228820 combined with dasatinib delayed CML onset in mice and improved their survival when compared with dasatinib alone.
These results suggest p38MAPK inhibitors and cefadroxil may be useful additions to TKI therapy in CML, the investigators said.
“Our proposed approach of using inhibitors to shut down a key nutrient uptake process specific to CML stem cells, in combination with TKI therapy, may thus provide concrete therapeutic benefits to patients with CML,” Dr Naka said. “It will open up a novel avenue for curative CML therapy.” ![]()
Preclinical research has revealed nutrients that support the activity of chronic myelogenous leukemia (CML) stem cells and suggests these nutrients may be promising targets for CML therapy.
Investigators discovered that CML stem cells accumulate high levels of certain dipeptide species, and these dipeptides act as nutrients for the cells.
When the team inhibited dipeptide uptake, they observed decreased CML stem cell activity.
Combining agents that inhibit dipeptide uptake with tyrosine kinase inhibitors (TKIs) proved more effective against CML than TKI treatment alone, both in vitro and in vivo.
Kazuhito Naka, PhD, of Hiroshima University in Japan, and his colleagues conducted this research and disclosed the results in Nature Communications.
The team began by analyzing CML stem cells isolated from a mouse model of the disease. They found that CML stem cells accumulate significantly higher levels of certain dipeptide species—such as Ala-Leu, Asp-Leu, Ser-Tyr, and Thr-Val—than normal hematopoietic stem cells.
Additional investigation revealed that CML stem cells take up dipeptides via the Slc15A2 transporter. And once internalized, the dipeptides act as nutrients and play a role in CML stem cell maintenance.
The dipeptides activate amino-acid signaling via a pathway involving p38MAPK and Smad3, and this promotes CML stem cell maintenance.
To build upon these findings, the investigators assessed the effects of cefadroxil, which inhibits Slc15A2-mediated nutrient signaling, in combination with imatinib as CML treatment.
In murine CML stem cell cultures, the 2 drugs in combination reduced colony formation more effectively than imatinib alone.
In vivo, mice with CML responded to imatinib alone but eventually experienced disease recurrence. And cefadroxil alone promoted disease development. But when cefadroxil was given in combination with imatinib, disease recurrence was significantly lower than in mice that received imatinib alone.
The investigators then showed that cefadroxil decreases the number of CML stem cells in CML-affected mice. And cefadroxil combined with imatinib reduces CML stem cell numbers more effectively than imatinib alone.
In serial transplantation experiments, CML stem cells isolated from cefadroxil-treated mice completely lost their ability to drive BCR-ABL1+ disease in new recipients. These animals survived for more than 90 days, whereas mice that received CML stem cells from vehicle-treated mice developed BCR-ABL1+ disease and died before 80 days.
The investigators also tested cefadroxil in stem cells derived from humans with chronic CML. Cefadroxil suppressed the colony-forming capacity of all 3 samples tested. And combining cefadroxil with imatinib or dasatinib reduced colony formation more effectively than either TKI alone.
Lastly, the team decided to test 3 clinical-grade p38MAPK inhibitors that are already approved for use in the US—Ly2228820 (ralimetinib), VX-702, and BIRB796 (doramapimod). When cultured with CML stem cells, each of these drugs significantly decreased colony formation.
In addition, Ly2228820 combined with dasatinib delayed CML onset in mice and improved their survival when compared with dasatinib alone.
These results suggest p38MAPK inhibitors and cefadroxil may be useful additions to TKI therapy in CML, the investigators said.
“Our proposed approach of using inhibitors to shut down a key nutrient uptake process specific to CML stem cells, in combination with TKI therapy, may thus provide concrete therapeutic benefits to patients with CML,” Dr Naka said. “It will open up a novel avenue for curative CML therapy.” ![]()