User login
Lactic acidosis: Clinical implications and management strategies
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
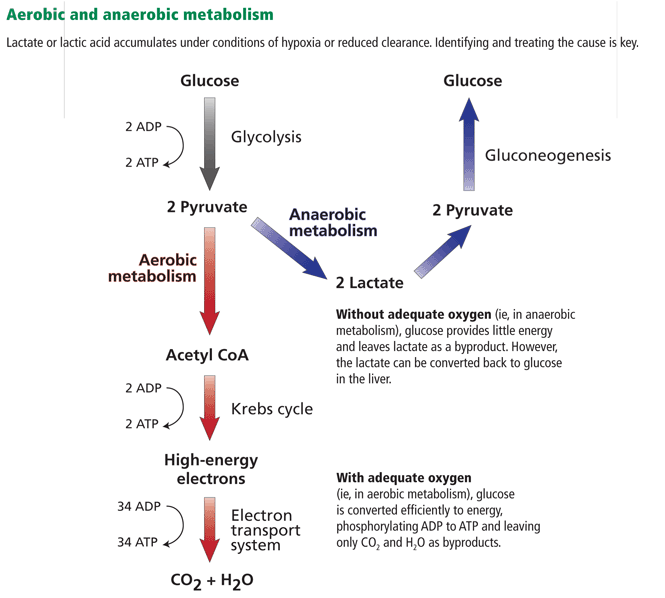
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.

The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
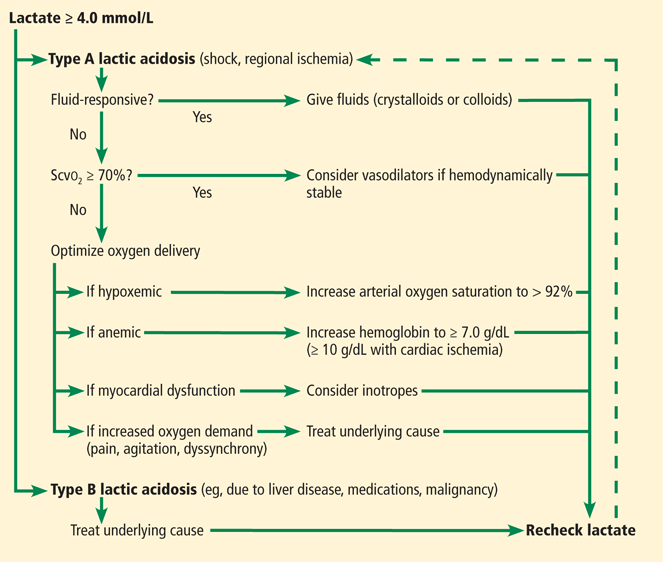
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
Physicians are paying more attention to serum lactate levels in hospitalized patients than in the past, especially with the advent of point-of-care testing. Elevated lactate levels are associated with tissue hypoxia and hypoperfusion but can also be found in a number of other conditions. Therefore, confusion can arise as to how to interpret elevated levels and subsequently manage these patients in a variety of settings.
In this review, we discuss the mechanisms underlying lactic acidosis, its prognostic implications, and its use as a therapeutic target in treating patients in septic shock and other serious disorders.
LACTATE IS A PRODUCT OF ANAEROBIC RESPIRATION
Lactate, or lactic acid, is produced from pyruvate as an end product of glycolysis under anaerobic conditions (Figure 1). It is produced in most tissues in the body, but primarily in skeletal muscle, brain, intestine, and red blood cells. During times of stress, lactate is also produced in the lungs, white blood cells, and splanchnic organs.
Most lactate in the blood is cleared by the liver, where it is the substrate for gluconeogenesis, and a small amount is cleared by the kidneys.1,2 The entire pathway by which lactate is produced and converted back to glucose is called the Cori cycle.
NORMAL LEVELS ARE LESS THAN ABOUT 2.0 MMOL/L
In this review, we will present lactate levels in the SI units of mmol/L (1 mmol/L = 9 mg/dL).
Basal lactate production is approximately 0.8 mmol/kg body weight/hour. The average normal arterial blood lactate level is approximately 0.620 mmol/L and the venous level is slightly higher at 0.997 mmol/L,3 but overall, arterial and venous lactate levels correlate well.
Normal lactate levels are less than 2 mmol/L,4 intermediate levels range from 2 to less than 4 mmol/L, and high levels are 4 mmol/L or higher.5
To minimize variations in measurement, blood samples should be drawn without a tourniquet into tubes containing fluoride, placed on ice, and processed quickly (ideally within 15 minutes).
INCREASED PRODUCTION, DECREASED CLEARANCE, OR BOTH
An elevated lactate level can be the result of increased production, decreased clearance, or both (as in liver dysfunction).
Type A lactic acidosis—due to hypoperfusion and hypoxia—occurs when there is a mismatch between oxygen delivery and consumption, with resultant anaerobic glycolysis.
The guidelines from the Surviving Sepsis Campaign6 emphasize using lactate levels to diagnose patients with sepsis-induced hypoperfusion. However, hyperlactatemia can indicate inadequate oxygen delivery due to any type of shock (Table 1).
Type B lactic acidosis—not due to hypoperfusion—occurs in a variety of conditions (Table 1), including liver disease, malignancy, use of certain medications (eg, metformin, epinephrine), total parenteral nutrition, human immunodeficiency virus infection, thiamine deficiency, mitochondrial myopathies, and congenital lactic acidosis.1–3,7 Yet other causes include trauma, excessive exercise, diabetic ketoacidosis, ethanol intoxication, dysfunction of the enzyme pyruvate dehydrogenase, and increased muscle degradation leading to increased production of pyruvate. In these latter scenarios, glucose metabolism exceeds the oxidation capacity of the mitochondria, and the rise in pyruvate concentration drives lactate production.8,9 Mitochondrial dysfunction and subsequent deficits in cellular oxygen use can also result in persistently high lactate levels.10
In some situations, patients with mildly elevated lactic acid levels in type B lactic acidosis can be monitored to ensure stability, rather than be treated aggressively.
HIGHER LEVELS AND LOWER CLEARANCE PREDICT DEATH
The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
Lactate levels and mortality rate
Shapiro et al11 showed that increases in lactate level are associated with proportional increases in the mortality rate. Mikkelsen et al12 showed that intermediate levels (2.0–3.9 mmol/L) and high levels (≥ 4 mmol/L) of serum lactate are associated with increased risk of death independent of organ failure and shock. Patients with mildly elevated and intermediate lactate levels and sepsis have higher rates of in-hospital and 30-day mortality, which correlate with the baseline lactate level.13
In a post hoc analysis of a randomized controlled trial, patients with septic shock who presented to the emergency department with hypotension and a lactate level higher than 2 mmol/L had a significantly higher in-hospital mortality rate than those who presented with hypotension and a lactate level of 2 mmol/L or less (26% vs 9%, P < .0001).14 These data suggest that elevated lactate levels may have a significant prognostic role, independent of blood pressure.
Slower clearance
The prognostic implications of lactate clearance (reductions in lactate levels over time, as opposed to a single value in time), have also been evaluated.
Lactate clearance of at least 10% at 6 hours after presentation has been associated with a lower mortality rate than nonclearance (19% vs 60%) in patients with sepsis or septic shock with elevated levels.15–17 Similar findings have been reported in a general intensive care unit population,18 as well as a surgical intensive care population.sup>19
Puskarich et al20 have also shown that lactate normalization to less than 2 mmol/L during early sepsis resuscitation is the strongest predictor of survival (odds ratio [OR] 5.2), followed by lactate clearance of 50% (OR 4.0) within the first 6 hours of presentation. Not only is lactate clearance associated with improved outcomes, but a faster rate of clearance after initial presentation is also beneficial.15,16,18
Lactate clearance over a longer period (> 6 hours) has not been studied in patients with septic shock. However, in the general intensive care unit population, therapy guided by lactate clearance for the first 8 hours after presentation has shown a reduction in mortality rate.18 There are no data available on outcomes of lactate-directed therapy beyond 8 hours, but lactate concentration and lactate clearance at 24 hours correlate with the 28-day mortality rate.21
Cryptic shock
Cryptic shock describes a state in a subgroup of patients who have elevated lactate levels and global tissue hypoxia despite being normotensive or even hypertensive. These patients have a higher mortality rate independent of blood pressure. Jansen et al18 found that patients with a lactate level higher than 4 mmol/L and preserved blood pressure had a mortality rate of 15%, while those without shock or hyperlactatemia had a mortality rate of 2.5%. In addition, patients with an elevated lactate level in the absence of hypotension have mortality rates similar to those in patients with high lactate levels and hypotension refractory to fluid boluses, suggesting the presence of tissue hypoxia even in these normotensive patients.6
HOW TO APPROACH AN ELEVATED LACTATE LEVEL
An elevated lactate level should prompt an evaluation for causes of decreased oxygen delivery, due either to a systemic low-flow state (as a result of decreased cardiac output) or severe anemia, or to regionally decreased perfusion, (eg, limb or mesenteric ischemia). If tissue hypoxia is ruled out after an exhaustive workup, consideration should be given to causes of hyperlactatemia without concomitant tissue hypoxia (type B acidosis).
Treatment differs depending on the underlying mechanism of the lactate elevation; nevertheless, treatment is mostly related to optimizing oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both (Figure 2). The specific treatment differs based on the shock state, but there are similarities that can guide the clinician.
FLUID SUPPORT
Giving fluids, with a goal of improving cardiac output, remains a cornerstone of initial therapy for most shock states.22,23
How much fluid?
Fluids should be given until the patient is no longer preload-dependent, although there is much debate about which assessment strategy should be used to determine if cardiac output will improve with more fluid (ie, fluid-responsiveness).24 In many cases, fluid resuscitation alone may be enough to restore hemodynamic stability, improve tissue perfusion, and reduce elevated lactate concentrations.25
The decision to give more fluids should not be made lightly, though, as a more positive fluid balance early in the course of septic shock and over 4 days has been associated with a higher mortality rate.26 Additionally, pushing fluids in patients with cardiogenic shock due to impaired left ventricular systolic function may lead to or worsen pulmonary edema. Therefore, the indiscriminate use of fluids should be avoided.
Which fluids?
Despite years of research, controversy persists about whether crystalloids or colloids are better for resuscitation. Randomized trials in heterogeneous intensive care unit patients have not detected differences in 28-day mortality rates between those allocated to crystalloids or 4% albumin27 and those allocated to crystalloids or hydroxyethyl starch.28
Hydroxyethyl starch may not be best. In a study of patients with severe sepsis, those randomized to receive hydroxyethyl starch had a higher 90-day mortality rate than patients randomized to crystalloids (51% vs 43%, P = .03).29 A sequential prospective before-and-after study did not detect a difference in the time to normalization (< 2.2 mmol/L) of lactate (P = .68) or cessation of vasopressors (P = .11) in patients with severe sepsis who received fluid resuscitation with crystalloids, gelatin, or hydroxyethyl starch. More patients who received hydroxyethyl starch in these studies developed acute kidney injury than those receiving crystalloids.28–30
Taken together, these data strongly suggest hydroxyethyl starch should not be used for fluid resuscitation in the intensive care unit.
Normal saline or albumin? Although some data suggest that albumin may be preferable to 0.9% sodium chloride in patients with severe sepsis,31,32 these analyses should be viewed as hypothesis-generating. There do not seem to be differences between fluid types in terms of subsequent serum lactate concentrations or achievement of lactate clearance goals.28–30 Until further studies are completed, both albumin and crystalloids are reasonable for resuscitation.
Caironi et al33 performed an open-label study comparing albumin replacement (with a goal serum albumin concentration of 3 g/dL) plus a crystalloid solution vs a crystalloid solution alone in patients with severe sepsis or septic shock. They detected no difference between the albumin and crystalloid groups in mortality rates at 28 days (31.8% vs 32.0%, P = .94) or 90 days (41.1% vs 43.6%, P = .29). However, patients in the albumin group had a shorter time to cessation of vasoactive agents (median 3 vs 4 days, P = .007) and lower cardiovascular Sequential Organ Failure Assessment subscores (median 1.20 vs 1.42, P = .03), and more frequently achieved a mean arterial pressure of at least 65 mm Hg within 6 hours of randomization (86.0% vs 82.5%, P = .04).
Although serum lactate levels were lower in the albumin group at baseline (1.7 mmol/L vs 1.8 mmol/L, P = .05), inspection of the data appears to show a similar daily lactate clearance rate between groups over the first 7 study days (although these data were not analyzed by the authors). Achievement of a lactate level lower than 2 mmol/L on the first day of therapy was not significantly different between groups (73.4% vs 72.5%, P = .11).33
In a post hoc subgroup analysis, patients with septic shock at baseline randomized to albumin had a lower 90-day mortality rate than patients randomized to crystalloid solutions (RR 0.87, 95% CI 0.77–0.99). There was no difference in the 90-day mortality rate in patients without septic shock (RR 1.13, 95% CI 0.92–1.39, P = .03 for heterogeneity).33
These data suggest that albumin replacement may not improve outcomes in patients with severe sepsis, but may have advantages in terms of hemodynamic variables (and potentially mortality) in patients with septic shock. The role of albumin replacement in patients with septic shock warrants further study.
VASOPRESSORS
Vasopressors, inotropes, or both should be given to patients who have signs of hypoperfusion (including elevated lactate levels) despite preload optimization or ongoing fluid administration. The most appropriate drug depends on the goal: vasopressors are used to increase systemic vascular resistance, while inotropes are used to improve cardiac output and oxygen delivery.
Blood pressure target
The Surviving Sepsis Campaign guidelines recommend a mean arterial blood pressure target of at least 65 mm Hg during initial resuscitation and when vasopressors are applied for patients with septic shock.22 This recommendation is based on small studies that did not show differences in serum lactate levels or regional blood flow when the mean arterial pressure was elevated above 65 mm Hg with norepinephrine.34,35 However, the campaign guidelines note that the mean arterial pressure goal must be individualized in order to achieve optimal perfusion.
A large, open-label trial36 detected no difference in 28-day mortality rates in patients with septic shock between those allocated to a mean arterial pressure goal of 80 to 85 mm Hg or 65 to 70 mm Hg (36.6% vs 34.0%, P = .57). Although lactate levels did not differ between groups, the incidence of new-onset atrial fibrillation was higher in the higher-target group (6.7% vs 2.8%, P = .02). Fewer patients with chronic hypertension needed renal replacement therapy in the higher pressure group, further emphasizing the need to individualize the mean arterial pressure goal for patients in shock.36
Which vasopressor agent?
Dopamine and norepinephrine have traditionally been the preferred initial vasopressors for patients with shock. Until recently there were few data to guide selection between the two, but this is changing.
In a 2010 study of 1,679 patients with shock requiring vasopressors, there was no difference in the 28-day mortality rate between patients randomized to dopamine or norepinephrine (53% vs 49%, P = .10).37 Patients allocated to dopamine, though, had a higher incidence of arrhythmias (24% vs 12%, P < .001) and more frequently required open-label norepinephrine (26% vs 20%, P < .001). Although lactate levels and the time to achievement of a mean arterial pressure of 65 mm Hg were similar between groups, patients allocated to norepinephrine had more vasopressor-free days through day 28.
An a priori-planned subgroup analysis evaluated the influence of the type of shock on patient outcome. Patients with cardiogenic shock randomized to dopamine had a higher mortality rate than those randomized to norepinephrine (P = .03). However, the overall effect of treatment did not differ among the shock subgroups (interaction P = .87), suggesting that the reported differences in mortality according to subgroup may be spurious.
In a 2012 meta-analysis of patients with septic shock, dopamine use was associated with a higher mortality rate than norepinephrine use.38
In light of these data, norepinephrine should be preferred over dopamine as the initial vasopressor in most types of shock.
Epinephrine does not offer an outcome advantage over norepinephrine and may be associated with a higher incidence of adverse events.39–42 Indeed, in a study of patients with septic shock, lactate concentrations on the first day after randomization were significantly higher in patients allocated to epinephrine than in patients allocated to norepinephrine plus dobutamine.39 Similar effects on lactate concentrations with epinephrine were seen in patients with various types of shock40 and in those with cardiogenic shock.42
These differences in lactate concentrations may be directly attributable to epinephrine. Epinephrine can increase lactate concentrations through glycolysis and pyruvate dehydrogenase activation by stimulation of sodium-potassium ATPase activity via beta-2 adrenergic receptors in skeletal muscles,43 as well as decrease splanchnic perfusion.42,44,45 These effects may preclude using lactate clearance as a resuscitation goal in patients receiving epinephrine. Epinephrine is likely best reserved for patients with refractory shock,22 particularly those in whom cardiac output is known to be low.
Phenylephrine, essentially a pure vasoconstrictor, should be avoided in low cardiac output states and is best reserved for patients who develop a tachyarrhythmia on norepinephrine.22
Vasopressin, also a pure vasoconstrictor that should be avoided in low cardiac output states, has been best studied in patients with vasodilatory shock. Although controversy exists on the mortality benefits of vasopressin in vasodilatory shock, it is a relatively safe drug with consistent norepinephrine-sparing effects when added to existing norepinephrine therapy.46,47 In patients with less severe septic shock, including those with low lactate concentrations, adding vasopressin to norepinephrine instead of continuing norepinephrine alone may confer a mortality advantage.48
OTHER MEASURES TO OPTIMIZE OXYGEN DELIVERY
In circulatory shock from any cause, tissue oxygen demand exceeds oxygen delivery. Once arterial oxygenation and hemoglobin levels (by packed red blood cell transfusion) have been optimized, cardiac output is the critical determinant of oxygen delivery. Cardiac output may be augmented by ensuring adequate preload (by fluid resuscitation) or by giving inotropes or vasodilators.
The optimal cardiac output is difficult to define, and the exact marker for determining when cardiac output should be augmented is unclear. A strategy of increasing cardiac output to predefined “supranormal” levels was not associated with a lower mortality rate.49 Therefore, the decision to augment cardiac output must be individualized and will likely vary in the same patient over time.23
A reasonable approach to determining when augmentation of cardiac output is necessary was proposed in a study by Rivers et al.50 In that study, in patients randomized to early goal-directed therapy, inotropes were recommended when the central venous oxygenation saturation (Scvo2) was below 70% despite adequate fluid resuscitation (central venous pressure ≥ 8 mm Hg) and hematocrits were higher than 30%.
When an inotrope is indicated to improve cardiac output, dobutamine is usually the preferred agent. Dobutamine has a shorter half-life (allowing for easier titration) and causes less hypotension (assuming preload has been optimized) than phosphodiesterase type III inhibitors such as milrinone.
Mechanical support devices, such as intra-aortic balloon counterpulsation, and vasodilators can also be used to improve tissue perfusion in selected patients with low cardiac output syndromes.
USING LACTATE LEVELS TO GUIDE THERAPY
Lactate levels above 4.0 mmol/L
Lactate may be a useful marker for determining whether organ dysfunction is present and, hence, what course of therapy should be given, especially in sepsis. A serum lactate level higher than 4.0 mmol/L has been used as the trigger to start aggressive resuscitation in patients with sepsis.50,51
Traditionally, as delineated by Rivers et al50 in their landmark study of early goal-directed therapy, this entailed placing an arterial line and a central line for hemodynamic monitoring, with specific interventions directed at increasing the central venous pressure, mean arterial pressure, and central venous oxygen saturation.50 However, a recent study in a similar population of patients with sepsis with elevated lactate found no significant advantage of protocol-based resuscitation over care provided according to physician judgment, and no significant benefit in central venous catheterization and hemodynamic monitoring in all patients.51
Lactate clearance: 10% or above at 8 hours?
Regardless of the approach chosen, decreasing lactate levels can be interpreted as an adequate response to the interventions provided. As a matter of fact, several groups of investigators have also demonstrated the merits of lactate clearance alone as a prognostic indicator in patients requiring hemodynamic support.
McNelis et al52 retrospectively evaluated 95 postsurgical patients who required hemodynamic monitoring.52,53 The authors found that the slower the lactate clearance, the higher the mortality rate.
Given the prognostic implications of lactate clearance, investigators have evaluated whether lactate clearance could be used as a surrogate resuscitation goal for optimizing oxygen delivery. Using lactate clearance may have significant practical advantages over using central venous oxygen saturation, since it does not require a central venous catheter or continuous oximetric monitoring.
In a study comparing these two resuscitation end points, patients were randomized to a goal of either central venous oxygen saturation of 70% or more or lactate clearance of 10% or more within the first 6 hours after presentation as a marker of oxygen delivery.53 Mortality rates were similar with either strategy. Of note, only 10% of the patients actually required therapies to improve their oxygen delivery. Furthermore, there were no differences in the treatments given (including fluids, vasopressors, inotropes, packed red blood cells) throughout the treatment period.
These findings provide several insights. First, few patients admitted to the emergency department with severe sepsis and treated with an initial quantitative resuscitation protocol require additional therapy for augmenting oxygen delivery. Second, lactate clearance, in a setting where initial resuscitation with fluids and vasopressors restores adequate oxygen delivery for the majority of patients, is likely as good a target for resuscitation as central venous oxygen saturation.
This study, however, does not address the question of whether lactate clearance is useful as an additional marker of oxygen delivery (in conjunction with central venous oxygen saturation). Indeed, caution should be taken to target central venous oxygen saturation goals alone, as patients with septic shock presenting with venous hyperoxia (central venous oxygen saturation > 89%) have been shown to have a higher mortality rate than patients with normoxia (central venous oxygen saturation 71%–89%).54
This was further demonstrated by Arnold et al in a study of patients presenting to the emergency department with severe sepsis.15 In this study, significant discordance between central venous oxygen saturation and lactate clearance was seen, where 79% of patients with less than 10% lactate clearance had concomitant central venous oxygen saturation of 70% or greater.
Jansen et al18 evaluated the role of targeting lactate clearance in conjunction with central venous oxygen saturation monitoring. In this study, critically ill patients with elevated lactate and inadequate lactate clearance were randomized to usual care or to resuscitation to adequate lactate clearance (20% or more). The therapies to optimize oxygen delivery were given according to the central venous oxygen saturation. Overall, after adjustment for predefined risk factors, the in-hospital mortality rate was lower in the lactate clearance group. This may signify that patients with sepsis and central venous oxygen saturation of 70% or more may continue to have poor lactate clearance, warranting further treatment.
Taken together, serum lactate may be helpful for prognostication, determination of course of therapy, and quantification for tissue hypoperfusion for targeted therapies. Figure 2 presents our approach to an elevated lactate level. As performed in the study by Jansen et al,18 it seems reasonable to measure lactate levels every 2 hours for the first 8 hours of resuscitation in patients with type A lactic acidosis. These levels should be interpreted in the context of lactate clearance (at least 10%, but preferably 20%) and normalization, and should be treated with an approach similar to the one outlined in Figure 2.
TREATING TYPE B LACTIC ACIDOSIS (NORMAL PERFUSION AND OXYGENATION)
Treating type B lactic acidosis is quite different because the goal is not to correct mismatches in oxygen consumption and delivery. Since most cases are due to underlying conditions such as malignancy or medications, treatment should be centered around eliminating the cause (eg, treat the malignancy, discontinue the offending medication). The main reason for treatment is to alleviate the harmful effects of acidosis. For example, acidosis can result in a negative inotropic effect.
Sodium bicarbonate, dichloroacetate, carbicarb, and tromethamine have all been studied in the management of type B lactic acidosis, with little success.55,56
Renal replacement therapy has had some success in drug-induced lactic acidosis.57,58
l-carnitine has had promising results in treating patients with human immunodeficiency virus infection, since these patients are carnitine-deficient and carnitine plays an important role in mitochondrial function.59
Thiamine and biotin deficiencies can occur in patients receiving total parenteral nutrition without vitamins and in patients who drink alcohol heavily and can cause lactic acidosis. These nutrients should be supplemented accordingly.
Treatment of mitochondrial disorders includes antioxidants (coenzyme Q10, vitamin C, vitamin E) and amino acids (l-arginine).60
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
- Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc 2013; 88:1127–1140.
- Fuller BM, Dellinger RP. Lactate as a hemodynamic marker in the critically ill. Curr Opin Crit Care 2012; 18:267–272.
- Fall PJ, Szerlip HM. Lactic acidosis: from sour milk to septic shock. J Intensive Care Med 2005; 20:255–271.
- Kruse O, Grunnet N, Barfod C. Blood lactate as a predictor for in-hospital mortality in patients admitted acutely to hospital: a systematic review. Scand J Trauma Resusc Emerg Med 2011;19:74.
- Howell MD, Donnino M, Clardy P, Talmor D, Shapiro NI. Occult hypoperfusion and mortality in patients with suspected infection. Intensive Care Med 2007; 33:1892–1899.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Outcomes of patients undergoing early sepsis resuscitation for cryptic shock compared with overt shock. Resuscitation 2011; 82:1289–1293.
- Bakker J, Nijsten MW, Jansen TC. Clinical use of lactate monitoring in critically ill patients. Ann Intensive Care 2013; 3:12.
- Levy B, Gibot S, Franck P, Cravoisy A, Bollaert PE. Relation between muscle Na+K+ ATPase activity and raised lactate concentrations in septic shock: a prospective study. Lancet 2005; 365:871–875.
- Vary TC. Sepsis-induced alterations in pyruvate dehydrogenase complex activity in rat skeletal muscle: effects on plasma lactate. Shock 1996; 6:89–94.
- Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002; 360:219–223.
- Shapiro NI, Howell MD, Talmor D, et al. Serum lactate as a predictor of mortality in emergency department patients with infection. Ann Emerg Med 2005; 45:524–528.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009; 37:1670–1677.
- Liu V, Morehouse JW, Soule J, Whippy A, Escobar GJ. Fluid volume, lactate values, and mortality in sepsis patients with intermediate lactate values. Ann Am Thorac Soc 2013; 10:466–473.
- Sterling SA, Puskarich MA, Shapiro NI, et al; Emergency Medicine Shock Research Network (EMShockNET). Characteristics and outcomes of patients with vasoplegic versus tissue dysoxic septic shock. Shock 2013; 40:11–14.
- Arnold RC, Shapiro NI, Jones AE, et al; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of early lactate clearance as a determinant of survival in patients with presumed sepsis. Shock 2009; 32:35–39.
- Jones AE. Lactate clearance for assessing response to resuscitation in severe sepsis. Acad Emerg Med 2013; 20:844–847.
- Nguyen HB, Rivers EP, Knoblich BP, et al. Early lactate clearance is associated with improved outcome in severe sepsis and septic shock. Crit Care Med 2004; 32:1637–1642.
- Jansen TC, van Bommel J, Schoonderbeek FJ, et al; LACTATE study group. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med 2010; 182:752–761.
- Husain FA, Martin MJ, Mullenix PS, Steele SR, Elliott DC. Serum lactate and base deficit as predictors of mortality and morbidity. Am J Surg 2003; 185:485–491.
- Puskarich MA, Trzeciak S, Shapiro NI, et al. Whole blood lactate kinetics in patients undergoing quantitative resuscitation for severe sepsis and septic shock. Chest 2013; 143:1548–1553.
- Marty P, Roquilly A, Vallee F, et al. Lactate clearance for death prediction in severe sepsis or septic shock patients during the first 24 hours in intensive care unit: an observational study. Ann Intensive Care 2013; 3:3.
- Dellinger RP, Levy MM, Rhodes A, et al; Surviving Sepsis Campaign Guidelines Committee including the Pediatric Subgroup. Surviving sepsis campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013; 41:580–637.
- Vincent JL, De Backer D. Circulatory shock. N Engl J Med 2013; 369:1726–1734.
- Durairaj L, Schmidt GA. Fluid therapy in resuscitated sepsis: less is more. Chest 2008; 133:252–263.
- Vincent JL, Dufaye P, Berré J, Leeman M, Degaute JP, Kahn RJ. Serial lactate determinations during circulatory shock. Crit Care Med 1983; 11:449–451.
- Boyd JH, Forbes J, Nakada TA, Walley KR, Russell JA. Fluid resuscitation in septic shock: a positive fluid balance and elevated central venous pressure are associated with increased mortality. Crit Care Med 2011; 39:259–265.
- Finfer S, Bellomo R, Boyce N, French J, Myburgh J, Norton R; SAFE Study Investigators. A comparison of albumin and saline for fluid resuscitation in the intensive care unit. N Engl J Med 2004; 350:2247–2256.
- Myburgh JA, Finfer S, Bellomo R, et al; CHEST Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group. Hydroxyethyl starch or saline for fluid resuscitation in intensive care. N Engl J Med 2012; 367:1901–1911.
- Perner A, Haase N, Guttormsen AB, et al; 6S Trial Group; Scandinavian Critical Care Trials Group. Hydroxyethyl starch 130/0.42 versus Ringer’s acetate in severe sepsis. N Engl J Med 2012; 367:124–134.
- Bayer O, Reinhart K, Kohl M, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis: a prospective sequential analysis. Crit Care Med 2012; 40:2543–2551.
- Delaney AP, Dan A, McCaffrey J, Finfer S. The role of albumin as a resuscitation fluid for patients with sepsis: a systematic review and meta-analysis. Crit Care Med 2011; 39:386–391.
- SAFE Study Investigators; Finfer S, McEvoy S, Bellomo R, McArthur C, Myburgh J, Norton R. Impact of albumin compared to saline on organ function and mortality of patients with severe sepsis. Intensive Care Med 2011; 37:86–96.
- Caironi P, Tognoni G, Masson S, et al; ALBIOS Study Investigators. Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421.
- Bourgoin A, Leone M, Delmas A, Garnier F, Albanèse J, Martin C. Increasing mean arterial pressure in patients with septic shock: effects on oxygen variables and renal function. Crit Care Med 2005; 33:780–786.
- LeDoux D, Astiz ME, Carpati CM, Rackow EC. Effects of perfusion pressure on tissue perfusion in septic shock. Crit Care Med 2000; 28:2729–2732.
- Asfar P, Meziani F, Hamel JF, et al; SEPSISPAM Investigators. High versus low blood-pressure target in patients with septic shock. N Engl J Med 2014; 370:1583–1593.
- De Backer D, Biston P, Devriendt J, et al; SOAP II Investigators. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789.
- De Backer D, Aldecoa C, Njimi H, Vincent JL. Dopamine versus norepinephrine in the treatment of septic shock: a meta-analysis. Crit Care Med 2012; 40:725–730.
- Annane D, Vignon P, Renault A, et al: CATS Study Group. Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Lancet 2007; 370:676–684.
- Myburgh JA, Higgins A, Jovanovska A, Lipman J, Ramakrishnan N, Santamaria J; CAT Study investigators. A comparison of epinephrine and norepinephrine in critically ill patients. Intensive Care Med 2008; 34:2226–2234.
- Schmittinger CA, Torgersen C, Luckner G, Schröder DC, Lorenz I, Dünser MW. Adverse cardiac events during catecholamine vasopressor therapy: a prospective observational study. Intensive Care Med 2012; 38:950–958.
- Levy B, Perez P, Perny J, Thivilier C, Gerard A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011; 39:450–455.
- Watt MJ, Howlett KF, Febbraio MA, Spriet LL, Hargreaves M. Adrenaline increases skeletal muscle glycogenolysis, pyruvate dehydrogenase activation and carbohydrate oxidation during moderate exercise in humans. J Physiol 2001; 534:269–278.
- De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med 2003; 31:1659–1667.
- Levy B, Bollaert PE, Charpentier C, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med 1997; 23:282–287.
- Polito A, Parisini E, Ricci Z, Picardo S, Annane D. Vasopressin for treatment of vasodilatory shock: an ESICM systematic review and meta-analysis. Intensive Care Med 2012; 38:9–19.
- Serpa Neto A, Nassar APJ, Cardoso SO, et al. Vasopressin and terlipressin in adult vasodilatory shock: a systematic review and meta-analysis of nine randomized controlled trials. Crit Care 2012; 16:R154.
- Russell JA, Walley KR, Singer J, et al; VASST Investigators. Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 2008; 358:877–887.
- Gattinoni L, Brazzi L, Pelosi P, et al; for the SvO2 Collaborative Group. A trial of goal-oriented hemodynamic therapy in critically ill patients. N Engl J Med 1995; 333:1025–1032.
- Rivers E, Nguyen B, Havstad S, et al; Early Goal-Directed Therapy Collaborative Group. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377.
- ProCESS Investigators; Yealy DM, Kellum JA, Huang DT, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693.
- McNelis J, Marini CP, Jurkiewicz A, et al. Prolonged lactate clearance is associated with increased mortality in the surgical intensive care unit. Am J Surg 2001; 182:481–485.
- Jones AE, Shapiro NI, Trzeciak S, Arnold RC, Claremont HA, Kline JA; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Lactate clearance vs central venous oxygen saturation as goals of early sepsis therapy: a randomized clinical trial. JAMA 2010; 303:739–746.
- Pope JV, Jones AE, Gaieski DF, Arnold RC, Trzeciak S, Shapiro NI; Emergency Medicine Shock Research Network (EMShockNet) Investigators. Multicenter study of central venous oxygen saturation (ScvO2) as a predictor of mortality in patients with sepsis. Ann Emerg Med 2010; 55:40–46.e1
- Kraut JA, Kurtz I. Use of base in the treatment of severe acidemic states. Am J Kidney Dis 2001; 38:703–727.
- Levraut J, Grimaud D. Treatment of metabolic acidosis. Curr Opin Crit Care 2003; 9:260–265.
- Orija AA, Jenks CL. Nucleoside analog reverse transcriptase inhibitor induced lactic acidosis treated with continuous renal replacement in the medical intensive care unit. Crit Care & Shock 2012; 15:9–11.
- Friesecke S, Abel P, Kraft M, Gerner A, Runge S. Combined renal replacement therapy for severe metformin-induced lactic acidosis. Nephrol Dial Transplant 2006; 21:2038–2039.
- Claessens YE, Cariou A, Monchi M, et al. Detecting life-threatening lactic acidosis related to nucleoside-analog treatment of human immunodeficiency virus-infected patients, and treatment with l-carnitine. Crit Care Med 2003; 31:1042–1047.
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R; Medicine Society TM. A modern approach to the treatment of mitochondrial disease. Curr Treat Options Neurol 2009; 11:414–430.
KEY POINTS
- Serum lactate levels can become elevated by a variety of underlying processes, categorized as increased production in conditions of hypoperfusion and hypoxia (type A lactic acidosis), or as increased production or decreased clearance not due to hypoperfusion and hypoxia (type B).
- The higher the lactate level and the slower the rate of normalization (lactate clearance), the higher the risk of death.
- Treatments differ depending on the underlying mechanism of the lactate elevation. Thus, identifying the reason for hyperlactatemia and differentiating between type A and B lactic acidosis are of the utmost importance.
- Treatment of type A lactic acidosis aims to improve perfusion and match oxygen consumption with oxygen delivery by giving fluids, packed red blood cells, and vasopressors or inotropic agents, or both.
- Treatment of type B involves more specific management, such as discontinuing offending medications or supplementing key cofactors for anaerobic metabolism.
Does noninvasive positive pressure ventilation have a role in managing hypercapnic respiratory failure due to an acute exacerbation of COPD?
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?

NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?
NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
Yes. In selected patients with hypercapnic respiratory failure due to an acute exacerbation of chronic obstructive pulmonary disease (COPD), noninvasive positive pressure ventilation (NIPPV) is an effective adjunct to usual medical therapy. In controlled trials, it reduced the need for endotracheal intubation, the length of hospital stay, and the risk of death.
Acute COPD exacerbations are responsible for more than 500,000 hospitalizations yearly in the United States, and 6% to 34% of patients die.1
Many patients need invasive ventilatory assistance via an endotracheal tube, but such therapy puts the patient at risk of ventilator-associated pneumonia, pneumothorax, and tracheal stenosis.
WHAT IS NONINVASIVE POSITIVE PRESSURE VENTILATION?
WHY IS IT BENEFICIAL?
Several mechanisms may explain why noninvasive positive pressure ventilation is beneficial in acute exacerbations of COPD.
Patients with decompensated respiratory failure lack sufficient alveolar ventilation, owing to abnormal respiratory mechanics and inspiratory muscle fatigue.10 For these patients, breathing faster does not fully compensate. Noninvasive positive pressure ventilation partially counteracts these factors by providing a larger tidal volume with the same inspiratory effort.10,11
Additionally, this treatment can decrease the work of breathing by partially overcoming auto-PEEP (positive end-expiratory pressure) in certain situations.2 Auto-PEEP is pressure greater than the atmospheric pressure remaining in the alveoli at the end of exhalation.12 This condition is related to limited expiratory flow and is common in those with severe COPD. Noninvasive positive pressure ventilation decreases the pressure difference between the atmosphere and the alveoli, thereby reducing the inspiratory force needed for initiation of inspiratory effort, which may reduce the work of breathing. However, caution should be used when using this therapy in tachypneic patients, in whom NIPPV may not fully overcome the auto-PEEP.
WHAT STUDIES SHOWED
Several randomized trials have shown NIPPV to be beneficial in acute hypercapnic COPD exacerbations. A recent meta-analysis of eight studies13 showed that, compared with usual care alone, this therapy was associated with:
- A lower mortality rate (relative risk 0.41; 95% confidence interval [CI] 0.26–0.64)
- Less need for endotracheal intubation (relative risk 0.42; 95% CI 0.31–0.59)
- A lower rate of treatment failure (relative risk 0.51; 95% CI 0.38–0.67)
- Greater improvements in the 1-hour post-treatment pH and PaCO2 levels
- A lower respiratory rate
- A shorter length of stay in the hospital.
WHICH PATIENTS SHOULD RECEIVE IT?
NIPPV is not suitable for all patients with hypercapnic respiratory failure. It should not be substituted for endotracheal intubation and mechanical ventilation if they are indicated, eg, in patients who are medically unstable because of hypotension, sepsis, hypoxia, or other life-threatening systemic illness. In addition, those who cannot protect the airway, who have had a worsening in mental status, or who have excessive secretions should not undergo NIPPV because they have a high risk of aspiration. Factors that predict that this therapy will fail include an Acute Physiology and Chronic Health Evaluation (APACHE) score of 29 or higher, a respiratory rate of 30 or higher, and a pH lower than 7.25 after 2 hours of this therapy.15
GENERAL WARD OR INTENSIVE CARE UNIT?
Mild to moderate COPD exacerbations (in which the pH is 7.30 or higher) can be effectively treated with NIPPV in a general ward if the staff has appropriate expertise.5,18 Keeping the patient in a general ward reduces cost and provides a favorable outcome in selected patients.5,19 However, if the patient’s hemodynamic or mental status deteriorates or if gas exchange, pH, respiratory rate, or dyspnea fail to improve, he or she should be transferred to an intensive care unit and endotracheal intubation should be considered.18 The use of NIPPV in general wards should always be approached with caution and should never be attempted without adequate patient supervision and an experienced respiratory therapy team.
TAKE-HOME MESSAGE
NIPPV has been shown to be an effective adjunct in the treatment of acute hypercapnic respiratory failure secondary to a COPD exacerbation, reducing the need for endotracheal intubation, the length of hospital stay, and the mortality rate. On the basis of controlled trials, NIPPV is now considered the ventilatory therapy of choice in selected patients with this condition. However, it should not be used as a substitute for intubation and mechanical ventilation if these are needed or if the patient is at risk of aspiration.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.
- Connors AF Jr, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and P for Outcomes and Risks of Treatments). Am J Respir Crit Care Med 1996; 154:959–967. (Erratum in: Am J Respir Crit Care Med 1997; 155:386).
- Mehta S, Hill NS. Noninvasive ventilation. Am J Respir Crit Care Med 2001; 163:540–577.
- Brochard L, Mancebo J, Wysocki M, et al. Noninvasive ventilation for acute exacerbations of chronic obstructive pulmonary disease. N Engl J Med 1995; 333:817–822.
- Kramer N, Meyer TJ, Meharg J, Cece RD, Hill NS. Randomized, prospective trial of noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 1995; 151:1799–1806.
- Plant PK, Owen JL, Elliott MW. Early use of non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease on general respiratory wards: a multicentre randomised controlled trial. Lancet 2000; 355:1931–1935.
- Wysocki M, Tric L, Wolff MA, Millet H, Herman B. Noninvasive pressure support ventilation in patients with acute respiratory failure. A randomized comparison with conventional therapy. Chest 1995; 107:761–768.
- Masip J, Betbesé AJ, Páez J, et al. Non-invasive pressure support ventilation versus conventional oxygen therapy in acute cardiogenic pulmonary oedema: a randomised trial. Lancet 2000; 356:2126–2132.
- Antonelli M, Conti G, Rocco M, et al. A comparison of noninvasive positive-pressure ventilation and conventional mechanical ventilation in patients with acute respiratory failure. N Engl J Med 1998; 339:429–435.
- Girault C, Daudenthun I, Chevron V, Tamion F, Leroy J, Bonmarchand G. Noninvasive ventilation as a systematic extubation and weaning technique in acute-on-chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med 1999; 160:86–92.
- Brochard L. Noninvasive ventilation for acute respiratory failure. JAMA 2002; 288:932–935.
- Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a face mask. N Engl J Med 1990; 323:1523–1530.
- Mughal MM, Culver DA, Minai OA, Arroliga AC. Auto-positive end-expiratory pressure: mechanisms and treatment. Cleve Clin J Med 2005; 72:801–809.
- Lightowler JV, Wedzicha JA, Elliott MW, Ram FS. Non-invasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ 2003; 326:185.
- British Thoracic Society Standards of Care Committee. Non-invasive ventilation in acute respiratory failure. Thorax 2002; 57:192–211.
- Confalonieri M, Garuti G, Cattaruzza MS, et al. A chart of failure risk for noninvasive ventilation in patients with COPD exacerbation. Eur Respir J 2005; 25:348–355.
- American Respiratory Care Foundation Consensus Conference. Non-invasive positive pressure ventilation. Respir Care 1997; 42:364–369.
- Celikel T, Sungur M, Ceyhan B, Karakurt S. Comparison of noninvasive positive pressure ventilation with standard medical therapy in hypercapnic acute respiratory failure. Chest 1998; 114:1636–1642.
- Organized jointly by the American Thoracic Society, the European Respiratory Society, the European Society of Intensive Care Medicine, and the Société de Réanimation de Langue Francaise, and approved by ATS Board of Directors, December 2000. International Consensus Conferences in Intensive Care Medicine: noninvasive positive pressure ventilation in acute respiratory failure. Am J Respir Crit Care Med 2001; 163:283–291.
- Plant PK, Owen JL, Parrott S, Elliott MW. Cost effectiveness of ward based non-invasive ventilation for acute exacerbations of chronic obstructive pulmonary disease: economic analysis of randomised controlled trial. BMJ 2003; 326:956.