User login
How a molecule turns B cells into macrophages
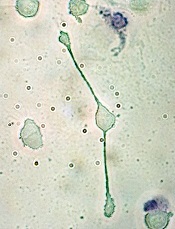
pseudopodia to engulf particles
The transcription factor C/EBPα reprograms B cells into macrophages by “short-circuiting” the cells so they re-express genes reserved for embryonic development, according to research published in Stem Cell Reports.
Over the past 28 years, researchers have shown that a number of specialized cell types can be forcibly converted into other cell types, but the science of how this change takes place is still emerging.
“For a long time, it was unclear whether forcing cell fate decisions by expressing transcription factors in the wrong cell type could teach us something about what happens normally during physiological differentiation,” said Thomas Graf, PhD, of the Center for Genomic Regulation in Barcelona, Spain.
“What we have now found is that the two processes are actually surprisingly similar.”
The researchers found that B-cell transdifferentiation takes place when C/EBPα binds to two regions of DNA that act as gene expression enhancers. One of these regions is normally active in immune cells, and the other is only turned on when macrophage precursors are ready to differentiate.
This indicates that the convergence of these two enhancer pathways can cause the B cell to act like a macrophage precursor, thus triggering the unnatural transdifferentiation.
“This has taught us a great deal about how a transcription factor can activate a new gene expression program (in our case, that of macrophages) but has left us in the dark about the other part of the equation; namely, how the factor silences the B-cell program, something that must happen if transdifferentiation is to work,” Dr Graf said. “This is one of the questions we are focusing on now.”
Dr Graf is interested in this pathway because C/EBPα-induced, B cell-to-macrophage transdifferentiation can convert both human B-cell lymphoma and leukemia cells into functional, non-cancerous macrophages.
He believes that induced transdifferentiation could become therapeutically relevant if a drug could be found that can replace the transcription factor. And understanding the mechanisms of the process could help labs that use this transdifferentiation approach to generate cells for regenerative purposes. ![]()
pseudopodia to engulf particles
The transcription factor C/EBPα reprograms B cells into macrophages by “short-circuiting” the cells so they re-express genes reserved for embryonic development, according to research published in Stem Cell Reports.
Over the past 28 years, researchers have shown that a number of specialized cell types can be forcibly converted into other cell types, but the science of how this change takes place is still emerging.
“For a long time, it was unclear whether forcing cell fate decisions by expressing transcription factors in the wrong cell type could teach us something about what happens normally during physiological differentiation,” said Thomas Graf, PhD, of the Center for Genomic Regulation in Barcelona, Spain.
“What we have now found is that the two processes are actually surprisingly similar.”
The researchers found that B-cell transdifferentiation takes place when C/EBPα binds to two regions of DNA that act as gene expression enhancers. One of these regions is normally active in immune cells, and the other is only turned on when macrophage precursors are ready to differentiate.
This indicates that the convergence of these two enhancer pathways can cause the B cell to act like a macrophage precursor, thus triggering the unnatural transdifferentiation.
“This has taught us a great deal about how a transcription factor can activate a new gene expression program (in our case, that of macrophages) but has left us in the dark about the other part of the equation; namely, how the factor silences the B-cell program, something that must happen if transdifferentiation is to work,” Dr Graf said. “This is one of the questions we are focusing on now.”
Dr Graf is interested in this pathway because C/EBPα-induced, B cell-to-macrophage transdifferentiation can convert both human B-cell lymphoma and leukemia cells into functional, non-cancerous macrophages.
He believes that induced transdifferentiation could become therapeutically relevant if a drug could be found that can replace the transcription factor. And understanding the mechanisms of the process could help labs that use this transdifferentiation approach to generate cells for regenerative purposes. ![]()
pseudopodia to engulf particles
The transcription factor C/EBPα reprograms B cells into macrophages by “short-circuiting” the cells so they re-express genes reserved for embryonic development, according to research published in Stem Cell Reports.
Over the past 28 years, researchers have shown that a number of specialized cell types can be forcibly converted into other cell types, but the science of how this change takes place is still emerging.
“For a long time, it was unclear whether forcing cell fate decisions by expressing transcription factors in the wrong cell type could teach us something about what happens normally during physiological differentiation,” said Thomas Graf, PhD, of the Center for Genomic Regulation in Barcelona, Spain.
“What we have now found is that the two processes are actually surprisingly similar.”
The researchers found that B-cell transdifferentiation takes place when C/EBPα binds to two regions of DNA that act as gene expression enhancers. One of these regions is normally active in immune cells, and the other is only turned on when macrophage precursors are ready to differentiate.
This indicates that the convergence of these two enhancer pathways can cause the B cell to act like a macrophage precursor, thus triggering the unnatural transdifferentiation.
“This has taught us a great deal about how a transcription factor can activate a new gene expression program (in our case, that of macrophages) but has left us in the dark about the other part of the equation; namely, how the factor silences the B-cell program, something that must happen if transdifferentiation is to work,” Dr Graf said. “This is one of the questions we are focusing on now.”
Dr Graf is interested in this pathway because C/EBPα-induced, B cell-to-macrophage transdifferentiation can convert both human B-cell lymphoma and leukemia cells into functional, non-cancerous macrophages.
He believes that induced transdifferentiation could become therapeutically relevant if a drug could be found that can replace the transcription factor. And understanding the mechanisms of the process could help labs that use this transdifferentiation approach to generate cells for regenerative purposes. ![]()
Newfound mechanism could be used to fight cancers
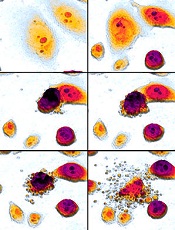
apoptosis in cancer cells
Researchers say they have identified a new mechanism by which the tumor suppressor protein p53 triggers apoptosis, and they believe this process could be harnessed to kill cancer cells.
The team discovered how p53 acts in the cytoplasm to trigger cell death by binding to and activating the BAX protein.
The process involves a shape change in one of p53’s amino acids that serves as the “switch” to activate BAX and trigger the apoptotic pathway.
The team also identified the enzyme in the cytoplasm that promotes the change that controls the “switch.”
Richard Kriwacki, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described these findings in Molecular Cell.
Like up to half of all proteins, p53 includes both structured and disordered regions. A disordered region is a segment that does not adopt a single shape but
remains flexible and constantly switches between different shapes until
it encounters a partner.
Dr Kriwacki and his colleagues showed that both structured and disordered regions of p53 play a role in BAX activation in the cytoplasm.
The process starts when a structured region of p53 known as the DNA-binding domain binds to BAX. That sets the stage for the unstructured region of p53 to form a second bond, which activates BAX and triggers apoptosis.
“There were no previous reports of this disordered region of p53 binding to BAX, so the finding that this region was the key to BAX activation was a total surprise,” Dr Kriwacki said.
The disordered p53 segment included the amino acid proline, which can change between two shapes, particularly in the presence of the enzyme Pin1.
Using NMR spectroscopy, the researchers showed that the proline shape change promotes p53 binding and activation of BAX.
“These results expand our understanding of the different ways p53 modulates cell behavior,” Dr Kriwacki said. “The findings also raise the possibility of killing tumor cells using small molecules to trigger BAX-dependent apoptosis.” ![]()
apoptosis in cancer cells
Researchers say they have identified a new mechanism by which the tumor suppressor protein p53 triggers apoptosis, and they believe this process could be harnessed to kill cancer cells.
The team discovered how p53 acts in the cytoplasm to trigger cell death by binding to and activating the BAX protein.
The process involves a shape change in one of p53’s amino acids that serves as the “switch” to activate BAX and trigger the apoptotic pathway.
The team also identified the enzyme in the cytoplasm that promotes the change that controls the “switch.”
Richard Kriwacki, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described these findings in Molecular Cell.
Like up to half of all proteins, p53 includes both structured and disordered regions. A disordered region is a segment that does not adopt a single shape but
remains flexible and constantly switches between different shapes until
it encounters a partner.
Dr Kriwacki and his colleagues showed that both structured and disordered regions of p53 play a role in BAX activation in the cytoplasm.
The process starts when a structured region of p53 known as the DNA-binding domain binds to BAX. That sets the stage for the unstructured region of p53 to form a second bond, which activates BAX and triggers apoptosis.
“There were no previous reports of this disordered region of p53 binding to BAX, so the finding that this region was the key to BAX activation was a total surprise,” Dr Kriwacki said.
The disordered p53 segment included the amino acid proline, which can change between two shapes, particularly in the presence of the enzyme Pin1.
Using NMR spectroscopy, the researchers showed that the proline shape change promotes p53 binding and activation of BAX.
“These results expand our understanding of the different ways p53 modulates cell behavior,” Dr Kriwacki said. “The findings also raise the possibility of killing tumor cells using small molecules to trigger BAX-dependent apoptosis.” ![]()
apoptosis in cancer cells
Researchers say they have identified a new mechanism by which the tumor suppressor protein p53 triggers apoptosis, and they believe this process could be harnessed to kill cancer cells.
The team discovered how p53 acts in the cytoplasm to trigger cell death by binding to and activating the BAX protein.
The process involves a shape change in one of p53’s amino acids that serves as the “switch” to activate BAX and trigger the apoptotic pathway.
The team also identified the enzyme in the cytoplasm that promotes the change that controls the “switch.”
Richard Kriwacki, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described these findings in Molecular Cell.
Like up to half of all proteins, p53 includes both structured and disordered regions. A disordered region is a segment that does not adopt a single shape but
remains flexible and constantly switches between different shapes until
it encounters a partner.
Dr Kriwacki and his colleagues showed that both structured and disordered regions of p53 play a role in BAX activation in the cytoplasm.
The process starts when a structured region of p53 known as the DNA-binding domain binds to BAX. That sets the stage for the unstructured region of p53 to form a second bond, which activates BAX and triggers apoptosis.
“There were no previous reports of this disordered region of p53 binding to BAX, so the finding that this region was the key to BAX activation was a total surprise,” Dr Kriwacki said.
The disordered p53 segment included the amino acid proline, which can change between two shapes, particularly in the presence of the enzyme Pin1.
Using NMR spectroscopy, the researchers showed that the proline shape change promotes p53 binding and activation of BAX.
“These results expand our understanding of the different ways p53 modulates cell behavior,” Dr Kriwacki said. “The findings also raise the possibility of killing tumor cells using small molecules to trigger BAX-dependent apoptosis.” ![]()
Companies ‘underinvest’ in long-term cancer research

for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
for a clinical trial
Photo by Esther Dyson
Pharmaceutical companies “underinvest” in long-term research to develop new anticancer drugs, according to a study published in American Economic Review.
Investigators used historical data to show that companies are more likely to develop drugs for late-stage cancers than early stage cancers or cancer prevention, and this is likely because late-stage cancer drugs can be brought to market faster.
The team found that late-stage drugs extend patient survival for shorter periods so that clinical trials for these drugs get wrapped up more quickly. This, in turn, gives drug manufacturers more time to control patented drugs in the marketplace.
“There is a pattern where we get more investment in drugs that take a short time to complete and less investment in drugs that take a longer time to complete,” said study author Heidi Williams, PhD, of the Massachusetts Institute of Technology in Cambridge.
To conduct this study, Dr Williams and her colleagues analyzed 4 decades of clinical trial data from a variety of sources, including the National Cancer Institute and the US Food and Drug Administration. The study encompassed more than 200 subcategories of cancers detected at different stages of development.
In analyzing the data, the investigators divided research and development (R&D) into 2 stages: invention (developing the basic idea for a product to the point where it is patentable) and commercialization (bringing an invented product to market).
They defined the “commercialization lag” of an R&D project as the amount of time between invention and commercialization.
The data showed that patient groups with longer commercialization lags (as proxied by longer survival times) tended to have lower levels of R&D investment than groups with shorter commercialization lags (and survival times).
The investigators also found that when surrogate endpoints (endpoints other than survival) were allowed, there were more trials and money poured into research. This supports the idea that companies are more likely to invest in drugs that will have shorter trials and take less time to develop.
Dr Williams and her colleagues used the surrogate endpoint variation to estimate improvements in cancer survival rates that would have been observed if commercialization lags were reduced. The team estimated that, among US cancer patients diagnosed in 2003, longer commercialization lags resulted in around 890,000 lost life-years.
The investigators noted that commercialization lags reduce both public and private R&D investments, but they found the commercialization lag-R&D correlation is significantly more negative for privately financed trials than publicly financed trials.
The team said that, due to either excessive discounting or the fixed patent term, private incentives decline more rapidly than public incentives, which is what gives rise to the distortion.
Based on their findings, Dr Williams and her colleagues devised 3 new policy approaches that could potentially spark the development of more drugs for early stage cancers or cancer prevention.
The first is expanded use of surrogate endpoints or more research to determine if wider use of surrogate endpoints is valid.
A second possible policy change is more public funding of R&D for anticancer drugs, since such funding is free of short-term, private-sector shareholder pressure to produce returns.
A third potential new policy would be changing the terms of drug patents, which typically run from the time of patent filing to when the drug hits the market. ![]()
Health Canada grants drug conditional approval for MCL

Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Photo courtesy of
Janssen Biotech, Inc.
Health Canada has granted conditional approval for the BTK inhibitor ibrutinib (Imbruvica) to treat patients with relapsed or refractory mantle cell lymphoma (MCL).
This approval was based on data from a phase 2 trial in which ibrutinib conferred an overall response rate of 68% in patients with relapsed/refractory MCL.
For ibrutinib to gain full approval, Health Canada must receive additional data confirming the drug provides a clinical benefit.
Ibrutinib was first approved in Canada in November 2014 for patients with chronic lymphocytic leukemia (CLL), including those with 17p deletion, who have received at least one prior therapy, or for the frontline treatment of patients with CLL with 17p deletion. For this use, ibrutinib was issued marketing authorization without conditions.
Now, Health Canada has issued ibrutinib conditional marketing authorization for the treatment of relapsed/refractory MCL. This decision was based on data from the phase 2 PCYC-1104 trial, which was presented at ASH 2012 and published in NEJM in August 2013.
The study included 111 MCL patients who had received at least one prior therapy. The primary endpoint of the study was overall response rate according to the revised International Working Group criteria for non-Hodgkin lymphoma.
The overall response rate was 68%, with a complete response rate of 21% and a partial response rate of 47%. With a median follow-up of 15.3 months, the median response duration was 17.5 months.
The estimated progression-free survival was 13.9 months, and the overall survival was not reached. The estimated rate of overall survival was 58% at 18 months.
Common nonhematologic adverse events included diarrhea (50%), fatigue (41%), nausea (31%), peripheral edema (28%), dyspnea (27%), constipation (25%), upper respiratory tract infection (23%), vomiting (23%), and decreased appetite (21%). The most common grade 3, 4, or 5 infection was pneumonia (6%).
Grade 3 and 4 hematologic adverse events included neutropenia (16%), thrombocytopenia (11%), and anemia (10%). Grade 3 bleeding events occurred in 5 patients.
Eight patients (7%) had an adverse event that led to treatment discontinuation.
Sixteen patients (14%) died during the trial, 12 due to disease progression and 4 due to an adverse event. Two patients died of pneumonia, 1 from sepsis, and 1 from a cardiac arrest that was not considered drug-related.
Ibrutinib is co-developed by Cilag GmbH International (a member of the Janssen Pharmaceutical Companies) and Pharmacyclics LLC, an AbbVie company. Janssen Inc. markets ibrutinib as Imbruvica in Canada. ![]()
Tool identifies optimal TKI for cancers

Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Photo courtesy of the
University of Colorado
Researchers say they have developed a tool that allows us to determine which tyrosine kinase inhibitor (TKI) will be most effective against a certain type of cancer.
The tool, known as the Kinase Addiction Ranker (KAR), predicts the genetic abnormalities that are driving the cancer in any population of cells and chooses the best TKI or combination of TKIs to target these abnormalities.
The researchers described the tool in Bioinformatics.
“A lot of [TKIs] inhibit a lot more than what they’re supposed to inhibit,” said study author Aik Choon Tan, PhD, of the University of Colorado Anschutz Medical Campus in Aurora.
“Maybe drug A was designed to inhibit kinase B, but it also inhibits kinase C and D as well. Our approach centers on exploiting the promiscuity of these drugs, the ‘drug spillover.’”
For each TKI, there is a signature describing the kinases each drug fully or partially inhibits. Dr Tan and his colleagues combined these kinase inhibition signatures with the results of high-throughput screening. They used the Genomics of Drug Sensitivity in Cancer database to determine which TKIs have already proven active against which cancer cell lines.
The result is KAR, which does 2 things. For any cancer cell line, the program ranks the kinases that are most important to the growth of the disease. Then, the program recommends the combination of existing TKIs that is likely to do the most good against the implicated kinases.
Dr Tan and his colleagues tested KAR using samples from 151 leukemia patients and found that, among the kinases analyzed, FLT3 had the highest variance in sensitivity to TKIs.
But EPHA5, EPHA3, and BTK were the kinases most commonly associated with drug sensitivity. They had significant associations in 72%, 58%, and 54% of the patient samples, respectively.
The researchers said the frequency of BTK dependence they observed is interesting given the fact that the BTK inhibitor ibrutinib produced favorable results in a phase 1b/2 trial of patients with chronic lymphocytic leukemia (CLL). The progression-free survival rate at 26 months was 75% in that trial.
Dr Tan and his colleagues said this was consistent with their findings, which showed that 70% of CLL patient data had a significant association between BTK inhibition and drug sensitivity.
The researchers also found that KAR could predict TKI sensitivity in 21 lung cancer cell lines. In addition, the tool was able to recommend a combination of TKIs that hindered proliferation in the lung cancer cell line H1581. KAR suggested ponatinib and the experimental anticancer agent AZD8055, and experiments showed that these drugs synergistically reduced proliferation in H1581.
KAR is available for download on the Tan lab’s website. ![]()
Analysis reveals ‘distinctive biology’ of CTCL
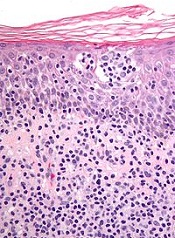
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
New research suggests cutaneous T-cell lymphoma (CTCL) is driven by a plethora of genetic mutations.
Investigators conducted a genomic analysis of normal and cancer cells from patients with CTCL and identified mutations in 17 genes that are implicated in CTCL pathogenesis.
They also found that somatic copy number variants (SCNVs) driving CTCL outnumbered somatic single-nucleotide variants (SSNVs) by more than 10 to 1.
The team reported these findings in Nature Genetics.
They performed exome and whole-genome DNA sequencing and RNA sequencing on purified CTCL cells and matched normal cells. And they identified genes implicated in CTCL pathogenesis by looking for:
- Genes with recurrent SSNVs altering the same amino acid more often than expected by chance
- Genes with SSNVs previously identified as recurrent mutations in other cancers
- Genes having a significantly increased burden of protein-altering SSNVs
- SCNVs that occurred more often than expected by chance.
This revealed mutations in 17 genes that are implicated in CTCL pathogenesis—TP53, ZEB1, ARID1A, DNMT3A, CDKN2A, FAS, NFKB2, CD28, RHOA, PLCG1, STAT5B, BRAF, ATM, CTCF, TNFAIP3, PRKCQ, and IRF4.
The investigators noted that these are genes involved in T-cell activation, apoptosis, NF-κB signaling, chromatin remodeling, and DNA damage response.
The team also discovered “a striking bias” for SCNVs as drivers of CTCL. They identified 12 statistically significant chromosome-arm SCNVs and 36 significant focal SCNVs.
Collectively, these SCNVs occurred 473 times in the CTCL samples analyzed—a mean of 7.5 focal deletions, 1.6 broad deletions, 1.0 focal amplification, and 1.8 broad amplifications per CTCL.
On the other hand, there were 38 SSNVs in CTCL driver genes—1.0 per tumor.
So, according to these data, SCNVs comprise 92% of all driver mutations in CTCL—a mean of 11.8 pathogenic SCNVs vs 1.0 SSNV per CTCL.
“This cancer has a very distinctive biology,” said Jaehyuk Choi, MD, PhD, of the Yale School of Medicine in New Haven, Connecticut.
And decoding this biology has revealed potential treatment approaches, according to Dr Choi and his colleagues.
For example, the presence of mutations activating the NF-κB pathway suggests NF-κB inhibitors such as bortezomib may have therapeutic potential in CTCL, and the presence of CD28 mutations suggests inhibitors such as abatacept may be effective against the disease. ![]()
Fertility preservation in young cancer patients
Image courtesy of NHS
Young patients with cancer, particularly females, may be uninformed about their options for preserving fertility, according to a study published in Cancer.
The research showed that males were both more likely to have discussed fertility preservation with their physicians and more likely to have taken steps to preserve fertility.
Other factors such as education and insurance status also appeared to have an impact on fertility preservation.
Margarett Shnorhavorian, MD, of the University of Washington in Seattle, and her colleagues conducted this research.
The team enlisted 459 adolescents and young adults who were diagnosed with cancer in 2007 or 2008, asking them to complete questionnaires on fertility preservation.
Eighty percent of males and 74% of females said they had been told that cancer therapy might affect their fertility. For females, multivariable analysis revealed no significant factors associated with this discussion.
However, multivariable analysis showed that males with an unknown treatment fertility risk were more likely to be uninformed of the potential risk (odds ratio [OR]= 2.73; 95% CI, 1.09-6.86), as were males who did not consult a medical oncologist (OR=2.28; 95% CI, 1.03-5.00).
Twenty-nine percent of males and 56.3% of females said they did not discuss fertility preservation with their doctors before they began cancer treatment. Males raising children younger than 18 were more likely than males without children to miss out on the discussion (OR=2.45; 95% CI, 1.24-4.85).
Males were also more likely to miss the discussion if they had a treatment fertility risk classified as “none/low” rather than “intermediate/high” (OR=3.39; 95% CI, 1.60-7.16) and if they had no insurance or government insurance rather than private insurance (OR=2.91; 95% CI, 1.41-5.97).
Males diagnosed in 2008 were less likely than those diagnosed in 2007 to miss out on the discussion (OR=0.43; 95% CI, 0.20-0.80).
Females raising children under 18 were more likely than females without children to say they did not discuss fertility preservation with their doctors (OR=3.38; 95% CI, 1.43-8.02). Females without private insurance were more likely to miss the discussion as well (OR=5.46; 95% CI, 1.59-18.72).
Females diagnosed in 2008 were less likely to miss the discussion than those diagnosed in 2007 (OR=0.36; 95% CI, 0.15-0.85).
Sixty-nine percent of males and 93.2% of females said they did not make fertility preservation arrangements. Men were more likely to lack arrangements if they were raising children younger than 18 years (OR=3.53; 95% CI, 1.63-7.65) or had less than a college degree (OR, 1.98; 95% CI, 1.00-3.97).
The researchers did not conduct a multivariable analysis for women because so few women made arrangements for fertility preservation.
“The access and health-related reasons for not making arrangements for fertility preservation reported by participants in this study further highlight the need for decreased cost, improved insurance coverage, and partnerships between cancer healthcare providers and fertility experts to develop strategies that increase awareness of fertility preservation options and decrease delays in cancer therapy as fertility preservation for adolescent and young adult cancer patients improves,” Dr Shnorhavorian concluded. ![]()
Image courtesy of NHS
Young patients with cancer, particularly females, may be uninformed about their options for preserving fertility, according to a study published in Cancer.
The research showed that males were both more likely to have discussed fertility preservation with their physicians and more likely to have taken steps to preserve fertility.
Other factors such as education and insurance status also appeared to have an impact on fertility preservation.
Margarett Shnorhavorian, MD, of the University of Washington in Seattle, and her colleagues conducted this research.
The team enlisted 459 adolescents and young adults who were diagnosed with cancer in 2007 or 2008, asking them to complete questionnaires on fertility preservation.
Eighty percent of males and 74% of females said they had been told that cancer therapy might affect their fertility. For females, multivariable analysis revealed no significant factors associated with this discussion.
However, multivariable analysis showed that males with an unknown treatment fertility risk were more likely to be uninformed of the potential risk (odds ratio [OR]= 2.73; 95% CI, 1.09-6.86), as were males who did not consult a medical oncologist (OR=2.28; 95% CI, 1.03-5.00).
Twenty-nine percent of males and 56.3% of females said they did not discuss fertility preservation with their doctors before they began cancer treatment. Males raising children younger than 18 were more likely than males without children to miss out on the discussion (OR=2.45; 95% CI, 1.24-4.85).
Males were also more likely to miss the discussion if they had a treatment fertility risk classified as “none/low” rather than “intermediate/high” (OR=3.39; 95% CI, 1.60-7.16) and if they had no insurance or government insurance rather than private insurance (OR=2.91; 95% CI, 1.41-5.97).
Males diagnosed in 2008 were less likely than those diagnosed in 2007 to miss out on the discussion (OR=0.43; 95% CI, 0.20-0.80).
Females raising children under 18 were more likely than females without children to say they did not discuss fertility preservation with their doctors (OR=3.38; 95% CI, 1.43-8.02). Females without private insurance were more likely to miss the discussion as well (OR=5.46; 95% CI, 1.59-18.72).
Females diagnosed in 2008 were less likely to miss the discussion than those diagnosed in 2007 (OR=0.36; 95% CI, 0.15-0.85).
Sixty-nine percent of males and 93.2% of females said they did not make fertility preservation arrangements. Men were more likely to lack arrangements if they were raising children younger than 18 years (OR=3.53; 95% CI, 1.63-7.65) or had less than a college degree (OR, 1.98; 95% CI, 1.00-3.97).
The researchers did not conduct a multivariable analysis for women because so few women made arrangements for fertility preservation.
“The access and health-related reasons for not making arrangements for fertility preservation reported by participants in this study further highlight the need for decreased cost, improved insurance coverage, and partnerships between cancer healthcare providers and fertility experts to develop strategies that increase awareness of fertility preservation options and decrease delays in cancer therapy as fertility preservation for adolescent and young adult cancer patients improves,” Dr Shnorhavorian concluded. ![]()
Image courtesy of NHS
Young patients with cancer, particularly females, may be uninformed about their options for preserving fertility, according to a study published in Cancer.
The research showed that males were both more likely to have discussed fertility preservation with their physicians and more likely to have taken steps to preserve fertility.
Other factors such as education and insurance status also appeared to have an impact on fertility preservation.
Margarett Shnorhavorian, MD, of the University of Washington in Seattle, and her colleagues conducted this research.
The team enlisted 459 adolescents and young adults who were diagnosed with cancer in 2007 or 2008, asking them to complete questionnaires on fertility preservation.
Eighty percent of males and 74% of females said they had been told that cancer therapy might affect their fertility. For females, multivariable analysis revealed no significant factors associated with this discussion.
However, multivariable analysis showed that males with an unknown treatment fertility risk were more likely to be uninformed of the potential risk (odds ratio [OR]= 2.73; 95% CI, 1.09-6.86), as were males who did not consult a medical oncologist (OR=2.28; 95% CI, 1.03-5.00).
Twenty-nine percent of males and 56.3% of females said they did not discuss fertility preservation with their doctors before they began cancer treatment. Males raising children younger than 18 were more likely than males without children to miss out on the discussion (OR=2.45; 95% CI, 1.24-4.85).
Males were also more likely to miss the discussion if they had a treatment fertility risk classified as “none/low” rather than “intermediate/high” (OR=3.39; 95% CI, 1.60-7.16) and if they had no insurance or government insurance rather than private insurance (OR=2.91; 95% CI, 1.41-5.97).
Males diagnosed in 2008 were less likely than those diagnosed in 2007 to miss out on the discussion (OR=0.43; 95% CI, 0.20-0.80).
Females raising children under 18 were more likely than females without children to say they did not discuss fertility preservation with their doctors (OR=3.38; 95% CI, 1.43-8.02). Females without private insurance were more likely to miss the discussion as well (OR=5.46; 95% CI, 1.59-18.72).
Females diagnosed in 2008 were less likely to miss the discussion than those diagnosed in 2007 (OR=0.36; 95% CI, 0.15-0.85).
Sixty-nine percent of males and 93.2% of females said they did not make fertility preservation arrangements. Men were more likely to lack arrangements if they were raising children younger than 18 years (OR=3.53; 95% CI, 1.63-7.65) or had less than a college degree (OR, 1.98; 95% CI, 1.00-3.97).
The researchers did not conduct a multivariable analysis for women because so few women made arrangements for fertility preservation.
“The access and health-related reasons for not making arrangements for fertility preservation reported by participants in this study further highlight the need for decreased cost, improved insurance coverage, and partnerships between cancer healthcare providers and fertility experts to develop strategies that increase awareness of fertility preservation options and decrease delays in cancer therapy as fertility preservation for adolescent and young adult cancer patients improves,” Dr Shnorhavorian concluded.
Less toxic chemo for HIV-positive Burkitt lymphoma
For HIV-positive patients with Burkitt lymphoma, a modified intensive chemotherapy regimen produced overall and progression-free survival rates comparable with those seen in HIV-free patients with Burkitt, with manageable toxicities, reported researchers in a multicenter clinical trial.
The AIDS Malignancy Consortium (AMC) 048 study looked at the use of a modified version of the dose intensive CODOX-M/IVAC regimen, consisting of cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, and high-dose cytarabine. Compared with the standard regimen, the investigators added rituximab, reduced and/or rescheduled cyclophosphamide and methotrexate, limited the use of vincristine, and used combination intrathecal chemotherapy to prevent central nervous system involvement.
The study included 34 HIV-positive patients (30 men and 4 women) with Burkitt, 26 of whom were also receiving highly active antiretroviral therapy (HAART). The patients ranged in age from 19-55 (median 42) years. Of the 34 patients, 25 had Ann Arbor stage IV disease, 2 had stage III, 1 had stage IIE, 2 had stage II, and 4 had stage I. Median age was 42 years (range, 19-55 years).
The median CD4 count was 195 cells/mL; five patients had fewer than 100 cells/mL
Progression-free survival at 1 year was 69%, and 1- and 2-year overall survival were 72% and 69%, respectively.
The modified CODOX-M/IVAC regimen was associated with a grade 3 to 4 toxicity rate of 79%, with no grade 3 or 4 mucositis reported. In contrast, virtually all patients who receive the unmodified regimen develop at least one grade 3 or greater toxicity.
In total, there were 20 hematologic, 14 infectious, and 6 metabolic toxicities. Five patients did not complete treatment because of adverse events.
There were 11 deaths, including 1 treatment-related death of a patient with encephalopathy, hepatic failure, hepatitis B, and pneumonia cited as contributing causes. Of the remaining 10 patients, 8 died from systemic disease progression, and 2 died during follow-up, 1 during remission from a fungal infection and 1 from nonmalignant complications of HIV.
The investigators say that the addition of rituximab may have contributed to the favorable outcomes, and that rescheduling and limiting the amount of high-dose methotrexate delivered likely contributed to lower incidences of both severe mucositis and neutropenic fever.
Although a separate trial is evaluating a different regimen (EPOCH-R; etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in Burkitt lymphoma, “the modified AMC 048 version of CODOX-M/IVAC-R may better serve patients who present with CNS disease or are at high risk for CNS relapse (e.g., patients with bone marrow, testicular, or multiple extranodal sites), because it contains high-dose cytarabine and methotrexate, drugs that cross the blood-brain barrier. Consequently, AMC048 represents a reasonable treatment option in the appropriate setting, possibly irrespective of HIV status.”
The study by Dr. Ariela Noy from the Memorial Sloan Kettering Cancer Center in New York and her colleagues, is published in Blood.
Although the results of AMC 048 are encouraging and demonstrate that intensive regimens for AIDS-related Burkitt lymphoma are both tolerable and efficacious, the question of whether multiagent dose intense regimens are needed remains unanswered. Using a short course of EPOCH (infusional etoposide, oral prednisone, infusional vincristine, bolus cyclophosphamide, and infusional doxorubicin) with a double dose of rituximab (SC-EPOCH-RR) to treat 11 patients with AIDS-related Burkitt lymphoma, researchers at the National Cancer Institute have observed progression-free survival of 100% and overall survival of 90% at a median follow-up of 73 months. This regimen omits systemic ifosfamide and high-dose methotrexate. Although both agents are thought to be important for disease control in Burkitt lymphoma, especially to treat and/or prevent lymphomatous CNS involvement, they also have substantial toxicities. In the NCI study, only 1 patient had CNS involvement at baseline and was successfully treated with intrathecal methotrexate alone. No patient relapsed in the CNS. However, given the small number of patients enrolled in this single institution study, there remains significant concern that omission of these agents will jeopardize disease control, specifically in high-risk patients. It will be interesting to see whether the results of the NCI study will be maintained in the ongoing larger cooperative group trial that currently evaluates dose-adjusted EPOCH-R.
Dr. Stefan K. Barta is with the Fox Chase Cancer Center/Temple University Health System in Philadelphia. He made his remarks in an editorial that accompanied the study.
Although the results of AMC 048 are encouraging and demonstrate that intensive regimens for AIDS-related Burkitt lymphoma are both tolerable and efficacious, the question of whether multiagent dose intense regimens are needed remains unanswered. Using a short course of EPOCH (infusional etoposide, oral prednisone, infusional vincristine, bolus cyclophosphamide, and infusional doxorubicin) with a double dose of rituximab (SC-EPOCH-RR) to treat 11 patients with AIDS-related Burkitt lymphoma, researchers at the National Cancer Institute have observed progression-free survival of 100% and overall survival of 90% at a median follow-up of 73 months. This regimen omits systemic ifosfamide and high-dose methotrexate. Although both agents are thought to be important for disease control in Burkitt lymphoma, especially to treat and/or prevent lymphomatous CNS involvement, they also have substantial toxicities. In the NCI study, only 1 patient had CNS involvement at baseline and was successfully treated with intrathecal methotrexate alone. No patient relapsed in the CNS. However, given the small number of patients enrolled in this single institution study, there remains significant concern that omission of these agents will jeopardize disease control, specifically in high-risk patients. It will be interesting to see whether the results of the NCI study will be maintained in the ongoing larger cooperative group trial that currently evaluates dose-adjusted EPOCH-R.
Dr. Stefan K. Barta is with the Fox Chase Cancer Center/Temple University Health System in Philadelphia. He made his remarks in an editorial that accompanied the study.
Although the results of AMC 048 are encouraging and demonstrate that intensive regimens for AIDS-related Burkitt lymphoma are both tolerable and efficacious, the question of whether multiagent dose intense regimens are needed remains unanswered. Using a short course of EPOCH (infusional etoposide, oral prednisone, infusional vincristine, bolus cyclophosphamide, and infusional doxorubicin) with a double dose of rituximab (SC-EPOCH-RR) to treat 11 patients with AIDS-related Burkitt lymphoma, researchers at the National Cancer Institute have observed progression-free survival of 100% and overall survival of 90% at a median follow-up of 73 months. This regimen omits systemic ifosfamide and high-dose methotrexate. Although both agents are thought to be important for disease control in Burkitt lymphoma, especially to treat and/or prevent lymphomatous CNS involvement, they also have substantial toxicities. In the NCI study, only 1 patient had CNS involvement at baseline and was successfully treated with intrathecal methotrexate alone. No patient relapsed in the CNS. However, given the small number of patients enrolled in this single institution study, there remains significant concern that omission of these agents will jeopardize disease control, specifically in high-risk patients. It will be interesting to see whether the results of the NCI study will be maintained in the ongoing larger cooperative group trial that currently evaluates dose-adjusted EPOCH-R.
Dr. Stefan K. Barta is with the Fox Chase Cancer Center/Temple University Health System in Philadelphia. He made his remarks in an editorial that accompanied the study.
For HIV-positive patients with Burkitt lymphoma, a modified intensive chemotherapy regimen produced overall and progression-free survival rates comparable with those seen in HIV-free patients with Burkitt, with manageable toxicities, reported researchers in a multicenter clinical trial.
The AIDS Malignancy Consortium (AMC) 048 study looked at the use of a modified version of the dose intensive CODOX-M/IVAC regimen, consisting of cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, and high-dose cytarabine. Compared with the standard regimen, the investigators added rituximab, reduced and/or rescheduled cyclophosphamide and methotrexate, limited the use of vincristine, and used combination intrathecal chemotherapy to prevent central nervous system involvement.
The study included 34 HIV-positive patients (30 men and 4 women) with Burkitt, 26 of whom were also receiving highly active antiretroviral therapy (HAART). The patients ranged in age from 19-55 (median 42) years. Of the 34 patients, 25 had Ann Arbor stage IV disease, 2 had stage III, 1 had stage IIE, 2 had stage II, and 4 had stage I. Median age was 42 years (range, 19-55 years).
The median CD4 count was 195 cells/mL; five patients had fewer than 100 cells/mL
Progression-free survival at 1 year was 69%, and 1- and 2-year overall survival were 72% and 69%, respectively.
The modified CODOX-M/IVAC regimen was associated with a grade 3 to 4 toxicity rate of 79%, with no grade 3 or 4 mucositis reported. In contrast, virtually all patients who receive the unmodified regimen develop at least one grade 3 or greater toxicity.
In total, there were 20 hematologic, 14 infectious, and 6 metabolic toxicities. Five patients did not complete treatment because of adverse events.
There were 11 deaths, including 1 treatment-related death of a patient with encephalopathy, hepatic failure, hepatitis B, and pneumonia cited as contributing causes. Of the remaining 10 patients, 8 died from systemic disease progression, and 2 died during follow-up, 1 during remission from a fungal infection and 1 from nonmalignant complications of HIV.
The investigators say that the addition of rituximab may have contributed to the favorable outcomes, and that rescheduling and limiting the amount of high-dose methotrexate delivered likely contributed to lower incidences of both severe mucositis and neutropenic fever.
Although a separate trial is evaluating a different regimen (EPOCH-R; etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in Burkitt lymphoma, “the modified AMC 048 version of CODOX-M/IVAC-R may better serve patients who present with CNS disease or are at high risk for CNS relapse (e.g., patients with bone marrow, testicular, or multiple extranodal sites), because it contains high-dose cytarabine and methotrexate, drugs that cross the blood-brain barrier. Consequently, AMC048 represents a reasonable treatment option in the appropriate setting, possibly irrespective of HIV status.”
The study by Dr. Ariela Noy from the Memorial Sloan Kettering Cancer Center in New York and her colleagues, is published in Blood.
For HIV-positive patients with Burkitt lymphoma, a modified intensive chemotherapy regimen produced overall and progression-free survival rates comparable with those seen in HIV-free patients with Burkitt, with manageable toxicities, reported researchers in a multicenter clinical trial.
The AIDS Malignancy Consortium (AMC) 048 study looked at the use of a modified version of the dose intensive CODOX-M/IVAC regimen, consisting of cyclophosphamide, vincristine, doxorubicin, high-dose methotrexate/ifosfamide, etoposide, and high-dose cytarabine. Compared with the standard regimen, the investigators added rituximab, reduced and/or rescheduled cyclophosphamide and methotrexate, limited the use of vincristine, and used combination intrathecal chemotherapy to prevent central nervous system involvement.
The study included 34 HIV-positive patients (30 men and 4 women) with Burkitt, 26 of whom were also receiving highly active antiretroviral therapy (HAART). The patients ranged in age from 19-55 (median 42) years. Of the 34 patients, 25 had Ann Arbor stage IV disease, 2 had stage III, 1 had stage IIE, 2 had stage II, and 4 had stage I. Median age was 42 years (range, 19-55 years).
The median CD4 count was 195 cells/mL; five patients had fewer than 100 cells/mL
Progression-free survival at 1 year was 69%, and 1- and 2-year overall survival were 72% and 69%, respectively.
The modified CODOX-M/IVAC regimen was associated with a grade 3 to 4 toxicity rate of 79%, with no grade 3 or 4 mucositis reported. In contrast, virtually all patients who receive the unmodified regimen develop at least one grade 3 or greater toxicity.
In total, there were 20 hematologic, 14 infectious, and 6 metabolic toxicities. Five patients did not complete treatment because of adverse events.
There were 11 deaths, including 1 treatment-related death of a patient with encephalopathy, hepatic failure, hepatitis B, and pneumonia cited as contributing causes. Of the remaining 10 patients, 8 died from systemic disease progression, and 2 died during follow-up, 1 during remission from a fungal infection and 1 from nonmalignant complications of HIV.
The investigators say that the addition of rituximab may have contributed to the favorable outcomes, and that rescheduling and limiting the amount of high-dose methotrexate delivered likely contributed to lower incidences of both severe mucositis and neutropenic fever.
Although a separate trial is evaluating a different regimen (EPOCH-R; etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) in Burkitt lymphoma, “the modified AMC 048 version of CODOX-M/IVAC-R may better serve patients who present with CNS disease or are at high risk for CNS relapse (e.g., patients with bone marrow, testicular, or multiple extranodal sites), because it contains high-dose cytarabine and methotrexate, drugs that cross the blood-brain barrier. Consequently, AMC048 represents a reasonable treatment option in the appropriate setting, possibly irrespective of HIV status.”
The study by Dr. Ariela Noy from the Memorial Sloan Kettering Cancer Center in New York and her colleagues, is published in Blood.
FROM BLOOD
Key clinical point: A modified form of a standard chemotherapy regimen for Burkitt lymphoma is effective in HIV-positive patients, with lower rates of adverse events.
Major finding: 1-year overall survival was 72%, and 2-year OS was 69%.
Data source: Open-label study of a modified chemotherapy regimen in 34 HIV-positive patients with Burkitt lymphoma.
Disclosures: The trial was supported by a grant from the National Cancer Institute, The authors and Dr. Barta declare no conflicts of interest.
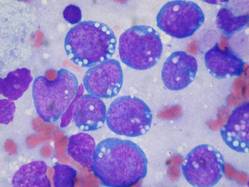
Early follicular lymphoma progression signals poor outcomes
For patients with follicular lymphoma treated with a rituximab-based combination chemotherapy regimen, early disease progression is associated with significantly worse overall survival, suggesting the need for additional interventions, according to results of a multicenter study.
Among 588 patients with stage 2-4 follicular lymphoma treated with first-line R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) and followed for a median of 7 years in the National LymphoCare Study, overall survival (OS) at 2 years was 68% for those who had disease progression within 2 years, compared with 97% for patients with no disease progression during that time.
Similarly, 5-year overall survival was 50% for patients with early progression of disease, compared with 90% for patients with no early progression, write Dr. Carla Casulo of the University of Rochester (N.Y.) Medical Center and colleagues. The study is in anearly online publication in the Journal of Clinical Oncology.
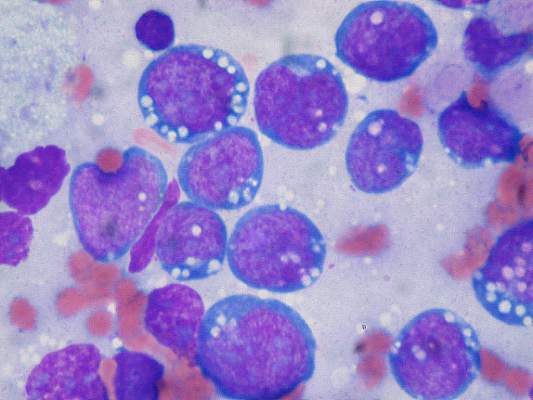
“Given our findings, early relapse after diagnosis in patients treated with first-line chemoimmunotherapy is a powerful prognostic indicator of outcome and should be used to stratify the risk of patients in studies of relapsed follicular lymphoma,” the authors wrote.
The findings were validated in an independent cohort of patients with follicular lymphoma treated with R-CHOP from the University of Iowa and Mayo Clinical Molecular Epidemiology Resource, and are consistent with findings from other studies of patients treated with different rituximab-based regimens, the investigators reported.
In unadjusted analysis, early disease progression was associated with a hazard ratio (HR) of 7.17 (95% confidence interval [CI] 4.83-10.65); the effect remained after adjustment for the Follicular Lymphoma International Prognostic Index (FLIPI) score (HR 6.44, 95% CI, 4.33-9.58).
Factors associated with early progression included age, Eastern Cooperative Oncology Group performance score, nodal sites, and disease stage.
Early use of aggressive salvage therapies or autologous stem-cell transplantation could improve outcomes in patients with early disease progression, the authors wrote. However, only 8 patients among the 110 with early progression went on to transplant, not a large enough sample for meaningful analysis, they added.
“This newly defined high-risk group of patients represents a distinct population in whom further study is warranted in both directed prospective clinical trials of follicular lymphoma biology and treatment. Moreover, we propose that 2-year progression-free survival may be a practical and meaningful clinical end point for trials involving a chemoimmunotherapy backbone,” they concluded.
If, in studying the immunologic and inflammatory host response to, and the genetic landscape of, these lymphomas, we are able to define this high-risk subgroup of patients with follicular lymphoma, the question becomes whether we could use this information to effectively treat these patients differently. Although high-dose chemotherapy and autologous stem-cell transplantation (HDC-ASCT) in first remission seems to have no effect on OS in all comers, results might be different for this cohort of high-risk patients. To study this would require an ability to identify these patients at diagnosis. Given that the efficacy of HDC-ASCT is maintained in the case of chemosensitive relapse, reserving HDC-ASCT for patients who relapse within the first 2 years of their initial therapy may be a more prudent strategy.
However, it may be that this is a particularly chemoresistant population and that, instead, attention should be paid to targeting the biologic and genetic factors that contribute to the poor prognosis of this group. Given the negative differential outcomes in patients with decreased tumor-infiltrating lymphocytes and increased monocyte/macrophage activation, immunologic approaches in the salvage setting, including immune checkpoint blockade drugs, chimeric antigen receptor T cells, and allogeneic transplantation may be biologically relevant.
Dr. Caron A. Jacobson and Dr. Arnold S. Freedman, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, made their remarks in an editorial accompanying the study.
If, in studying the immunologic and inflammatory host response to, and the genetic landscape of, these lymphomas, we are able to define this high-risk subgroup of patients with follicular lymphoma, the question becomes whether we could use this information to effectively treat these patients differently. Although high-dose chemotherapy and autologous stem-cell transplantation (HDC-ASCT) in first remission seems to have no effect on OS in all comers, results might be different for this cohort of high-risk patients. To study this would require an ability to identify these patients at diagnosis. Given that the efficacy of HDC-ASCT is maintained in the case of chemosensitive relapse, reserving HDC-ASCT for patients who relapse within the first 2 years of their initial therapy may be a more prudent strategy.
However, it may be that this is a particularly chemoresistant population and that, instead, attention should be paid to targeting the biologic and genetic factors that contribute to the poor prognosis of this group. Given the negative differential outcomes in patients with decreased tumor-infiltrating lymphocytes and increased monocyte/macrophage activation, immunologic approaches in the salvage setting, including immune checkpoint blockade drugs, chimeric antigen receptor T cells, and allogeneic transplantation may be biologically relevant.
Dr. Caron A. Jacobson and Dr. Arnold S. Freedman, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, made their remarks in an editorial accompanying the study.
If, in studying the immunologic and inflammatory host response to, and the genetic landscape of, these lymphomas, we are able to define this high-risk subgroup of patients with follicular lymphoma, the question becomes whether we could use this information to effectively treat these patients differently. Although high-dose chemotherapy and autologous stem-cell transplantation (HDC-ASCT) in first remission seems to have no effect on OS in all comers, results might be different for this cohort of high-risk patients. To study this would require an ability to identify these patients at diagnosis. Given that the efficacy of HDC-ASCT is maintained in the case of chemosensitive relapse, reserving HDC-ASCT for patients who relapse within the first 2 years of their initial therapy may be a more prudent strategy.
However, it may be that this is a particularly chemoresistant population and that, instead, attention should be paid to targeting the biologic and genetic factors that contribute to the poor prognosis of this group. Given the negative differential outcomes in patients with decreased tumor-infiltrating lymphocytes and increased monocyte/macrophage activation, immunologic approaches in the salvage setting, including immune checkpoint blockade drugs, chimeric antigen receptor T cells, and allogeneic transplantation may be biologically relevant.
Dr. Caron A. Jacobson and Dr. Arnold S. Freedman, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, made their remarks in an editorial accompanying the study.
For patients with follicular lymphoma treated with a rituximab-based combination chemotherapy regimen, early disease progression is associated with significantly worse overall survival, suggesting the need for additional interventions, according to results of a multicenter study.
Among 588 patients with stage 2-4 follicular lymphoma treated with first-line R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) and followed for a median of 7 years in the National LymphoCare Study, overall survival (OS) at 2 years was 68% for those who had disease progression within 2 years, compared with 97% for patients with no disease progression during that time.
Similarly, 5-year overall survival was 50% for patients with early progression of disease, compared with 90% for patients with no early progression, write Dr. Carla Casulo of the University of Rochester (N.Y.) Medical Center and colleagues. The study is in anearly online publication in the Journal of Clinical Oncology.
“Given our findings, early relapse after diagnosis in patients treated with first-line chemoimmunotherapy is a powerful prognostic indicator of outcome and should be used to stratify the risk of patients in studies of relapsed follicular lymphoma,” the authors wrote.
The findings were validated in an independent cohort of patients with follicular lymphoma treated with R-CHOP from the University of Iowa and Mayo Clinical Molecular Epidemiology Resource, and are consistent with findings from other studies of patients treated with different rituximab-based regimens, the investigators reported.
In unadjusted analysis, early disease progression was associated with a hazard ratio (HR) of 7.17 (95% confidence interval [CI] 4.83-10.65); the effect remained after adjustment for the Follicular Lymphoma International Prognostic Index (FLIPI) score (HR 6.44, 95% CI, 4.33-9.58).
Factors associated with early progression included age, Eastern Cooperative Oncology Group performance score, nodal sites, and disease stage.
Early use of aggressive salvage therapies or autologous stem-cell transplantation could improve outcomes in patients with early disease progression, the authors wrote. However, only 8 patients among the 110 with early progression went on to transplant, not a large enough sample for meaningful analysis, they added.
“This newly defined high-risk group of patients represents a distinct population in whom further study is warranted in both directed prospective clinical trials of follicular lymphoma biology and treatment. Moreover, we propose that 2-year progression-free survival may be a practical and meaningful clinical end point for trials involving a chemoimmunotherapy backbone,” they concluded.
For patients with follicular lymphoma treated with a rituximab-based combination chemotherapy regimen, early disease progression is associated with significantly worse overall survival, suggesting the need for additional interventions, according to results of a multicenter study.
Among 588 patients with stage 2-4 follicular lymphoma treated with first-line R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone) and followed for a median of 7 years in the National LymphoCare Study, overall survival (OS) at 2 years was 68% for those who had disease progression within 2 years, compared with 97% for patients with no disease progression during that time.
Similarly, 5-year overall survival was 50% for patients with early progression of disease, compared with 90% for patients with no early progression, write Dr. Carla Casulo of the University of Rochester (N.Y.) Medical Center and colleagues. The study is in anearly online publication in the Journal of Clinical Oncology.
“Given our findings, early relapse after diagnosis in patients treated with first-line chemoimmunotherapy is a powerful prognostic indicator of outcome and should be used to stratify the risk of patients in studies of relapsed follicular lymphoma,” the authors wrote.
The findings were validated in an independent cohort of patients with follicular lymphoma treated with R-CHOP from the University of Iowa and Mayo Clinical Molecular Epidemiology Resource, and are consistent with findings from other studies of patients treated with different rituximab-based regimens, the investigators reported.
In unadjusted analysis, early disease progression was associated with a hazard ratio (HR) of 7.17 (95% confidence interval [CI] 4.83-10.65); the effect remained after adjustment for the Follicular Lymphoma International Prognostic Index (FLIPI) score (HR 6.44, 95% CI, 4.33-9.58).
Factors associated with early progression included age, Eastern Cooperative Oncology Group performance score, nodal sites, and disease stage.
Early use of aggressive salvage therapies or autologous stem-cell transplantation could improve outcomes in patients with early disease progression, the authors wrote. However, only 8 patients among the 110 with early progression went on to transplant, not a large enough sample for meaningful analysis, they added.
“This newly defined high-risk group of patients represents a distinct population in whom further study is warranted in both directed prospective clinical trials of follicular lymphoma biology and treatment. Moreover, we propose that 2-year progression-free survival may be a practical and meaningful clinical end point for trials involving a chemoimmunotherapy backbone,” they concluded.
FROM JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: Disease progression within 2 years of chemotherapy for follicular lymphoma is associated with poor outcomes.
Major finding: Five-year overall survival was 50% for patients with follicular lymphoma with disease progression within 2-years of R-CHOP, vs. 90% for patients with no early progression.
Data source: Retrospective review involving 588 patients in the longitudinal National LymphoCare Study.
Disclosures: Genentech and F. Hoffmann-La Roche supported the study. Dr. Casulo and Dr. Jacobson reported no relevant disclosures. Dr. Freedman reported ties with UpToDate, Axio, and Immunogen.
Evolution drives cancer development, scientists say

Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer.
Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer.
Photo courtesy of University
of Colorado Cancer Center
Oncogenesis does not depend only on the accumulation of mutations but on evolutionary pressures acting on cell populations, according to a paper published in PNAS.
The authors say the ecosystem of a healthy tissue landscape lets healthy cells outcompete cells with cancerous mutations.
It is when the tissue ecosystem changes due to aging, smoking, or other stressors that cells with cancerous mutations can suddenly find themselves the most fit.
And this allows the cell population to expand over generations of natural selection.
This model of oncogenesis has profound implications for cancer therapy and drug design, according to the authors.
“We’ve been trying to make drugs that target mutations in cancer cells,” said author James DeGregori, PhD, of University of Colorado School of Medicine in Aurora.
“But if it’s the ecosystem of the body, and not only cancer-causing mutations, that allows the growth of cancer, we should also be prioritizing interventions and lifestyle choices that promote the fitness of healthy cells in order to suppress the emergence of cancer.”
The proposed model helps to answer a long-standing question known as Peto’s Paradox. If cancer is due to random activating mutation, larger animals with more cells should be at greater risk of developing cancer earlier in their lives. Why then do mammals of vastly different sizes and lifespans all seem to develop cancer mostly late in life?
“Blue whales have more than a million times more cells and live about 50 times longer than a mouse, but the whale has no more risk than a mouse of developing cancer over its lifespan,” Dr DeGregori noted.
The answer he and colleague Andrii Rozhok, PhD, propose is that, in addition to activating mutations, cancer may require age-associated changes to the tissue landscape in order for evolution to favor the survival and growth of cancer cells over the competition of healthy cells.
“Healthy cells are optimized for the ecosystem of the healthy body,” Dr DeGregori said. “But when the tissue ecosystem changes, such as with aging or smoking, cancer-causing mutations are often very good at exploiting the conditions of a damaged tissue landscape.”
This model is supported by studies showing that mutations that can cause cancer do not necessarily increase a cell’s fitness.
“In fact, healthy cells are so optimized to the healthy tissue landscape that almost any mutation makes them less fit,” Dr DeGregori said.
For example, some cancer cells mutate in a way that allows them to survive in the oxygen-poor tissue environments found in the center of developing tumors. But this adaptation only confers a fitness benefit in oxygen-poor tissues.
In a healthy, oxygen-rich tissue, this mutation would not confer this advantage. In healthy tissue, cells with this mutation lose the evolutionary race to the healthy cells. Cancer cells are outcompeted and die, or, at least, their population is held in check and remains insignificantly small.
But what happens when the tissue landscape changes?
“When the body changes due to aging, smoking, inherited genetic differences, or other factors, it changes the tissue ecosystem, allowing a new kind of cell to replace the healthy ones,” Dr DeGregori said.
Certainly, cancer development requires mutations and other genetic alterations. But how do these mutations cause cancer?
It may not be that these mutations create accidental “super cells” that immediately run amok. Instead, it may be that oncogenic mutations are often or always present in the body but are kept at bay by selection pressures set against them.
That is, until the tissue ecosystem and its pressures change in ways that make cells with cancerous mutations more likely to survive than healthy cells. Over time, this allows the population of cancer cells to overcome that of healthy cells.
People can avoid some of these tissue changes by lifestyle choices, Dr DeGregori noted, but aging cannot be stopped. Still, there may be features of the tissue landscape that, with new therapies and new understanding, could be reinforced in ways to resist cancer better for longer.