User login
Drug granted breakthrough, orphan designation for cGVHD

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
CHMP rejects ofatumumab as maintenance

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
Obinutuzumab approved to treat FL

The European Commission (EC) has approved the use of obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, in patients with follicular lymphoma (FL).
The approval means obinutuzumab can be given, first in combination with bendamustine and then alone as maintenance therapy, to FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Obinutuzumab was previously granted approval by the EC for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche. The drug is marketed as Gazyvaro in the European Union and Switzerland but as Gazyva in the rest of the world.
GADOLIN trial
The EC’s approval of obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by the IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% (18.7% CR, 56% PR) for those receiving bendamustine alone, as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Commission (EC) has approved the use of obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, in patients with follicular lymphoma (FL).
The approval means obinutuzumab can be given, first in combination with bendamustine and then alone as maintenance therapy, to FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Obinutuzumab was previously granted approval by the EC for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche. The drug is marketed as Gazyvaro in the European Union and Switzerland but as Gazyva in the rest of the world.
GADOLIN trial
The EC’s approval of obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by the IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% (18.7% CR, 56% PR) for those receiving bendamustine alone, as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Commission (EC) has approved the use of obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, in patients with follicular lymphoma (FL).
The approval means obinutuzumab can be given, first in combination with bendamustine and then alone as maintenance therapy, to FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Obinutuzumab was previously granted approval by the EC for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche. The drug is marketed as Gazyvaro in the European Union and Switzerland but as Gazyva in the rest of the world.
GADOLIN trial
The EC’s approval of obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by the IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% (18.7% CR, 56% PR) for those receiving bendamustine alone, as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
CHMP advises against approving MM drug

multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors. ![]()
multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors. ![]()
multiple myeloma
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has advised the European Commission not to approve ixazomib (Ninlaro), an oral proteasome inhibitor, as a treatment for patients with relapsed and/or refractory multiple myeloma (MM).
Takeda Pharmaceutical Company Limited, the company developing ixazomib, said it intends to appeal this opinion and request a re-examination by the CHMP.
“We are disappointed by the CHMP’s opinion,” said Christophe Bianchi, MD, president of Takeda Oncology. “With the support of European key medical experts, we will continue our efforts working closely with the CHMP to make Ninlaro—the first oral proteasome inhibitor—available for patients in Europe.”
“We stand behind the TOURMALINE-MM1 trial data, which were recently published in the New England Journal of Medicine and demonstrated a significant extension in progression-free survival for Ninlaro plus lenalidomide and dexamethasone versus placebo plus lenalidomide and dexamethasone and a favorable benefit-risk profile.”
TOURMALINE-MM1
The trial enrolled 722 patients with relapsed (77%), refractory (11%), relapsed and refractory (12%), or primary refractory (6%) MM.
The patients were randomized to receive ixazomib, lenalidomide, and dexamethasone (IRd, n=360) or placebo, lenalidomide, and dexamethasone (Rd, n=362). Baseline patient characteristics were similar between the treatment arms.
The study’s primary endpoint was progression-free survival, which was significantly longer in the IRd arm than the Rd arm. The median progression-free survival was 20.6 months and 14.7 months, respectively. The hazard ratio was 0.74 (P=0.01).
At a median follow-up of about 23 months, the median overall survival had not been reached in either treatment arm.
Adverse events (AEs) occurred in 98% of patients in the IRd arm and 99% in the Rd arm. Grade 3 or higher AEs occurred in 74% and 69% of patients, respectively; serious AEs occurred in 47% and 49%, respectively; and on-study deaths occurred in 4% and 6%, respectively.
Grade 3 and 4 thrombocytopenia, rash, and gastrointestinal AEs were more frequent in the IRd arm than the Rd arm.
The incidence of peripheral neuropathy was similar in the 2 arms, as was the percentage patients who developed new primary malignant tumors. ![]()
CHMP recommends extending brentuximab approval

Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
Photo from Business Wire
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended extending the current conditional approval of brentuximab vedotin (Adcetris) to include the treatment of adults with CD30+ Hodgkin lymphoma (HL) at increased risk of relapse or progression following autologous stem cell transplant (ASCT).
The CHMP’s recommendation will now be reviewed by the European Commission (EC).
If the recommendation is formally adopted by the EC, brentuximab vedotin will be approved for the aforementioned indication in the 28 member states of the European Union as well as Norway, Liechtenstein, and Iceland.
Brentuximab vedotin already has conditional marketing authorization from the EC for 2 indications:
- To treat adults with relapsed or refractory CD30+ HL after ASCT or following at least 2 prior therapies when ASCT or multi-agent chemotherapy is not a treatment option
- To treat adults with relapsed or refractory systemic anaplastic large-cell lymphoma (sALCL).
In January 2016, the EC approved a Type II variation to include data on the retreatment of adult patients with HL or sALCL who previously responded to brentuximab vedotin and later relapsed.
Brentuximab vedotin is under joint development by Seattle Genetics and Takeda Pharmaceutical Company Limited.
AETHERA trial
The CHMP’s recommendation to extend the approval of brentuximab vedotin is based on results from the phase 3 AETHERA trial.
The trial was designed to compare brentuximab vedotin to placebo, both administered for up to 16 cycles (approximately 1 year) every 3 weeks following ASCT. Results from the trial were published in The Lancet in March 2015 and presented at the 2014 ASH Annual Meeting.
The study enrolled 329 HL patients at risk of relapse or progression, including 165 on the brentuximab vedotin arm and 164 on the placebo arm.
Patients were eligible for enrollment if they had a history of primary refractory HL, relapsed within a year of receiving frontline chemotherapy, and/or had disease outside of the lymph nodes at the time of pre-ASCT relapse.
Brentuximab vedotin conferred a significant increase in progression-free survival over placebo, with a hazard ratio of 0.57 (P=0.001). The median progression-free survival was 43 months for patients who received brentuximab vedotin and 24 months for those who received placebo.
The most common adverse events (≥20%), of any grade and regardless of causality, in the brentuximab vedotin arm were neutropenia (78%), peripheral sensory neuropathy (56%), thrombocytopenia (41%), anemia (27%), upper respiratory tract infection (26%), fatigue (24%), peripheral motor neuropathy (23%), nausea (22%), cough (21%), and diarrhea (20%).
The most common adverse events (≥20%), of any grade and regardless of causality, in the placebo arm were neutropenia (34%), upper respiratory tract infection (23%), and thrombocytopenia (20%).
In all, 67% of patients on the brentuximab vedotin arm experienced peripheral neuropathy. Of those patients, 85% had resolution (59%) or partial improvement (26%) in symptoms at the time of their last evaluation, with a median time to improvement of 23 weeks (range, 0.1-138). ![]()
FDA approves FVIII single-chain therapy for hemophilia A

The US Food and Drug Administration (FDA) has approved lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, for use in adults and children with hemophilia A.
The product is approved for routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding.
Lonoctocog alfa is the first and only single-chain product for hemophilia A that is specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing.
The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa should be available in the US early this summer, according to CSL Behring, the company developing the drug.
Clinical trials
The approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which included a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate (ABR) was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly. Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
The US Food and Drug Administration (FDA) has approved lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, for use in adults and children with hemophilia A.
The product is approved for routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding.
Lonoctocog alfa is the first and only single-chain product for hemophilia A that is specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing.
The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa should be available in the US early this summer, according to CSL Behring, the company developing the drug.
Clinical trials
The approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which included a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate (ABR) was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly. Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
The US Food and Drug Administration (FDA) has approved lonoctocog alfa (Afstyla), a recombinant factor VIII (FVIII) single-chain therapy, for use in adults and children with hemophilia A.
The product is approved for routine prophylaxis to reduce the frequency of bleeding episodes, for on-demand treatment and control of bleeding episodes, and for perioperative management of bleeding.
Lonoctocog alfa is the first and only single-chain product for hemophilia A that is specifically designed to provide long-lasting protection from bleeds with 2- to 3-times weekly dosing.
The product uses a covalent bond that forms one structural entity, a single polypeptide-chain, to improve the stability of FVIII and provide longer-lasting FVIII activity.
Lonoctocog alfa should be available in the US early this summer, according to CSL Behring, the company developing the drug.
Clinical trials
The approval of lonoctocog alfa is based on results from the AFFINITY clinical development program, which included a trial of children (n=84) and a trial of adolescents and adults (n=175).
Among patients who received lonoctocog alfa prophylactically, the median annualized bleeding rate (ABR) was 1.14 in the adults and adolescents and 3.69 in children younger than 12.
In all, there were 1195 bleeding events—848 in the adults/adolescents and 347 in the children.
Ninety-four percent of bleeds in adults/adolescents and 96% of bleeds in pediatric patients were effectively controlled with no more than 2 infusions of lonoctocog alfa weekly. Eighty-one percent of bleeds in adults/adolescents and 86% of bleeds in pediatric patients were controlled by a single infusion.
Researchers assessed safety in 258 patients from both studies. Adverse reactions occurred in 14 patients and included hypersensitivity (n=4), dizziness (n=2), paresthesia (n=1), rash (n=1), erythema (n=1), pruritus (n=1), pyrexia (n=1), injection-site pain (n=1), chills (n=1), and feeling hot (n=1).
One patient withdrew from treatment due to hypersensitivity.
None of the patients developed neutralizing antibodies to FVIII or antibodies to host cell proteins. There were no reports of anaphylaxis or thrombosis. ![]()
PD-1 inhibitor granted accelerated approval for cHL

Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Reversal agent granted conditional approval in Canada

to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events.
FDA warns of counterfeit BiCNU
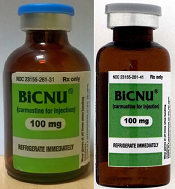
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing [email protected].
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
EC approves FC fusion protein for hemophilia B

Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.