User login
Lenalidomide + rituximab combo effective in recurrent follicular lymphoma
The combination of lenalidomide and rituximab was more active in patients with recurrent follicular lymphoma, compared with lenalidomide alone, and significantly increased the overall response rate, according to new data published online Aug. 24 in the Journal of Clinical Oncology.
Although both lenalidomide and rituximab are active agents in follicular lymphoma, their combined use in recurrent follicular lymphoma has not been previously evaluated in randomized clinical trials, said Dr. John P. Leonard of Cornell University, New York, and his colleagues.
The overall response rate of patients receiving the combination regimen was significantly higher than that of patients who received lenalidomide alone (P = .029). In the cohort receiving lenalidomide alone, 24 patients (53%) achieved an objective response (9 complete responses [20%]), while 35 patients (76%) in the lenalidomide/rituximab group were responders (18 complete responses [39%]).
At a median follow-up of 2.5 years (range, 0.1-4.8 years), the addition of rituximab to lenalidomide in this population also significantly increased the median time to progression: 1.1 year for lenalidomide alone versus 2 years for the combined therapy (P = .002).
Overall survival was 4.5 years for lenalidomide alone and has not yet been reached for the combination arm (P = .149.
This trial helps to establish the safety profile of single-agent lenalidomide in follicular lymphoma, while its “randomized nature also allows a direct assessment of potential toxicity resulting from the addition of rituximab to lenalidomide,” wrote Dr. Leonard and his associates (J Clin Oncol. 2015 Aug 24. doi: 10.1200/JCO.2014.59.9258).
“There was no evidence of increased toxicity from the lenalidomide/rituximab combination compared with lenalidomide alone,” they pointed out.
Both lenalidomide alone and lenalidomide/rituximab were well tolerated, with grade 3-4 adverse events occurring in 58% and 53% of patients, respectively, with 9% and 11% of patients experiencing grade 4 toxicity, respectively. The most common grade 3-4 adverse events included neutropenia (16% vs. 20%), fatigue (9% vs. 13%), and rash (4% vs. 4%).
The study was supported in part by grants from the National Cancer Institute to the Alliance for Clinical Trials in Oncology. Dr. Leonard reported financial relationships with Celgene and Genentech, and several coauthors also reported relationships with industry.
The combination of lenalidomide and rituximab was more active in patients with recurrent follicular lymphoma, compared with lenalidomide alone, and significantly increased the overall response rate, according to new data published online Aug. 24 in the Journal of Clinical Oncology.
Although both lenalidomide and rituximab are active agents in follicular lymphoma, their combined use in recurrent follicular lymphoma has not been previously evaluated in randomized clinical trials, said Dr. John P. Leonard of Cornell University, New York, and his colleagues.
The overall response rate of patients receiving the combination regimen was significantly higher than that of patients who received lenalidomide alone (P = .029). In the cohort receiving lenalidomide alone, 24 patients (53%) achieved an objective response (9 complete responses [20%]), while 35 patients (76%) in the lenalidomide/rituximab group were responders (18 complete responses [39%]).
At a median follow-up of 2.5 years (range, 0.1-4.8 years), the addition of rituximab to lenalidomide in this population also significantly increased the median time to progression: 1.1 year for lenalidomide alone versus 2 years for the combined therapy (P = .002).
Overall survival was 4.5 years for lenalidomide alone and has not yet been reached for the combination arm (P = .149.
This trial helps to establish the safety profile of single-agent lenalidomide in follicular lymphoma, while its “randomized nature also allows a direct assessment of potential toxicity resulting from the addition of rituximab to lenalidomide,” wrote Dr. Leonard and his associates (J Clin Oncol. 2015 Aug 24. doi: 10.1200/JCO.2014.59.9258).
“There was no evidence of increased toxicity from the lenalidomide/rituximab combination compared with lenalidomide alone,” they pointed out.
Both lenalidomide alone and lenalidomide/rituximab were well tolerated, with grade 3-4 adverse events occurring in 58% and 53% of patients, respectively, with 9% and 11% of patients experiencing grade 4 toxicity, respectively. The most common grade 3-4 adverse events included neutropenia (16% vs. 20%), fatigue (9% vs. 13%), and rash (4% vs. 4%).
The study was supported in part by grants from the National Cancer Institute to the Alliance for Clinical Trials in Oncology. Dr. Leonard reported financial relationships with Celgene and Genentech, and several coauthors also reported relationships with industry.
The combination of lenalidomide and rituximab was more active in patients with recurrent follicular lymphoma, compared with lenalidomide alone, and significantly increased the overall response rate, according to new data published online Aug. 24 in the Journal of Clinical Oncology.
Although both lenalidomide and rituximab are active agents in follicular lymphoma, their combined use in recurrent follicular lymphoma has not been previously evaluated in randomized clinical trials, said Dr. John P. Leonard of Cornell University, New York, and his colleagues.
The overall response rate of patients receiving the combination regimen was significantly higher than that of patients who received lenalidomide alone (P = .029). In the cohort receiving lenalidomide alone, 24 patients (53%) achieved an objective response (9 complete responses [20%]), while 35 patients (76%) in the lenalidomide/rituximab group were responders (18 complete responses [39%]).
At a median follow-up of 2.5 years (range, 0.1-4.8 years), the addition of rituximab to lenalidomide in this population also significantly increased the median time to progression: 1.1 year for lenalidomide alone versus 2 years for the combined therapy (P = .002).
Overall survival was 4.5 years for lenalidomide alone and has not yet been reached for the combination arm (P = .149.
This trial helps to establish the safety profile of single-agent lenalidomide in follicular lymphoma, while its “randomized nature also allows a direct assessment of potential toxicity resulting from the addition of rituximab to lenalidomide,” wrote Dr. Leonard and his associates (J Clin Oncol. 2015 Aug 24. doi: 10.1200/JCO.2014.59.9258).
“There was no evidence of increased toxicity from the lenalidomide/rituximab combination compared with lenalidomide alone,” they pointed out.
Both lenalidomide alone and lenalidomide/rituximab were well tolerated, with grade 3-4 adverse events occurring in 58% and 53% of patients, respectively, with 9% and 11% of patients experiencing grade 4 toxicity, respectively. The most common grade 3-4 adverse events included neutropenia (16% vs. 20%), fatigue (9% vs. 13%), and rash (4% vs. 4%).
The study was supported in part by grants from the National Cancer Institute to the Alliance for Clinical Trials in Oncology. Dr. Leonard reported financial relationships with Celgene and Genentech, and several coauthors also reported relationships with industry.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Lenalidomide combined with rituximab is more active in recurrent follicular lymphoma, compared with lenalidomide monotherapy.
Major finding: Overall response rate was 53% (20% complete response) for lenalidomide alone versus 76% (39% complete response) for lenalidomide and rituximab (P = .029).
Data source: Randomized phase II clinical trial of 91 patients with follicular lymphoma who were assigned to receive lenalidomide alone or lenalidomide combined with rituximab.
Disclosures: The study was supported in part by grants from the National Cancer Institute to the Alliance for Clinical Trials in Oncology. Dr. Leonard reported financial relationships with Celgene and Genentech, and several coauthors also reported relationships with industry.
Pathway appears key to fighting adenovirus

Using an animal model they developed, researchers have identified a pathway that inhibits replication of the adenovirus.
The team generated a new strain of Syrian hamster, a model in which human adenovirus replicates and causes illness similar to that observed in humans.
Experiments with this model suggested the Type I interferon pathway plays a key role in inhibiting adenovirus replication.
“[L]ike many other viruses, adenovirus can replicate at will when a patient’s immune system is suppressed,” said William Wold, PhD, of Saint Louis University in Missouri.
“Adenovirus can become very dangerous, such as for a child who is undergoing a bone marrow transplant to treat leukemia.”
Previously, Dr Wold led a research team that identified the Syrian hamster as an appropriate animal model to study adenovirus because species C human adenoviruses replicate in these animals.
For the current study, which was published in PLOS Pathogens, Dr Wold and his colleagues conducted experiments with a new Syrian hamster strain. In these animals, the STAT2 gene was functionally knocked out by site-specific gene targeting.
The researchers found that STAT2-knockout hamsters were extremely sensitive to infection with type 5 human adenovirus (Ad5).
The team infected both STAT2-knockout hamsters and wild-type controls with Ad5. Knockout hamsters had 100 to 1000 times the viral load of controls.
The knockout hamsters also had pathology characteristic of advanced adenovirus infection—yellow, mottled livers and enlarged gall bladders—whereas controls did not.
The adaptive immune response to Ad5 remained intact in the STAT2-knockout hamsters, as surviving animals were able to clear the virus.
However, the Type 1 interferon response was hampered in these animals. Knocking out STAT2 disrupted the Type 1 interferon pathway by interrupting the cascade of cell signaling.
The researchers said their findings suggest the disrupted Type I interferon pathway contributed to the increased Ad5 replication in the STAT2-knockout hamsters.
“Besides providing an insight into adenovirus infection in humans, our results are also interesting from the perspective of the animal model,” Dr Wold said. “The STAT2-knockout Syrian hamster may also be an important animal model for studying other viral infections, including Ebola, hanta, and dengue viruses.”
The model was created by Zhongde Wang, PhD, and his colleagues at Utah State University in Logan, Utah. Dr Wang’s lab is the first to develop gene-targeting technologies in the Syrian hamster.
“The success we achieved in conducting gene-targeting in the Syrian hamster has provided the opportunity to create models for many of the human diseases for which there are either no existent animal models or severe limitations in the available animal models,” Dr Wang said. ![]()
Using an animal model they developed, researchers have identified a pathway that inhibits replication of the adenovirus.
The team generated a new strain of Syrian hamster, a model in which human adenovirus replicates and causes illness similar to that observed in humans.
Experiments with this model suggested the Type I interferon pathway plays a key role in inhibiting adenovirus replication.
“[L]ike many other viruses, adenovirus can replicate at will when a patient’s immune system is suppressed,” said William Wold, PhD, of Saint Louis University in Missouri.
“Adenovirus can become very dangerous, such as for a child who is undergoing a bone marrow transplant to treat leukemia.”
Previously, Dr Wold led a research team that identified the Syrian hamster as an appropriate animal model to study adenovirus because species C human adenoviruses replicate in these animals.
For the current study, which was published in PLOS Pathogens, Dr Wold and his colleagues conducted experiments with a new Syrian hamster strain. In these animals, the STAT2 gene was functionally knocked out by site-specific gene targeting.
The researchers found that STAT2-knockout hamsters were extremely sensitive to infection with type 5 human adenovirus (Ad5).
The team infected both STAT2-knockout hamsters and wild-type controls with Ad5. Knockout hamsters had 100 to 1000 times the viral load of controls.
The knockout hamsters also had pathology characteristic of advanced adenovirus infection—yellow, mottled livers and enlarged gall bladders—whereas controls did not.
The adaptive immune response to Ad5 remained intact in the STAT2-knockout hamsters, as surviving animals were able to clear the virus.
However, the Type 1 interferon response was hampered in these animals. Knocking out STAT2 disrupted the Type 1 interferon pathway by interrupting the cascade of cell signaling.
The researchers said their findings suggest the disrupted Type I interferon pathway contributed to the increased Ad5 replication in the STAT2-knockout hamsters.
“Besides providing an insight into adenovirus infection in humans, our results are also interesting from the perspective of the animal model,” Dr Wold said. “The STAT2-knockout Syrian hamster may also be an important animal model for studying other viral infections, including Ebola, hanta, and dengue viruses.”
The model was created by Zhongde Wang, PhD, and his colleagues at Utah State University in Logan, Utah. Dr Wang’s lab is the first to develop gene-targeting technologies in the Syrian hamster.
“The success we achieved in conducting gene-targeting in the Syrian hamster has provided the opportunity to create models for many of the human diseases for which there are either no existent animal models or severe limitations in the available animal models,” Dr Wang said. ![]()
Using an animal model they developed, researchers have identified a pathway that inhibits replication of the adenovirus.
The team generated a new strain of Syrian hamster, a model in which human adenovirus replicates and causes illness similar to that observed in humans.
Experiments with this model suggested the Type I interferon pathway plays a key role in inhibiting adenovirus replication.
“[L]ike many other viruses, adenovirus can replicate at will when a patient’s immune system is suppressed,” said William Wold, PhD, of Saint Louis University in Missouri.
“Adenovirus can become very dangerous, such as for a child who is undergoing a bone marrow transplant to treat leukemia.”
Previously, Dr Wold led a research team that identified the Syrian hamster as an appropriate animal model to study adenovirus because species C human adenoviruses replicate in these animals.
For the current study, which was published in PLOS Pathogens, Dr Wold and his colleagues conducted experiments with a new Syrian hamster strain. In these animals, the STAT2 gene was functionally knocked out by site-specific gene targeting.
The researchers found that STAT2-knockout hamsters were extremely sensitive to infection with type 5 human adenovirus (Ad5).
The team infected both STAT2-knockout hamsters and wild-type controls with Ad5. Knockout hamsters had 100 to 1000 times the viral load of controls.
The knockout hamsters also had pathology characteristic of advanced adenovirus infection—yellow, mottled livers and enlarged gall bladders—whereas controls did not.
The adaptive immune response to Ad5 remained intact in the STAT2-knockout hamsters, as surviving animals were able to clear the virus.
However, the Type 1 interferon response was hampered in these animals. Knocking out STAT2 disrupted the Type 1 interferon pathway by interrupting the cascade of cell signaling.
The researchers said their findings suggest the disrupted Type I interferon pathway contributed to the increased Ad5 replication in the STAT2-knockout hamsters.
“Besides providing an insight into adenovirus infection in humans, our results are also interesting from the perspective of the animal model,” Dr Wold said. “The STAT2-knockout Syrian hamster may also be an important animal model for studying other viral infections, including Ebola, hanta, and dengue viruses.”
The model was created by Zhongde Wang, PhD, and his colleagues at Utah State University in Logan, Utah. Dr Wang’s lab is the first to develop gene-targeting technologies in the Syrian hamster.
“The success we achieved in conducting gene-targeting in the Syrian hamster has provided the opportunity to create models for many of the human diseases for which there are either no existent animal models or severe limitations in the available animal models,” Dr Wang said. ![]()
Secondary CNS lymphoma regimen linked to 41% survival at 5 years
Treatment with high doses of antimetabolites followed by rituximab plus high-dose sequential chemoimmunotherapy and autologous stem-cell transplantation was feasible and effective in a multicenter phase II study of 38 patients with aggressive B-cell lymphoma and secondary central nervous system involvement.
The patients, aged 18 to 70 years with Eastern Cooperative Oncology Group performance status of 3 or less at enrollment, were treated with high doses of methotrexate and cytarabine, followed by rituximab plus high-dose sequential chemoimmunotherapy (R-HDS) consisting of cylcophosphamide, cytarabine, and etoposide supported by autologous stem-cell transplantation in eligible patients.
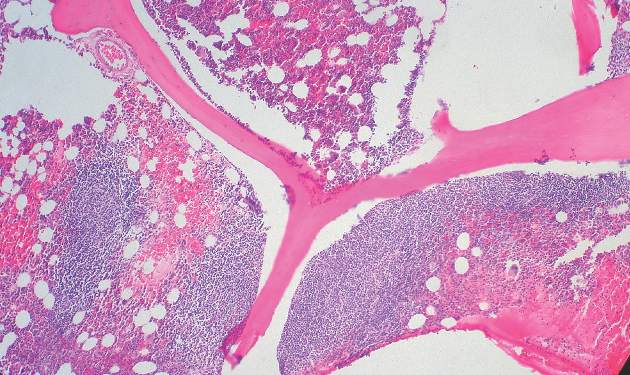
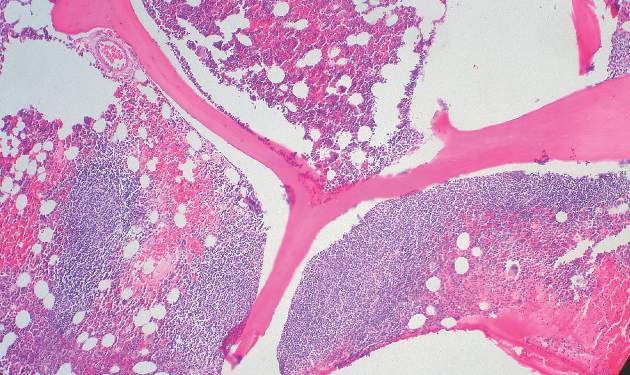
Toxicity was typically manageable, but 30 treatment courses were complicated by grade 3 or 4 febrile neutropenia and/or infections; 4 patients died because of toxicity, Dr. Andres J. M. Ferreri of San Raffaele Scientific Institute, Milan, Italy and colleagues reported online Aug. 17 in the Journal of Clinical Oncology.
The complete response rate was 63% (24 patients), and 17 patients remained relapse free at a median follow-up of 48 months, with a 2-year event-free survival rate of 50%, and a 5-year survival rate of 41%, the investigators said (J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.61.1236).
This novel radiotherapy-free regimen, developed based on encouraging outcomes with antimetabolites in patients with primary CNS lymphoma and with R-HDS in relapsed aggressive lymphoma, appears safe and effective in the setting of secondary CNS lymphoma (SCNSL).
“We propose this strategy as the standard of care for patients with SCNSL and as a comparison control regimen for future trials,” they concluded.
Dr. Ferreri reported having no disclosures. One co-author, Dr. Federico Caligaris-Cappio, reported serving in a consulting or advisory role for Janssen and Pharmacyclics.
The findings of Ferreri et al highlight the significant progress that has been made toward finding a cure for aggressive B-cell lymphoma with concomitant or subsequent CNS involvement.
Just a few years ago, this condition was fatal in nearly all cases, but in light of these and other recent findings, needed improvements in treatment seem possible.
Among other strategies, complete elimination of non-[blood-brain barrier]-crossing agents and administration of more than two cycles of induction chemotherapy might prove to be of value. In addition, oral targeted therapies with small molecules, most of which easily cross the BBB, hold promise.
Dr. Norbert Schmitz and Huei-Shan Wuthey are with Asklepios Hospital St. Georg, Hamburg, Germany. They made their remarks in an editorial(J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.63.1143) that accompanied the study. Dr. Schmitz reported serving in a consulting or advisory role for Roche and receiving research funding from Roche. Huei-Shan Wu reported having no disclosures.
The findings of Ferreri et al highlight the significant progress that has been made toward finding a cure for aggressive B-cell lymphoma with concomitant or subsequent CNS involvement.
Just a few years ago, this condition was fatal in nearly all cases, but in light of these and other recent findings, needed improvements in treatment seem possible.
Among other strategies, complete elimination of non-[blood-brain barrier]-crossing agents and administration of more than two cycles of induction chemotherapy might prove to be of value. In addition, oral targeted therapies with small molecules, most of which easily cross the BBB, hold promise.
Dr. Norbert Schmitz and Huei-Shan Wuthey are with Asklepios Hospital St. Georg, Hamburg, Germany. They made their remarks in an editorial(J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.63.1143) that accompanied the study. Dr. Schmitz reported serving in a consulting or advisory role for Roche and receiving research funding from Roche. Huei-Shan Wu reported having no disclosures.
The findings of Ferreri et al highlight the significant progress that has been made toward finding a cure for aggressive B-cell lymphoma with concomitant or subsequent CNS involvement.
Just a few years ago, this condition was fatal in nearly all cases, but in light of these and other recent findings, needed improvements in treatment seem possible.
Among other strategies, complete elimination of non-[blood-brain barrier]-crossing agents and administration of more than two cycles of induction chemotherapy might prove to be of value. In addition, oral targeted therapies with small molecules, most of which easily cross the BBB, hold promise.
Dr. Norbert Schmitz and Huei-Shan Wuthey are with Asklepios Hospital St. Georg, Hamburg, Germany. They made their remarks in an editorial(J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.63.1143) that accompanied the study. Dr. Schmitz reported serving in a consulting or advisory role for Roche and receiving research funding from Roche. Huei-Shan Wu reported having no disclosures.
Treatment with high doses of antimetabolites followed by rituximab plus high-dose sequential chemoimmunotherapy and autologous stem-cell transplantation was feasible and effective in a multicenter phase II study of 38 patients with aggressive B-cell lymphoma and secondary central nervous system involvement.
The patients, aged 18 to 70 years with Eastern Cooperative Oncology Group performance status of 3 or less at enrollment, were treated with high doses of methotrexate and cytarabine, followed by rituximab plus high-dose sequential chemoimmunotherapy (R-HDS) consisting of cylcophosphamide, cytarabine, and etoposide supported by autologous stem-cell transplantation in eligible patients.
Toxicity was typically manageable, but 30 treatment courses were complicated by grade 3 or 4 febrile neutropenia and/or infections; 4 patients died because of toxicity, Dr. Andres J. M. Ferreri of San Raffaele Scientific Institute, Milan, Italy and colleagues reported online Aug. 17 in the Journal of Clinical Oncology.
The complete response rate was 63% (24 patients), and 17 patients remained relapse free at a median follow-up of 48 months, with a 2-year event-free survival rate of 50%, and a 5-year survival rate of 41%, the investigators said (J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.61.1236).
This novel radiotherapy-free regimen, developed based on encouraging outcomes with antimetabolites in patients with primary CNS lymphoma and with R-HDS in relapsed aggressive lymphoma, appears safe and effective in the setting of secondary CNS lymphoma (SCNSL).
“We propose this strategy as the standard of care for patients with SCNSL and as a comparison control regimen for future trials,” they concluded.
Dr. Ferreri reported having no disclosures. One co-author, Dr. Federico Caligaris-Cappio, reported serving in a consulting or advisory role for Janssen and Pharmacyclics.
Treatment with high doses of antimetabolites followed by rituximab plus high-dose sequential chemoimmunotherapy and autologous stem-cell transplantation was feasible and effective in a multicenter phase II study of 38 patients with aggressive B-cell lymphoma and secondary central nervous system involvement.
The patients, aged 18 to 70 years with Eastern Cooperative Oncology Group performance status of 3 or less at enrollment, were treated with high doses of methotrexate and cytarabine, followed by rituximab plus high-dose sequential chemoimmunotherapy (R-HDS) consisting of cylcophosphamide, cytarabine, and etoposide supported by autologous stem-cell transplantation in eligible patients.
Toxicity was typically manageable, but 30 treatment courses were complicated by grade 3 or 4 febrile neutropenia and/or infections; 4 patients died because of toxicity, Dr. Andres J. M. Ferreri of San Raffaele Scientific Institute, Milan, Italy and colleagues reported online Aug. 17 in the Journal of Clinical Oncology.
The complete response rate was 63% (24 patients), and 17 patients remained relapse free at a median follow-up of 48 months, with a 2-year event-free survival rate of 50%, and a 5-year survival rate of 41%, the investigators said (J Clin Oncol. 2015 Aug 17. doi: 10.1200/JCO.2015.61.1236).
This novel radiotherapy-free regimen, developed based on encouraging outcomes with antimetabolites in patients with primary CNS lymphoma and with R-HDS in relapsed aggressive lymphoma, appears safe and effective in the setting of secondary CNS lymphoma (SCNSL).
“We propose this strategy as the standard of care for patients with SCNSL and as a comparison control regimen for future trials,” they concluded.
Dr. Ferreri reported having no disclosures. One co-author, Dr. Federico Caligaris-Cappio, reported serving in a consulting or advisory role for Janssen and Pharmacyclics.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: High doses of antimetabolites followed by R-HDS and autologous stem-cell transplantation was feasible and effective in 38 patients with aggressive B-cell lymphoma and secondary central nervous system involvement.
Major finding: The 2-year event-free survival rate was 50%, and the 5-year survival rate was 41%
Data source: A multicenter phase II study of 38 adults.
Disclosures: Dr. Ferreri reported having no disclosures. One co-author, Dr. Federico Caligaris-Cappio, reported serving in a consulting or advisory role for Janssen and Pharmacyclics.
T-cell finding may have broad implications

resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
resolution microscope
Image courtesy of UNSW
Early exposure to inflammatory cytokines can “paralyze” CD4 T cells, according to research published in Immunity.
The study suggests this mechanism may act as a firewall, shutting down the immune response before it gets out of hand.
According to researchers, this discovery could lead to more effective immunotherapies for cancer and reduce the need for immunosuppressants in transplant patients, among other applications.
“There’s a 3-signal process to activate T cells, of which each component is essential for proper activation,” said study author Gail Sckisel, PhD, of the University of California, Davis in Sacramento.
“But no one had really looked at what happens if they are delivered out of sequence. If the third signal—cytokines—is given prematurely, it basically paralyzes CD4 T cells.”
To be activated, T cells must first recognize an antigen, receive appropriate costimulatory signals, and then encounter inflammatory cytokines to expand the immune response. Until now, no one realized that sending the third signal early—as is done with some immunotherapies—could actually hamper overall immunity.
“These stimulatory immunotherapies are designed to activate the immune system,” Dr Sckisel explained. “But considering how T cells respond, that approach could damage a patient’s ability to fight off pathogens. While immunotherapies might fight cancer, they may also open the door to opportunistic infections.”
She and her colleagues demonstrated this principle in mice. After they received systemic immunotherapy, the animals had trouble mounting a primary T-cell response.
The researchers confirmed this finding in samples from patients receiving high-dose interleukin 2 to treat metastatic melanoma.
“We need to be very careful because immunotherapy could be generating both short-term gain and long-term loss,” said study author William Murphy, PhD, also of the University of California, Davis.
“The patients who were receiving immunotherapy were totally shut down, which shows how profoundly we were suppressing the immune system.”
In addition to illuminating how T cells respond to cancer immunotherapy, the study also provides insights into autoimmune disorders. The researchers believe this CD4 paralysis mechanism could play a role in preventing autoimmunity, a hypothesis they supported by testing immunotherapy in a multiple sclerosis model.
By shutting down CD4 T cells, immune stimulation prevented an autoimmune response. This provides the opportunity to paralyze the immune system to prevent autoimmunity or modulate it to accept transplanted cells or entire organs.
“Transplant patients go on immunosuppressants for the rest of their lives, but if we could safely induce paralysis just prior to surgery, it’s possible that patients could develop tolerance,” Dr Sckisel said.
CD4 paralysis may also be co-opted by pathogens, such as HIV, which could use this chronic inflammation response to disable the immune system.
“This really highlights the importance of CD4 T cells,” Dr Murphy said. “The fact that they’re regulated and suppressed means they are definitely the orchestrators we need to take into account. It also shows how smart HIV is. The virus has been telling us CD4 T cells are critical because that’s what it attacks.”
The team’s next step is to continue this research in older mice. Age can bring a measurable loss in immune function, and inflammation may play a role in that process.
“For elderly people who have flu or pneumonia, their immune systems are activated, but maybe they can’t fight anything else,” Dr Murphy said. “This could change how we treat people who are very sick. If we can block pathways that suppress the immune response, we may be able to better fight infection.” ![]()
FDA approves brentuximab vedotin as consolidation

Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The US Food and Drug Administration (FDA) has approved brentuximab vedotin (Adcetris) for use as consolidation treatment after autologous hematopoietic stem cell transplant (auto-HSCT) in patients with classical Hodgkin lymphoma (HL) at high risk of relapse or progression.
The drug is the first FDA-approved consolidation option available to these patients.
The approval was based on results of the phase 3 AETHERA trial.
Results from this trial also served to convert a prior accelerated approval of brentuximab vedotin to regular approval. The drug is now fully approved for the treatment of classical HL patients who have failed auto-HSCT and those who have failed at least 2 prior multi-agent chemotherapy regimens and are not candidates for auto-HSCT.
Brentuximab vedotin is also FDA-approved to treat patients with systemic anaplastic large-cell lymphoma who have failed at least 1 prior multi-agent chemotherapy regimen. The drug has accelerated approval for this indication based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
AETHERA trial
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following auto-HSCT. Results from the trial were published in The Lancet in March and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment in the AETHERA trial if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-auto-HSCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for patients who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
FDA approves new formulation of pain patch for cancer patients

receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
Continuous therapy improved outcomes in multiple myeloma
Compared with fixed duration therapy for patients with multiple myeloma, continuous therapy significantly prolonged median progression by approximately 1 year and improved overall survival by about 10%, investigators reported online Aug. 17 in the Journal of Clinical Oncology.
The goal of continuous therapy is to maintain the results of first-line therapy by keeping the patient free of symptoms and preventing or delaying disease progression. There has been some concern, however, that patients who progress while on continuous therapy may become resistant to at least that particular regimen.
In the current study, Dr. Antonio Palumbo of the University of Torino (Italy) and his colleagues pooled the results of three randomized phase III trials to evaluate the impact of continuous therapy, compared with fixed duration of therapy on time-to-event endpoints. In addition to overall survival, they evaluated progression-free survival (PFS) at two different time points in the course of therapy.
PFS1 was defined as the time from random assignment until the first disease progression or death, while PFS2 was defined as the time randomization until the second disease progression or death. Using both of these endpoints more accurately estimated the impact of both first- and second-line therapies on patient outcomes, the investigators said.
The pooled analysis of the three trials included 604 patients who were randomly assigned to continuous therapy and 614 who were randomized to fixed duration therapy. The median follow-up was 52 months. In the intent-to-treat population, continuous therapy (n = 417), compared with fixed duration (n = 410) significantly improved PFS1 (median, 32 vs. 16 months, respectively; hazard ratio, 0.47; 95% confidence interval, 0.40-0.56; P less than .001), as well as PFS2 (median, 55 vs. 40 months, respectively; HR, 0.61; 95% CI, 0.50-0.75; P less than .001). Overall survival also was improved in the continuous therapy group (4-year overall survival, 69% vs. 60%, respectively; HR, 0.69; 95% CI, 0.54-0.88; P = .003) (J Clin Oncol. 2015 Aug. 17 doi: 10.1200/JCO.2014.60.2466).
“Our findings suggest that most of the PFS1 advantage associated with CT [continuous therapy] up front is maintained after first relapse and that CT does not induce a significant chemotherapy resistance,” Dr. Palumbo and his associates wrote. “The improvement in PFS2 suggests that most of the benefit observed during the first remission is not affected by a short second remission.”
The GIMEMA-MM-03-05 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Janssen-Cilag and Celgene. The RV-MM-PI-209 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Celgene. The CC-5013-MM-015 study was sponsored and supported by Celgene. Several of the coauthors reported financial relationships with industry, including Celgene and Janssen-Citag.
Compared with fixed duration therapy for patients with multiple myeloma, continuous therapy significantly prolonged median progression by approximately 1 year and improved overall survival by about 10%, investigators reported online Aug. 17 in the Journal of Clinical Oncology.
The goal of continuous therapy is to maintain the results of first-line therapy by keeping the patient free of symptoms and preventing or delaying disease progression. There has been some concern, however, that patients who progress while on continuous therapy may become resistant to at least that particular regimen.
In the current study, Dr. Antonio Palumbo of the University of Torino (Italy) and his colleagues pooled the results of three randomized phase III trials to evaluate the impact of continuous therapy, compared with fixed duration of therapy on time-to-event endpoints. In addition to overall survival, they evaluated progression-free survival (PFS) at two different time points in the course of therapy.
PFS1 was defined as the time from random assignment until the first disease progression or death, while PFS2 was defined as the time randomization until the second disease progression or death. Using both of these endpoints more accurately estimated the impact of both first- and second-line therapies on patient outcomes, the investigators said.
The pooled analysis of the three trials included 604 patients who were randomly assigned to continuous therapy and 614 who were randomized to fixed duration therapy. The median follow-up was 52 months. In the intent-to-treat population, continuous therapy (n = 417), compared with fixed duration (n = 410) significantly improved PFS1 (median, 32 vs. 16 months, respectively; hazard ratio, 0.47; 95% confidence interval, 0.40-0.56; P less than .001), as well as PFS2 (median, 55 vs. 40 months, respectively; HR, 0.61; 95% CI, 0.50-0.75; P less than .001). Overall survival also was improved in the continuous therapy group (4-year overall survival, 69% vs. 60%, respectively; HR, 0.69; 95% CI, 0.54-0.88; P = .003) (J Clin Oncol. 2015 Aug. 17 doi: 10.1200/JCO.2014.60.2466).
“Our findings suggest that most of the PFS1 advantage associated with CT [continuous therapy] up front is maintained after first relapse and that CT does not induce a significant chemotherapy resistance,” Dr. Palumbo and his associates wrote. “The improvement in PFS2 suggests that most of the benefit observed during the first remission is not affected by a short second remission.”
The GIMEMA-MM-03-05 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Janssen-Cilag and Celgene. The RV-MM-PI-209 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Celgene. The CC-5013-MM-015 study was sponsored and supported by Celgene. Several of the coauthors reported financial relationships with industry, including Celgene and Janssen-Citag.
Compared with fixed duration therapy for patients with multiple myeloma, continuous therapy significantly prolonged median progression by approximately 1 year and improved overall survival by about 10%, investigators reported online Aug. 17 in the Journal of Clinical Oncology.
The goal of continuous therapy is to maintain the results of first-line therapy by keeping the patient free of symptoms and preventing or delaying disease progression. There has been some concern, however, that patients who progress while on continuous therapy may become resistant to at least that particular regimen.
In the current study, Dr. Antonio Palumbo of the University of Torino (Italy) and his colleagues pooled the results of three randomized phase III trials to evaluate the impact of continuous therapy, compared with fixed duration of therapy on time-to-event endpoints. In addition to overall survival, they evaluated progression-free survival (PFS) at two different time points in the course of therapy.
PFS1 was defined as the time from random assignment until the first disease progression or death, while PFS2 was defined as the time randomization until the second disease progression or death. Using both of these endpoints more accurately estimated the impact of both first- and second-line therapies on patient outcomes, the investigators said.
The pooled analysis of the three trials included 604 patients who were randomly assigned to continuous therapy and 614 who were randomized to fixed duration therapy. The median follow-up was 52 months. In the intent-to-treat population, continuous therapy (n = 417), compared with fixed duration (n = 410) significantly improved PFS1 (median, 32 vs. 16 months, respectively; hazard ratio, 0.47; 95% confidence interval, 0.40-0.56; P less than .001), as well as PFS2 (median, 55 vs. 40 months, respectively; HR, 0.61; 95% CI, 0.50-0.75; P less than .001). Overall survival also was improved in the continuous therapy group (4-year overall survival, 69% vs. 60%, respectively; HR, 0.69; 95% CI, 0.54-0.88; P = .003) (J Clin Oncol. 2015 Aug. 17 doi: 10.1200/JCO.2014.60.2466).
“Our findings suggest that most of the PFS1 advantage associated with CT [continuous therapy] up front is maintained after first relapse and that CT does not induce a significant chemotherapy resistance,” Dr. Palumbo and his associates wrote. “The improvement in PFS2 suggests that most of the benefit observed during the first remission is not affected by a short second remission.”
The GIMEMA-MM-03-05 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Janssen-Cilag and Celgene. The RV-MM-PI-209 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Celgene. The CC-5013-MM-015 study was sponsored and supported by Celgene. Several of the coauthors reported financial relationships with industry, including Celgene and Janssen-Citag.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Continuous therapy improved outcomes in multiple myeloma, compared with fixed duration therapy.
Major finding: Overall survival was improved (4-year OS, 69% v 60%, respectively; HR, 0.69; P = .003) as well as progression-free survival.
Data source: Pooled analysis of the three trials that included 604 patients randomly assigned to continuous therapy and 614 to fixed duration therapy.
Disclosures: The GIMEMA-MM-03-05 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Janssen-Cilag and Celgene. The RV-MM-PI-209 study was sponsored by Fondazione Neoplasie Sangue Onlus and supported by Celgene. The CC-5013-MM-015 study was sponsored and supported by Celgene. Several of the coauthors reported financial relationships with industry, including Celgene and Janssen-Citag.
How malaria increases the risk of Burkitt lymphoma

Image by Ed Uthman
A link between malaria and Burkitt lymphoma was first described more than 50 years ago, but how the parasitic infection promotes lymphomagenesis has remained a mystery.
Now, research in mice has revealed that B-cell DNA becomes vulnerable to cancer-causing mutations during prolonged combat against the malaria parasite.
Davide Robbiani, MD, PhD, of The Rockefeller University in New York, New York, and his colleagues described this research in Cell.
The team infected mice with the malaria parasite Plasmodium chabaudi and, immediately, the mice experienced an increase in germinal center (GC) B lymphocytes, which can give rise to Burkitt lymphoma.
“In malaria-infected mice, these cells divide very rapidly over the course of months,” Dr Robbiani said.
As the GC B lymphocytes proliferate, they also express high levels of activation-induced cytidine deaminase (AID), which induces mutations in their DNA. As a result, these cells can diversify to generate a wide range of antibodies.
But in addition to beneficial mutations in antibody genes, AID can cause off-target damage and shuffling of cancer-causing genes.
“In mice infected with the malaria parasite, these so-called chromosomal rearrangements occur very frequently in GC lymphocytes,” Dr Robbiani said. “And at least some of the changes are due to AID.”
To further investigate this phenomenon, the researchers bred mice lacking the p53 gene, which is known to protect cells from Burkitt lymphoma. All of the mice that expressed AID but not p53 ultimately developed lymphoma.
And when these mice were infected with the malaria parasite, they developed lymphomas specifically in mature B cells, similar to what happens in Burkitt lymphoma.
“This finding sheds new light on a long-standing mystery of why two seemingly different diseases are associated with each other,” Dr Robbiani said.
Researchers are now attempting to determine how AID causes its off-target damage to DNA, which could lead to new treatments.
“If we could somehow limit this collateral damage to cancer-causing genes without reducing the infection-fighting powers of B cells, that could be very useful,” Dr Robbiani said. “But first, we have to find out how the collateral DNA damage occurs in the first place.”
Dr Robbiani noted that hepatitis C virus and Helicobacter pylori infections, as well as some autoimmune diseases, are also linked with
chronic B lymphocyte activation and an increased risk of lymphoma.
Therefore,
strategies aimed at reducing unintended DNA damage caused by AID might
also help reduce the risk of lymphoma in patients with these conditions.
“It’s possible that AID also plays a role in the association between these other infections and cancer,” Dr Robbiani said. “This is purely a speculation at this point, though highly suggestive.” ![]()
Image by Ed Uthman
A link between malaria and Burkitt lymphoma was first described more than 50 years ago, but how the parasitic infection promotes lymphomagenesis has remained a mystery.
Now, research in mice has revealed that B-cell DNA becomes vulnerable to cancer-causing mutations during prolonged combat against the malaria parasite.
Davide Robbiani, MD, PhD, of The Rockefeller University in New York, New York, and his colleagues described this research in Cell.
The team infected mice with the malaria parasite Plasmodium chabaudi and, immediately, the mice experienced an increase in germinal center (GC) B lymphocytes, which can give rise to Burkitt lymphoma.
“In malaria-infected mice, these cells divide very rapidly over the course of months,” Dr Robbiani said.
As the GC B lymphocytes proliferate, they also express high levels of activation-induced cytidine deaminase (AID), which induces mutations in their DNA. As a result, these cells can diversify to generate a wide range of antibodies.
But in addition to beneficial mutations in antibody genes, AID can cause off-target damage and shuffling of cancer-causing genes.
“In mice infected with the malaria parasite, these so-called chromosomal rearrangements occur very frequently in GC lymphocytes,” Dr Robbiani said. “And at least some of the changes are due to AID.”
To further investigate this phenomenon, the researchers bred mice lacking the p53 gene, which is known to protect cells from Burkitt lymphoma. All of the mice that expressed AID but not p53 ultimately developed lymphoma.
And when these mice were infected with the malaria parasite, they developed lymphomas specifically in mature B cells, similar to what happens in Burkitt lymphoma.
“This finding sheds new light on a long-standing mystery of why two seemingly different diseases are associated with each other,” Dr Robbiani said.
Researchers are now attempting to determine how AID causes its off-target damage to DNA, which could lead to new treatments.
“If we could somehow limit this collateral damage to cancer-causing genes without reducing the infection-fighting powers of B cells, that could be very useful,” Dr Robbiani said. “But first, we have to find out how the collateral DNA damage occurs in the first place.”
Dr Robbiani noted that hepatitis C virus and Helicobacter pylori infections, as well as some autoimmune diseases, are also linked with
chronic B lymphocyte activation and an increased risk of lymphoma.
Therefore,
strategies aimed at reducing unintended DNA damage caused by AID might
also help reduce the risk of lymphoma in patients with these conditions.
“It’s possible that AID also plays a role in the association between these other infections and cancer,” Dr Robbiani said. “This is purely a speculation at this point, though highly suggestive.” ![]()
Image by Ed Uthman
A link between malaria and Burkitt lymphoma was first described more than 50 years ago, but how the parasitic infection promotes lymphomagenesis has remained a mystery.
Now, research in mice has revealed that B-cell DNA becomes vulnerable to cancer-causing mutations during prolonged combat against the malaria parasite.
Davide Robbiani, MD, PhD, of The Rockefeller University in New York, New York, and his colleagues described this research in Cell.
The team infected mice with the malaria parasite Plasmodium chabaudi and, immediately, the mice experienced an increase in germinal center (GC) B lymphocytes, which can give rise to Burkitt lymphoma.
“In malaria-infected mice, these cells divide very rapidly over the course of months,” Dr Robbiani said.
As the GC B lymphocytes proliferate, they also express high levels of activation-induced cytidine deaminase (AID), which induces mutations in their DNA. As a result, these cells can diversify to generate a wide range of antibodies.
But in addition to beneficial mutations in antibody genes, AID can cause off-target damage and shuffling of cancer-causing genes.
“In mice infected with the malaria parasite, these so-called chromosomal rearrangements occur very frequently in GC lymphocytes,” Dr Robbiani said. “And at least some of the changes are due to AID.”
To further investigate this phenomenon, the researchers bred mice lacking the p53 gene, which is known to protect cells from Burkitt lymphoma. All of the mice that expressed AID but not p53 ultimately developed lymphoma.
And when these mice were infected with the malaria parasite, they developed lymphomas specifically in mature B cells, similar to what happens in Burkitt lymphoma.
“This finding sheds new light on a long-standing mystery of why two seemingly different diseases are associated with each other,” Dr Robbiani said.
Researchers are now attempting to determine how AID causes its off-target damage to DNA, which could lead to new treatments.
“If we could somehow limit this collateral damage to cancer-causing genes without reducing the infection-fighting powers of B cells, that could be very useful,” Dr Robbiani said. “But first, we have to find out how the collateral DNA damage occurs in the first place.”
Dr Robbiani noted that hepatitis C virus and Helicobacter pylori infections, as well as some autoimmune diseases, are also linked with
chronic B lymphocyte activation and an increased risk of lymphoma.
Therefore,
strategies aimed at reducing unintended DNA damage caused by AID might
also help reduce the risk of lymphoma in patients with these conditions.
“It’s possible that AID also plays a role in the association between these other infections and cancer,” Dr Robbiani said. “This is purely a speculation at this point, though highly suggestive.” ![]()
Nanoparticle-based vaccine could prevent EBV

Photo by Sakura Midori
Researchers say they have developed a nanoparticle-based vaccine against Epstein-Barr virus (EBV) that can induce potent neutralizing antibodies in mice and monkeys.
These results suggest that using a structure-based vaccine design and self-assembling nanoparticles to deliver a viral protein that prompts an immune response could be a promising approach for developing an EBV vaccine for humans.
Most efforts to develop a preventive EBV vaccine have focused on glycoprotein 350 (gp350), a molecule on the surface of EBV that helps the virus attach to B cells. EBV gp350 is thought to be a key target for antibodies capable of preventing viral infection.
Previously, researchers showed that vaccinating monkeys with gp350 protected the animals from developing lymphomas after exposure to a high dose of EBV.
However, in the only large human trial of an experimental EBV vaccine conducted to date, the EBV gp350 vaccine did not prevent EBV infection, although it did reduce the rate of infectious mononucleosis by 78%.
With this in mind, Masaru Kanekiyo, DVM, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues set out to create a better vaccine.
They described their work in a paper published in Cell.
The team designed a nanoparticle-based vaccine that expressed the cell-binding portion of gp350. In tests, the experimental vaccine induced potent neutralizing antibodies in both mice and cynomolgus macaques (Macaca fascicularis).
In fact, compared with soluble gp350, the nanoparticle-based vaccine induced 10- to 100-fold higher levels of neutralizing antibodies in mice.
The researchers believe the nanoparticle vaccine design could be used to create or redesign vaccines against other pathogens as well. ![]()
Photo by Sakura Midori
Researchers say they have developed a nanoparticle-based vaccine against Epstein-Barr virus (EBV) that can induce potent neutralizing antibodies in mice and monkeys.
These results suggest that using a structure-based vaccine design and self-assembling nanoparticles to deliver a viral protein that prompts an immune response could be a promising approach for developing an EBV vaccine for humans.
Most efforts to develop a preventive EBV vaccine have focused on glycoprotein 350 (gp350), a molecule on the surface of EBV that helps the virus attach to B cells. EBV gp350 is thought to be a key target for antibodies capable of preventing viral infection.
Previously, researchers showed that vaccinating monkeys with gp350 protected the animals from developing lymphomas after exposure to a high dose of EBV.
However, in the only large human trial of an experimental EBV vaccine conducted to date, the EBV gp350 vaccine did not prevent EBV infection, although it did reduce the rate of infectious mononucleosis by 78%.
With this in mind, Masaru Kanekiyo, DVM, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues set out to create a better vaccine.
They described their work in a paper published in Cell.
The team designed a nanoparticle-based vaccine that expressed the cell-binding portion of gp350. In tests, the experimental vaccine induced potent neutralizing antibodies in both mice and cynomolgus macaques (Macaca fascicularis).
In fact, compared with soluble gp350, the nanoparticle-based vaccine induced 10- to 100-fold higher levels of neutralizing antibodies in mice.
The researchers believe the nanoparticle vaccine design could be used to create or redesign vaccines against other pathogens as well. ![]()
Photo by Sakura Midori
Researchers say they have developed a nanoparticle-based vaccine against Epstein-Barr virus (EBV) that can induce potent neutralizing antibodies in mice and monkeys.
These results suggest that using a structure-based vaccine design and self-assembling nanoparticles to deliver a viral protein that prompts an immune response could be a promising approach for developing an EBV vaccine for humans.
Most efforts to develop a preventive EBV vaccine have focused on glycoprotein 350 (gp350), a molecule on the surface of EBV that helps the virus attach to B cells. EBV gp350 is thought to be a key target for antibodies capable of preventing viral infection.
Previously, researchers showed that vaccinating monkeys with gp350 protected the animals from developing lymphomas after exposure to a high dose of EBV.
However, in the only large human trial of an experimental EBV vaccine conducted to date, the EBV gp350 vaccine did not prevent EBV infection, although it did reduce the rate of infectious mononucleosis by 78%.
With this in mind, Masaru Kanekiyo, DVM, PhD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues set out to create a better vaccine.
They described their work in a paper published in Cell.
The team designed a nanoparticle-based vaccine that expressed the cell-binding portion of gp350. In tests, the experimental vaccine induced potent neutralizing antibodies in both mice and cynomolgus macaques (Macaca fascicularis).
In fact, compared with soluble gp350, the nanoparticle-based vaccine induced 10- to 100-fold higher levels of neutralizing antibodies in mice.
The researchers believe the nanoparticle vaccine design could be used to create or redesign vaccines against other pathogens as well. ![]()
Tool that lets patients report AEs proves reliable

receiving chemotherapy
Photo by Rhoda Baer
Results of a multicenter study indicate that a tool cancer patients can use to report adverse events (AEs) is as accurate as other, established patient-reported and clinical measures.
The tool is the National Cancer Institute’s Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE).
Study investigators were able to validate 119 of 124 PRO-CTCAE questions against 2 established measurement tools.
The 5 questions that were not validated could not be evaluated due to underrepresentation in the study population.
This research was published in JAMA Oncology.
“In most cancer clinical trials, information on side effects is collected by providers who have limited time with their patients, and current patient questionnaires are limited in scope and depth,” said study author Amylou Dueck, PhD, of the Mayo Clinic in Scottsdale, Arizona.
“PRO-CTCAE is a library of items for patients to directly report on the level of each of their symptoms, to enhance the reporting of side effects in cancer clinical trials, which is normally based on information from providers. The study itself is unprecedented, as more than 100 distinct questions about symptomatic adverse events were validated simultaneously.”
To assess the PRO-CTCAE, Dr Dueck and her colleagues recruited 975 cancer patients from 9 clinical practices across the US, including 7 cancer centers.
The patients had a range of cancers and were undergoing outpatient chemotherapy and/or radiation therapy. The investigators said these participants reflected the geographic, ethnic, racial, and economic diversity in cancer clinical trials.
The patients were asked to fill out the PRO-CTCAE questionnaire before appointments. The investigators then compared patient reports to clinician-reported Eastern Cooperative Oncology Group (ECOG) performance status and the European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire (QLQ-C30).
A majority of patients completed items on the PRO-CTCAE questionnaire at their first visit (96.4%, 940/975) and second visit (90.6%, 852/940).
Most patients (99.8%, 938/940) reported having at least 1 symptomatic AE, with 81.7% (768/940) reporting at least 1 AE as frequent, severe, and/or interfering “quite a bit” with daily activities.
To gauge the accuracy of the PRO-CTCAE, the investigators assessed construct validity, test-retest reliability, and responsiveness of PRO-CTCAE items.
Construct validity
The investigators explained that construct validity reflects the association between a new measurement tool and an established measure.
Construct validity is often investigated through convergent validity, which determines whether the new tool moves in the same direction as an established instrument, and known-groups validity, which determines whether the tool can distinguish between groups of patients who are thought to be distinct.
When the investigators considered all QLQ-C30 functioning/global scales, they found that all 124 items on the PRO-CTCAE questionnaire were associated in the expected direction with 1 or more scales. One hundred and fourteen of the PRO-CTCAE items demonstrated a meaningful correlation (Pearson r≥0.1), and 111 of them were statistically significant (P<0.05 for all).
Scores for 94 of 124 PRO-CTCAE items were higher among patients with an ECOG performance status of 2 to 4 (17.1% of patients) than among patients with a score of 0 to 1. The difference was significant for 58 of the items (P<0.05 for all).
Test-retest reliability and responsiveness
The investigators said they estimated test-retest reliability using the intraclass correlation coefficient (ICC), based on a 1-way analysis of variance model with an ICC of 0.7 or greater interpreted as high.
Test-retest reliability was 0.7 or greater for 36 of 49 prespecified PRO-CTCAE items. The median ICC was 0.76 [range, 0.53-0.96).
The investigators assessed the responsiveness of PRO-CTCAE items by comparing any change from the first visit to the second visit in 27 items that were selected a priori.
Correlations between PRO-CTCAE item changes and corresponding QLQ-C30 scale changes were significant for all 27 items (P≤0.006 for all).
“This is a landmark study demonstrating that meaningful information about adverse events can be elicited from patients themselves, which is a major step for advancing the patient-centeredness of clinical trials,” said study author Ethan Basch, MD, of the Lineberger Cancer Center of the University of North Carolina in Chapel Hill. ![]()
receiving chemotherapy
Photo by Rhoda Baer
Results of a multicenter study indicate that a tool cancer patients can use to report adverse events (AEs) is as accurate as other, established patient-reported and clinical measures.
The tool is the National Cancer Institute’s Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE).
Study investigators were able to validate 119 of 124 PRO-CTCAE questions against 2 established measurement tools.
The 5 questions that were not validated could not be evaluated due to underrepresentation in the study population.
This research was published in JAMA Oncology.
“In most cancer clinical trials, information on side effects is collected by providers who have limited time with their patients, and current patient questionnaires are limited in scope and depth,” said study author Amylou Dueck, PhD, of the Mayo Clinic in Scottsdale, Arizona.
“PRO-CTCAE is a library of items for patients to directly report on the level of each of their symptoms, to enhance the reporting of side effects in cancer clinical trials, which is normally based on information from providers. The study itself is unprecedented, as more than 100 distinct questions about symptomatic adverse events were validated simultaneously.”
To assess the PRO-CTCAE, Dr Dueck and her colleagues recruited 975 cancer patients from 9 clinical practices across the US, including 7 cancer centers.
The patients had a range of cancers and were undergoing outpatient chemotherapy and/or radiation therapy. The investigators said these participants reflected the geographic, ethnic, racial, and economic diversity in cancer clinical trials.
The patients were asked to fill out the PRO-CTCAE questionnaire before appointments. The investigators then compared patient reports to clinician-reported Eastern Cooperative Oncology Group (ECOG) performance status and the European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire (QLQ-C30).
A majority of patients completed items on the PRO-CTCAE questionnaire at their first visit (96.4%, 940/975) and second visit (90.6%, 852/940).
Most patients (99.8%, 938/940) reported having at least 1 symptomatic AE, with 81.7% (768/940) reporting at least 1 AE as frequent, severe, and/or interfering “quite a bit” with daily activities.
To gauge the accuracy of the PRO-CTCAE, the investigators assessed construct validity, test-retest reliability, and responsiveness of PRO-CTCAE items.
Construct validity
The investigators explained that construct validity reflects the association between a new measurement tool and an established measure.
Construct validity is often investigated through convergent validity, which determines whether the new tool moves in the same direction as an established instrument, and known-groups validity, which determines whether the tool can distinguish between groups of patients who are thought to be distinct.
When the investigators considered all QLQ-C30 functioning/global scales, they found that all 124 items on the PRO-CTCAE questionnaire were associated in the expected direction with 1 or more scales. One hundred and fourteen of the PRO-CTCAE items demonstrated a meaningful correlation (Pearson r≥0.1), and 111 of them were statistically significant (P<0.05 for all).
Scores for 94 of 124 PRO-CTCAE items were higher among patients with an ECOG performance status of 2 to 4 (17.1% of patients) than among patients with a score of 0 to 1. The difference was significant for 58 of the items (P<0.05 for all).
Test-retest reliability and responsiveness
The investigators said they estimated test-retest reliability using the intraclass correlation coefficient (ICC), based on a 1-way analysis of variance model with an ICC of 0.7 or greater interpreted as high.
Test-retest reliability was 0.7 or greater for 36 of 49 prespecified PRO-CTCAE items. The median ICC was 0.76 [range, 0.53-0.96).
The investigators assessed the responsiveness of PRO-CTCAE items by comparing any change from the first visit to the second visit in 27 items that were selected a priori.
Correlations between PRO-CTCAE item changes and corresponding QLQ-C30 scale changes were significant for all 27 items (P≤0.006 for all).
“This is a landmark study demonstrating that meaningful information about adverse events can be elicited from patients themselves, which is a major step for advancing the patient-centeredness of clinical trials,” said study author Ethan Basch, MD, of the Lineberger Cancer Center of the University of North Carolina in Chapel Hill. ![]()
receiving chemotherapy
Photo by Rhoda Baer
Results of a multicenter study indicate that a tool cancer patients can use to report adverse events (AEs) is as accurate as other, established patient-reported and clinical measures.
The tool is the National Cancer Institute’s Patient Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE).
Study investigators were able to validate 119 of 124 PRO-CTCAE questions against 2 established measurement tools.
The 5 questions that were not validated could not be evaluated due to underrepresentation in the study population.
This research was published in JAMA Oncology.
“In most cancer clinical trials, information on side effects is collected by providers who have limited time with their patients, and current patient questionnaires are limited in scope and depth,” said study author Amylou Dueck, PhD, of the Mayo Clinic in Scottsdale, Arizona.
“PRO-CTCAE is a library of items for patients to directly report on the level of each of their symptoms, to enhance the reporting of side effects in cancer clinical trials, which is normally based on information from providers. The study itself is unprecedented, as more than 100 distinct questions about symptomatic adverse events were validated simultaneously.”
To assess the PRO-CTCAE, Dr Dueck and her colleagues recruited 975 cancer patients from 9 clinical practices across the US, including 7 cancer centers.
The patients had a range of cancers and were undergoing outpatient chemotherapy and/or radiation therapy. The investigators said these participants reflected the geographic, ethnic, racial, and economic diversity in cancer clinical trials.
The patients were asked to fill out the PRO-CTCAE questionnaire before appointments. The investigators then compared patient reports to clinician-reported Eastern Cooperative Oncology Group (ECOG) performance status and the European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire (QLQ-C30).
A majority of patients completed items on the PRO-CTCAE questionnaire at their first visit (96.4%, 940/975) and second visit (90.6%, 852/940).
Most patients (99.8%, 938/940) reported having at least 1 symptomatic AE, with 81.7% (768/940) reporting at least 1 AE as frequent, severe, and/or interfering “quite a bit” with daily activities.
To gauge the accuracy of the PRO-CTCAE, the investigators assessed construct validity, test-retest reliability, and responsiveness of PRO-CTCAE items.
Construct validity
The investigators explained that construct validity reflects the association between a new measurement tool and an established measure.
Construct validity is often investigated through convergent validity, which determines whether the new tool moves in the same direction as an established instrument, and known-groups validity, which determines whether the tool can distinguish between groups of patients who are thought to be distinct.
When the investigators considered all QLQ-C30 functioning/global scales, they found that all 124 items on the PRO-CTCAE questionnaire were associated in the expected direction with 1 or more scales. One hundred and fourteen of the PRO-CTCAE items demonstrated a meaningful correlation (Pearson r≥0.1), and 111 of them were statistically significant (P<0.05 for all).
Scores for 94 of 124 PRO-CTCAE items were higher among patients with an ECOG performance status of 2 to 4 (17.1% of patients) than among patients with a score of 0 to 1. The difference was significant for 58 of the items (P<0.05 for all).
Test-retest reliability and responsiveness
The investigators said they estimated test-retest reliability using the intraclass correlation coefficient (ICC), based on a 1-way analysis of variance model with an ICC of 0.7 or greater interpreted as high.
Test-retest reliability was 0.7 or greater for 36 of 49 prespecified PRO-CTCAE items. The median ICC was 0.76 [range, 0.53-0.96).
The investigators assessed the responsiveness of PRO-CTCAE items by comparing any change from the first visit to the second visit in 27 items that were selected a priori.
Correlations between PRO-CTCAE item changes and corresponding QLQ-C30 scale changes were significant for all 27 items (P≤0.006 for all).
“This is a landmark study demonstrating that meaningful information about adverse events can be elicited from patients themselves, which is a major step for advancing the patient-centeredness of clinical trials,” said study author Ethan Basch, MD, of the Lineberger Cancer Center of the University of North Carolina in Chapel Hill.