User login
Inhibitor shows promise for hematologic disorders

Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()
Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()
Photo courtesy of EHA
MILAN—The IDH2 inhibitor AG-221 is well-tolerated and exhibits durable clinical activity in patients with hematologic disorders, results of a phase 1 study suggest.
The drug prompted responses in patients with myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML).
Fourteen of 25 patients achieved a response, and 12 of those responses are ongoing.
Most adverse events (AEs) were grade 1 or 2 in nature. However, 4 patients did have serious AEs that were possibly related to treatment.
Stéphane de Botton, MD, PhD, of Institut Gustave Roussy in Villejuif, France, presented these results at the 19th Annual Congress of the European Hematology Association (EHA) as abstract LB2434.
Dr de Botton and his colleagues enrolled 35 patients who had a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at 30 mg BID (n=7), 50 mg BID (n=7), 75 mg BID (n=6), 100 mg QD (n=5), 100 mg BID (n=5), or 150 mg QD (n=5). Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
Safety data
“AG-221 was remarkable well-tolerated, and the [maximum tolerated dose] has not been reached,” Dr de Botton said. “The majority of adverse events were grade 1 and 2.”
Eighteen patients were evaluable for safety. AEs of all grades included nausea (n=4), pyrexia (n=4), thrombocytopenia (n=4), anemia (n=3), dizziness (n=3), febrile neutropenia (n=3), peripheral edema (n=3), sepsis (n=3), cough (n=2), diarrhea (n=2), fatigue (n=2), leukocytosis (n=2), neutropenia (n=2), petechiae (n=2), and rash (n=2).
Grade 3 or higher AEs included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1). Dr de Botton noted that diarrhea and rash were not expected events.
Four patients had serious AEs possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Seven patients died within 30 days of study drug termination: 4 in the 30-mg cohort, 2 in the 50-mg cohort, and 1 in the 100-mg-BID cohort.
Five deaths were due to complications of disease-related sepsis (all in cycle 1), 1 complication of a humeral fracture, and 1 complication of a stroke.
Activity and response data
The researchers observed high AG-221 accumulation after multiple doses. And results were “really very similar” between the 30-mg-BID cohort and the 100-mg-QD cohort, Dr de Botton noted.
He also said AG-221 was “very efficient” at inhibiting 2-HG in the plasma. 2-HG was inhibited up to 100% in subjects with R140Q mutations and up to 60% in subjects with R172K mutations.
Twenty-five patients were evaluable for response. The remaining 10 patients did not have day-28 marrow assessments, either due to early termination (n=7) or receiving less than 28 days of treatment although they were still on the study (n=3).
In all, there were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery (CRps), 1 CR with incomplete hematologic recovery (CRi), and 5 partial responses (PRs). Five patients had stable disease (SD), and 6 had progressive disease (PD).
The most responses occurred in the 50-mg group, which had 3 CRs, 1 CRi, and 1 PR. This was followed by the 30-mg group, which had 2 CRs, 1 CRp, and 1 PR.
“The majority of responses occurred in cycle 1,” Dr de Botton noted, “except in the first cohort [30 mg], where responses occurred late, at the end of cycle 3 and cycle 4.”
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CRp, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with SD remain on study.
This study is sponsored by Celgene Corporation and Agios Pharmaceuticals Inc., the companies developing AG-221. ![]()
Ruxolitinib improves disease control in PV

©ASCO/Phil McCarten
CHICAGO—The JAK1/2 inhibitor ruxolitinib may be a “valuable new treatment option” for patients with polycythemia vera (PV) who cannot tolerate or are resistant to hydroxyurea, according to a speaker at the 2014 ASCO Annual Meeting.
Results of the phase 3 RESPONSE trial showed that ruxolitinib can reduce spleen size in these patients and improve hematocrit control without the need for phlebotomy.
And the drug was generally well-tolerated.
“Patients with PV may not have their disease controlled with existing therapies, increasing their risk for cardiovascular complications,” said Srdan Verstovsek, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
“In the RESPONSE trial, patients treated with ruxolitinib showed better disease control and improved symptom management compared to [patients who received] current therapies.”
Dr Verstovsek presented results from the trial at ASCO as abstract 7026. RESPONSE was funded by Incyte Corporation, the company developing ruxolitinib.
The study has enrolled 222 patients with PV that is resistant to or intolerant of hydroxyurea. Patients were randomized to receive either ruxolitinib at a starting dose of 10 mg twice-daily or best available therapy (BAT). The ruxolitinib dose was adjusted as needed throughout the study.
At week 32, 77% of patients on ruxolitinib and 20% on BAT achieved hematocrit control or spleen reduction.
The primary endpoint of the study was the proportion of patients whose hematocrit was controlled without phlebotomy from week 8 through 32 and whose spleen volume was reduced by 35% or more from baseline as assessed by imaging at 32 weeks.
Twenty-one percent of patients in the ruxolitinib arm achieved this endpoint, compared to 1% of patients in the BAT arm.
“Ninety-one percent of patients who achieved the primary end point had a confirmed response at 48 weeks,” Dr Verstovsek said.
He also noted that nearly half of ruxolitinib-treated patients had a 50% or greater reduction in debilitating PV symptoms, compared to 5% of those on BAT.
Patients treated with ruxolitinib also experienced a reduction in night sweats and itchiness. In addition, a greater proportion of patients in the ruxolitinib arm achieved complete hematologic response when compared to the BAT arm—24% and 9%, respectively.
Ruxolitinib was generally well-tolerated, Dr Verstovsek said, noting that 85% of patients were still receiving the drug at a median follow-up of 81 weeks.
However, 3.6% of patients on ruxolitinib discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
Most adverse events observed in the ruxolitinib arm were grade 1 or 2 and were consistent with those previously seen in ruxolitinib studies in PV and myelofibrosis.
Few patients developed grade 3 or 4 cytopenias. Within the first 32 weeks of treatment, grade 3 or 4 hematologic adverse events in the ruxolitinib treatment arm were anemia (1.8%) and thrombocytopenia (5.5%). Fewer patients treated with ruxolitinib experienced thromboembolic events when compared to those who received BAT.
The most common non-hematologic adverse events were headache, diarrhea, and fatigue, which were mainly grade 1 or 2. Dr Verstovsek noted that herpes zoster infection was higher in the ruxolitinib arm than in the BAT arm.
Ruxolitinib is currently approved in more than 60 countries for patients with myelofibrosis. Global regulatory filings for PV are underway based on the RESPONSE data. ![]()
©ASCO/Phil McCarten
CHICAGO—The JAK1/2 inhibitor ruxolitinib may be a “valuable new treatment option” for patients with polycythemia vera (PV) who cannot tolerate or are resistant to hydroxyurea, according to a speaker at the 2014 ASCO Annual Meeting.
Results of the phase 3 RESPONSE trial showed that ruxolitinib can reduce spleen size in these patients and improve hematocrit control without the need for phlebotomy.
And the drug was generally well-tolerated.
“Patients with PV may not have their disease controlled with existing therapies, increasing their risk for cardiovascular complications,” said Srdan Verstovsek, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
“In the RESPONSE trial, patients treated with ruxolitinib showed better disease control and improved symptom management compared to [patients who received] current therapies.”
Dr Verstovsek presented results from the trial at ASCO as abstract 7026. RESPONSE was funded by Incyte Corporation, the company developing ruxolitinib.
The study has enrolled 222 patients with PV that is resistant to or intolerant of hydroxyurea. Patients were randomized to receive either ruxolitinib at a starting dose of 10 mg twice-daily or best available therapy (BAT). The ruxolitinib dose was adjusted as needed throughout the study.
At week 32, 77% of patients on ruxolitinib and 20% on BAT achieved hematocrit control or spleen reduction.
The primary endpoint of the study was the proportion of patients whose hematocrit was controlled without phlebotomy from week 8 through 32 and whose spleen volume was reduced by 35% or more from baseline as assessed by imaging at 32 weeks.
Twenty-one percent of patients in the ruxolitinib arm achieved this endpoint, compared to 1% of patients in the BAT arm.
“Ninety-one percent of patients who achieved the primary end point had a confirmed response at 48 weeks,” Dr Verstovsek said.
He also noted that nearly half of ruxolitinib-treated patients had a 50% or greater reduction in debilitating PV symptoms, compared to 5% of those on BAT.
Patients treated with ruxolitinib also experienced a reduction in night sweats and itchiness. In addition, a greater proportion of patients in the ruxolitinib arm achieved complete hematologic response when compared to the BAT arm—24% and 9%, respectively.
Ruxolitinib was generally well-tolerated, Dr Verstovsek said, noting that 85% of patients were still receiving the drug at a median follow-up of 81 weeks.
However, 3.6% of patients on ruxolitinib discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
Most adverse events observed in the ruxolitinib arm were grade 1 or 2 and were consistent with those previously seen in ruxolitinib studies in PV and myelofibrosis.
Few patients developed grade 3 or 4 cytopenias. Within the first 32 weeks of treatment, grade 3 or 4 hematologic adverse events in the ruxolitinib treatment arm were anemia (1.8%) and thrombocytopenia (5.5%). Fewer patients treated with ruxolitinib experienced thromboembolic events when compared to those who received BAT.
The most common non-hematologic adverse events were headache, diarrhea, and fatigue, which were mainly grade 1 or 2. Dr Verstovsek noted that herpes zoster infection was higher in the ruxolitinib arm than in the BAT arm.
Ruxolitinib is currently approved in more than 60 countries for patients with myelofibrosis. Global regulatory filings for PV are underway based on the RESPONSE data. ![]()
©ASCO/Phil McCarten
CHICAGO—The JAK1/2 inhibitor ruxolitinib may be a “valuable new treatment option” for patients with polycythemia vera (PV) who cannot tolerate or are resistant to hydroxyurea, according to a speaker at the 2014 ASCO Annual Meeting.
Results of the phase 3 RESPONSE trial showed that ruxolitinib can reduce spleen size in these patients and improve hematocrit control without the need for phlebotomy.
And the drug was generally well-tolerated.
“Patients with PV may not have their disease controlled with existing therapies, increasing their risk for cardiovascular complications,” said Srdan Verstovsek, MD, PhD, of MD Anderson Cancer Center in Houston, Texas.
“In the RESPONSE trial, patients treated with ruxolitinib showed better disease control and improved symptom management compared to [patients who received] current therapies.”
Dr Verstovsek presented results from the trial at ASCO as abstract 7026. RESPONSE was funded by Incyte Corporation, the company developing ruxolitinib.
The study has enrolled 222 patients with PV that is resistant to or intolerant of hydroxyurea. Patients were randomized to receive either ruxolitinib at a starting dose of 10 mg twice-daily or best available therapy (BAT). The ruxolitinib dose was adjusted as needed throughout the study.
At week 32, 77% of patients on ruxolitinib and 20% on BAT achieved hematocrit control or spleen reduction.
The primary endpoint of the study was the proportion of patients whose hematocrit was controlled without phlebotomy from week 8 through 32 and whose spleen volume was reduced by 35% or more from baseline as assessed by imaging at 32 weeks.
Twenty-one percent of patients in the ruxolitinib arm achieved this endpoint, compared to 1% of patients in the BAT arm.
“Ninety-one percent of patients who achieved the primary end point had a confirmed response at 48 weeks,” Dr Verstovsek said.
He also noted that nearly half of ruxolitinib-treated patients had a 50% or greater reduction in debilitating PV symptoms, compared to 5% of those on BAT.
Patients treated with ruxolitinib also experienced a reduction in night sweats and itchiness. In addition, a greater proportion of patients in the ruxolitinib arm achieved complete hematologic response when compared to the BAT arm—24% and 9%, respectively.
Ruxolitinib was generally well-tolerated, Dr Verstovsek said, noting that 85% of patients were still receiving the drug at a median follow-up of 81 weeks.
However, 3.6% of patients on ruxolitinib discontinued treatment due to adverse events, compared to 1.8% of patients on BAT.
Most adverse events observed in the ruxolitinib arm were grade 1 or 2 and were consistent with those previously seen in ruxolitinib studies in PV and myelofibrosis.
Few patients developed grade 3 or 4 cytopenias. Within the first 32 weeks of treatment, grade 3 or 4 hematologic adverse events in the ruxolitinib treatment arm were anemia (1.8%) and thrombocytopenia (5.5%). Fewer patients treated with ruxolitinib experienced thromboembolic events when compared to those who received BAT.
The most common non-hematologic adverse events were headache, diarrhea, and fatigue, which were mainly grade 1 or 2. Dr Verstovsek noted that herpes zoster infection was higher in the ruxolitinib arm than in the BAT arm.
Ruxolitinib is currently approved in more than 60 countries for patients with myelofibrosis. Global regulatory filings for PV are underway based on the RESPONSE data. ![]()
Studies confirm importance of CALR mutation in PMF
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
MILAN—Two new studies appear to confirm the prognostic significance of CALR mutations in patients with primary myelofibrosis (PMF).
One study showed that PMF patients with mutated CALR had prolonged overall survival (OS) compared to patients with wild-type CALR. And additional subclonal mutations did not impair the positive impact of CALR mutations.
Another study suggested that indels in exon 9 of CALR are founding driver mutations in PMF. These mutations are independent predictors of clinical course, disease progression, and OS.
Both studies were presented at the 19th Congress of the European Hematology Association (EHA).
Paola Guglielmelli, MD, PhD, of the University of Florence in Italy, presented data on CALR mutations in the context of additional mutations (abstract S1355).
And Elisa Rumi, MD, of the University of Pavia in Italy, presented information on mutated CALR and other founding driver mutations in PMF (abstract S1356).
CALR & other subclonal mutations in PMF
To investigate the prognostic role of CALR mutations in relation to additional subclonal mutations, Dr Guglielmelli and her colleagues analyzed 274 samples from PMF patients.
The team genotyped the samples for mutations in 11 genes: JAK2, CALR, MPL, EZH2, ASXL1, SRSF2, IDH1, IDH2, CBL, TET2, and DNMT3A.
Two hundred and fifty-six patients (93.4%) presented with at least 1 somatic mutation, and 104 (38%) presented with at least 2.
The median follow-up was 3.8 years (range, 0.52-29.20 years). Among all patients, the median OS was 12.2 years (range, 5.6-18.8 years). Eighty-four patients died (30.7%), 44 (16.1%) of them due to leukemia.
The presence of CALR mutations was associated with better OS, independent of IPSS and molecular risk categories (hazard ratio [HR] 0.51, P=0.03). But CALR mutations did not impact the risk of progression to acute leukemia.
Among patients with a low- to intermediate-1-risk IPSS score, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR 0.4, P=0.02). Among patients with intermediate-2 to high risk, those with CALR mutations lived a median of 4.2 years, and those without lived a median of 2.6 years (HR=0.5, P=0.09).
Among patients with high molecular risk, those with CALR mutations lived a median of 17.7 years, and those without lived a median of 4.3 years, (HR=0.3, P=0.008). High molecular risk was defined as at least 1 mutation in ASXL1, EZH2, SRSF2, or IDH1/2.
Among patients in the low-molecular-risk group, those with CALR mutations lived a median of 27.7 years, and those without lived a median of 21.7 years (HR=0.6, P=0.048).
Dr Guglielmelli said these results confirm the association between CALR mutation and favorable outcomes in PMF. They also show that additional subclonal mutations do not impair the positive impact of CALR mutation, thereby reinforcing the idea that CALR-mutated PMF is a distinct entity in terms of prognosis.
Founding driver mutations in PMF
In another presentation at the EHA Congress, Dr Rumi presented data on founding driver mutations in PMF. She and her colleagues analyzed 617 PMF patients, screening them for JAK2 V617F mutations, indels of CALR exon 9, and MPL exon 10 mutations.
The researchers assessed the impact of these mutations on thrombosis, progression to leukemia, and OS.
Their analysis suggested CALR-mutated PMF patients have a lower risk of thrombosis than JAK2-mutated patients (P=0.021). And this difference retained significance after adjusting for age.
Triple-negative PMF patients had a higher risk of leukemic evolution than CALR-mutated patients (P=0.016) and JAK2-mutated patients (P=0.043).
After adjusting for age, the risk remained significantly higher in triple-negative patients compared to JAK2-mutated patients (P=0.04) and retained borderline significance compared to CALR-mutated patients (P=0.052).
Patients with CALR-mutated PMF had a better OS than JAK2-mutated patients (P<0.001), MPL-mutated patients (P=0.009), and triple-negative patients (P<0.001).
In a multivariate analysis, CALR-mutated patients maintained a better OS than JAK2-mutated patients (P=0.019) and triple-negative patients (P<0.001).
Based on these results, Dr Rumi concluded that mutations in JAK2, CALR, and MPL are independent predictors of clinical course, disease progression, and OS in PMF. So screening patients for these mutations can likely improve upon the risk stratification provided by IPSS. ![]()
FDA removes partial clinical hold on imetelstat
The US Food and Drug Administration (FDA) has removed the partial clinical hold on the investigator-sponsored trial of imetelstat in myelofibrosis, which the agency imposed in March.
The partial hold was placed because of a safety signal of liver toxicity in myelofibrosis patients receiving the drug.
The principal investigator of the trial, Ayalew Tefferi, MD, of Mayo Clinic in Rochester, Minnesota, provided follow-up information to the FDA regarding the reversibility of the hepatotoxicity.
Upon review of the additional data, the FDA allowed the myelofibrosis trial to proceed.
Imetelstat is a lipid-conjugated oligonucleotide that binds with high affinity to the RNA template of telomerase, thereby inhibiting telomerase activity.
Most cancers are characterized by short telomeres and a high level of telomerase activity, which makes telomerase a rational target for the treatment of cancer.
Imetelstat is also being investigated in essential thrombocythemia and other myeloid hematologic malignancies.
The trial in myelofibrosis enrolled 33 high-risk or intermediate-2 risk patients with either primary or secondary myelofibrosis. Dr Teferi presented the data at the annual meeting of the American Society of Hematology in 2013 as abstract 662.
At the time of the meeting, myelosuppression appeared to be the major dose-limiting toxicity.
Geron’s Investigational New Drug application to the FDA for imetelstat remains on full clinical hold. This affects clinical trials in essential thrombocythemia, polycythemia vera, and multiple myeloma. ![]()
The US Food and Drug Administration (FDA) has removed the partial clinical hold on the investigator-sponsored trial of imetelstat in myelofibrosis, which the agency imposed in March.
The partial hold was placed because of a safety signal of liver toxicity in myelofibrosis patients receiving the drug.
The principal investigator of the trial, Ayalew Tefferi, MD, of Mayo Clinic in Rochester, Minnesota, provided follow-up information to the FDA regarding the reversibility of the hepatotoxicity.
Upon review of the additional data, the FDA allowed the myelofibrosis trial to proceed.
Imetelstat is a lipid-conjugated oligonucleotide that binds with high affinity to the RNA template of telomerase, thereby inhibiting telomerase activity.
Most cancers are characterized by short telomeres and a high level of telomerase activity, which makes telomerase a rational target for the treatment of cancer.
Imetelstat is also being investigated in essential thrombocythemia and other myeloid hematologic malignancies.
The trial in myelofibrosis enrolled 33 high-risk or intermediate-2 risk patients with either primary or secondary myelofibrosis. Dr Teferi presented the data at the annual meeting of the American Society of Hematology in 2013 as abstract 662.
At the time of the meeting, myelosuppression appeared to be the major dose-limiting toxicity.
Geron’s Investigational New Drug application to the FDA for imetelstat remains on full clinical hold. This affects clinical trials in essential thrombocythemia, polycythemia vera, and multiple myeloma. ![]()
The US Food and Drug Administration (FDA) has removed the partial clinical hold on the investigator-sponsored trial of imetelstat in myelofibrosis, which the agency imposed in March.
The partial hold was placed because of a safety signal of liver toxicity in myelofibrosis patients receiving the drug.
The principal investigator of the trial, Ayalew Tefferi, MD, of Mayo Clinic in Rochester, Minnesota, provided follow-up information to the FDA regarding the reversibility of the hepatotoxicity.
Upon review of the additional data, the FDA allowed the myelofibrosis trial to proceed.
Imetelstat is a lipid-conjugated oligonucleotide that binds with high affinity to the RNA template of telomerase, thereby inhibiting telomerase activity.
Most cancers are characterized by short telomeres and a high level of telomerase activity, which makes telomerase a rational target for the treatment of cancer.
Imetelstat is also being investigated in essential thrombocythemia and other myeloid hematologic malignancies.
The trial in myelofibrosis enrolled 33 high-risk or intermediate-2 risk patients with either primary or secondary myelofibrosis. Dr Teferi presented the data at the annual meeting of the American Society of Hematology in 2013 as abstract 662.
At the time of the meeting, myelosuppression appeared to be the major dose-limiting toxicity.
Geron’s Investigational New Drug application to the FDA for imetelstat remains on full clinical hold. This affects clinical trials in essential thrombocythemia, polycythemia vera, and multiple myeloma. ![]()
Mutations implicated in hematologic disorders
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
Credit: Jeremy L. Grisham
An analysis of more than 30,000 individuals has revealed several genetic mutations that appear to play roles in hematologic disorders.
Investigators discovered variants that showed correlations with platelet counts, white blood cell (WBC) counts, hemoglobin concentration, and hematocrit levels.
The group believes these findings could have implications for a range of conditions, including cytopenias, myeloproliferative neoplasms, and stroke.
Guillaume Lettre, PhD, of Université de Montréal and the Montreal Heart Institute in Canada, and his colleagues recounted their discoveries in a letter to Nature Genetics.
The investigators analyzed hemoglobin concentration, hematocrit levels, WBC counts, and platelet counts in 31,340 individuals genotyped on an exome array.
This revealed several missense variants in CXCR2 that were associated with a decreased WBC count. And in a resequencing study, the team identified a CXCR2 frameshift mutation that was associated with congenital neutropenia.
The group also discovered several missense and splice-site variants in genes known to regulate hematopoiesis—TFR2, HBB, TUBB1, SH2B3, and EPO.
A TFR2 mutation (rs139178017) was independently associated with higher hematocrit levels and hemoglobin concentration.
An HBB variant (rs33971440) and an EPO variant (rs62483572), on the other hand, were associated with lower hematocrit levels and hemoglobin concentrations. Further analyses confirmed that having these mutations increased a person’s risk of anemia, with odds ratios of 36.1 and 1.7, respectively.
A TUBB1 missense variant (rs41303899) was associated with decreased platelet count, while 2 missense variants of SH2B3 (rs148636776 and rs72650673) were associated with increased platelet counts.
Lastly, a mutation in JAK2 (rs77375493) was associated with increases in platelets, WBCs, hemoglobin, and hematocrit. And further analyses suggested that individuals with this variant had early stage myeloproliferative neoplasms.
“[T]hese donors also had a higher risk of having a stroke during their lifetime,” said study author Jean-Claude Tardif, MD, of Université de Montréal and the Montreal Heart Institute.
He and his colleagues believe these findings are encouraging, as they provide additional insight into hematologic disorders. But the results also suggest the experimental approach used in this study can be applied to other diseases as well. ![]()
New insight into TPO and platelet production

Credit: Walter and Eliza Hall
Institute of Medical Research
Investigators say they’ve determined how thrombopoietin (TPO) stimulates platelet production, and their findings may have implications for myeloproliferative neoplasms.
Researchers have long known that TPO is responsible for signaling cells in the bone marrow to produce platelets, but precisely which cells respond to TPO’s signals has been unclear.
Now, a group of investigators studying the TPO receptor Mpl have pinpointed those cells and made an unexpected discovery.
“Thrombopoietin did not directly stimulate the platelet’s ‘parent’ cells—the megakaryocytes—to make more platelets,” said study author Ashley Ng, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“Thrombopoietin signals actually acted on stem cells and progenitor cells, several generations back.”
Dr Ng and his colleagues reported these findings in PNAS.
The researchers had generated mice that express the Mpl receptor normally on stem and progenitor cells but lack expression on megakaryocytes and platelets. And these mice exhibited “profound” megakaryocytosis and thrombocytosis, as well as “remarkable” expansion of megakaryocyte-committed and multipotent progenitor cells.
“The progenitor and stem cells in the bone marrow began massively expanding and effectively turned the bone marrow into a megakaryocyte-making machine,” Dr Ng said.
Furthermore, although the progenitor cells showed signs of chronic TPO overstimulation, TPO levels were normal. This suggests that stem and progenitor cells expressing Mpl were responsible for TPO clearance, according to the investigators.
“Our findings support a theory whereby megakaryocytes and platelets control platelet numbers by ‘mopping up’ excess amounts of thrombopoietin in the bone marrow,” Dr Ng said. “In fact, we show this ‘mopping up’ action is absolutely essential in preventing blood disease where too many megakaryocytes and platelets are produced.”
So the researchers believe these findings will have implications for myeloproliferative neoplasms, particularly essential thrombocythemia.
“[P]revious studies have shown megakaryocytes and platelets in people with essential thrombocythemia have fewer Mpl receptors, which fits our model for excessive platelet production,” Dr Ng said.
To add support to their model, the investigators compared the progenitor cells responsible for overproducing megakaryocytes in their model to progenitor cells from patients with essential thrombocythemia. Both sets of cells showed a TPO stimulation signature.
“We think this study now provides a comprehensive model of how thrombopoietin controls platelet production,” Dr Ng said, “and perhaps gives some insight into the biology and mechanism behind specific myeloproliferative disorders.” ![]()
Credit: Walter and Eliza Hall
Institute of Medical Research
Investigators say they’ve determined how thrombopoietin (TPO) stimulates platelet production, and their findings may have implications for myeloproliferative neoplasms.
Researchers have long known that TPO is responsible for signaling cells in the bone marrow to produce platelets, but precisely which cells respond to TPO’s signals has been unclear.
Now, a group of investigators studying the TPO receptor Mpl have pinpointed those cells and made an unexpected discovery.
“Thrombopoietin did not directly stimulate the platelet’s ‘parent’ cells—the megakaryocytes—to make more platelets,” said study author Ashley Ng, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“Thrombopoietin signals actually acted on stem cells and progenitor cells, several generations back.”
Dr Ng and his colleagues reported these findings in PNAS.
The researchers had generated mice that express the Mpl receptor normally on stem and progenitor cells but lack expression on megakaryocytes and platelets. And these mice exhibited “profound” megakaryocytosis and thrombocytosis, as well as “remarkable” expansion of megakaryocyte-committed and multipotent progenitor cells.
“The progenitor and stem cells in the bone marrow began massively expanding and effectively turned the bone marrow into a megakaryocyte-making machine,” Dr Ng said.
Furthermore, although the progenitor cells showed signs of chronic TPO overstimulation, TPO levels were normal. This suggests that stem and progenitor cells expressing Mpl were responsible for TPO clearance, according to the investigators.
“Our findings support a theory whereby megakaryocytes and platelets control platelet numbers by ‘mopping up’ excess amounts of thrombopoietin in the bone marrow,” Dr Ng said. “In fact, we show this ‘mopping up’ action is absolutely essential in preventing blood disease where too many megakaryocytes and platelets are produced.”
So the researchers believe these findings will have implications for myeloproliferative neoplasms, particularly essential thrombocythemia.
“[P]revious studies have shown megakaryocytes and platelets in people with essential thrombocythemia have fewer Mpl receptors, which fits our model for excessive platelet production,” Dr Ng said.
To add support to their model, the investigators compared the progenitor cells responsible for overproducing megakaryocytes in their model to progenitor cells from patients with essential thrombocythemia. Both sets of cells showed a TPO stimulation signature.
“We think this study now provides a comprehensive model of how thrombopoietin controls platelet production,” Dr Ng said, “and perhaps gives some insight into the biology and mechanism behind specific myeloproliferative disorders.” ![]()
Credit: Walter and Eliza Hall
Institute of Medical Research
Investigators say they’ve determined how thrombopoietin (TPO) stimulates platelet production, and their findings may have implications for myeloproliferative neoplasms.
Researchers have long known that TPO is responsible for signaling cells in the bone marrow to produce platelets, but precisely which cells respond to TPO’s signals has been unclear.
Now, a group of investigators studying the TPO receptor Mpl have pinpointed those cells and made an unexpected discovery.
“Thrombopoietin did not directly stimulate the platelet’s ‘parent’ cells—the megakaryocytes—to make more platelets,” said study author Ashley Ng, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“Thrombopoietin signals actually acted on stem cells and progenitor cells, several generations back.”
Dr Ng and his colleagues reported these findings in PNAS.
The researchers had generated mice that express the Mpl receptor normally on stem and progenitor cells but lack expression on megakaryocytes and platelets. And these mice exhibited “profound” megakaryocytosis and thrombocytosis, as well as “remarkable” expansion of megakaryocyte-committed and multipotent progenitor cells.
“The progenitor and stem cells in the bone marrow began massively expanding and effectively turned the bone marrow into a megakaryocyte-making machine,” Dr Ng said.
Furthermore, although the progenitor cells showed signs of chronic TPO overstimulation, TPO levels were normal. This suggests that stem and progenitor cells expressing Mpl were responsible for TPO clearance, according to the investigators.
“Our findings support a theory whereby megakaryocytes and platelets control platelet numbers by ‘mopping up’ excess amounts of thrombopoietin in the bone marrow,” Dr Ng said. “In fact, we show this ‘mopping up’ action is absolutely essential in preventing blood disease where too many megakaryocytes and platelets are produced.”
So the researchers believe these findings will have implications for myeloproliferative neoplasms, particularly essential thrombocythemia.
“[P]revious studies have shown megakaryocytes and platelets in people with essential thrombocythemia have fewer Mpl receptors, which fits our model for excessive platelet production,” Dr Ng said.
To add support to their model, the investigators compared the progenitor cells responsible for overproducing megakaryocytes in their model to progenitor cells from patients with essential thrombocythemia. Both sets of cells showed a TPO stimulation signature.
“We think this study now provides a comprehensive model of how thrombopoietin controls platelet production,” Dr Ng said, “and perhaps gives some insight into the biology and mechanism behind specific myeloproliferative disorders.” ![]()
FDA places imetelstat trials on hold
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold. ![]()
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold. ![]()
The US Food and Drug Administration (FDA) has issued a full clinical hold for the telomerase inhibitor imetelstat, citing concerns that long-term exposure to the drug may pose a risk of chronic liver injury.
The hold temporarily suspends all ongoing clinical trials of imetelstat sponsored by the drug’s maker, Geron Corporation.
It’s possible that other studies of imetelstat, such as ongoing investigator-sponsored trials, may be placed on hold as well.
At present, the hold affects the remaining 8 patients enrolled on Geron’s phase 2 study of imetelstat in essential thrombocythemia and polycythemia vera.
It also affects the remaining 2 patients in the company’s phase 2 study of the drug in previously treated multiple myeloma. And Geron believes its planned phase 2 trial of imetelstat in myelofibrosis will likely be delayed as well.
The FDA has not yet issued a written notice of the hold but has given Geron verbal notice.
The agency indicated that the hold is due to persistent low-grade liver function test abnormalities observed in the study of imetelstat in patients with essential thrombocythemia or polycythemia vera and the potential risk of chronic liver injury following long-term exposure to imetelstat.
The FDA expressed concerns about whether these abnormalities are reversible. Geron said it plans to work with the FDA to put an end to the hold. ![]()
Nanoparticle therapy active in B-cell malignancies

An experimental nanoparticle therapy has shown preclinical activity against a range of B-cell malignancies, researchers have reported.
The therapy, SNS01-T, inhibited tumor growth and improved survival in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma.
The treatment exhibited single-agent activity in these mice but also demonstrated synergy with lenalidomide and bortezomib.
Sarah Francis, PhD, of the University of Waterloo in Ontario, Canada, and her colleagues conducted this research and recounted the results in Molecular Therapy.
SNS01-T consists of 3 components: small interfering RNA targeting the native eukaryotic translation initiation factor 5A (eIF5A), plasmids expressing a pro-apoptotic mutant of elF5A under the control of a B-cell specific promoter, and a synthetic cationic polymer polyethylenimine, which serves as the delivery vehicle.
The small interfering RNA component of SNS01-T suppresses elF5A expression, thereby interfering with translation of eIF5A and reducing levels of hypusinated elF5A in cancer cells. This inhibits activation of NF-kB and induces apoptosis.
The B-cell specific plasmid component of SNS01-T expresses an arginine substituted form of eIF5A—eIF5AK50R—that cannot be hypusinated, which leads to selective induction of apoptosis in B cells.
Dr Francis and her colleagues found that SNS01-T is preferentially taken up by malignant B cells. In myeloma cells, uptake of SNS01-T was up to 5-fold higher than uptake by normal, naïve B cells.
Uptake into myeloma cells induced 45% cell death within 24 hours, but there was almost no measureable death of normal, naïve B cells.
SNS01-T also prompted dose-dependent inhibition of tumor growth in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma, with up to 90% inhibition at the highest doses.
When administered at doses of 0.18 mg/kg or more, SNS01-T significantly extended the life span of treated mice. The researchers observed a reduction in the pro-survival form of the eIF5A protein in tumor tissue, which was consistent with drug activity.
SNS01-T also demonstrated synergy with bortezomib and lenalidomide. Mice that received SNS01-T and lenalidomide in combination had a 100% survival rate, compared to 60% for mice that received SNS01-T alone and 20% for mice that received lenalidomide alone.
A single 6-week cycle of SNS01-T and lenalidomide eradicated tumors in 67% of mice, and there was no regrowth after an additional 8 weeks without further treatment.
Similarly, combination SNS01-T and bortezomib inhibited tumor growth by 89%, compared to 59% for SNS01-T alone and 39% for bortezomib alone.
This research was supported by Senesco Technologies, Inc., the company developing SNS01-T.![]()
An experimental nanoparticle therapy has shown preclinical activity against a range of B-cell malignancies, researchers have reported.
The therapy, SNS01-T, inhibited tumor growth and improved survival in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma.
The treatment exhibited single-agent activity in these mice but also demonstrated synergy with lenalidomide and bortezomib.
Sarah Francis, PhD, of the University of Waterloo in Ontario, Canada, and her colleagues conducted this research and recounted the results in Molecular Therapy.
SNS01-T consists of 3 components: small interfering RNA targeting the native eukaryotic translation initiation factor 5A (eIF5A), plasmids expressing a pro-apoptotic mutant of elF5A under the control of a B-cell specific promoter, and a synthetic cationic polymer polyethylenimine, which serves as the delivery vehicle.
The small interfering RNA component of SNS01-T suppresses elF5A expression, thereby interfering with translation of eIF5A and reducing levels of hypusinated elF5A in cancer cells. This inhibits activation of NF-kB and induces apoptosis.
The B-cell specific plasmid component of SNS01-T expresses an arginine substituted form of eIF5A—eIF5AK50R—that cannot be hypusinated, which leads to selective induction of apoptosis in B cells.
Dr Francis and her colleagues found that SNS01-T is preferentially taken up by malignant B cells. In myeloma cells, uptake of SNS01-T was up to 5-fold higher than uptake by normal, naïve B cells.
Uptake into myeloma cells induced 45% cell death within 24 hours, but there was almost no measureable death of normal, naïve B cells.
SNS01-T also prompted dose-dependent inhibition of tumor growth in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma, with up to 90% inhibition at the highest doses.
When administered at doses of 0.18 mg/kg or more, SNS01-T significantly extended the life span of treated mice. The researchers observed a reduction in the pro-survival form of the eIF5A protein in tumor tissue, which was consistent with drug activity.
SNS01-T also demonstrated synergy with bortezomib and lenalidomide. Mice that received SNS01-T and lenalidomide in combination had a 100% survival rate, compared to 60% for mice that received SNS01-T alone and 20% for mice that received lenalidomide alone.
A single 6-week cycle of SNS01-T and lenalidomide eradicated tumors in 67% of mice, and there was no regrowth after an additional 8 weeks without further treatment.
Similarly, combination SNS01-T and bortezomib inhibited tumor growth by 89%, compared to 59% for SNS01-T alone and 39% for bortezomib alone.
This research was supported by Senesco Technologies, Inc., the company developing SNS01-T.![]()
An experimental nanoparticle therapy has shown preclinical activity against a range of B-cell malignancies, researchers have reported.
The therapy, SNS01-T, inhibited tumor growth and improved survival in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma.
The treatment exhibited single-agent activity in these mice but also demonstrated synergy with lenalidomide and bortezomib.
Sarah Francis, PhD, of the University of Waterloo in Ontario, Canada, and her colleagues conducted this research and recounted the results in Molecular Therapy.
SNS01-T consists of 3 components: small interfering RNA targeting the native eukaryotic translation initiation factor 5A (eIF5A), plasmids expressing a pro-apoptotic mutant of elF5A under the control of a B-cell specific promoter, and a synthetic cationic polymer polyethylenimine, which serves as the delivery vehicle.
The small interfering RNA component of SNS01-T suppresses elF5A expression, thereby interfering with translation of eIF5A and reducing levels of hypusinated elF5A in cancer cells. This inhibits activation of NF-kB and induces apoptosis.
The B-cell specific plasmid component of SNS01-T expresses an arginine substituted form of eIF5A—eIF5AK50R—that cannot be hypusinated, which leads to selective induction of apoptosis in B cells.
Dr Francis and her colleagues found that SNS01-T is preferentially taken up by malignant B cells. In myeloma cells, uptake of SNS01-T was up to 5-fold higher than uptake by normal, naïve B cells.
Uptake into myeloma cells induced 45% cell death within 24 hours, but there was almost no measureable death of normal, naïve B cells.
SNS01-T also prompted dose-dependent inhibition of tumor growth in mouse models of multiple myeloma, mantle cell lymphoma, and diffuse large B-cell lymphoma, with up to 90% inhibition at the highest doses.
When administered at doses of 0.18 mg/kg or more, SNS01-T significantly extended the life span of treated mice. The researchers observed a reduction in the pro-survival form of the eIF5A protein in tumor tissue, which was consistent with drug activity.
SNS01-T also demonstrated synergy with bortezomib and lenalidomide. Mice that received SNS01-T and lenalidomide in combination had a 100% survival rate, compared to 60% for mice that received SNS01-T alone and 20% for mice that received lenalidomide alone.
A single 6-week cycle of SNS01-T and lenalidomide eradicated tumors in 67% of mice, and there was no regrowth after an additional 8 weeks without further treatment.
Similarly, combination SNS01-T and bortezomib inhibited tumor growth by 89%, compared to 59% for SNS01-T alone and 39% for bortezomib alone.
This research was supported by Senesco Technologies, Inc., the company developing SNS01-T.
Report reveals detailed data of ruxolitinib in MF
Ruxolitinib can ease the symptoms of myelofibrosis and improve patient survival, according to results of the COMFORT-I study.
Detailed data from this phase 3 trial, which led to the US approval of ruxolitinib in November, appear in the March 1 edition of The New England Journal of Medicine. Some of these data were also presented at the recent ASH meeting.
“The phase 1/2 clinical trial showed that ruxolitinib improves quality of life for many patients with myelofibrosis, and now this phase 3 study indicates that the drug extends survival in a patient population that has lacked effective treatments,” said principal investigator Srdan Verstovsek, MD, PhD, of The University of Texas MD Anderson Cancer Center.
The trial, which was funded by the maker of ruxolitinib, enrolled 309 patients from 89 US centers. The researchers randomized patients to receive ruxolitinib (n=155) or placebo (n=154) orally twice daily.
Patients recorded their symptoms over the course of the 24-week study using an electronic diary. And the investigators monitored patients’ spleen volumes via MRI. The primary endpoint of the study was a 35% reduction in spleen volume.
Most patients on placebo experienced progressive splenomegaly and a worsening of myelofibrosis-related symptoms. For this reason, 38 patients in the placebo arm withdrew from the study, and 111 patients crossed over to the ruxolitinib arm.
The median time to cross over was 41 weeks. Only 2 patients remained on placebo at the time of analysis.
Reduction in splenomegaly
“It quickly became apparent who was getting the placebo and who was getting the drug,” said study author Jason Gotlib, MD, of Stanford University Medical Center.
Dr Gotlib noted that the spleen volume in patients receiving the drug began to decrease within 1 to 2 weeks. In fact, 41.9% of treated patients experienced at least 35% shrinkage in their spleen volume. And the spleen remained smaller in 67% of those responders for 48 weeks or longer.
In comparison, 0.7% of patients in the placebo group experienced spleen reduction by 35%. At week 24, the ruxolitinib group had an average reduction in spleen volume of 31.6%, while the placebo group experienced an average increase in spleen volume of 8.1%.
Symptom improvement
Every night, patients completed the Myelofibrosis Symptom Assessment Form, an electronic diary. They evaluated the intensity of night sweats, itching, abdominal discomfort, pain under the ribs on the left side, a feeling of fullness, muscle and bone pain, and inactivity.
In the treated group, 45.9% of patients reported a reduction of 50% or more in their total symptom score over 24 weeks, compared to 5.3% in the placebo group. In addition, treated patients gained weight, while patients in the placebo group lost weight.
Improvements usually occurred within the first 4 weeks of treatment and were not limited to patients who also experienced reduction in spleen size, Dr Verstovsek said.
Adverse events and other drawbacks
Nonhematologic adverse events occurred at similar rates in both treatment arms. And roughly 11% of patients in each arm withdrew from the study due to adverse events.
However, anemia and thrombocytopenia were more common among patients who received ruxolitinib. One patient discontinued the drug due to anemia and one due to thrombocytopenia.
Other shortcomings of the drug are that it failed to reverse bone marrow damage and did not have a lasting effect on splenomegaly. Although many patients maintained a smaller spleen volume for at least 48 weeks while on ruxolitinib, their spleens began to enlarge again if they stopped taking the drug.
“Ruxolitinib doesn’t cure the disease,” Dr Gotlib said. “But the degree of benefit is clinically meaningful and substantial and allows many patients to re-engage in their daily activities.”
Mysteries of survival and mechanism
At a median follow-up of 51 weeks, there had been 13 deaths (8.4%) in the ruxolitinib group, compared with 24 (15.7%) in the placebo group. This represents a nearly 50% reduction in mortality, but Dr Gotlib indicated that the long-term implications of these data are not clear.
Another puzzler is how, exactly, ruxolitinib works. The drug is an inhibitor of JAK1 and JAK2, but patients responded to the drug whether or not they had the JAK2V617F mutation.
“Ruxolitinib is inhibiting overactive JAK/STAT intracellular signaling pathway no matter what,” Dr Verstovsek said. ![]()
Ruxolitinib can ease the symptoms of myelofibrosis and improve patient survival, according to results of the COMFORT-I study.
Detailed data from this phase 3 trial, which led to the US approval of ruxolitinib in November, appear in the March 1 edition of The New England Journal of Medicine. Some of these data were also presented at the recent ASH meeting.
“The phase 1/2 clinical trial showed that ruxolitinib improves quality of life for many patients with myelofibrosis, and now this phase 3 study indicates that the drug extends survival in a patient population that has lacked effective treatments,” said principal investigator Srdan Verstovsek, MD, PhD, of The University of Texas MD Anderson Cancer Center.
The trial, which was funded by the maker of ruxolitinib, enrolled 309 patients from 89 US centers. The researchers randomized patients to receive ruxolitinib (n=155) or placebo (n=154) orally twice daily.
Patients recorded their symptoms over the course of the 24-week study using an electronic diary. And the investigators monitored patients’ spleen volumes via MRI. The primary endpoint of the study was a 35% reduction in spleen volume.
Most patients on placebo experienced progressive splenomegaly and a worsening of myelofibrosis-related symptoms. For this reason, 38 patients in the placebo arm withdrew from the study, and 111 patients crossed over to the ruxolitinib arm.
The median time to cross over was 41 weeks. Only 2 patients remained on placebo at the time of analysis.
Reduction in splenomegaly
“It quickly became apparent who was getting the placebo and who was getting the drug,” said study author Jason Gotlib, MD, of Stanford University Medical Center.
Dr Gotlib noted that the spleen volume in patients receiving the drug began to decrease within 1 to 2 weeks. In fact, 41.9% of treated patients experienced at least 35% shrinkage in their spleen volume. And the spleen remained smaller in 67% of those responders for 48 weeks or longer.
In comparison, 0.7% of patients in the placebo group experienced spleen reduction by 35%. At week 24, the ruxolitinib group had an average reduction in spleen volume of 31.6%, while the placebo group experienced an average increase in spleen volume of 8.1%.
Symptom improvement
Every night, patients completed the Myelofibrosis Symptom Assessment Form, an electronic diary. They evaluated the intensity of night sweats, itching, abdominal discomfort, pain under the ribs on the left side, a feeling of fullness, muscle and bone pain, and inactivity.
In the treated group, 45.9% of patients reported a reduction of 50% or more in their total symptom score over 24 weeks, compared to 5.3% in the placebo group. In addition, treated patients gained weight, while patients in the placebo group lost weight.
Improvements usually occurred within the first 4 weeks of treatment and were not limited to patients who also experienced reduction in spleen size, Dr Verstovsek said.
Adverse events and other drawbacks
Nonhematologic adverse events occurred at similar rates in both treatment arms. And roughly 11% of patients in each arm withdrew from the study due to adverse events.
However, anemia and thrombocytopenia were more common among patients who received ruxolitinib. One patient discontinued the drug due to anemia and one due to thrombocytopenia.
Other shortcomings of the drug are that it failed to reverse bone marrow damage and did not have a lasting effect on splenomegaly. Although many patients maintained a smaller spleen volume for at least 48 weeks while on ruxolitinib, their spleens began to enlarge again if they stopped taking the drug.
“Ruxolitinib doesn’t cure the disease,” Dr Gotlib said. “But the degree of benefit is clinically meaningful and substantial and allows many patients to re-engage in their daily activities.”
Mysteries of survival and mechanism
At a median follow-up of 51 weeks, there had been 13 deaths (8.4%) in the ruxolitinib group, compared with 24 (15.7%) in the placebo group. This represents a nearly 50% reduction in mortality, but Dr Gotlib indicated that the long-term implications of these data are not clear.
Another puzzler is how, exactly, ruxolitinib works. The drug is an inhibitor of JAK1 and JAK2, but patients responded to the drug whether or not they had the JAK2V617F mutation.
“Ruxolitinib is inhibiting overactive JAK/STAT intracellular signaling pathway no matter what,” Dr Verstovsek said. ![]()
Ruxolitinib can ease the symptoms of myelofibrosis and improve patient survival, according to results of the COMFORT-I study.
Detailed data from this phase 3 trial, which led to the US approval of ruxolitinib in November, appear in the March 1 edition of The New England Journal of Medicine. Some of these data were also presented at the recent ASH meeting.
“The phase 1/2 clinical trial showed that ruxolitinib improves quality of life for many patients with myelofibrosis, and now this phase 3 study indicates that the drug extends survival in a patient population that has lacked effective treatments,” said principal investigator Srdan Verstovsek, MD, PhD, of The University of Texas MD Anderson Cancer Center.
The trial, which was funded by the maker of ruxolitinib, enrolled 309 patients from 89 US centers. The researchers randomized patients to receive ruxolitinib (n=155) or placebo (n=154) orally twice daily.
Patients recorded their symptoms over the course of the 24-week study using an electronic diary. And the investigators monitored patients’ spleen volumes via MRI. The primary endpoint of the study was a 35% reduction in spleen volume.
Most patients on placebo experienced progressive splenomegaly and a worsening of myelofibrosis-related symptoms. For this reason, 38 patients in the placebo arm withdrew from the study, and 111 patients crossed over to the ruxolitinib arm.
The median time to cross over was 41 weeks. Only 2 patients remained on placebo at the time of analysis.
Reduction in splenomegaly
“It quickly became apparent who was getting the placebo and who was getting the drug,” said study author Jason Gotlib, MD, of Stanford University Medical Center.
Dr Gotlib noted that the spleen volume in patients receiving the drug began to decrease within 1 to 2 weeks. In fact, 41.9% of treated patients experienced at least 35% shrinkage in their spleen volume. And the spleen remained smaller in 67% of those responders for 48 weeks or longer.
In comparison, 0.7% of patients in the placebo group experienced spleen reduction by 35%. At week 24, the ruxolitinib group had an average reduction in spleen volume of 31.6%, while the placebo group experienced an average increase in spleen volume of 8.1%.
Symptom improvement
Every night, patients completed the Myelofibrosis Symptom Assessment Form, an electronic diary. They evaluated the intensity of night sweats, itching, abdominal discomfort, pain under the ribs on the left side, a feeling of fullness, muscle and bone pain, and inactivity.
In the treated group, 45.9% of patients reported a reduction of 50% or more in their total symptom score over 24 weeks, compared to 5.3% in the placebo group. In addition, treated patients gained weight, while patients in the placebo group lost weight.
Improvements usually occurred within the first 4 weeks of treatment and were not limited to patients who also experienced reduction in spleen size, Dr Verstovsek said.
Adverse events and other drawbacks
Nonhematologic adverse events occurred at similar rates in both treatment arms. And roughly 11% of patients in each arm withdrew from the study due to adverse events.
However, anemia and thrombocytopenia were more common among patients who received ruxolitinib. One patient discontinued the drug due to anemia and one due to thrombocytopenia.
Other shortcomings of the drug are that it failed to reverse bone marrow damage and did not have a lasting effect on splenomegaly. Although many patients maintained a smaller spleen volume for at least 48 weeks while on ruxolitinib, their spleens began to enlarge again if they stopped taking the drug.
“Ruxolitinib doesn’t cure the disease,” Dr Gotlib said. “But the degree of benefit is clinically meaningful and substantial and allows many patients to re-engage in their daily activities.”
Mysteries of survival and mechanism
At a median follow-up of 51 weeks, there had been 13 deaths (8.4%) in the ruxolitinib group, compared with 24 (15.7%) in the placebo group. This represents a nearly 50% reduction in mortality, but Dr Gotlib indicated that the long-term implications of these data are not clear.
Another puzzler is how, exactly, ruxolitinib works. The drug is an inhibitor of JAK1 and JAK2, but patients responded to the drug whether or not they had the JAK2V617F mutation.
“Ruxolitinib is inhibiting overactive JAK/STAT intracellular signaling pathway no matter what,” Dr Verstovsek said. ![]()
JAK inhibitor ruxolitinib improves treatment landscape in MF
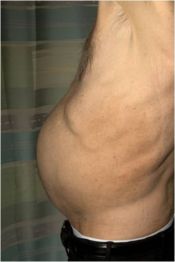
CHICAGO—Two phase 3 studies demonstrate the effectiveness of the investigational Janus kinase (JAK) inhibitor INC424 (ruxolitinib) in treating patients with myelofibrosis (MF), according to presentations at the 2011 ASCO Annual Meeting.
Ruxolitinib has the potential to change the treatment landscape in MF, said Alessandro Vannucchi, MD, of the University of Florence, who reported the results of the COMFORT-II trial.
Srdan Verostovsek, MD, PhD, of the MD Anderson Cancer Center, presented results from COMFORT-I.
COMFORT-II
In the COMFORT-II trial, ruxolitinib produced a volumetric spleen size reduction of 35% or greater in 28.5% of MF patients at 48 weeks. None of the patients receiving best available therapy (BAT) experienced such a reduction, Dr Vannucchi said.
At week 24, 31.9% of ruxolitinib-treated patients had a 35% or greater volumetric spleen size reduction, compared to none of the BAT-treated patients. Ruxolitinib also showed a marked improvement in overall quality of life measures, functioning, and symptoms relative to the BAT arm.
This open-label, phase 3 study included 146 patients randomized to ruxolitinib starting at doses of 15 or 20 mg twice daily and 73 patients randomized to BAT, which was administered at doses and schedules determined by the investigator.
Two-thirds of the BAT patients received at least one medication, and one-third received no medication. The most commonly administered agents were hydroxyurea (47% of patients) and glucocorticoids (16% of patients).
All study participants had intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The safety profile of ruxolitinib was consistent with previous studies, Dr Vannucchi said. The most common grade 3 or higher adverse events in the ruxolitinib arm were anemia and thrombocytopenia. In the BAT arm, the most common events were anemia, thrombocytopenia, pneumonia, and dyspnea.
COMFORT-I
The COMFORT-I trial enrolled 309 patients with intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. They were randomly assigned to twice-daily oral ruxolitinib (155 patients) or placebo (154 patients).
Ruxolitinib was dosed at 15 mg or 20 mg, depending on the baseline platelet count. Patients in the placebo arm could cross over to the ruxolitinib arm upon disease progression, and nearly a quarter of patients did cross over (11% prior to week 24 and 13% after week 24).
The median follow-up was 32 weeks. A significantly higher proportion of patients on the ruxolitinib arm (41.9%) attained the primary endpoint of at least a 35% reduction in spleen volume after 24 weeks of therapy compared with placebo patients (0.7%), Dr Verstovsek reported.
Ruxolitinib was also more effective than placebo for improving symptom burden, as 45.9% of patients in the ruxolitinib had at least a 50% improvement in symptom burden, compared to 5.3% of placebo-treated patients.
Symptoms that significantly improved with ruxolitinib included abdominal discomfort, pain under the left ribs, early satiety, night sweats, itching, bone or muscle pain, and inactivity.
“Data from the COMFORT studies indicate that ruxolitinib has the potential to significantly improve the current treatment landscape for most patients with MF,” Dr Vannucchi said. ![]()
CHICAGO—Two phase 3 studies demonstrate the effectiveness of the investigational Janus kinase (JAK) inhibitor INC424 (ruxolitinib) in treating patients with myelofibrosis (MF), according to presentations at the 2011 ASCO Annual Meeting.
Ruxolitinib has the potential to change the treatment landscape in MF, said Alessandro Vannucchi, MD, of the University of Florence, who reported the results of the COMFORT-II trial.
Srdan Verostovsek, MD, PhD, of the MD Anderson Cancer Center, presented results from COMFORT-I.
COMFORT-II
In the COMFORT-II trial, ruxolitinib produced a volumetric spleen size reduction of 35% or greater in 28.5% of MF patients at 48 weeks. None of the patients receiving best available therapy (BAT) experienced such a reduction, Dr Vannucchi said.
At week 24, 31.9% of ruxolitinib-treated patients had a 35% or greater volumetric spleen size reduction, compared to none of the BAT-treated patients. Ruxolitinib also showed a marked improvement in overall quality of life measures, functioning, and symptoms relative to the BAT arm.
This open-label, phase 3 study included 146 patients randomized to ruxolitinib starting at doses of 15 or 20 mg twice daily and 73 patients randomized to BAT, which was administered at doses and schedules determined by the investigator.
Two-thirds of the BAT patients received at least one medication, and one-third received no medication. The most commonly administered agents were hydroxyurea (47% of patients) and glucocorticoids (16% of patients).
All study participants had intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The safety profile of ruxolitinib was consistent with previous studies, Dr Vannucchi said. The most common grade 3 or higher adverse events in the ruxolitinib arm were anemia and thrombocytopenia. In the BAT arm, the most common events were anemia, thrombocytopenia, pneumonia, and dyspnea.
COMFORT-I
The COMFORT-I trial enrolled 309 patients with intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. They were randomly assigned to twice-daily oral ruxolitinib (155 patients) or placebo (154 patients).
Ruxolitinib was dosed at 15 mg or 20 mg, depending on the baseline platelet count. Patients in the placebo arm could cross over to the ruxolitinib arm upon disease progression, and nearly a quarter of patients did cross over (11% prior to week 24 and 13% after week 24).
The median follow-up was 32 weeks. A significantly higher proportion of patients on the ruxolitinib arm (41.9%) attained the primary endpoint of at least a 35% reduction in spleen volume after 24 weeks of therapy compared with placebo patients (0.7%), Dr Verstovsek reported.
Ruxolitinib was also more effective than placebo for improving symptom burden, as 45.9% of patients in the ruxolitinib had at least a 50% improvement in symptom burden, compared to 5.3% of placebo-treated patients.
Symptoms that significantly improved with ruxolitinib included abdominal discomfort, pain under the left ribs, early satiety, night sweats, itching, bone or muscle pain, and inactivity.
“Data from the COMFORT studies indicate that ruxolitinib has the potential to significantly improve the current treatment landscape for most patients with MF,” Dr Vannucchi said. ![]()
CHICAGO—Two phase 3 studies demonstrate the effectiveness of the investigational Janus kinase (JAK) inhibitor INC424 (ruxolitinib) in treating patients with myelofibrosis (MF), according to presentations at the 2011 ASCO Annual Meeting.
Ruxolitinib has the potential to change the treatment landscape in MF, said Alessandro Vannucchi, MD, of the University of Florence, who reported the results of the COMFORT-II trial.
Srdan Verostovsek, MD, PhD, of the MD Anderson Cancer Center, presented results from COMFORT-I.
COMFORT-II
In the COMFORT-II trial, ruxolitinib produced a volumetric spleen size reduction of 35% or greater in 28.5% of MF patients at 48 weeks. None of the patients receiving best available therapy (BAT) experienced such a reduction, Dr Vannucchi said.
At week 24, 31.9% of ruxolitinib-treated patients had a 35% or greater volumetric spleen size reduction, compared to none of the BAT-treated patients. Ruxolitinib also showed a marked improvement in overall quality of life measures, functioning, and symptoms relative to the BAT arm.
This open-label, phase 3 study included 146 patients randomized to ruxolitinib starting at doses of 15 or 20 mg twice daily and 73 patients randomized to BAT, which was administered at doses and schedules determined by the investigator.
Two-thirds of the BAT patients received at least one medication, and one-third received no medication. The most commonly administered agents were hydroxyurea (47% of patients) and glucocorticoids (16% of patients).
All study participants had intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The safety profile of ruxolitinib was consistent with previous studies, Dr Vannucchi said. The most common grade 3 or higher adverse events in the ruxolitinib arm were anemia and thrombocytopenia. In the BAT arm, the most common events were anemia, thrombocytopenia, pneumonia, and dyspnea.
COMFORT-I
The COMFORT-I trial enrolled 309 patients with intermediate-2 or high-risk primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF. They were randomly assigned to twice-daily oral ruxolitinib (155 patients) or placebo (154 patients).
Ruxolitinib was dosed at 15 mg or 20 mg, depending on the baseline platelet count. Patients in the placebo arm could cross over to the ruxolitinib arm upon disease progression, and nearly a quarter of patients did cross over (11% prior to week 24 and 13% after week 24).
The median follow-up was 32 weeks. A significantly higher proportion of patients on the ruxolitinib arm (41.9%) attained the primary endpoint of at least a 35% reduction in spleen volume after 24 weeks of therapy compared with placebo patients (0.7%), Dr Verstovsek reported.
Ruxolitinib was also more effective than placebo for improving symptom burden, as 45.9% of patients in the ruxolitinib had at least a 50% improvement in symptom burden, compared to 5.3% of placebo-treated patients.
Symptoms that significantly improved with ruxolitinib included abdominal discomfort, pain under the left ribs, early satiety, night sweats, itching, bone or muscle pain, and inactivity.
“Data from the COMFORT studies indicate that ruxolitinib has the potential to significantly improve the current treatment landscape for most patients with MF,” Dr Vannucchi said. ![]()