User login
FDA approves obinutuzumab for FL

The US Food and Drug Administration (FDA) has approved obinutuzumab (Gazyva) for certain patients with previously treated follicular lymphoma (FL).
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug was previously approved by the FDA for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Now, obinutuzumab is approved for use in combination with bendamustine, followed by obinutuzumab alone, to treat patients with FL who did not respond to a rituximab-containing regimen or whose FL returned after such treatment.
The recommended dose and schedule for the regimen is:
- Obinutuzumab at 1000 mg by intravenous infusion on days 1, 8, and 15 of cycle 1; on day 1 of cycles 2-6 (28-day cycles); then every 2 months for 2 years.
- Bendamustine at 90 mg/m2 by intravenous infusion on days 1 and 2 of cycles 1-6.
Full prescribing information for obinutuzumab is available on the FDA website or at www.Gazyva.com.
Phase 3 study
The approval for obinutuzumab in FL is based on results from the phase 3 GADOLIN study. The trial included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS as assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
Best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%).
About obinutuzumab
Obinutuzumab is being studied in a large clinical program, including the phase 3 GOYA and GALLIUM studies.
In GOYA, researchers are comparing obinutuzumab head-to-head with rituximab plus CHOP chemotherapy in first-line diffuse large B-cell lymphoma. In GALLIUM, researchers are comparing obinutuzumab plus chemotherapy head-to-head with rituximab plus chemotherapy in first-line indolent non-Hodgkin lymphoma.
Additional combination studies investigating obinutuzumab with other approved or investigational medicines, including cancer immunotherapies and small-molecule inhibitors, are planned or underway across a range of blood cancers.
Obinutuzumab was discovered by Roche Glycart AG, a wholly owned, independent research unit of Roche. In the US, obinutuzumab is part of a collaboration between Genentech and Biogen.
Genentech has a patient assistance program, Genentech Access Solutions, that can help qualifying patients access obinutuzumab and other Genentech medications.
The program is designed to help people navigate the access and reimbursement process and provide assistance to eligible patients in the US who are uninsured or cannot afford the out-of-pocket costs for their medicine. For more information, visit www.Genentech-Access.com. ![]()
The US Food and Drug Administration (FDA) has approved obinutuzumab (Gazyva) for certain patients with previously treated follicular lymphoma (FL).
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug was previously approved by the FDA for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Now, obinutuzumab is approved for use in combination with bendamustine, followed by obinutuzumab alone, to treat patients with FL who did not respond to a rituximab-containing regimen or whose FL returned after such treatment.
The recommended dose and schedule for the regimen is:
- Obinutuzumab at 1000 mg by intravenous infusion on days 1, 8, and 15 of cycle 1; on day 1 of cycles 2-6 (28-day cycles); then every 2 months for 2 years.
- Bendamustine at 90 mg/m2 by intravenous infusion on days 1 and 2 of cycles 1-6.
Full prescribing information for obinutuzumab is available on the FDA website or at www.Gazyva.com.
Phase 3 study
The approval for obinutuzumab in FL is based on results from the phase 3 GADOLIN study. The trial included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS as assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
Best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%).
About obinutuzumab
Obinutuzumab is being studied in a large clinical program, including the phase 3 GOYA and GALLIUM studies.
In GOYA, researchers are comparing obinutuzumab head-to-head with rituximab plus CHOP chemotherapy in first-line diffuse large B-cell lymphoma. In GALLIUM, researchers are comparing obinutuzumab plus chemotherapy head-to-head with rituximab plus chemotherapy in first-line indolent non-Hodgkin lymphoma.
Additional combination studies investigating obinutuzumab with other approved or investigational medicines, including cancer immunotherapies and small-molecule inhibitors, are planned or underway across a range of blood cancers.
Obinutuzumab was discovered by Roche Glycart AG, a wholly owned, independent research unit of Roche. In the US, obinutuzumab is part of a collaboration between Genentech and Biogen.
Genentech has a patient assistance program, Genentech Access Solutions, that can help qualifying patients access obinutuzumab and other Genentech medications.
The program is designed to help people navigate the access and reimbursement process and provide assistance to eligible patients in the US who are uninsured or cannot afford the out-of-pocket costs for their medicine. For more information, visit www.Genentech-Access.com. ![]()
The US Food and Drug Administration (FDA) has approved obinutuzumab (Gazyva) for certain patients with previously treated follicular lymphoma (FL).
Obinutuzumab is a glycoengineered, humanized, monoclonal antibody that selectively binds to the extracellular domain of the CD20 antigen on B cells.
The drug was previously approved by the FDA for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia.
Now, obinutuzumab is approved for use in combination with bendamustine, followed by obinutuzumab alone, to treat patients with FL who did not respond to a rituximab-containing regimen or whose FL returned after such treatment.
The recommended dose and schedule for the regimen is:
- Obinutuzumab at 1000 mg by intravenous infusion on days 1, 8, and 15 of cycle 1; on day 1 of cycles 2-6 (28-day cycles); then every 2 months for 2 years.
- Bendamustine at 90 mg/m2 by intravenous infusion on days 1 and 2 of cycles 1-6.
Full prescribing information for obinutuzumab is available on the FDA website or at www.Gazyva.com.
Phase 3 study
The approval for obinutuzumab in FL is based on results from the phase 3 GADOLIN study. The trial included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS as assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
Best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%).
About obinutuzumab
Obinutuzumab is being studied in a large clinical program, including the phase 3 GOYA and GALLIUM studies.
In GOYA, researchers are comparing obinutuzumab head-to-head with rituximab plus CHOP chemotherapy in first-line diffuse large B-cell lymphoma. In GALLIUM, researchers are comparing obinutuzumab plus chemotherapy head-to-head with rituximab plus chemotherapy in first-line indolent non-Hodgkin lymphoma.
Additional combination studies investigating obinutuzumab with other approved or investigational medicines, including cancer immunotherapies and small-molecule inhibitors, are planned or underway across a range of blood cancers.
Obinutuzumab was discovered by Roche Glycart AG, a wholly owned, independent research unit of Roche. In the US, obinutuzumab is part of a collaboration between Genentech and Biogen.
Genentech has a patient assistance program, Genentech Access Solutions, that can help qualifying patients access obinutuzumab and other Genentech medications.
The program is designed to help people navigate the access and reimbursement process and provide assistance to eligible patients in the US who are uninsured or cannot afford the out-of-pocket costs for their medicine. For more information, visit www.Genentech-Access.com. ![]()
Orphan designation recommended for BTK inhibitor

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) is recommending orphan designation for the second-generation BTK inhibitor acalabrutinib (ACP-196) for 3 indications.
The COMP is recommending the drug receive orphan designation as a treatment for chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL), mantle cell lymphoma, and Waldenström’s macroglobulinemia.
The COMP adopts an opinion on orphan drug designation, and that opinion is submitted to the European Commission (EC) for endorsement.
To be granted orphan designation by the EC, a medicine must be intended for the treatment, prevention, or diagnosis of a disease that is life-threatening and has a prevalence of up to 5 in 10,000 in the European Union. Additionally, the medicine must aim to provide significant benefit to those affected by the condition.
Orphan designation provides companies with development and market exclusivity incentives for designated compounds and medicines.
About acalabrutinib
Acalabrutinib is under development by AstraZeneca and Acerta Pharma BV. The drug is currently being evaluated in trials of patients with CLL/SLL, mantle cell lymphoma, Waldentröm’s macroglobulinemia, and a range of other hematologic malignancies and solid tumor cancers.
Data from a phase 1/2 trial of acalabrutinib in CLL were presented at the 2015 ASH Annual Meeting and simultaneously published in NEJM.
The researchers reported on 61 patients with relapsed CLL who had a median age of 62 (range, 44-84) and a median of 3 prior therapies (range, 1-13).
Patients enrolled in the phase 1 portion of the study received escalating doses of acalabrutinib, with a maximum dose of 400 mg once daily. Patients involved in the phase 2 portion of the study were treated with a 100 mg dose twice daily.
At a median follow-up of 14.3 months (range, 0.5 to 20), 53 patients were still receiving treatment.
The most common adverse events of all grades (occurring in at least 20% of patients) were headache (43%), diarrhea (39%), increased weight (26%), pyrexia (23%), upper respiratory tract infection (23%), fatigue (21%), peripheral edema (21%), hypertension (20%), and nausea (20%).
Grade 3/4 adverse events included diarrhea (2%), increased weight (2%), pyrexia (3%), fatigue (3%), hypertension (7%), and arthralgia (2%).
The overall response rate among the 60 evaluable patients was 95%. This included partial responses in 85% of patients and partial responses with lymphocytosis in 10%. The rate of stable disease was 5%.
The researchers noted that responses occurred in all dosing cohorts, and the response rate increased over time. ![]()
FDA investigates issues with rivaroxaban trial

The US Food and Drug Administration (FDA) is investigating issues surrounding a defect in the device that was used to test international normalized ratios (INRs) in the ROCKET AF trial.
ROCKET AF was used to support approval for the direct oral anticoagulant rivaroxaban (Xarelto) in the US and European Union.
According to a legal brief filed in federal court earlier this week, the FDA has asked rivaroxaban’s manufacturer, Johnson & Johnson, if there was evidence of the defect in the INR-measuring device while ROCKET AF was underway.
The trial was a comparison of rivaroxaban and warfarin in patients with nonvalvular atrial fibrillation.
Results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
However, an article recently published in The BMJ questioned these results because the Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips), which was used to measure patients’ INRs during the trial, was recalled in December 2014 after giving falsely low test results.
The article suggested the system could have provided falsely low INR values in some patients in the warfarin arm. The lower values could have led investigators to give incorrect doses of warfarin, increasing the risk of bleeding for those patients and therefore giving a false impression of the comparative safety of rivaroxaban.
Two days after The BMJ article was published, the European Medicines Agency released a statement saying the defect with the INRatio system does not change the overall conclusions of ROCKET AF.
Researchers at the Duke Clinical Research Institute, which oversaw ROCKET AF, also assessed the impact of the INRatio defect. Their findings, published in NEJM, suggested the defect did not affect the trial’s outcome.
However, it appears the FDA still has some concerns. The agency has asked Johnson & Johnson whether there was evidence of the INRatio defect during ROCKET AF, according to a legal brief filed by lawyers representing individuals claiming injuries resulting from rivaroxaban.
The brief also cited internal emails that, according to the lawyers, showed some ROCKET AF investigators questioned the accuracy of the INRatio system during the trial. In fact, the lawyers said a special program was set up to investigate the malfunctions, but Johnson & Johnson never disclosed this information to the FDA. ![]()
The US Food and Drug Administration (FDA) is investigating issues surrounding a defect in the device that was used to test international normalized ratios (INRs) in the ROCKET AF trial.
ROCKET AF was used to support approval for the direct oral anticoagulant rivaroxaban (Xarelto) in the US and European Union.
According to a legal brief filed in federal court earlier this week, the FDA has asked rivaroxaban’s manufacturer, Johnson & Johnson, if there was evidence of the defect in the INR-measuring device while ROCKET AF was underway.
The trial was a comparison of rivaroxaban and warfarin in patients with nonvalvular atrial fibrillation.
Results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
However, an article recently published in The BMJ questioned these results because the Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips), which was used to measure patients’ INRs during the trial, was recalled in December 2014 after giving falsely low test results.
The article suggested the system could have provided falsely low INR values in some patients in the warfarin arm. The lower values could have led investigators to give incorrect doses of warfarin, increasing the risk of bleeding for those patients and therefore giving a false impression of the comparative safety of rivaroxaban.
Two days after The BMJ article was published, the European Medicines Agency released a statement saying the defect with the INRatio system does not change the overall conclusions of ROCKET AF.
Researchers at the Duke Clinical Research Institute, which oversaw ROCKET AF, also assessed the impact of the INRatio defect. Their findings, published in NEJM, suggested the defect did not affect the trial’s outcome.
However, it appears the FDA still has some concerns. The agency has asked Johnson & Johnson whether there was evidence of the INRatio defect during ROCKET AF, according to a legal brief filed by lawyers representing individuals claiming injuries resulting from rivaroxaban.
The brief also cited internal emails that, according to the lawyers, showed some ROCKET AF investigators questioned the accuracy of the INRatio system during the trial. In fact, the lawyers said a special program was set up to investigate the malfunctions, but Johnson & Johnson never disclosed this information to the FDA. ![]()
The US Food and Drug Administration (FDA) is investigating issues surrounding a defect in the device that was used to test international normalized ratios (INRs) in the ROCKET AF trial.
ROCKET AF was used to support approval for the direct oral anticoagulant rivaroxaban (Xarelto) in the US and European Union.
According to a legal brief filed in federal court earlier this week, the FDA has asked rivaroxaban’s manufacturer, Johnson & Johnson, if there was evidence of the defect in the INR-measuring device while ROCKET AF was underway.
The trial was a comparison of rivaroxaban and warfarin in patients with nonvalvular atrial fibrillation.
Results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
However, an article recently published in The BMJ questioned these results because the Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips), which was used to measure patients’ INRs during the trial, was recalled in December 2014 after giving falsely low test results.
The article suggested the system could have provided falsely low INR values in some patients in the warfarin arm. The lower values could have led investigators to give incorrect doses of warfarin, increasing the risk of bleeding for those patients and therefore giving a false impression of the comparative safety of rivaroxaban.
Two days after The BMJ article was published, the European Medicines Agency released a statement saying the defect with the INRatio system does not change the overall conclusions of ROCKET AF.
Researchers at the Duke Clinical Research Institute, which oversaw ROCKET AF, also assessed the impact of the INRatio defect. Their findings, published in NEJM, suggested the defect did not affect the trial’s outcome.
However, it appears the FDA still has some concerns. The agency has asked Johnson & Johnson whether there was evidence of the INRatio defect during ROCKET AF, according to a legal brief filed by lawyers representing individuals claiming injuries resulting from rivaroxaban.
The brief also cited internal emails that, according to the lawyers, showed some ROCKET AF investigators questioned the accuracy of the INRatio system during the trial. In fact, the lawyers said a special program was set up to investigate the malfunctions, but Johnson & Johnson never disclosed this information to the FDA. ![]()
Adjunct T-cell therapy granted orphan designation
The US Food and Drug Administration (FDA) has granted orphan drug designation for BPX-501, an adjunct T-cell therapy.
The designation is for the combination of BPX-501 genetically modified T cells and the activator agent rimiducid as replacement T-cell therapy for the treatment of immunodeficiency and graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplant (HSCT).
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. Infusion of rimiducid is designed to trigger activation of this domain of caspase-9 (iCasp9), which leads to selective apoptosis of the CaspaCIDe-containing cells.
This technology is intended to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs, ostensibly enabling physicians to more safely perform haploidentical HSCTs by adding back the BPX-501 genetically engineered T cells to speed immune reconstitution and provide control over viral infections.
Following an allogeneic HSCT, a lack of sufficient mature T cells constitutes immune deficiency that can contribute to infections, viral reactivation, and relapse.
The ability to correct this immune deficiency by adding back mature donor T cells, without raising the risk of uncontrollable GVHD, has the potential to change the risk profile of allogeneic transplant, according to Bellicum Pharmaceuticals, the company developing BPX-501.
BPX-501 is being evaluated in multiple phase 1/2 trials in adults and pediatric patients with leukemias, lymphomas, and genetic blood diseases in the US and Europe.
About orphan designation
The FDA’s Office of Orphan Products Development grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent rare diseases and disorders that affect fewer than 200,000 people in the US.
Orphan designation qualifies a company for various development incentives, including tax credits for qualified clinical testing and marketing exclusivity for a period of 7 years. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for BPX-501, an adjunct T-cell therapy.
The designation is for the combination of BPX-501 genetically modified T cells and the activator agent rimiducid as replacement T-cell therapy for the treatment of immunodeficiency and graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplant (HSCT).
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. Infusion of rimiducid is designed to trigger activation of this domain of caspase-9 (iCasp9), which leads to selective apoptosis of the CaspaCIDe-containing cells.
This technology is intended to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs, ostensibly enabling physicians to more safely perform haploidentical HSCTs by adding back the BPX-501 genetically engineered T cells to speed immune reconstitution and provide control over viral infections.
Following an allogeneic HSCT, a lack of sufficient mature T cells constitutes immune deficiency that can contribute to infections, viral reactivation, and relapse.
The ability to correct this immune deficiency by adding back mature donor T cells, without raising the risk of uncontrollable GVHD, has the potential to change the risk profile of allogeneic transplant, according to Bellicum Pharmaceuticals, the company developing BPX-501.
BPX-501 is being evaluated in multiple phase 1/2 trials in adults and pediatric patients with leukemias, lymphomas, and genetic blood diseases in the US and Europe.
About orphan designation
The FDA’s Office of Orphan Products Development grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent rare diseases and disorders that affect fewer than 200,000 people in the US.
Orphan designation qualifies a company for various development incentives, including tax credits for qualified clinical testing and marketing exclusivity for a period of 7 years. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for BPX-501, an adjunct T-cell therapy.
The designation is for the combination of BPX-501 genetically modified T cells and the activator agent rimiducid as replacement T-cell therapy for the treatment of immunodeficiency and graft-versus-host disease (GVHD) after allogeneic hematopoietic stem cell transplant (HSCT).
BPX-501 consists of genetically modified donor T cells incorporating the CaspaCIDe safety switch, which is designed to eliminate cells in the event of toxicity.
The CaspaCIDe switch consists of the CID-binding domain coupled to the signaling domain of caspase-9, an enzyme that is part of the apoptotic pathway. Infusion of rimiducid is designed to trigger activation of this domain of caspase-9 (iCasp9), which leads to selective apoptosis of the CaspaCIDe-containing cells.
This technology is intended to provide a safety net to eliminate BPX-501 alloreactive T cells if severe GVHD occurs, ostensibly enabling physicians to more safely perform haploidentical HSCTs by adding back the BPX-501 genetically engineered T cells to speed immune reconstitution and provide control over viral infections.
Following an allogeneic HSCT, a lack of sufficient mature T cells constitutes immune deficiency that can contribute to infections, viral reactivation, and relapse.
The ability to correct this immune deficiency by adding back mature donor T cells, without raising the risk of uncontrollable GVHD, has the potential to change the risk profile of allogeneic transplant, according to Bellicum Pharmaceuticals, the company developing BPX-501.
BPX-501 is being evaluated in multiple phase 1/2 trials in adults and pediatric patients with leukemias, lymphomas, and genetic blood diseases in the US and Europe.
About orphan designation
The FDA’s Office of Orphan Products Development grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent rare diseases and disorders that affect fewer than 200,000 people in the US.
Orphan designation qualifies a company for various development incentives, including tax credits for qualified clinical testing and marketing exclusivity for a period of 7 years. ![]()
Product approved to treat hemophilia A in EU

The European Commission has approved a full-length recombinant factor VIII product for the treatment and prevention of bleeding in hemophilia A patients of all ages.
The product, Kovaltry (formerly BAY 81-8973), will be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The approval of Kovaltry is based on results from the LEOPOLD trials—3 multinational trials of patients with severe hemophilia A.
The trials were supported by Bayer HealthCare AG, the company developing Kovaltry.
LEOPOLD I
LEOPOLD I is an open-label, cross-over, phase 3 study of males, ages 12 to 65, with severe hemophilia A. Sixty-two patients were assigned to either 2- or 3-times-weekly dosing with Kovaltry, based on each patient’s phenotype, prior bleeding history, and other factors.
The median annualized bleeding rate (ABR) was 1.0 for all the patients who received Kovaltry prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD II
LEOPOLD II is a randomized, cross-over, open-label trial conducted in males ages 12 to 65. In this phase 3 study, 80 subjects were randomized to receive Kovaltry as a low-dose prophylaxis regimen (n=28) twice per week, high-dose prophylaxis (n=31) 3 times a week, or on-demand treatment (n=21).
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD Kids
LEOPOLD Kids is an open-label, non-randomized, phase 3 study designed to evaluate Kovaltry in children age 12 and younger. The study is divided into 2 parts. Part A enrolled only previously treated children, and part B, which is ongoing, includes only untreated children.
For part A, 51 children received Kovaltry twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days. The median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with at least a reasonable suspected causal relationship to Kovaltry.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
The European Commission has approved a full-length recombinant factor VIII product for the treatment and prevention of bleeding in hemophilia A patients of all ages.
The product, Kovaltry (formerly BAY 81-8973), will be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The approval of Kovaltry is based on results from the LEOPOLD trials—3 multinational trials of patients with severe hemophilia A.
The trials were supported by Bayer HealthCare AG, the company developing Kovaltry.
LEOPOLD I
LEOPOLD I is an open-label, cross-over, phase 3 study of males, ages 12 to 65, with severe hemophilia A. Sixty-two patients were assigned to either 2- or 3-times-weekly dosing with Kovaltry, based on each patient’s phenotype, prior bleeding history, and other factors.
The median annualized bleeding rate (ABR) was 1.0 for all the patients who received Kovaltry prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD II
LEOPOLD II is a randomized, cross-over, open-label trial conducted in males ages 12 to 65. In this phase 3 study, 80 subjects were randomized to receive Kovaltry as a low-dose prophylaxis regimen (n=28) twice per week, high-dose prophylaxis (n=31) 3 times a week, or on-demand treatment (n=21).
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD Kids
LEOPOLD Kids is an open-label, non-randomized, phase 3 study designed to evaluate Kovaltry in children age 12 and younger. The study is divided into 2 parts. Part A enrolled only previously treated children, and part B, which is ongoing, includes only untreated children.
For part A, 51 children received Kovaltry twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days. The median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with at least a reasonable suspected causal relationship to Kovaltry.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
The European Commission has approved a full-length recombinant factor VIII product for the treatment and prevention of bleeding in hemophilia A patients of all ages.
The product, Kovaltry (formerly BAY 81-8973), will be marketed for this indication in the 28 member countries of the European Union, as well as Iceland, Liechtenstein, and Norway.
The approval of Kovaltry is based on results from the LEOPOLD trials—3 multinational trials of patients with severe hemophilia A.
The trials were supported by Bayer HealthCare AG, the company developing Kovaltry.
LEOPOLD I
LEOPOLD I is an open-label, cross-over, phase 3 study of males, ages 12 to 65, with severe hemophilia A. Sixty-two patients were assigned to either 2- or 3-times-weekly dosing with Kovaltry, based on each patient’s phenotype, prior bleeding history, and other factors.
The median annualized bleeding rate (ABR) was 1.0 for all the patients who received Kovaltry prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD II
LEOPOLD II is a randomized, cross-over, open-label trial conducted in males ages 12 to 65. In this phase 3 study, 80 subjects were randomized to receive Kovaltry as a low-dose prophylaxis regimen (n=28) twice per week, high-dose prophylaxis (n=31) 3 times a week, or on-demand treatment (n=21).
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
LEOPOLD Kids
LEOPOLD Kids is an open-label, non-randomized, phase 3 study designed to evaluate Kovaltry in children age 12 and younger. The study is divided into 2 parts. Part A enrolled only previously treated children, and part B, which is ongoing, includes only untreated children.
For part A, 51 children received Kovaltry twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days. The median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with at least a reasonable suspected causal relationship to Kovaltry.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
Antiplatelet agent approved for long-term use

Photo courtesy of the CDC
The European Commission has approved use of the antiplatelet agent ticagrelor (Brilique) at a 60 mg dose to treat patients beyond the first year after a heart attack who are at high risk of developing a further atherothrombotic event.
The treatment may be used as continuation therapy after an initial 1-year treatment with 90 mg ticagrelor plus aspirin or after a year of other dual antiplatelet therapy.
This approval is applicable to all 28 European Union (EU) member countries plus Iceland, Norway, and Liechtenstein.
Ticagrelor at a 90 mg dose is already approved in the EU for the prevention of atherothrombotic events in adults with acute coronary syndrome (ACS). In the management of ACS, the recommended maintenance dose of ticagrelor is 90 mg twice daily during the first year after an ACS event.
Now, after the first year, patients with a history of heart attack can continue to be treated with ticagrelor at 60 mg twice daily, which should be taken with a daily maintenance dose of aspirin at 75 mg to 150 mg.
Trial results
The latest EU approval of ticagrelor was based on results from the PEGASUS TIMI-54 study. This trial, which involved more than 21,000 patients, was presented at the American College of Cardiology Congress in March 2015 and simultaneously published in NEJM.
Investigators compared ticagrelor (at 60 mg or 90 mg) plus low-dose aspirin to placebo plus low-dose aspirin in patients who had experienced a heart attack 1 to 3 years prior to study enrollment.
The primary efficacy endpoint was a composite of cardiovascular death, myocardial infarction, or stroke.
The investigators found that patients in either ticagrelor arm were significantly less likely to achieve this endpoint than placebo-treated patients.
At 3 years, the proportion of patients meeting the primary endpoint was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
Patients receiving ticagrelor also had a significantly higher incidence of major bleeding and dyspnea. The rate of TIMI major bleeding was 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rate of dyspnea was 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for both comparisons). The rate of dyspnea leading to treatment discontinuation was 6.5%, 4.55%, and 0.79%, respectively (P<0.001 for both comparisons).
Ticagrelor has been approved in more than 100 countries. The drug is under development by AstraZeneca.![]()
Photo courtesy of the CDC
The European Commission has approved use of the antiplatelet agent ticagrelor (Brilique) at a 60 mg dose to treat patients beyond the first year after a heart attack who are at high risk of developing a further atherothrombotic event.
The treatment may be used as continuation therapy after an initial 1-year treatment with 90 mg ticagrelor plus aspirin or after a year of other dual antiplatelet therapy.
This approval is applicable to all 28 European Union (EU) member countries plus Iceland, Norway, and Liechtenstein.
Ticagrelor at a 90 mg dose is already approved in the EU for the prevention of atherothrombotic events in adults with acute coronary syndrome (ACS). In the management of ACS, the recommended maintenance dose of ticagrelor is 90 mg twice daily during the first year after an ACS event.
Now, after the first year, patients with a history of heart attack can continue to be treated with ticagrelor at 60 mg twice daily, which should be taken with a daily maintenance dose of aspirin at 75 mg to 150 mg.
Trial results
The latest EU approval of ticagrelor was based on results from the PEGASUS TIMI-54 study. This trial, which involved more than 21,000 patients, was presented at the American College of Cardiology Congress in March 2015 and simultaneously published in NEJM.
Investigators compared ticagrelor (at 60 mg or 90 mg) plus low-dose aspirin to placebo plus low-dose aspirin in patients who had experienced a heart attack 1 to 3 years prior to study enrollment.
The primary efficacy endpoint was a composite of cardiovascular death, myocardial infarction, or stroke.
The investigators found that patients in either ticagrelor arm were significantly less likely to achieve this endpoint than placebo-treated patients.
At 3 years, the proportion of patients meeting the primary endpoint was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
Patients receiving ticagrelor also had a significantly higher incidence of major bleeding and dyspnea. The rate of TIMI major bleeding was 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rate of dyspnea was 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for both comparisons). The rate of dyspnea leading to treatment discontinuation was 6.5%, 4.55%, and 0.79%, respectively (P<0.001 for both comparisons).
Ticagrelor has been approved in more than 100 countries. The drug is under development by AstraZeneca.![]()
Photo courtesy of the CDC
The European Commission has approved use of the antiplatelet agent ticagrelor (Brilique) at a 60 mg dose to treat patients beyond the first year after a heart attack who are at high risk of developing a further atherothrombotic event.
The treatment may be used as continuation therapy after an initial 1-year treatment with 90 mg ticagrelor plus aspirin or after a year of other dual antiplatelet therapy.
This approval is applicable to all 28 European Union (EU) member countries plus Iceland, Norway, and Liechtenstein.
Ticagrelor at a 90 mg dose is already approved in the EU for the prevention of atherothrombotic events in adults with acute coronary syndrome (ACS). In the management of ACS, the recommended maintenance dose of ticagrelor is 90 mg twice daily during the first year after an ACS event.
Now, after the first year, patients with a history of heart attack can continue to be treated with ticagrelor at 60 mg twice daily, which should be taken with a daily maintenance dose of aspirin at 75 mg to 150 mg.
Trial results
The latest EU approval of ticagrelor was based on results from the PEGASUS TIMI-54 study. This trial, which involved more than 21,000 patients, was presented at the American College of Cardiology Congress in March 2015 and simultaneously published in NEJM.
Investigators compared ticagrelor (at 60 mg or 90 mg) plus low-dose aspirin to placebo plus low-dose aspirin in patients who had experienced a heart attack 1 to 3 years prior to study enrollment.
The primary efficacy endpoint was a composite of cardiovascular death, myocardial infarction, or stroke.
The investigators found that patients in either ticagrelor arm were significantly less likely to achieve this endpoint than placebo-treated patients.
At 3 years, the proportion of patients meeting the primary endpoint was 7.85% in the 90 mg group, 7.77% in the 60 mg group, and 9.04% in the placebo group (P=0.008 for 90 mg vs placebo and P=0.004 for 60 mg vs placebo).
Patients receiving ticagrelor also had a significantly higher incidence of major bleeding and dyspnea. The rate of TIMI major bleeding was 2.60% in the 90 mg group, 2.30% in the 60 mg group, and 1.06% in the placebo group (P<0.001 for each ticagrelor dose vs placebo).
The rate of dyspnea was 18.93% in the 90 mg group, 15.84% in 60 mg group, and 6.38% in the placebo group (P<0.001 for both comparisons). The rate of dyspnea leading to treatment discontinuation was 6.5%, 4.55%, and 0.79%, respectively (P<0.001 for both comparisons).
Ticagrelor has been approved in more than 100 countries. The drug is under development by AstraZeneca.![]()
Drug granted breakthrough designation for AML
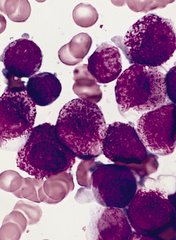
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for midostaurin (PKC412) to treat acute myeloid leukemia (AML).
Midostaurin is a multi-targeted kinase inhibitor being developed for adults with newly diagnosed AML who are FLT3-positive, as detected by an FDA-approved test, and who are eligible to receive standard induction and consolidation chemotherapy.
Breakthrough therapy designation is intended to expedite the development and review of new medicines intended to treat serious or life-threatening conditions. The therapy must demonstrate substantial improvement over an available therapy on at least one clinically significant endpoint.
The designation includes all of the fast track program features, as well as more intensive FDA guidance on an efficient drug development program.
Phase 3 trial
The breakthrough designation for midostaurin is primarily based on the results of the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 717 patients with newly diagnosed, FLT3-positive AML who were younger than 60 at enrollment. All of the patients received standard induction and consolidation therapy. Roughly half also received midostaurin (n=360), while the other half received placebo (n=357).
Patients who received midostaurin experienced a significant improvement in overall survival (hazard ratio=0.77, P=0.0074). The median overall survival was 74.4 months in the midostaurin arm and 25.6 months in the placebo arm.
The median event-free survival was 8 months in the midostaurin arm and 3.6 months in the placebo arm (P=0.0032). The 5-year event-free survival was 27.5% for midostaurin and 19.3% for placebo.
There was no significant difference between the treatment arms with regard to most non-hematologic grade 3/4 adverse events. The exception was rash/desquamation, which occurred in 13% of patients in the midostaurin arm and 8% of patients in the placebo arm (P=0.02).
Other grade 3/4 non-hematologic events occurring in 10% of patients or more included, in the midostaurin and placebo arms, respectively: febrile neutropenia (81%, 82%), infection (40%, 38%), diarrhea (15%, 16%), hypokalemia (13%, 17%), pain (13%, 13%), other infection (12%, 12%), ALT/SGPT (12%, 9%), and fatigue (9%, 11%).
There were 18 deaths (5%) in the midostaurin arm and 19 (5.3%) in the placebo arm during induction and consolidation.
Midostaurin development
Novartis has opened a Global Individual Patient Program (compassionate use program) and a US Expanded Treatment Protocol (ETP) to enable midostaurin access. Patients 18 years of age and older with newly diagnosed FLT3-mutated AML who are able to receive standard induction and consolidation therapy will be considered.
To help identify patients who may have a FLT3 mutation and potentially benefit from treatment with midostaurin, Novartis is collaborating with Invivoscribe Technologies, Inc. which is leading regulatory submissions for a companion diagnostic.
Midostaurin is also being investigated for the treatment of aggressive systemic mastocytosis/mast cell leukemia. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for midostaurin (PKC412) to treat acute myeloid leukemia (AML).
Midostaurin is a multi-targeted kinase inhibitor being developed for adults with newly diagnosed AML who are FLT3-positive, as detected by an FDA-approved test, and who are eligible to receive standard induction and consolidation chemotherapy.
Breakthrough therapy designation is intended to expedite the development and review of new medicines intended to treat serious or life-threatening conditions. The therapy must demonstrate substantial improvement over an available therapy on at least one clinically significant endpoint.
The designation includes all of the fast track program features, as well as more intensive FDA guidance on an efficient drug development program.
Phase 3 trial
The breakthrough designation for midostaurin is primarily based on the results of the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 717 patients with newly diagnosed, FLT3-positive AML who were younger than 60 at enrollment. All of the patients received standard induction and consolidation therapy. Roughly half also received midostaurin (n=360), while the other half received placebo (n=357).
Patients who received midostaurin experienced a significant improvement in overall survival (hazard ratio=0.77, P=0.0074). The median overall survival was 74.4 months in the midostaurin arm and 25.6 months in the placebo arm.
The median event-free survival was 8 months in the midostaurin arm and 3.6 months in the placebo arm (P=0.0032). The 5-year event-free survival was 27.5% for midostaurin and 19.3% for placebo.
There was no significant difference between the treatment arms with regard to most non-hematologic grade 3/4 adverse events. The exception was rash/desquamation, which occurred in 13% of patients in the midostaurin arm and 8% of patients in the placebo arm (P=0.02).
Other grade 3/4 non-hematologic events occurring in 10% of patients or more included, in the midostaurin and placebo arms, respectively: febrile neutropenia (81%, 82%), infection (40%, 38%), diarrhea (15%, 16%), hypokalemia (13%, 17%), pain (13%, 13%), other infection (12%, 12%), ALT/SGPT (12%, 9%), and fatigue (9%, 11%).
There were 18 deaths (5%) in the midostaurin arm and 19 (5.3%) in the placebo arm during induction and consolidation.
Midostaurin development
Novartis has opened a Global Individual Patient Program (compassionate use program) and a US Expanded Treatment Protocol (ETP) to enable midostaurin access. Patients 18 years of age and older with newly diagnosed FLT3-mutated AML who are able to receive standard induction and consolidation therapy will be considered.
To help identify patients who may have a FLT3 mutation and potentially benefit from treatment with midostaurin, Novartis is collaborating with Invivoscribe Technologies, Inc. which is leading regulatory submissions for a companion diagnostic.
Midostaurin is also being investigated for the treatment of aggressive systemic mastocytosis/mast cell leukemia. ![]()
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for midostaurin (PKC412) to treat acute myeloid leukemia (AML).
Midostaurin is a multi-targeted kinase inhibitor being developed for adults with newly diagnosed AML who are FLT3-positive, as detected by an FDA-approved test, and who are eligible to receive standard induction and consolidation chemotherapy.
Breakthrough therapy designation is intended to expedite the development and review of new medicines intended to treat serious or life-threatening conditions. The therapy must demonstrate substantial improvement over an available therapy on at least one clinically significant endpoint.
The designation includes all of the fast track program features, as well as more intensive FDA guidance on an efficient drug development program.
Phase 3 trial
The breakthrough designation for midostaurin is primarily based on the results of the phase 3 RATIFY trial, which were presented at the 2015 ASH Annual Meeting.
The trial included 717 patients with newly diagnosed, FLT3-positive AML who were younger than 60 at enrollment. All of the patients received standard induction and consolidation therapy. Roughly half also received midostaurin (n=360), while the other half received placebo (n=357).
Patients who received midostaurin experienced a significant improvement in overall survival (hazard ratio=0.77, P=0.0074). The median overall survival was 74.4 months in the midostaurin arm and 25.6 months in the placebo arm.
The median event-free survival was 8 months in the midostaurin arm and 3.6 months in the placebo arm (P=0.0032). The 5-year event-free survival was 27.5% for midostaurin and 19.3% for placebo.
There was no significant difference between the treatment arms with regard to most non-hematologic grade 3/4 adverse events. The exception was rash/desquamation, which occurred in 13% of patients in the midostaurin arm and 8% of patients in the placebo arm (P=0.02).
Other grade 3/4 non-hematologic events occurring in 10% of patients or more included, in the midostaurin and placebo arms, respectively: febrile neutropenia (81%, 82%), infection (40%, 38%), diarrhea (15%, 16%), hypokalemia (13%, 17%), pain (13%, 13%), other infection (12%, 12%), ALT/SGPT (12%, 9%), and fatigue (9%, 11%).
There were 18 deaths (5%) in the midostaurin arm and 19 (5.3%) in the placebo arm during induction and consolidation.
Midostaurin development
Novartis has opened a Global Individual Patient Program (compassionate use program) and a US Expanded Treatment Protocol (ETP) to enable midostaurin access. Patients 18 years of age and older with newly diagnosed FLT3-mutated AML who are able to receive standard induction and consolidation therapy will be considered.
To help identify patients who may have a FLT3 mutation and potentially benefit from treatment with midostaurin, Novartis is collaborating with Invivoscribe Technologies, Inc. which is leading regulatory submissions for a companion diagnostic.
Midostaurin is also being investigated for the treatment of aggressive systemic mastocytosis/mast cell leukemia.
Health Canada approves ruxolitinib for PV

Image courtesy of AFIP
Health Canada has approved the JAK1/2 inhibitor ruxolitinib (Jakavi) for the control of hematocrit in adult patients with polycythemia vera (PV) that is resistant to or intolerant of a cytoreductive agent.
Ruxolitinib is the first targeted treatment approved to treat PV in Canada.
The approval is based on results of the phase 3 RESPONSE trial, which showed that ruxolitinib could provide hematocrit control without phlebotomy in patients with PV.
For RESPONSE, researchers compared ruxolitinib to best available therapy (BAT) for PV. The trial was sponsored by Incyte Corporation and Novartis Pharmaceuticals, the companies developing ruxolitinib.
The study’s primary endpoint was the proportion of patients who achieved hematocrit control and were not eligible for phlebotomy from weeks 8 through 32 (with no more than 1 instance of phlebotomy eligibility between randomization and week 8) and who saw a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
The primary endpoint was met by significantly more patients in the ruxolitinib arm than the BAT arm— 20.9% and 0.9%, respectively (P<0.0001).
Sixty percent of patients in the ruxolitinib arm achieved hematocrit control, as did 19.6% of patients in the BAT arm. The percentage of patients who had at least a 35% reduction in spleen volume was 38.2% in the ruxolitinib arm and 0.9% in the BAT arm.
The proportion of patients achieving a complete hematologic remission at week 32 was 23.6% in the ruxolitinib arm and 8.9% in the BAT arm (P=0.0028). The proportion of patients achieving a durable primary response at week 48 was 19.1% in the ruxolitinib arm and 0.9% in the BAT arm (P<0.0001).
At 80 weeks, the most common adverse events in the ruxolitinib arm were headache (22%), diarrhea (20%), pruritus (20%), and fatigue (17%). Grade 3 or 4 anemia and thrombocytopenia occurred in 2% and 6% of patients, respectively. Five percent of patients discontinued ruxolitinib due to adverse events.
Image courtesy of AFIP
Health Canada has approved the JAK1/2 inhibitor ruxolitinib (Jakavi) for the control of hematocrit in adult patients with polycythemia vera (PV) that is resistant to or intolerant of a cytoreductive agent.
Ruxolitinib is the first targeted treatment approved to treat PV in Canada.
The approval is based on results of the phase 3 RESPONSE trial, which showed that ruxolitinib could provide hematocrit control without phlebotomy in patients with PV.
For RESPONSE, researchers compared ruxolitinib to best available therapy (BAT) for PV. The trial was sponsored by Incyte Corporation and Novartis Pharmaceuticals, the companies developing ruxolitinib.
The study’s primary endpoint was the proportion of patients who achieved hematocrit control and were not eligible for phlebotomy from weeks 8 through 32 (with no more than 1 instance of phlebotomy eligibility between randomization and week 8) and who saw a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
The primary endpoint was met by significantly more patients in the ruxolitinib arm than the BAT arm— 20.9% and 0.9%, respectively (P<0.0001).
Sixty percent of patients in the ruxolitinib arm achieved hematocrit control, as did 19.6% of patients in the BAT arm. The percentage of patients who had at least a 35% reduction in spleen volume was 38.2% in the ruxolitinib arm and 0.9% in the BAT arm.
The proportion of patients achieving a complete hematologic remission at week 32 was 23.6% in the ruxolitinib arm and 8.9% in the BAT arm (P=0.0028). The proportion of patients achieving a durable primary response at week 48 was 19.1% in the ruxolitinib arm and 0.9% in the BAT arm (P<0.0001).
At 80 weeks, the most common adverse events in the ruxolitinib arm were headache (22%), diarrhea (20%), pruritus (20%), and fatigue (17%). Grade 3 or 4 anemia and thrombocytopenia occurred in 2% and 6% of patients, respectively. Five percent of patients discontinued ruxolitinib due to adverse events.
Image courtesy of AFIP
Health Canada has approved the JAK1/2 inhibitor ruxolitinib (Jakavi) for the control of hematocrit in adult patients with polycythemia vera (PV) that is resistant to or intolerant of a cytoreductive agent.
Ruxolitinib is the first targeted treatment approved to treat PV in Canada.
The approval is based on results of the phase 3 RESPONSE trial, which showed that ruxolitinib could provide hematocrit control without phlebotomy in patients with PV.
For RESPONSE, researchers compared ruxolitinib to best available therapy (BAT) for PV. The trial was sponsored by Incyte Corporation and Novartis Pharmaceuticals, the companies developing ruxolitinib.
The study’s primary endpoint was the proportion of patients who achieved hematocrit control and were not eligible for phlebotomy from weeks 8 through 32 (with no more than 1 instance of phlebotomy eligibility between randomization and week 8) and who saw a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
The primary endpoint was met by significantly more patients in the ruxolitinib arm than the BAT arm— 20.9% and 0.9%, respectively (P<0.0001).
Sixty percent of patients in the ruxolitinib arm achieved hematocrit control, as did 19.6% of patients in the BAT arm. The percentage of patients who had at least a 35% reduction in spleen volume was 38.2% in the ruxolitinib arm and 0.9% in the BAT arm.
The proportion of patients achieving a complete hematologic remission at week 32 was 23.6% in the ruxolitinib arm and 8.9% in the BAT arm (P=0.0028). The proportion of patients achieving a durable primary response at week 48 was 19.1% in the ruxolitinib arm and 0.9% in the BAT arm (P<0.0001).
At 80 weeks, the most common adverse events in the ruxolitinib arm were headache (22%), diarrhea (20%), pruritus (20%), and fatigue (17%). Grade 3 or 4 anemia and thrombocytopenia occurred in 2% and 6% of patients, respectively. Five percent of patients discontinued ruxolitinib due to adverse events.
Development of myelofibrosis drug on hold
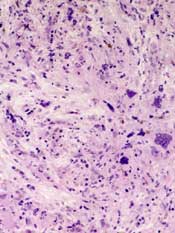
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold.
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold.
The US Food and Drug Administration (FDA) has placed a full clinical hold on trials conducted under the investigational new drug application for pacritinib, a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The hold means all patients currently on pacritinib must stop taking the drug immediately, and no patients can be enrolled on a pacritinib trial or start pacritinib as initial or crossover treatment.
In addition, CTI BioPharma has withdrawn the new drug application for pacritinib while the company reviews data from the phase 3 PERSIST-2 trial.
The FDA’s decision to place a full clinical hold on pacritinib trials was due to interim results from PERSIST-2. The aim of this trial was to compare pacritinib to best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
The overall survival results from PERSIST-2 indicate that pacritinib had a detrimental effect on survival, which is consistent with results from the PERSIST-1 trial. The deaths in pacritinib-treated patients on PERSIST-2 include intracranial hemorrhage, cardiac failure, and cardiac arrest.
Based on these results, the FDA has made recommendations for CTI BioPharma that supersede the agency’s previous recommendations.
On February 4, 2016, the FDA placed a partial clinical hold on pacritinib trials and made related recommendations for CTI BioPharma, advising that the company modify trial protocols and take other actions in compliance with the partial clinical hold.
Now that pacritinib trials are on full clinical hold, the FDA is recommending that CTI BioPharma conduct dose exploration studies for pacritinib in patients with MF and submit final study reports and datasets for PERSIST-1 and PERSIST-2.
The FDA is also recommending that CTI BioPharma provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, make certain modifications to protocols, and request a meeting with the FDA prior to submitting a response to the full clinical hold.
CTI BioPharma said all clinical investigators worldwide have been notified of the hold.
MF drug trials placed on partial clinical hold

The US Food and Drug Administration (FDA) has placed a partial clinical hold on trials conducted under the investigational new drug
(IND) application for pacritinib.
Pacritinib is a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The partial clinical hold impacts part of the clinical work currently being conducted under the pacritinib IND and will also affect planned clinical trials.
The FDA said the reasons for the partial clinical hold are excess mortality and other adverse events in pacritinib-treated patients (compared to the control arm) in the PERSIST-1 trial.
The excess mortality was most evident during the non-randomized crossover period following the initial 24 weeks of randomized treatment, during which patients in the control arm could switch to pacritinib treatment.
Under the partial clinical hold, investigators may not enroll new patients or start pacritinib as initial or crossover treatment, and patients not deriving benefit after 30 weeks of pacritinib treatment must stop using pacritinib.
In addition, the FDA has recommended that CTI BioPharma make certain modifications to protocols, provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, and take certain other actions.
CTI BioPharma said it intends to implement the FDA’s recommendations, and all clinical investigators worldwide have been notified of the partial clinical hold.
Just before the FDA notified CTI BioPharma of the partial clinical hold, the company completed enrollment in the phase 3 PERSIST-2 trial.
In PERSIST-2, researchers are comparing the efficacy and safety of pacritinib and best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Under the partial clinical hold, patients on this trial who are currently receiving pacritinib may continue to do so unless they are not deriving benefit after 30 weeks of pacritinib treatment, and crossover of patients from the control arm to the pacritinib arm will not be allowed.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on trials conducted under the investigational new drug
(IND) application for pacritinib.
Pacritinib is a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The partial clinical hold impacts part of the clinical work currently being conducted under the pacritinib IND and will also affect planned clinical trials.
The FDA said the reasons for the partial clinical hold are excess mortality and other adverse events in pacritinib-treated patients (compared to the control arm) in the PERSIST-1 trial.
The excess mortality was most evident during the non-randomized crossover period following the initial 24 weeks of randomized treatment, during which patients in the control arm could switch to pacritinib treatment.
Under the partial clinical hold, investigators may not enroll new patients or start pacritinib as initial or crossover treatment, and patients not deriving benefit after 30 weeks of pacritinib treatment must stop using pacritinib.
In addition, the FDA has recommended that CTI BioPharma make certain modifications to protocols, provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, and take certain other actions.
CTI BioPharma said it intends to implement the FDA’s recommendations, and all clinical investigators worldwide have been notified of the partial clinical hold.
Just before the FDA notified CTI BioPharma of the partial clinical hold, the company completed enrollment in the phase 3 PERSIST-2 trial.
In PERSIST-2, researchers are comparing the efficacy and safety of pacritinib and best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Under the partial clinical hold, patients on this trial who are currently receiving pacritinib may continue to do so unless they are not deriving benefit after 30 weeks of pacritinib treatment, and crossover of patients from the control arm to the pacritinib arm will not be allowed.
The US Food and Drug Administration (FDA) has placed a partial clinical hold on trials conducted under the investigational new drug
(IND) application for pacritinib.
Pacritinib is a JAK2/FLT3 inhibitor being developed by CTI BioPharma for the treatment of myelofibrosis (MF).
The partial clinical hold impacts part of the clinical work currently being conducted under the pacritinib IND and will also affect planned clinical trials.
The FDA said the reasons for the partial clinical hold are excess mortality and other adverse events in pacritinib-treated patients (compared to the control arm) in the PERSIST-1 trial.
The excess mortality was most evident during the non-randomized crossover period following the initial 24 weeks of randomized treatment, during which patients in the control arm could switch to pacritinib treatment.
Under the partial clinical hold, investigators may not enroll new patients or start pacritinib as initial or crossover treatment, and patients not deriving benefit after 30 weeks of pacritinib treatment must stop using pacritinib.
In addition, the FDA has recommended that CTI BioPharma make certain modifications to protocols, provide certain notifications, revise relevant statements in the related investigator’s brochure and informed consent documents, and take certain other actions.
CTI BioPharma said it intends to implement the FDA’s recommendations, and all clinical investigators worldwide have been notified of the partial clinical hold.
Just before the FDA notified CTI BioPharma of the partial clinical hold, the company completed enrollment in the phase 3 PERSIST-2 trial.
In PERSIST-2, researchers are comparing the efficacy and safety of pacritinib and best available therapy in patients with thrombocytopenia and primary MF, post-polycythemia vera MF, or post-essential thrombocythemia MF.
Under the partial clinical hold, patients on this trial who are currently receiving pacritinib may continue to do so unless they are not deriving benefit after 30 weeks of pacritinib treatment, and crossover of patients from the control arm to the pacritinib arm will not be allowed.