User login
Therapy granted PIM designation for CTCL

mycosis fungoides
The UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) has granted SGX301 (synthetic hypericin) Promising Innovative Medicine (PIM) designation for the treatment of cutaneous T-cell lymphoma (CTCL).
The PIM designation is the first step toward inclusion in the Early Access to Medicines Scheme (EAMS).
EAMS provides early access to new medicines for patients with life-threatening and seriously debilitating conditions.
PIM status is awarded following an assessment of early nonclinical and clinical data by the MHRA.
PIM designation has been created as an early signal to companies that a product’s development plan is appropriate and indicates that a product could be a candidate for the second phase of the EAMS scheme once further development work has been conducted.
In the second phase, the product is made available to UK patients before a marketing authorization is approved.
The requirements for PIM designation are:
- The condition should be life-threatening or seriously debilitating with a high unmet medical need (ie, there is no method of treatment, diagnosis, or prevention available, or existing methods have serious limitations).
- The medicinal product is likely to offer a major advantage over methods currently used in the UK.
- The potential adverse effects of the medicinal product are likely to be outweighed by the benefits, allowing for the reasonable expectation of a positive benefit-risk balance. A positive benefit-risk balance should be based on preliminary scientific evidence, as justified by the applicant.
About SGX301
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is currently recruiting patients. SGX301 is under development by Soligenix, Inc. ![]()
mycosis fungoides
The UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) has granted SGX301 (synthetic hypericin) Promising Innovative Medicine (PIM) designation for the treatment of cutaneous T-cell lymphoma (CTCL).
The PIM designation is the first step toward inclusion in the Early Access to Medicines Scheme (EAMS).
EAMS provides early access to new medicines for patients with life-threatening and seriously debilitating conditions.
PIM status is awarded following an assessment of early nonclinical and clinical data by the MHRA.
PIM designation has been created as an early signal to companies that a product’s development plan is appropriate and indicates that a product could be a candidate for the second phase of the EAMS scheme once further development work has been conducted.
In the second phase, the product is made available to UK patients before a marketing authorization is approved.
The requirements for PIM designation are:
- The condition should be life-threatening or seriously debilitating with a high unmet medical need (ie, there is no method of treatment, diagnosis, or prevention available, or existing methods have serious limitations).
- The medicinal product is likely to offer a major advantage over methods currently used in the UK.
- The potential adverse effects of the medicinal product are likely to be outweighed by the benefits, allowing for the reasonable expectation of a positive benefit-risk balance. A positive benefit-risk balance should be based on preliminary scientific evidence, as justified by the applicant.
About SGX301
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is currently recruiting patients. SGX301 is under development by Soligenix, Inc. ![]()
mycosis fungoides
The UK’s Medicines and Healthcare Products Regulatory Agency (MHRA) has granted SGX301 (synthetic hypericin) Promising Innovative Medicine (PIM) designation for the treatment of cutaneous T-cell lymphoma (CTCL).
The PIM designation is the first step toward inclusion in the Early Access to Medicines Scheme (EAMS).
EAMS provides early access to new medicines for patients with life-threatening and seriously debilitating conditions.
PIM status is awarded following an assessment of early nonclinical and clinical data by the MHRA.
PIM designation has been created as an early signal to companies that a product’s development plan is appropriate and indicates that a product could be a candidate for the second phase of the EAMS scheme once further development work has been conducted.
In the second phase, the product is made available to UK patients before a marketing authorization is approved.
The requirements for PIM designation are:
- The condition should be life-threatening or seriously debilitating with a high unmet medical need (ie, there is no method of treatment, diagnosis, or prevention available, or existing methods have serious limitations).
- The medicinal product is likely to offer a major advantage over methods currently used in the UK.
- The potential adverse effects of the medicinal product are likely to be outweighed by the benefits, allowing for the reasonable expectation of a positive benefit-risk balance. A positive benefit-risk balance should be based on preliminary scientific evidence, as justified by the applicant.
About SGX301
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is currently recruiting patients. SGX301 is under development by Soligenix, Inc. ![]()
Cheap manufacture of generic cancer drugs is feasible, study shows

Photo courtesy of FDA
AMSTERDAM—New research suggests some generic cancer drugs could be manufactured for less than 1% of the prices currently charged in the US and UK.
For example, researchers calculated that manufacturing a 400 mg tablet of imatinib costs $0.92.
Charging $1.04 per tablet would cover costs and allow for a 10% profit margin.
However, the current price of imatinib is $84.36 per tablet in the UK and $247.74 per tablet in the US.
Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, reported these findings at ECCO 2017: European Cancer Congress (abstract 1032).
Barber and her colleagues collected data on per-kilogram costs of exported active pharmaceutical ingredients (APIs) from an online database of Indian export logs.
The team then estimated generic prices for tablets through an established costing algorithm. They calculated per-dose API costs and added excipient costs of $2.63 per kg of finished pharmaceutical product and per-tablet costs of production of $0.01, plus a 10% profit margin accounting for a 26.6% average tax on profits (assuming manufacture in India.)
Finally, the researchers compared the calculated price to current unit prices in the US, UK, Spain, and India.
For imatinib, the team determined the cost of the API to be $2284 per kg and the API cost per tablet to be $0.91. They then added excipient cost ($0.002 per tablet), conversion cost ($0.01 per tablet), and a 10% profit margin accounting for a 26.6% tax on profits.
This resulted in the estimated generic price of $1.04 per tablet. The per-tablet price is below the estimated price in India ($0.22) but much higher than the estimated price in Spain ($57.53), the UK ($84.36), and the US ($247.74).
Barber noted that, according to her group’s calculations, imatinib could be produced for $54 a month.
Another drug that could be produced for a low cost is etoposide. Barber and her colleagues calculated a generic price for etoposide of $0.97 per 100 mg tablet.
However, the per-tablet price is $1.50 in India, $8.65 in Spain, $11.34 in the UK, and $87.14 in the US.
The researchers calculated a generic price for mercaptopurine of $0.03 per 50 mg tablet, which is the same as the per-tablet price in India. However, a 50 mg mercaptopurine tablet costs $3.14 in Spain, $2.56 in the UK, and $0.40 in the US.
“Showing that certain cancers could be treated for very low prices could transform the future of people with these cancers in very low-income countries where there are usually few or no treatment options,” Barber said. ![]()
Photo courtesy of FDA
AMSTERDAM—New research suggests some generic cancer drugs could be manufactured for less than 1% of the prices currently charged in the US and UK.
For example, researchers calculated that manufacturing a 400 mg tablet of imatinib costs $0.92.
Charging $1.04 per tablet would cover costs and allow for a 10% profit margin.
However, the current price of imatinib is $84.36 per tablet in the UK and $247.74 per tablet in the US.
Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, reported these findings at ECCO 2017: European Cancer Congress (abstract 1032).
Barber and her colleagues collected data on per-kilogram costs of exported active pharmaceutical ingredients (APIs) from an online database of Indian export logs.
The team then estimated generic prices for tablets through an established costing algorithm. They calculated per-dose API costs and added excipient costs of $2.63 per kg of finished pharmaceutical product and per-tablet costs of production of $0.01, plus a 10% profit margin accounting for a 26.6% average tax on profits (assuming manufacture in India.)
Finally, the researchers compared the calculated price to current unit prices in the US, UK, Spain, and India.
For imatinib, the team determined the cost of the API to be $2284 per kg and the API cost per tablet to be $0.91. They then added excipient cost ($0.002 per tablet), conversion cost ($0.01 per tablet), and a 10% profit margin accounting for a 26.6% tax on profits.
This resulted in the estimated generic price of $1.04 per tablet. The per-tablet price is below the estimated price in India ($0.22) but much higher than the estimated price in Spain ($57.53), the UK ($84.36), and the US ($247.74).
Barber noted that, according to her group’s calculations, imatinib could be produced for $54 a month.
Another drug that could be produced for a low cost is etoposide. Barber and her colleagues calculated a generic price for etoposide of $0.97 per 100 mg tablet.
However, the per-tablet price is $1.50 in India, $8.65 in Spain, $11.34 in the UK, and $87.14 in the US.
The researchers calculated a generic price for mercaptopurine of $0.03 per 50 mg tablet, which is the same as the per-tablet price in India. However, a 50 mg mercaptopurine tablet costs $3.14 in Spain, $2.56 in the UK, and $0.40 in the US.
“Showing that certain cancers could be treated for very low prices could transform the future of people with these cancers in very low-income countries where there are usually few or no treatment options,” Barber said. ![]()
Photo courtesy of FDA
AMSTERDAM—New research suggests some generic cancer drugs could be manufactured for less than 1% of the prices currently charged in the US and UK.
For example, researchers calculated that manufacturing a 400 mg tablet of imatinib costs $0.92.
Charging $1.04 per tablet would cover costs and allow for a 10% profit margin.
However, the current price of imatinib is $84.36 per tablet in the UK and $247.74 per tablet in the US.
Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, reported these findings at ECCO 2017: European Cancer Congress (abstract 1032).
Barber and her colleagues collected data on per-kilogram costs of exported active pharmaceutical ingredients (APIs) from an online database of Indian export logs.
The team then estimated generic prices for tablets through an established costing algorithm. They calculated per-dose API costs and added excipient costs of $2.63 per kg of finished pharmaceutical product and per-tablet costs of production of $0.01, plus a 10% profit margin accounting for a 26.6% average tax on profits (assuming manufacture in India.)
Finally, the researchers compared the calculated price to current unit prices in the US, UK, Spain, and India.
For imatinib, the team determined the cost of the API to be $2284 per kg and the API cost per tablet to be $0.91. They then added excipient cost ($0.002 per tablet), conversion cost ($0.01 per tablet), and a 10% profit margin accounting for a 26.6% tax on profits.
This resulted in the estimated generic price of $1.04 per tablet. The per-tablet price is below the estimated price in India ($0.22) but much higher than the estimated price in Spain ($57.53), the UK ($84.36), and the US ($247.74).
Barber noted that, according to her group’s calculations, imatinib could be produced for $54 a month.
Another drug that could be produced for a low cost is etoposide. Barber and her colleagues calculated a generic price for etoposide of $0.97 per 100 mg tablet.
However, the per-tablet price is $1.50 in India, $8.65 in Spain, $11.34 in the UK, and $87.14 in the US.
The researchers calculated a generic price for mercaptopurine of $0.03 per 50 mg tablet, which is the same as the per-tablet price in India. However, a 50 mg mercaptopurine tablet costs $3.14 in Spain, $2.56 in the UK, and $0.40 in the US.
“Showing that certain cancers could be treated for very low prices could transform the future of people with these cancers in very low-income countries where there are usually few or no treatment options,” Barber said. ![]()
Company withdraws MAA for pegfilgrastim biosimilar
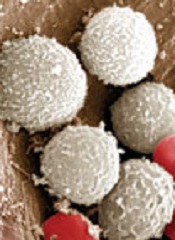
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has announced that Sandoz GmbH withdrew its marketing authorization application (MAA) for Zioxtenzo.
The active ingredient of Zioxtenzo is pegfilgrastim, and the product was intended to be biosimilar to Amgen’s Neulasta.
The intended use for Zioxtenzo was to reduce the duration of neutropenia and the occurrence of febrile neutropenia in cancer patients.
In its application for Zioxtenzo, Sandoz presented results of studies designed to show the product is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
In addition, there were 2 studies comparing the safety and effectiveness of Zioxtenzo and Neulasta in patients receiving cancer drugs.
Sandoz withdrew the MAA for Zioxtenzo after the CHMP had evaluated the initial documentation provided by the company and formulated a list of questions. The company had not responded to the questions at the time of the withdrawal.
Based on a review of the data, at the time of the withdrawal, the CHMP had 2 main concerns and was of the provisional opinion that Zioxtenzo could not have been approved as a biosimilar of Neulasta.
One concern was that study results were not able to show that the concentrations of pegfilgrastim in blood were the same after taking Zioxtenzo and Neulasta.
The other concern was the lack of a certificate of Good Manufacturing Practice for Zioxtenzo’s manufacturing site. An inspection of the site would therefore be needed before the drug could be approved.
At the time of the MAA withdrawal, Sandoz had not demonstrated that Zioxtenzo is highly similar to Neulasta, and an inspection to confirm that Zioxtenzo was being manufactured according to Good Manufacturing Practice standards had not yet taken place.
In its letter notifying the CHMP of the MAA withdrawal, Sandoz said it would not be able to provide the additional data required by the CHMP within the timeframe allowed for the procedure.
The company also said the withdrawal of Zioxtenzo will not impact ongoing clinical trials, and there are no compassionate use programs for Zioxtenzo. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has announced that Sandoz GmbH withdrew its marketing authorization application (MAA) for Zioxtenzo.
The active ingredient of Zioxtenzo is pegfilgrastim, and the product was intended to be biosimilar to Amgen’s Neulasta.
The intended use for Zioxtenzo was to reduce the duration of neutropenia and the occurrence of febrile neutropenia in cancer patients.
In its application for Zioxtenzo, Sandoz presented results of studies designed to show the product is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
In addition, there were 2 studies comparing the safety and effectiveness of Zioxtenzo and Neulasta in patients receiving cancer drugs.
Sandoz withdrew the MAA for Zioxtenzo after the CHMP had evaluated the initial documentation provided by the company and formulated a list of questions. The company had not responded to the questions at the time of the withdrawal.
Based on a review of the data, at the time of the withdrawal, the CHMP had 2 main concerns and was of the provisional opinion that Zioxtenzo could not have been approved as a biosimilar of Neulasta.
One concern was that study results were not able to show that the concentrations of pegfilgrastim in blood were the same after taking Zioxtenzo and Neulasta.
The other concern was the lack of a certificate of Good Manufacturing Practice for Zioxtenzo’s manufacturing site. An inspection of the site would therefore be needed before the drug could be approved.
At the time of the MAA withdrawal, Sandoz had not demonstrated that Zioxtenzo is highly similar to Neulasta, and an inspection to confirm that Zioxtenzo was being manufactured according to Good Manufacturing Practice standards had not yet taken place.
In its letter notifying the CHMP of the MAA withdrawal, Sandoz said it would not be able to provide the additional data required by the CHMP within the timeframe allowed for the procedure.
The company also said the withdrawal of Zioxtenzo will not impact ongoing clinical trials, and there are no compassionate use programs for Zioxtenzo. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has announced that Sandoz GmbH withdrew its marketing authorization application (MAA) for Zioxtenzo.
The active ingredient of Zioxtenzo is pegfilgrastim, and the product was intended to be biosimilar to Amgen’s Neulasta.
The intended use for Zioxtenzo was to reduce the duration of neutropenia and the occurrence of febrile neutropenia in cancer patients.
In its application for Zioxtenzo, Sandoz presented results of studies designed to show the product is highly similar to Neulasta in terms of chemical structure, purity, the way it works, and how the body handles the drug.
In addition, there were 2 studies comparing the safety and effectiveness of Zioxtenzo and Neulasta in patients receiving cancer drugs.
Sandoz withdrew the MAA for Zioxtenzo after the CHMP had evaluated the initial documentation provided by the company and formulated a list of questions. The company had not responded to the questions at the time of the withdrawal.
Based on a review of the data, at the time of the withdrawal, the CHMP had 2 main concerns and was of the provisional opinion that Zioxtenzo could not have been approved as a biosimilar of Neulasta.
One concern was that study results were not able to show that the concentrations of pegfilgrastim in blood were the same after taking Zioxtenzo and Neulasta.
The other concern was the lack of a certificate of Good Manufacturing Practice for Zioxtenzo’s manufacturing site. An inspection of the site would therefore be needed before the drug could be approved.
At the time of the MAA withdrawal, Sandoz had not demonstrated that Zioxtenzo is highly similar to Neulasta, and an inspection to confirm that Zioxtenzo was being manufactured according to Good Manufacturing Practice standards had not yet taken place.
In its letter notifying the CHMP of the MAA withdrawal, Sandoz said it would not be able to provide the additional data required by the CHMP within the timeframe allowed for the procedure.
The company also said the withdrawal of Zioxtenzo will not impact ongoing clinical trials, and there are no compassionate use programs for Zioxtenzo. ![]()
Recent price hikes for generic cancer meds exceed 100%

Photo by Steven Harbour
AMSTERDAM—The UK has seen substantial price increases for some generic cancer drugs over the last few years, according to a study presented at ECCO 2017: European Cancer Congress (abstract 966).
Of the 89 drugs analyzed in this study, 21 of them—including 17 generics—had price increases from 2011 to 2016.
Fourteen of the generic cancer drugs had price increases over 100%, and 2 of the drugs had increases exceeding 1000%.
“We were surprised to find several companies consistently raising the prices of cancer treatment,” said study investigator Andrew Hill, PhD, of the University of Liverpool in the UK.
“Twenty treatments have shown rises of over 100% in the last 5 years, and in 2—busulfan (used to treat leukemia) and tamoxifen (breast cancer)—prices have increased by over 1000%. We have found that some companies take over the supply of some generic cancer medicines and then raise the price progressively.”
Dr Hill and his co-investigator Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, analyzed prices for 190 formulations of 89 cancer drugs.
Twenty-eight formulations of 21 drugs had price increases from 2011 to 2016. Seventeen of these 21 drugs were generic in 2016.
Twenty formulations of 14 generic cancer drugs had price increases exceeding 100%.
For example, the cost per tablet or injection increased for:
- Ifosfamide (2 g vial)—from £89 to £180, or 103%.
- Melphalan (50 mg vial)—from £33 to £137, or 315%.
- Chlorambucil (2 mg)—from £0.33 to £1.62, or 390%.
- Cyclophosphamide (50 mg)—from £0.20 to £1.39, or 695%.
- Busulfan (2 mg)—from £0.21 to £2.61, or 1227%.
Dr Hill said the UK’s Department of Health is aware of this issue and has introduced the Health Services Medical Supplies (Costs) Bill to enable price regulation in the future.
Companies found to be raising prices with no clear justification will be referred to the Competition and Markets Authority, and they could face fines.
However, Dr Hill and Barber said they found large price increases for generic cancer drugs in other European countries as well.
In Spain and Italy, failure to accept the high prices demanded for some generic drugs has led to warnings from companies that they could stop the supply of these drugs.
For instance, Italy fined the generic company Aspen €5 million after a 1500% increase in the price of cancer drugs, including melphalan and chlorambucil. Aspen then threatened Italy with drug shortages unless higher prices were accepted.
In Spain, Aspen demanded a 4000% increase in melphalan prices.
“We hope that, by explaining what we have found in the UK, other European countries will take note and protect themselves against these kinds of price rises,” Dr Hill said. “At a time when cancer patients are living longer and better lives due to effective treatments, this situation is particularly worrying.” ![]()
Photo by Steven Harbour
AMSTERDAM—The UK has seen substantial price increases for some generic cancer drugs over the last few years, according to a study presented at ECCO 2017: European Cancer Congress (abstract 966).
Of the 89 drugs analyzed in this study, 21 of them—including 17 generics—had price increases from 2011 to 2016.
Fourteen of the generic cancer drugs had price increases over 100%, and 2 of the drugs had increases exceeding 1000%.
“We were surprised to find several companies consistently raising the prices of cancer treatment,” said study investigator Andrew Hill, PhD, of the University of Liverpool in the UK.
“Twenty treatments have shown rises of over 100% in the last 5 years, and in 2—busulfan (used to treat leukemia) and tamoxifen (breast cancer)—prices have increased by over 1000%. We have found that some companies take over the supply of some generic cancer medicines and then raise the price progressively.”
Dr Hill and his co-investigator Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, analyzed prices for 190 formulations of 89 cancer drugs.
Twenty-eight formulations of 21 drugs had price increases from 2011 to 2016. Seventeen of these 21 drugs were generic in 2016.
Twenty formulations of 14 generic cancer drugs had price increases exceeding 100%.
For example, the cost per tablet or injection increased for:
- Ifosfamide (2 g vial)—from £89 to £180, or 103%.
- Melphalan (50 mg vial)—from £33 to £137, or 315%.
- Chlorambucil (2 mg)—from £0.33 to £1.62, or 390%.
- Cyclophosphamide (50 mg)—from £0.20 to £1.39, or 695%.
- Busulfan (2 mg)—from £0.21 to £2.61, or 1227%.
Dr Hill said the UK’s Department of Health is aware of this issue and has introduced the Health Services Medical Supplies (Costs) Bill to enable price regulation in the future.
Companies found to be raising prices with no clear justification will be referred to the Competition and Markets Authority, and they could face fines.
However, Dr Hill and Barber said they found large price increases for generic cancer drugs in other European countries as well.
In Spain and Italy, failure to accept the high prices demanded for some generic drugs has led to warnings from companies that they could stop the supply of these drugs.
For instance, Italy fined the generic company Aspen €5 million after a 1500% increase in the price of cancer drugs, including melphalan and chlorambucil. Aspen then threatened Italy with drug shortages unless higher prices were accepted.
In Spain, Aspen demanded a 4000% increase in melphalan prices.
“We hope that, by explaining what we have found in the UK, other European countries will take note and protect themselves against these kinds of price rises,” Dr Hill said. “At a time when cancer patients are living longer and better lives due to effective treatments, this situation is particularly worrying.” ![]()
Photo by Steven Harbour
AMSTERDAM—The UK has seen substantial price increases for some generic cancer drugs over the last few years, according to a study presented at ECCO 2017: European Cancer Congress (abstract 966).
Of the 89 drugs analyzed in this study, 21 of them—including 17 generics—had price increases from 2011 to 2016.
Fourteen of the generic cancer drugs had price increases over 100%, and 2 of the drugs had increases exceeding 1000%.
“We were surprised to find several companies consistently raising the prices of cancer treatment,” said study investigator Andrew Hill, PhD, of the University of Liverpool in the UK.
“Twenty treatments have shown rises of over 100% in the last 5 years, and in 2—busulfan (used to treat leukemia) and tamoxifen (breast cancer)—prices have increased by over 1000%. We have found that some companies take over the supply of some generic cancer medicines and then raise the price progressively.”
Dr Hill and his co-investigator Melissa Barber, of the London School of Hygiene and Tropical Medicine in the UK, analyzed prices for 190 formulations of 89 cancer drugs.
Twenty-eight formulations of 21 drugs had price increases from 2011 to 2016. Seventeen of these 21 drugs were generic in 2016.
Twenty formulations of 14 generic cancer drugs had price increases exceeding 100%.
For example, the cost per tablet or injection increased for:
- Ifosfamide (2 g vial)—from £89 to £180, or 103%.
- Melphalan (50 mg vial)—from £33 to £137, or 315%.
- Chlorambucil (2 mg)—from £0.33 to £1.62, or 390%.
- Cyclophosphamide (50 mg)—from £0.20 to £1.39, or 695%.
- Busulfan (2 mg)—from £0.21 to £2.61, or 1227%.
Dr Hill said the UK’s Department of Health is aware of this issue and has introduced the Health Services Medical Supplies (Costs) Bill to enable price regulation in the future.
Companies found to be raising prices with no clear justification will be referred to the Competition and Markets Authority, and they could face fines.
However, Dr Hill and Barber said they found large price increases for generic cancer drugs in other European countries as well.
In Spain and Italy, failure to accept the high prices demanded for some generic drugs has led to warnings from companies that they could stop the supply of these drugs.
For instance, Italy fined the generic company Aspen €5 million after a 1500% increase in the price of cancer drugs, including melphalan and chlorambucil. Aspen then threatened Italy with drug shortages unless higher prices were accepted.
In Spain, Aspen demanded a 4000% increase in melphalan prices.
“We hope that, by explaining what we have found in the UK, other European countries will take note and protect themselves against these kinds of price rises,” Dr Hill said. “At a time when cancer patients are living longer and better lives due to effective treatments, this situation is particularly worrying.” ![]()
CHMP recommends hybrid drug for ALL, other disorders

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended marketing authorization for an oral formulation of methotrexate (Jylamvo) as a treatment for acute lymphoblastic leukemia (ALL) and other disorders.
Jylamvo is a hybrid medicine of Methotrexat “Lederle” 25 mg-Stechampulle and Methotrexate “Lederle” 2.5 mg tablets, which have been authorized in the European Union since 1984 and 1959, respectively.
Jylamvo contains the same active substance as these reference medicines—the antineoplastic and immunomodulating agent methotrexate—but is given by mouth as a solution (2 mg/mL).
Jylamvo is intended for use as maintenance treatment in ALL patients age 3 and older.
The drug is also intended to treat:
- Active rheumatoid arthritis in adults
- Polyarthritic forms of active, severe juvenile idiopathic arthritis in adolescents and children age 3 and older when the response to non-steroidal anti-inflammatory drugs has been inadequate
- Severe, treatment-refractory, disabling psoriasis that does not respond sufficiently to other forms of treatment (such as phototherapy, retinoids, and psoralen and ultraviolet A radiation therapy) and severe psoriatic arthritis in adults.
The applicant for Jylamvo is Therakind Limited. Applications for hybrid medicines rely, in part, on the results of preclinical tests and clinical trials for a reference product and, in part, on new data.
The CHMP said studies have demonstrated the satisfactory quality of Jylamvo and its bioequivalence to Methotrexate “Lederle” 2.5 mg tablets and a third product, Ebetrexat 10 mg tablets, which is authorized for similar indications.
The CHMP has proposed that Jylamvo be prescribed by physicians with experience of the various properties of the medicinal product and its mode of action.
Detailed recommendations for the use of Jylamvo will be described in the summary of product characteristics, which will be published in the European public assessment report and made available in all official European Union languages if the European Commission grants marketing authorization for Jylamvo.
The European Commission typically makes a decision on a product within 67 days of the time the CHMP adopts its opinion. ![]()
Combo granted orphan designation for DLBCL

The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the combination of TG-1101 (ublituximab), an anti-CD20 monoclonal antibody, and TGR-1202, a PI3K delta inhibitor, in the treatment of diffuse large B-cell lymphoma (DLBCL).
The combination is currently being evaluated in patients with relapsed or refractory DLBCL in the phase 2b UNITY-DLBCL trial.
Ublituximab and TGR-1202 are both products of TG Therapeutics, Inc.
Updated results from a phase 1 study of ublituximab and TGR-1202 in patients with DLBCL and other malignancies were presented at the 21st Congress of the European Hematology Association.
The data included 165 patients treated with varying doses of TGR-1202 alone (n=90) or in combination with ublituximab (n=75). The patients were heavily pretreated, with the majority having 3 or more prior lines of therapy.
There were 7 evaluable patients with DLBCL who received the combination at the phase 3 doses— ublituximab at 900 mg and TGR-1202 at 800 mg micronized.
The overall response rate for this group was 57%. Of the 4 responders, 1 patient had a complete response, and 3 had a partial response. Two patients had stable disease, and 1 progressed.
In the overall study population, the most common adverse events were diarrhea (47%), nausea (45%), fatigue (37%), vomiting (27%), and neutropenia (21%). The most common grade 3/4 adverse events were neutropenia (18%) and anemia (5%).
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to
treat, diagnose, or prevent rare diseases/disorders affecting fewer than
200,000 people in the US.
Orphan designation provides companies
with certain incentives to develop products for rare diseases. This
includes a 50% tax break on research and development, a fee waiver,
access to federal grants, and 7 years of market exclusivity if the
product is approved. ![]()
Health Canada expands indication for lenalidomide

Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.” ![]()
Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.” ![]()
Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.”
CMA report reveals successes and shortcomings

The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program.
The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program.
The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program.
FDA approves ibrutinib to treat rel/ref MZL

Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities.
Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities.
Photo courtesy of
Janssen Biotech, Inc.
The US Food and Drug Administration (FDA) has approved the Bruton’s tyrosine kinase inhibitor ibrutinib (Imbruvica®) for the treatment of marginal zone lymphoma (MZL).
The drug is now approved to treat patients with relapsed/refractory MZL who require systemic therapy and have received at least 1 prior anti-CD20-based therapy.
Ibrutinib has accelerated approval for this indication, based on the overall response rate the drug produced in a phase 2 trial.
Continued approval of ibrutinib as a treatment for MZL may be contingent upon verification and description of clinical benefit in a confirmatory trial.
The FDA’s approval of ibrutinib for MZL makes it the first treatment approved specifically for patients with this disease. It also marks the seventh FDA approval and fifth disease indication for ibrutinib since the drug was first approved in 2013.
Ibrutinib is also FDA-approved to treat chronic lymphocytic leukemia/small lymphocytic lymphoma, patients with mantle cell lymphoma who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia. The approval for mantle cell lymphoma is an accelerated approval.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
Phase 2 trial
The FDA’s approval of ibrutinib for MZL is based on data from the phase 2, single-arm PCYC-1121 study, in which researchers evaluated the drug in MZL patients who required systemic therapy and had received at least 1 prior anti-CD20-based therapy.
Results from this study were presented at the 2016 ASH Annual Meeting (abstract 1213).
The efficacy analysis included 63 patients with 3 subtypes of MZL: mucosa-associated lymphoid tissue (n=32), nodal (n=17), and splenic (n=14).
The overall response rate was 46%, with a partial response rate of 42.9% and a complete response rate of 3.2%. Responses were observed across all 3 MZL subtypes.
The median time to response was 4.5 months (range, 2.3-16.4 months). And the median duration of response was not reached (range, 16.7 months to not reached).
Overall, the safety data from this study was consistent with the known safety profile of ibrutinib in B-cell malignancies.
The most common adverse events of all grades (occurring in >20% of patients) were thrombocytopenia (49%), fatigue (44%), anemia (43%), diarrhea (43%), bruising (41%), musculoskeletal pain (40%), hemorrhage (30%), rash (29%), nausea (25%), peripheral edema (24%), arthralgia (24%), neutropenia (22%), cough (22%), dyspnea (21%), and upper respiratory tract infection (21%).
The most common (>10%) grade 3 or 4 events were decreases in hemoglobin and neutrophils (13% each) and pneumonia (10%).
The risks associated with ibrutinib as listed in the Warnings and Precautions section of the prescribing information are hemorrhage, infections, cytopenias, atrial fibrillation, hypertension, secondary primary malignancies, tumor lysis syndrome, and embryo fetal toxicities.
Pharma is gaming the system for orphan drugs, investigation suggests
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.
Photo courtesy of the FDA
An investigation by Kaiser Health News (KHN) suggests some pharmaceutical companies are using the Orphan Drug Act to create monopolies and charge high prices for drugs that are already approved for mass market use in the US.
The US Food and Drug Administration (FDA) grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent conditions that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This includes a 50% tax break on research and development, a fee waiver, access to federal grants, and 7 years of market exclusivity if the product is approved.
However, the KHN investigation showed that some companies have been applying for—and obtaining—orphan designation for drugs already used to treat large populations.
The report states that more than 70 drugs that currently have orphan status were first approved by the FDA for mass market use.
In fact, 7 of the 10 best-selling drugs of 2015 were also orphan drugs. Included on this list are Rituxan (rituximab), Neulasta (pegfilgrastim), and Revlimid (lenalidomide).
The report also states that more than 80 drugs with orphan designation have been approved to treat more than one rare disease. For example, Gleevec (imatinib) has 9 orphan designations.
The KHN investigation revealed that, overall, about a third of orphan designations granted since the Orphan Drug Act was passed in 1983 have been either for repurposed mass market drugs or drugs that received multiple orphan designations. (Roughly 450 orphan drugs have been brought to market since 1983, according to the report.)
For each orphan designation, a drug’s developer qualifies for “a fresh batch of incentives,” the report notes.
The exclusivity incentive means the FDA won’t approve another version of an orphan drug to treat the rare disease(s) in question for 7 years, even if the company’s patent on the brand-name drug has expired.
For example, generic versions of imatinib are being used to treat chronic myeloid leukemia in the US because the patent for Gleevec has expired. However, because of an orphan designation, Novartis still has exclusivity for Gleevec (and will until 2020) as a treatment for patients with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia who are also on chemotherapy.
The KHN report notes that exclusivity can be “a potent pricing tool” due to a lack of competition. And this means orphan drugs may “come with astronomical price tags.”
For instance, there are 33 orphan drugs that cost at least $28,000 for a 30-day supply and 4 orphan drugs that cost more than $70,000 per month.
According to the KHN report, the FDA is planning to investigate this issue.
Gayatri Rao, MD, director of the FDA’s Office of Orphan Products Development, has asked for a review of all orphan designations granted in 2010 and 2015. She said the review will not extend further because the FDA does not have the resources to review all orphan drugs.