User login
Chemo quadruples risk for myeloid cancers
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.

“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
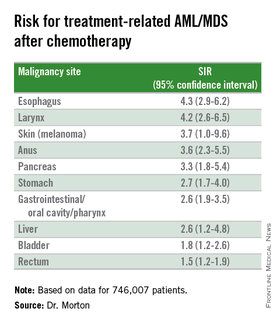
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
AT ASH 2015
Key clinical point: This study quantifies the risks for treatment-related myeloid cancers after chemotherapy.
Major finding: Chemotherapy is associated with a fourfold risk for treatment-related AML/MDS, compared with the general population.
Data source: Retrospective review of data on 746,007 adults treated with chemotherapy for a first primary malignancy.
Disclosures: The National Cancer Institute supported the study. Dr. Morton reported having no conflicts of interest to disclose.

VIDEO: High-intensity conditioning stands ground in MDS, AML
ORLANDO – Reduced-intensity conditioning regimens failed to show a significant survival benefit over high-intensity regimens in myelodysplastic syndrome or acute myeloid leukemia in the phase III MAVERICK trial.
Pretransplant reduced intensity conditioning (RIC) also resulted in a significantly higher risk of relapse and inferior relapse-free survival vs. myeloablative conditioning (MAC).
The findings, reported in the late-breaking abstract (LBA-8) session at the annual meeting of the American Society of Hematology, generated a lively debate over the study construct and whether its conclusion that MAC remain “the treatment of choice” in appropriate candidates should be applied to both diseases.
Session comoderator Dr. David P. Steensma, a myelodysplasia physician at the Dana-Farber Cancer Institute in Boston, said in an interview that the data will not change his practice and that physicians should continue to “push the envelope” and provide as intense a conditioning regimen as their patients can tolerate.
“The take-home message is that using a reduced conditioning regimen whatever your choice might be, even though it is gentler on the patient and may be easier for them to go through the transplant, the biggest risk is still the disease, the underlying leukemia or myelodysplasia coming back. And the benefit from reduced intensity is not enough to outweigh that risk.”
To sort out this complex trial, we spoke with study author Dr. Bart L. Scott of the Fred Hutchinson Cancer Research Center at the University of Washington, Seattle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Reduced-intensity conditioning regimens failed to show a significant survival benefit over high-intensity regimens in myelodysplastic syndrome or acute myeloid leukemia in the phase III MAVERICK trial.
Pretransplant reduced intensity conditioning (RIC) also resulted in a significantly higher risk of relapse and inferior relapse-free survival vs. myeloablative conditioning (MAC).
The findings, reported in the late-breaking abstract (LBA-8) session at the annual meeting of the American Society of Hematology, generated a lively debate over the study construct and whether its conclusion that MAC remain “the treatment of choice” in appropriate candidates should be applied to both diseases.
Session comoderator Dr. David P. Steensma, a myelodysplasia physician at the Dana-Farber Cancer Institute in Boston, said in an interview that the data will not change his practice and that physicians should continue to “push the envelope” and provide as intense a conditioning regimen as their patients can tolerate.
“The take-home message is that using a reduced conditioning regimen whatever your choice might be, even though it is gentler on the patient and may be easier for them to go through the transplant, the biggest risk is still the disease, the underlying leukemia or myelodysplasia coming back. And the benefit from reduced intensity is not enough to outweigh that risk.”
To sort out this complex trial, we spoke with study author Dr. Bart L. Scott of the Fred Hutchinson Cancer Research Center at the University of Washington, Seattle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ORLANDO – Reduced-intensity conditioning regimens failed to show a significant survival benefit over high-intensity regimens in myelodysplastic syndrome or acute myeloid leukemia in the phase III MAVERICK trial.
Pretransplant reduced intensity conditioning (RIC) also resulted in a significantly higher risk of relapse and inferior relapse-free survival vs. myeloablative conditioning (MAC).
The findings, reported in the late-breaking abstract (LBA-8) session at the annual meeting of the American Society of Hematology, generated a lively debate over the study construct and whether its conclusion that MAC remain “the treatment of choice” in appropriate candidates should be applied to both diseases.
Session comoderator Dr. David P. Steensma, a myelodysplasia physician at the Dana-Farber Cancer Institute in Boston, said in an interview that the data will not change his practice and that physicians should continue to “push the envelope” and provide as intense a conditioning regimen as their patients can tolerate.
“The take-home message is that using a reduced conditioning regimen whatever your choice might be, even though it is gentler on the patient and may be easier for them to go through the transplant, the biggest risk is still the disease, the underlying leukemia or myelodysplasia coming back. And the benefit from reduced intensity is not enough to outweigh that risk.”
To sort out this complex trial, we spoke with study author Dr. Bart L. Scott of the Fred Hutchinson Cancer Research Center at the University of Washington, Seattle.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ASH 2015

FDA approves generic imatinib

Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
Photo by Steven Harbour
The US Food and Drug Administration (FDA) has approved the use of imatinib mesylate, a generic version of Novartis’s Gleevec being developed by a subsidiary of Sun Pharmaceuticals Limited.
Under the terms of a settlement agreement with Novartis, the Sun Pharma subsidiary is allowed to launch its generic imatinib in the US on February 1, 2016.
The drug will be available in 100 mg and 400 mg tablets.
The Sun Pharma subsidiary was the first company to file an abbreviated new drug application for generic imatinib with a para IV certification and is therefore eligible for 180 days of marketing exclusivity in the US.
Sun Pharma’s imatinib mesylate is approved for the following indications:
- Newly diagnosed adult and pediatric patients with Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase
- Patients with (Ph+ CML) in blast crisis, accelerated phase, or in chronic phase after failure of interferon-alpha therapy
- Adult patients with relapsed or refractory Ph+ acute lymphoblastic leukemia
- Adult patients with myelodysplastic/myeloproliferative diseases associated with PDGFR gene re-arrangements
- Adult patients with aggressive systemic mastocytosis without the D816V c-Kit mutation or with c-Kit mutational status unknown
- Adult patients with hypereosinophilic syndrome (HES) and/or chronic eosinophilic leukemia (CEL) who have the FIP1L1-PDGFRα fusion kinase and for patients with HES and/or CEL who are FIP1L1- PDGFRα fusion kinase negative or unknown
- Adult patients with unresectable, recurrent and/or metastatic dermatofibrosarcoma protuberans.
The drug is not approved to treat patients with KIT (CD117)-positive unresectable and/or metastatic malignant gastrointestinal stromal tumors. ![]()
A new category of cytopenias

New research suggests that clonality may underlie the cytopenias observed in patients with idiopathic cytopenias of undetermined significance (ICUS).
So researchers say these patients should be classified as having clonal cytopenias of undetermined significance (CCUS).
The team believes that recognition of CCUS as a clinical entity would help identify it as a formally defined diagnostic group and could aid future studies.
“We don’t know to what extent patients who have low blood counts and mutations are at increased risk of developing an overt malignancy,” said Rafael Bejar, MD, PhD, of the UCSD Moores Cancer Center in La Jolla, California.
“We hope that by defining CCUS, future studies will follow these patients to learn what these mutations mean for their future, as their genetically abnormal cells may represent early stages of subsequent blood cancers.”
Dr Bejar and his colleagues defined CCUS in Blood. Their research was funded, in part, by Genoptix Medical Laboratory, and several study authors are employees of Genoptix.
The researchers prospectively examined bone marrow samples from 144 patients with unexplained cytopenias. Seventeen percent of these patients were diagnosed with myelodysplastic syndromes (MDS), 15% with ICUS and some evidence of dysplasia, and 69% with ICUS and no dysplasia.
The team then sequenced patient samples looking for mutations in 22 frequently mutated myeloid malignancy genes. They identified somatic mutations in 71% of MDS patients, 62% of patients with ICUS and some dysplasia, and 20% of ICUS patients with no dysplasia.
Overall, 35% of ICUS patients carried a somatic mutation or chromosomal abnormality indicative of clonal hematopoiesis.
To build upon these findings, the researchers performed a retrospective analysis of data from 91 patients with lower-risk MDS and 249 patients with ICUS.
The team identified mutations in 79% of patients with MDS, 45% of patients with ICUS and dysplasia, and 17% of patients with ICUS and no dysplasia.
The researchers said the spectrum of mutated genes was similar among the 3 groups. The exception was SF3B1, which was rarely mutated in patients without dysplasia.
The team added that variant allele fractions were comparable between clonal ICUS (CCUS) and MDS, and the same was true for the patients’ mean age and blood counts. But CCUS was a more frequent diagnosis than MDS.
Dr Bejar and his colleagues said larger, carefully controlled studies will be needed to confirm the findings of this research. ![]()
New research suggests that clonality may underlie the cytopenias observed in patients with idiopathic cytopenias of undetermined significance (ICUS).
So researchers say these patients should be classified as having clonal cytopenias of undetermined significance (CCUS).
The team believes that recognition of CCUS as a clinical entity would help identify it as a formally defined diagnostic group and could aid future studies.
“We don’t know to what extent patients who have low blood counts and mutations are at increased risk of developing an overt malignancy,” said Rafael Bejar, MD, PhD, of the UCSD Moores Cancer Center in La Jolla, California.
“We hope that by defining CCUS, future studies will follow these patients to learn what these mutations mean for their future, as their genetically abnormal cells may represent early stages of subsequent blood cancers.”
Dr Bejar and his colleagues defined CCUS in Blood. Their research was funded, in part, by Genoptix Medical Laboratory, and several study authors are employees of Genoptix.
The researchers prospectively examined bone marrow samples from 144 patients with unexplained cytopenias. Seventeen percent of these patients were diagnosed with myelodysplastic syndromes (MDS), 15% with ICUS and some evidence of dysplasia, and 69% with ICUS and no dysplasia.
The team then sequenced patient samples looking for mutations in 22 frequently mutated myeloid malignancy genes. They identified somatic mutations in 71% of MDS patients, 62% of patients with ICUS and some dysplasia, and 20% of ICUS patients with no dysplasia.
Overall, 35% of ICUS patients carried a somatic mutation or chromosomal abnormality indicative of clonal hematopoiesis.
To build upon these findings, the researchers performed a retrospective analysis of data from 91 patients with lower-risk MDS and 249 patients with ICUS.
The team identified mutations in 79% of patients with MDS, 45% of patients with ICUS and dysplasia, and 17% of patients with ICUS and no dysplasia.
The researchers said the spectrum of mutated genes was similar among the 3 groups. The exception was SF3B1, which was rarely mutated in patients without dysplasia.
The team added that variant allele fractions were comparable between clonal ICUS (CCUS) and MDS, and the same was true for the patients’ mean age and blood counts. But CCUS was a more frequent diagnosis than MDS.
Dr Bejar and his colleagues said larger, carefully controlled studies will be needed to confirm the findings of this research. ![]()
New research suggests that clonality may underlie the cytopenias observed in patients with idiopathic cytopenias of undetermined significance (ICUS).
So researchers say these patients should be classified as having clonal cytopenias of undetermined significance (CCUS).
The team believes that recognition of CCUS as a clinical entity would help identify it as a formally defined diagnostic group and could aid future studies.
“We don’t know to what extent patients who have low blood counts and mutations are at increased risk of developing an overt malignancy,” said Rafael Bejar, MD, PhD, of the UCSD Moores Cancer Center in La Jolla, California.
“We hope that by defining CCUS, future studies will follow these patients to learn what these mutations mean for their future, as their genetically abnormal cells may represent early stages of subsequent blood cancers.”
Dr Bejar and his colleagues defined CCUS in Blood. Their research was funded, in part, by Genoptix Medical Laboratory, and several study authors are employees of Genoptix.
The researchers prospectively examined bone marrow samples from 144 patients with unexplained cytopenias. Seventeen percent of these patients were diagnosed with myelodysplastic syndromes (MDS), 15% with ICUS and some evidence of dysplasia, and 69% with ICUS and no dysplasia.
The team then sequenced patient samples looking for mutations in 22 frequently mutated myeloid malignancy genes. They identified somatic mutations in 71% of MDS patients, 62% of patients with ICUS and some dysplasia, and 20% of ICUS patients with no dysplasia.
Overall, 35% of ICUS patients carried a somatic mutation or chromosomal abnormality indicative of clonal hematopoiesis.
To build upon these findings, the researchers performed a retrospective analysis of data from 91 patients with lower-risk MDS and 249 patients with ICUS.
The team identified mutations in 79% of patients with MDS, 45% of patients with ICUS and dysplasia, and 17% of patients with ICUS and no dysplasia.
The researchers said the spectrum of mutated genes was similar among the 3 groups. The exception was SF3B1, which was rarely mutated in patients without dysplasia.
The team added that variant allele fractions were comparable between clonal ICUS (CCUS) and MDS, and the same was true for the patients’ mean age and blood counts. But CCUS was a more frequent diagnosis than MDS.
Dr Bejar and his colleagues said larger, carefully controlled studies will be needed to confirm the findings of this research. ![]()
Blood cancer drugs set to be removed from CDF

Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Photo courtesy of CDC
England’s National Health Service (NHS) plans to remove several drugs used to treat hematologic malignancies from the Cancer Drugs Fund (CDF).
The plan is that, as of November 4, 2015, pomalidomide, lenalidomide, ibrutinib, dasatinib, brentuximab, bosutinib, and bendamustine will no longer be funded via the CDF for certain indications.
Ofatumumab was removed from the CDF list yesterday but is now available through the NHS.
Drugs used to treat solid tumor malignancies are set to be de-funded through CDF in November as well.
However, the NHS said the proposal to remove a drug from the CDF is not necessarily a final decision.
In cases where a drug offers enough clinical benefit, the pharmaceutical company developing that drug has the opportunity to reduce the price they are asking the NHS to pay to ensure that it achieves a satisfactory level of value for money. The NHS said a number of such negotiations are underway.
In addition, patients who are currently receiving the drugs set to be removed from the CDF will continue to have access to those drugs.
About the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England and NICE are planning to consult on a proposed new system for commissioning cancer drugs. The NHS said the new system will be designed to provide the agency with a more systematic approach to getting the best price for cancer drugs.
Reason for drug removals
The NHS previously increased the budget for the CDF from £200 million in 2013/14, to £280 million in 2014/15, and £340 million from April 2015. This represents a total increase of 70% since August 2014.
However, current projections suggest that spending would rise to around £410 million for this year, an over-spend of £70 million, in the absence of further prioritization. The NHS said this money could be used for other aspects of cancer treatment or NHS services for other patient groups.
Therefore, some drugs are set to be removed from the CDF. The NHS said all decisions on drugs to be maintained in the CDF were based on the advice of clinicians, the best available evidence, and the cost of the treatment.
“There is no escaping the fact that we face a difficult set of choices, but it is our duty to ensure we get maximum value from every penny available on behalf of patients,” said Peter Clark, chair of the CDF.
“We must ensure we invest in those treatments that offer the most benefit, based on rigorous evidence-based clinical analysis and an assessment of the cost of those treatments.”
While de-funding certain drugs will reduce costs, the CDF is not expected to be back on budget this financial year. The NHS does expect the CDF will be operating within its budget during 2016/17.
Blood cancer drugs to be removed
The following drugs are currently on the CDF list for the following indications, but they are set to be de-listed on November 4, 2015.
Bendamustine
For the treatment of chronic lymphocytic leukemia (CLL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- CLL (not licensed in this indication)
- Second-line indication, third-line indication, or fourth-line indication
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
For the treatment of relapsed mantle cell lymphoma (MCL) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MCL
- Option for second- or subsequent-line chemotherapy
- No previous treatment with bendamustine
- To be used within the treating Trust’s governance framework, as bendamustine is not licensed in this indication
*Bendamustine will remain on the CDF for other indications.
Bosutinib
For the treatment of refractory, chronic phase chronic myeloid leukemia (CML) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Chronic phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
For the treatment of refractory, accelerated phase CML where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Refractory to nilotinib or dasatinib (if dasatinib accessed via a clinical trial or via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
For the treatment of accelerated phase CML where there is intolerance of treatments and where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Accelerated phase CML
- Significant intolerance to dasatinib (grade 3 or 4 adverse events; if dasatinib accessed via its current approved CDF indication)
- Significant intolerance to nilotinib (grade 3 or 4 events)
*Bosutinib will still be available through the CDF for patients with chronic phase CML that is intolerant of other treatments.
Brentuximab
For the treatment of refractory, systemic anaplastic lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory systemic anaplastic large-cell lymphoma
For the treatment of relapsed or refractory CD30+ Hodgkin lymphoma where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Relapsed or refractory CD30+ Hodgkin lymphoma
- Following autologous stem cell transplant or following at least 2 prior therapies when autologous stem cell transplant or multi-agent chemotherapy is not an option
Dasatinib
For the treatment of Philadelphia-chromosome-positive (Ph+) acute lymphoblastic leukemia where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Refractory or significant intolerance or resistance to prior therapy including imatinib (grade 3 or 4 adverse events)
- Second-line indication or third-line indication
*Dasatinib will still be available for chronic phase and accelerated phase CML.
Ibrutinib
For the treatment of relapsed/refractory CLL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed CLL
- Must have received at least 1 prior therapy for CLL
- Considered not appropriate for treatment or retreatment with purine-analogue-based therapy due to:
- Failure to respond to chemo-immunotherapy or
- A progression-free interval of less than 3 years or
- Age of 70 years or more or
- Age of 65 years or more plus the presence of comorbidities or
- A 17p or TP53 deletion
- ECOG performance status of 0-2
- A neutrophil count of ≥0.75 x 10⁹/L
- A platelet count of ≥30 x 10⁹/L
- Patient not on warfarin or CYP3A4/5 inhibitors
- No prior treatment with idelalisib
For the treatment of relapsed/refractory MCL where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- Confirmed MCL with cyclin D1 overexpression or translocation breakpoints at t(11;14)
- Failure to achieve at least partial response with, or documented disease progression disease after, the most recent treatment regimen
- ECOG performance status of 0-2
- At least 1 but no more than 5 previous lines of treatment
Lenalidomide
For the second-line treatment of multiple myeloma (MM) where all the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically trained and accredited in the use of systemic anticancer therapy
- MM
- Second-line indication
- Contraindication to bortezomib or previously received bortezomib in the first-line setting
*Lenalidomide will still be available for patients with myelodysplastic syndromes with 5q deletion.
Pomalidomide
For the treatment of relapsed and refractory MM where the following criteria are met:
- Application made by and first cycle of systemic anticancer therapy to be prescribed by a consultant specialist specifically
- MM
- Performance status of 0-2
- Previously received treatment with adequate trials of at least all of the following options of therapy: bortezomib, lenalidomide, and alkylating agents
- Failed treatment with bortezomib or lenalidomide, as defined by: progression on or before 60 days of treatment, progressive disease 6 months or less after achieving a partial response, or intolerance to bortezomib
- Refractory disease to previous treatment
- No resistance to high-dose dexamethasone used in the last line of therapy
- No peripheral neuropathy of grade 2 or more
A complete list of proposed changes to the CDF, as well as the drugs that were de-listed on March 12, 2015, is available on the NHS website. ![]()
Heme Themes: Transplant timing for myelofibrosis in the era of JAK2 inhibitors
ALEXANDRIA, VA. – How are mutation analysis gene panels affecting risk stratification and decision making regarding stem cell transplants in myelofibrosis patients? How are the results of the Dynamic International Prognostic Scoring System (DIPSS) and performance status improvements seen with JAK2 inhibitors influencing who is a candidate for transplant and the timing of transplants?
Watch the conversation between Dr. Vikas Gupta of the leukemia and bone marrow transplant programs at the Princess Margaret Cancer Centre, Toronto, and associate professor of medicine at the University of Toronto, and Dr. Rami S. Komrokji of the Moffitt Cancer Center, Tampa, Fla., as they discuss their individual approaches that consider patient wishes and goals, type of donor, and disease risk in their decisions to perform stem cell transplants upfront or to delay them until after JAK2 inhibitor therapy.
Have an insight to share? Post a comment.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ALEXANDRIA, VA. – How are mutation analysis gene panels affecting risk stratification and decision making regarding stem cell transplants in myelofibrosis patients? How are the results of the Dynamic International Prognostic Scoring System (DIPSS) and performance status improvements seen with JAK2 inhibitors influencing who is a candidate for transplant and the timing of transplants?
Watch the conversation between Dr. Vikas Gupta of the leukemia and bone marrow transplant programs at the Princess Margaret Cancer Centre, Toronto, and associate professor of medicine at the University of Toronto, and Dr. Rami S. Komrokji of the Moffitt Cancer Center, Tampa, Fla., as they discuss their individual approaches that consider patient wishes and goals, type of donor, and disease risk in their decisions to perform stem cell transplants upfront or to delay them until after JAK2 inhibitor therapy.
Have an insight to share? Post a comment.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ALEXANDRIA, VA. – How are mutation analysis gene panels affecting risk stratification and decision making regarding stem cell transplants in myelofibrosis patients? How are the results of the Dynamic International Prognostic Scoring System (DIPSS) and performance status improvements seen with JAK2 inhibitors influencing who is a candidate for transplant and the timing of transplants?
Watch the conversation between Dr. Vikas Gupta of the leukemia and bone marrow transplant programs at the Princess Margaret Cancer Centre, Toronto, and associate professor of medicine at the University of Toronto, and Dr. Rami S. Komrokji of the Moffitt Cancer Center, Tampa, Fla., as they discuss their individual approaches that consider patient wishes and goals, type of donor, and disease risk in their decisions to perform stem cell transplants upfront or to delay them until after JAK2 inhibitor therapy.
Have an insight to share? Post a comment.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT U.S. FOCUS ON MPN AND MDS
Heme Themes: Challenges in averting the progression of MPN, MDS
ALEXANDRIA, VA. – What are the emerging combination therapies for slowing disease progression and improving therapeutic tolerability in myeloproliferative neoplasms and myelodysplastic syndromes?
Join Dr. Jaroslaw Maciejewski, chairman of the department of translational hematology and oncology research at the Taussig Cancer Institute, Cleveland Clinic, and professor of medicine at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Dr. Ruben A. Mesa, chair of the Division of Hematology/Oncology, department of internal medicine, Mayo Clinic, Scottsdale, Arizona, for their one-on-one discussion of emerging treatment approaches. Then join the conversation by posting your comments and recommendations for other Heme Themes.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ALEXANDRIA, VA. – What are the emerging combination therapies for slowing disease progression and improving therapeutic tolerability in myeloproliferative neoplasms and myelodysplastic syndromes?
Join Dr. Jaroslaw Maciejewski, chairman of the department of translational hematology and oncology research at the Taussig Cancer Institute, Cleveland Clinic, and professor of medicine at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Dr. Ruben A. Mesa, chair of the Division of Hematology/Oncology, department of internal medicine, Mayo Clinic, Scottsdale, Arizona, for their one-on-one discussion of emerging treatment approaches. Then join the conversation by posting your comments and recommendations for other Heme Themes.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
ALEXANDRIA, VA. – What are the emerging combination therapies for slowing disease progression and improving therapeutic tolerability in myeloproliferative neoplasms and myelodysplastic syndromes?
Join Dr. Jaroslaw Maciejewski, chairman of the department of translational hematology and oncology research at the Taussig Cancer Institute, Cleveland Clinic, and professor of medicine at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, and Dr. Ruben A. Mesa, chair of the Division of Hematology/Oncology, department of internal medicine, Mayo Clinic, Scottsdale, Arizona, for their one-on-one discussion of emerging treatment approaches. Then join the conversation by posting your comments and recommendations for other Heme Themes.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM U.S. FOCUS ON MPN & MDS
Imetelstat elicits response in myelofibrosis, thrombocythemia
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The results of both of these studies are compelling and certainly warrant further research, given the limited treatment options for myeloproliferative disorders.
Although imetelstat’s mechanism of action remains to be elucidated, both studies hint at the possibility that the agent may actually change the natural history of these debilitating disorders.
More important, assessing imetelstat’s long-term safety profile is a vital next step for researchers.
Dr. Mary Armanios and Carol W. Greider, Ph.D., are at Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore. Dr. Armanios reported having no relevant disclosures; Dr. Greider reported patents related to an RNA component of telomerase and telomerase-associated proteins. Dr. Armanios and Dr. Greider made these remarks in an editorial accompanying the two reports on imetelstat (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMe1508740).
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
The telomerase inhibitor imetelstat showed promise against advanced myelofibrosis and essential thrombocythemia in two industry-funded preliminary studies, according to separate reports published online Sept. 3 in the New England Journal of Medicine.
In previous in vitro and animal studies, imetelstat inhibited the proliferation of various types of malignant cells but was not active in normal somatic tissue. Researchers assessed the agent for advanced myelofibrosis in part because, at present, only one available treatment – allogeneic stem-cell transplantation (ASCT) – sometimes induces long-term remission. ASCT carries a relatively high rate of treatment-related death and complications, and is contraindicated in many older patients.
In the first report, researchers conducted a small, single-center cohort study to collect preliminary data on the agent’s efficacy and safety in 33 patients with primary myelofibrosis (18 participants), myelofibrosis that was related to polycythemia (10 participants), or myelofibrosis associated with essential thrombocytopenia (10 participants). Imetelstat was administered in 2-hour intravenous infusions given in 3-week cycles, said Dr. Ayalew Tefferi of the division of hematology, Mayo Clinic, Rochester Minn.
The median duration of treatment was 8.6 months (range, 1.4-21.7 months). Seven patients (21%) had either a complete or partial response; the 4 patients with a complete response had documented complete reversal of bone marrow fibrosis. The time to onset of response was 3.5 months (range, 1.4-7.2 months), and the median duration of response was 18 months (range, 13-20 months).
These remissions “confirm selective anticlonal activity, which has not previously been documented in drug treatment of myelofibrosis,” noted Dr. Tefferi and his associates (N Engl J Med 2015 Sep 3. doi:10.1056/NEJMoa1310523). Three of the seven patients who responded to imetelstat “had been heavily dependent on red-cell transfusions at study entry and became transfusion-independent and sustained a hemoglobin level of more than 10 g/dL for a minimum of 3 months during therapy,” they noted.
In addition, 8 of 10 patients who had marked leukocytosis at baseline had either a complete resolution (3 patients) or a reduction of at least 50% in white-cell counts (5 patients). All 11 participants who had thrombocytosis at baseline had either complete resolution (10 patients) or a reduction in platelet count of at least 50% (1 patient). Of the 27 participants who had leukoerythroblastosis at baseline, 22 showed either complete resolution (13 patients) or a reduction of at least 50% in the percentage of immature myeloid cells and nucleated red cells (9 patients). Also, 17 of the 21 participants who had at least 1% circulating blasts at baseline had either a complete disappearance of circulating blasts (14 patients) or a reduction of at least 50% (3 patients).
The most clinically significant adverse effect of imetelstat, myelosuppression, occurred in 22 patients (67%) and often necessitated dose reductions. Low-grade elevations in liver enzymes also were a concern. One patient died from an intracranial hemorrhage that the treating physician attributed to drug-induced grade 4 thrombocytopenia. Other adverse events that may or may not have been treatment related included fever, epistaxis, bruising, hematoma, lung infection, skin infection, and upper-GI hemorrhage.
These findings not only identify imetelstat as a possible treatment for myelofibrosis, they also suggest that other telomerase-targeting strategies may be beneficial in this disease, Dr. Tefferi and his associates added.
In the second report, researchers performing a phase-II study at seven medical centers in the United States, Germany, and Switzerland found that imetelstat produced rapid and durable hematologic and molecular responses in all 18 patients in their study of essential thrombocythemia refractory to other treatments. This result is particularly encouraging because current standard therapies “induce nonspecific reductions in platelet counts but do not typically eliminate or alter the biologic characteristics of the disease,” said Dr. Gabriela M. Baerlocher of the department of hematology and the Stem Cell Molecular Diagnostics Laboratory, University of Bern, Switzerland.
These study participants had either failed to respond to hydroxyurea, anagrelide, and interferon therapy or were forced to discontinue these agents because of adverse effects. After weekly treatment with imetelstat at one of two doses, 100% of the patients achieved a hematologic response, attaining platelet counts of 250,000-300,000 per cc. Sixteen participants (89%) achieved a complete hematologic response. The median time to complete response was 6.1 weeks, Dr. Baerlocher and her associates said (N Engl J Med. 2015 Sep 3. doi:10.1056/NEJMoa1503479).
After a median follow-up of 17 months on a maintenance dose of imetelstat, 10 patients were still receiving treatment. The median duration of response had not been reached as of press time (range, 5-30 months).
The most important adverse events were neutropenia (15 patients) and abnormal results on liver-function tests (14 patients). The treating physicians attributed 18 adverse events of grade 3 or higher to the study drug, including headache, anemia, and one syncopal episode. Other adverse events included fatigue, nausea, diarrhea, infections, and rash.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: The telomerase inhibitor imetelstat showed promise in separate preliminary studies for treatment of myelofibrosis and thrombocythemia.
Major finding: A complete or partial response to imetelstat was seen in 7 of 33 patients with advanced myelofibrosis and 18 of 18 with thrombocythemia.
Data source: An international phase-II open-label study involving 18 patients with essential thrombocythemia and a single-center observational cohort study involving 33 patients with myelofibrosis.
Disclosures: Both studies were funded by Geron.
New HMA shows early promise for MDS/AML

Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy. ![]()
Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy. ![]()
Image by Christoph Bock
Investigators say a novel hypomethylating agent (HMA) is safe and clinically active in patients with myelodysplastic syndromes (MDS) or acute myelogenous leukemia (AML) who have failed standard therapy.
The HMA, guadecitabine (SGI-110), reverses aberrant DNA methylation by inhibiting DNA methyltransferase enzymes.
The investigators tested guadecitabine in a phase 1 study of patients with relapsed or refractory AML or MDS.
They reported the results in The Lancet Oncology. The study was sponsored by Astex Pharmaceuticals, the company developing guadecitabine.
“In this study, we observed induced clinical responses in heavily pretreated patients, including prior treatment with current HMAs,” said study author Hagop Kantarjian, MD, of The University of Texas MD Anderson Cancer Center in Houston.
“Together with the results of a large phase 2 study to be published later, these data support further investigation, including the recently commenced global phase 3 study in treatment-naïve AML patients”.
Dr Kantarjian and his colleagues enrolled 93 patients in the phase 1 study, 74 with AML and 19 with MDS. The patients had received 1 to 9 prior treatment regimens, and most had received prior azacitidine or decitabine.
The trial had a 3+3 dose-escalation design. Patients received guadecitabine doses ranging from 3 mg/m2 to 125 mg/m2.
The patients were also randomized to receive guadecitabine either once-daily for 5 consecutive days (35 AML, 9 MDS) or once-weekly (28 AML, 6 MDS) for 3 weeks in a 28-day treatment cycle. A twice-weekly treatment schedule was added to the study after a protocol amendment (11 AML, 4 MDS).
The investigators said the 3 treatment groups were well balanced with regard to baseline characteristics. However, the initial median bone marrow blast percentage in the daily × 5 group was twice that of the once-weekly and twice-weekly groups—42%, 19%, and 20%, respectively.
Safety and efficacy
The investigators said the treatment was well-tolerated. The most common grade 3 or higher adverse events were febrile neutropenia (41%), pneumonia (29%), thrombocytopenia (25%), anemia (25%), and sepsis (17%).
The most common serious adverse events were febrile neutropenia (31%), pneumonia (28%), and sepsis (17%).
There were 2 dose-limiting toxicities in MDS patients at the 125 mg/m2 daily × 5 dose. So the maximum tolerated dose for these patients was 90 mg/m2 daily × 5. The maximum tolerated dose was not reached in patients with AML.
Six patients with AML and 6 with MDS had a clinical response to guadecitabine. The investigators said potent, dose-related DNA demethylation occurred on the daily × 5 regimen, reaching a plateau at 60 mg/m2. So the team recommended this as the phase 2 dose.
A phase 2 study of guadecitabine is ongoing. The study enrolled more than 300 patients with treatment-naïve or relapsed/refractory AML or MDS.
Investigators recently began an 800-patient, phase 3 study (ASTRAL-1), in which guadecitabine is being compared with physician’s choice of decitabine, azacitidine, or low-dose cytarabine in treatment-naïve AML patients who are not candidates for intensive induction chemotherapy. ![]()
Lenalidomide can treat pulmonary sarcoidosis in MDS

Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide. ![]()
Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide. ![]()
Treatment with lenalidomide can have a significant effect on pulmonary sarcoidosis in myelodysplastic syndrome (MDS), according to a case study.
The case was a 71-year-old woman with newly diagnosed 5q-MDS and a long-standing history of refractory pulmonary sarcoidosis.
After 2 cycles of treatment with lenalidomide, the patient had substantial improvements in lung function, fatigue, daily activity, and quality of life.
This case is the first of its kind to show the potential effects of lenalidomide as a therapeutic option in patients with pulmonary sarcoidosis.
Ali Bazargan, MD, of St. Vincent’s Hospital in Melbourne, Victoria, Australia, and his colleagues described this case in CHEST.
The patient had a 12-year history of stage IV pulmonary sarcoidosis with no extrapulmonary organ involvement. She had never smoked but had a history of hypertension that was managed with perindopril.
The patient presented with refractory and worsening dyspnea, despite receiving long-term therapy with methotrexate and inhaled and systemic corticosteroids. Before she began receiving lenalidomide, the patient was taking 15 mg of prednisolone and 400 mg of inhaled budesonide daily.
Blood tests revealed the patient had macrocytic anemia (hemoglobin level, 81 g/L; mean corpuscular volume, 114 fL).
A subsequent bone marrow biopsy revealed hypocellular marrow with trilineage dysplasia consistent with 5q-MDS but no evidence of noncaseating granulomas. So the patient began receiving lenalidomide at 10 mg daily.
While the researchers were trying to establish her diagnosis of 5q-MDS, the patient became transfusion-dependent and experienced severe dyspnea, fatigue, and a considerable decline in quality of life.
A chest CT scan revealed irregular masses in her lung, with bibasal alveolar infiltrates that had developed within a 12-month period.
However, after 2 cycles of lenalidomide, the patient had significant improvements in dyspnea, fatigue, daily activity, and quality of life. Lung function testing showed an increase in vital capacity from 1.73 L to 1.93 L.
And a chest CT scan performed 4 months after the patient began taking lenalidomide showed that the bibasal alveolar infiltrates had completely cleared.
During this period, the patient’s dose of prednisolone was reduced from 15 mg daily to 5 mg on alternate days, but she continues to receive the same dose of lenalidomide. ![]()