User login
Do Erythropoiesis-Stimulating Agents Have a Risk Evaluation and Mitigation Strategy? (FULL)
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
Epoetin alfa and darbepoetin alfa are erythropoiesis-stimulating agents (ESAs), approved for the treatment of anemia (low red blood cells [RBCs]) resulting from chronic kidney disease, chemotherapy, and certain treatments for HIV. These ESAs also are used to reduce the number of blood transfusions during and after certain major surgeries. Erythropoiesis-stimulating agents work like the human protein erythropoietin, which stimulates bone marrow to make RBCs. Epoetin alfa (marketed as Procrit and Epogen) and darbepoetin alfa (marketed as Aranesp) are manufactured by Amgen, Inc. (Thousand Oaks, CA).
In 1989 epoetin alfa was approved for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 1993 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy. Epoetin alfa also is indicated for anemia due to zidovudine in patients with HIV and reduction of RBC transfusions during certain surgeries.
Darbepoetin alfa was approved in 2001 for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, and in 2006 for the treatment of anemia due to the effects of concomitant myelosuppressive chemotherapy.
Risk Evaluation and Mitigation Strategies
Both epoetin alfa and darbepoetin alfa increase the risk of death, myocardial infarction, stroke, venous thromboembolism, and thrombosis of vascular access and tumor progression or recurrence. Epoetin alfa also can lead to an increase in adverse cardiovascular events, hypertension, seizures, and severe anemia.
In 2008, the FDA determined that Risk Evaluation and Mitigation Strategies (REMS) were necessary for ESAs (darbopoetin alfa and epoetin alfa), to ensure that the benefits for use as treatment for anemia associated with myelosuppressive chemotherapy outweigh the risk of shortened overall survival (OS) and/or the increased risk of tumor progression or recurrence in patients with cancer. The REMS was approved in 2010.
Under the ESA REMS program, referred to as the ESA APPRISE Oncology Program, health care providers (HCPs) that prescribed and/or dispensed darbopoetin alfa to patients with cancer and hospitals that dispensed darbopoetin alfa to patients with cancer were required to enroll and become certified in the ESA REMS. The ESA REMS also required the completion of a Patient and Healthcare Provider Acknowledgement Form for each patient with cancer before the new ESA treatment course to ensure patients were counseled about the benefits and risks of these products.
In April 2017, the FDA determined that the ESA REMS that was limited to the use of epoetin alfa and darbopoetin alfa to treat patients with anemia due to associated myelosuppressive chemotherapy was no longer necessary; the benefits of ESAs outweighed the risks of shortened OS and/or increased risk of tumor progression or recurrence in patients with cancer. 1 The FDA recognized the burden that some REMS can place on HCPs and patients. The agency has authority to modify or remove the REMS to minimize the burden on the health care delivery system of complying with the strategy.
Data
The FDA discontinued the REMS based on an evaluation of the results of the REMS Assessments submitted by Amgen and additional FDA analyses to understand the impact of the various regulatory and other actions on the use of ESAs. The REMS Assessment showed the following:
- The results from surveyed prescribers demonstrated acceptable knowledge of the product risks of decreased survival and/or the increased risk of tumor progression or recurrence and the need to counsel patients about these risks; and
- The drug utilization data indicated appropriate prescribing of ESAs consistent with the intended use as a treatment alternative to RBC transfusion for anemia associated with myelosuppressive chemotherapy.
The FDA also conducted an evaluation of the impact of multiple actions, including the ESA REMS, on the use of the ESAs using sponsor-submitted data from outpatient oncology practices between 2006 and 2014. During 2004 to 2009, the FDA took multiple regulatory actions, including labeling changes. In 2007, the Center for Medicare and Medicaid Services (CMS) made a National Coverage Determination (NCD) to limit coverage of ESAs for nonrenal disease indications. These actions coincided with the following:
- A decrease in the proportion of patients receiving chemotherapy using ESAs;
- An increase in the proportion of patients receiving chemotherapy who initiate ESAs at a hemoglobin level < 10 g/dL; and
- An increase in the proportion of patients who initiate ESAs at a dosage consistent with product prescribing information.
Full implementation of the ESA REMS in 2011 had minimal impact on trends in these 3 ESA utilization metrics beyond the changes observed after the CMS coverage determination and multiple other FDA regulatory actions.
This information led the FDA to conclude that it was no longer necessary to require the certification of prescribers and hospitals that prescribe and/or dispense ESAs to patients with cancer in order to ensure that the benefits outweigh the risks.
The FDA has released the REMS requirements for the epoetin alfa and darbopoetin alfa ESA products, and the risks can be communicated by the current product prescribing information. The appropriate use of ESAs is supported by the CMS NCD, the American Society of Clinical Oncology, and American Society of Hematology clinical guidelines, which are evidence-based guidelines intended to provide a basis for the standard of care in clinical oncology.
Education
While the REMS is no longer necessary to ensure the benefits outweigh the risks, the serious risks of shortened OS and/or increased risk of tumor progression or recurrence associated with these drugs remain. The boxed warning language remains as follows: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE. Health care providers are encouraged to discuss the risks and benefits of using ESAs with each patient before initiating use.
Click here to read the digital edition.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
1. U.S. Food & Drug Administration. Information on erythropoiesis-stimulating agents (ESA) epoetin alfa (marketed as Procrit, Epogen), darbepoetin alfa (marketed as Aranesp). https://www.fda.gov/Drugs/DrugSafety/ucm109375.htm. Updated April 13, 2017. Accessed July 13, 2017.
Abuse-Deterrent Opioids: What Practitioners Need to Know
Opioid Abuse-Deterrent Formulations
The meaning of the term abuse-deterrent is often misunderstood to mean abuse-proof. The FDA defines abuse-deterrent properties as those properties expected to meaningfully deter abuse even if they do not fully prevent abuse. Abuse-deterrent properties make certain types of abuse, such as crushing in order to snort or dissolving in order to inject, more difficult or less rewarding. However, this does not mean that the product is impossible to abuse or that these properties will necessarily prevent addiction, overdose, or death.
Of note, currently marketed abuse-deterrent formulation technologies do not effectively deter one of the most common forms of opioid abuse—simply swallowing a number of intact tablets or capsules. Abuse-deterrent opioids do not reduce the risk for opioid addiction, and they carry the same warnings about the risk for addiction as do conventional opioids.
Abuse and Misuse Data
The FDA is encouraging pharmaceutical industry efforts to develop pain medicines that are more difficult to abuse and to prioritize the need for data and study methods that will help evaluate the impact of abuse-deterrent opioids on misuse and abuse in the community. To collect this important information, the FDA requires that all companies that have brand-name opioids with labeling describing abuse-deterrent properties conduct postmarketing studies to determine the impact of abuse-deterrent formulation technologies in the real world. Each company is given a time line to which they must adhere. These types of studies take several years to conduct and analyze. Data collected will include the amount prescribed for each product; adverse events related to the use, abuse, and misuse of the products; and epidemiologic data on the rates of abuse and misuse and their consequences (addiction, overdose, and death). These studies should allow the FDA to assess the impact in the community, if any, attributable to the abuse-deterrent properties.
The science of abuse deterrence is relatively new, and both the formulation technologies and the analytical, clinical, and statistical methods for evaluating those technologies ar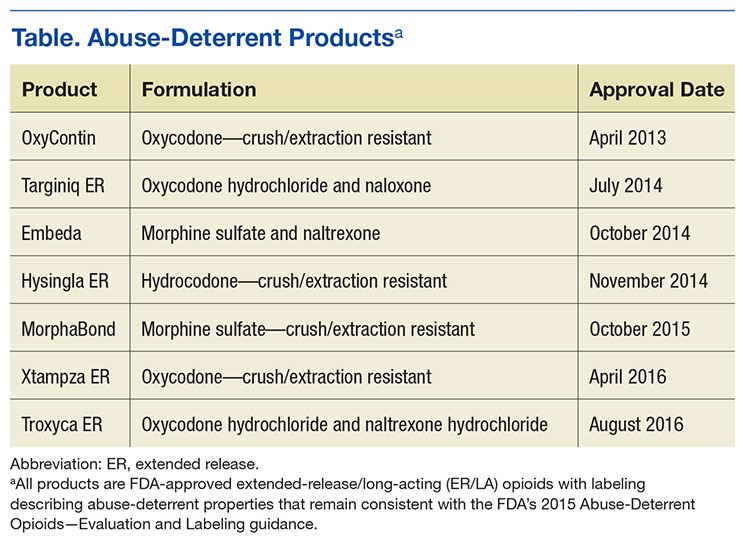
Key Points for Practitioners
The FDA’s work to facilitate the safe use of opioids is taking place within a larger policy framework aimed at addressing opioid abuse while ensuring appropriate access to pain treatment. The FDA has undertaken several efforts helpful to clinicians. The FDA’s Extended-Release and Long-Acting Opioid Analgesics Risk Evaluation and Mitigation Strategy (ER/LA REMS) Program is required for all companies who make these products. The program’s goal is to reduce serious adverse outcomes of inappropriate prescribing, misuse, and abuse of ER/LA opioid analgesics while maintaining patient access to pain medications. Adverse outcomes of concern include addiction, unintentional overdose, and death.
As part of the REMS, all ER/LA opioid analgesic pharmaceutical companies must provide education for prescribers of their medications through accredited continuing education activities that are supported by independent educational grants. Companies must also provide information that prescribers can use when counseling patients about the risks and benefits associated with ER/LA opioid analgesic use.
The FDA has developed core messages that are communicated to prescribers in the Blueprint for Prescriber Education. The Blueprint is directed to prescribers of ER/LA opioid analgesics but also may be relevant for other health care professionals (eg, pharmacists). Companies involved in the ER/LA Opioid Analgesics REMS Program have collaborated to implement a single shared REMS. This group provides a list of REMS-compliant continuing education activities, which can be found at http://www.er-la-opioidrems.com.
It is important for practitioners to understand that all currently approved abuse-deterrent opioid products still can be abused, and as scheduled controlled substances, they are addictive. The abuse-deterrent properties are expected to deter but do not wholly prevent abuse. Because in the end opioid medications must be able to deliver the opioid to the patient, there probably always will be potential for abuse of these products. Consequently, practitioners should counsel their patients on the following:
- Keep medicines in a secure location out of the reach and out of sight of children and pets. Put away medicines after every use. Accidental exposure to medicine in the home is a major source of unintentional poisonings in the U.S.
- If medicines are no longer needed, dispose of them properly. Disposing of all unused opioid analgesics reduces access to these medications by family members and household guests seeking opioids for abuse.
- The FDA recommends returning most prescription medications through a local or U.S. Drug Enforcement Administration (DEA)-sponsored take-back program or DEA-authorized collector. For opioid analgesics, the FDA recommends immediate removal from the home by flushing them down the toilet or sink.
Opioids Action Plan
In February 2016, FDA Commissioner Robert Califf (then the deputy commissioner for medical products and tobacco) announced the FDA Opioids Action Plan. The plan focuses on policies aimed at reversing the opioid epidemic while still providing patients in pain access to effective pain relief. The FDA actions include:
- Convening an expert advisory committee before approving any new drug application for an opioid that does not have abuse-deterrent properties;
- Consulting with the Pediatric Advisory Committee about a framework for pediatric opioid labeling before any new labeling is approved;
- Updating the REMS requirements for ER/LA opioid analgesics after considering the advisory committee’s recommendations from a meeting held in May 2016 and reviewing existing requirements;
- Improving access to naloxone (by facilitating the development of an over-the-counter version of naloxone, which is currently available only by prescription, thereby making it more accessible to treat opioid overdose), and medication-assisted treatment options for patients with opioid use disorders; and
- Supporting better pain management options, including alternative, nonaddictive treatments for pain.
The FDA is conducting research on pain measurements for conditions such as chronic low back pain, osteoarthritis, diabetic neuropathy, postherpetic neuralgia, and fibromyalgia. The FDA is also working to support the development of nonopioid options for these patients.
Consistent with the plan, in March 2016, the FDA announced that it was requiring changes to the labeling on immediate-release opioids, including additional warnings and safety information that incorporate elements similar to the ER/LA opioid analgesics labeling. Furthermore, among other steps, the FDA has contracted with the National Academy of Medicine to provide advice on how to incorporate current evidence about the public health impact of opioid use (for patients who are prescribed opioids as well as for nonpatients) into regulatory activities concerning opioids.
The FDA shares the responsibility of keeping patients safe. Working with the health care community and federal and state partners to help reduce opioid misuse and abuse and improve appropriate opioid prescribing while ensuring that patients in pain continue to have appropriate access to opioid analgesics is a top priority for the FDA and part of the targeted approach of the HHS focused on prevention, treatment, and intervention.
Opioid Abuse-Deterrent Formulations
The meaning of the term abuse-deterrent is often misunderstood to mean abuse-proof. The FDA defines abuse-deterrent properties as those properties expected to meaningfully deter abuse even if they do not fully prevent abuse. Abuse-deterrent properties make certain types of abuse, such as crushing in order to snort or dissolving in order to inject, more difficult or less rewarding. However, this does not mean that the product is impossible to abuse or that these properties will necessarily prevent addiction, overdose, or death.
Of note, currently marketed abuse-deterrent formulation technologies do not effectively deter one of the most common forms of opioid abuse—simply swallowing a number of intact tablets or capsules. Abuse-deterrent opioids do not reduce the risk for opioid addiction, and they carry the same warnings about the risk for addiction as do conventional opioids.
Abuse and Misuse Data
The FDA is encouraging pharmaceutical industry efforts to develop pain medicines that are more difficult to abuse and to prioritize the need for data and study methods that will help evaluate the impact of abuse-deterrent opioids on misuse and abuse in the community. To collect this important information, the FDA requires that all companies that have brand-name opioids with labeling describing abuse-deterrent properties conduct postmarketing studies to determine the impact of abuse-deterrent formulation technologies in the real world. Each company is given a time line to which they must adhere. These types of studies take several years to conduct and analyze. Data collected will include the amount prescribed for each product; adverse events related to the use, abuse, and misuse of the products; and epidemiologic data on the rates of abuse and misuse and their consequences (addiction, overdose, and death). These studies should allow the FDA to assess the impact in the community, if any, attributable to the abuse-deterrent properties.
The science of abuse deterrence is relatively new, and both the formulation technologies and the analytical, clinical, and statistical methods for evaluating those technologies ar
Key Points for Practitioners
The FDA’s work to facilitate the safe use of opioids is taking place within a larger policy framework aimed at addressing opioid abuse while ensuring appropriate access to pain treatment. The FDA has undertaken several efforts helpful to clinicians. The FDA’s Extended-Release and Long-Acting Opioid Analgesics Risk Evaluation and Mitigation Strategy (ER/LA REMS) Program is required for all companies who make these products. The program’s goal is to reduce serious adverse outcomes of inappropriate prescribing, misuse, and abuse of ER/LA opioid analgesics while maintaining patient access to pain medications. Adverse outcomes of concern include addiction, unintentional overdose, and death.
As part of the REMS, all ER/LA opioid analgesic pharmaceutical companies must provide education for prescribers of their medications through accredited continuing education activities that are supported by independent educational grants. Companies must also provide information that prescribers can use when counseling patients about the risks and benefits associated with ER/LA opioid analgesic use.
The FDA has developed core messages that are communicated to prescribers in the Blueprint for Prescriber Education. The Blueprint is directed to prescribers of ER/LA opioid analgesics but also may be relevant for other health care professionals (eg, pharmacists). Companies involved in the ER/LA Opioid Analgesics REMS Program have collaborated to implement a single shared REMS. This group provides a list of REMS-compliant continuing education activities, which can be found at http://www.er-la-opioidrems.com.
It is important for practitioners to understand that all currently approved abuse-deterrent opioid products still can be abused, and as scheduled controlled substances, they are addictive. The abuse-deterrent properties are expected to deter but do not wholly prevent abuse. Because in the end opioid medications must be able to deliver the opioid to the patient, there probably always will be potential for abuse of these products. Consequently, practitioners should counsel their patients on the following:
- Keep medicines in a secure location out of the reach and out of sight of children and pets. Put away medicines after every use. Accidental exposure to medicine in the home is a major source of unintentional poisonings in the U.S.
- If medicines are no longer needed, dispose of them properly. Disposing of all unused opioid analgesics reduces access to these medications by family members and household guests seeking opioids for abuse.
- The FDA recommends returning most prescription medications through a local or U.S. Drug Enforcement Administration (DEA)-sponsored take-back program or DEA-authorized collector. For opioid analgesics, the FDA recommends immediate removal from the home by flushing them down the toilet or sink.
Opioids Action Plan
In February 2016, FDA Commissioner Robert Califf (then the deputy commissioner for medical products and tobacco) announced the FDA Opioids Action Plan. The plan focuses on policies aimed at reversing the opioid epidemic while still providing patients in pain access to effective pain relief. The FDA actions include:
- Convening an expert advisory committee before approving any new drug application for an opioid that does not have abuse-deterrent properties;
- Consulting with the Pediatric Advisory Committee about a framework for pediatric opioid labeling before any new labeling is approved;
- Updating the REMS requirements for ER/LA opioid analgesics after considering the advisory committee’s recommendations from a meeting held in May 2016 and reviewing existing requirements;
- Improving access to naloxone (by facilitating the development of an over-the-counter version of naloxone, which is currently available only by prescription, thereby making it more accessible to treat opioid overdose), and medication-assisted treatment options for patients with opioid use disorders; and
- Supporting better pain management options, including alternative, nonaddictive treatments for pain.
The FDA is conducting research on pain measurements for conditions such as chronic low back pain, osteoarthritis, diabetic neuropathy, postherpetic neuralgia, and fibromyalgia. The FDA is also working to support the development of nonopioid options for these patients.
Consistent with the plan, in March 2016, the FDA announced that it was requiring changes to the labeling on immediate-release opioids, including additional warnings and safety information that incorporate elements similar to the ER/LA opioid analgesics labeling. Furthermore, among other steps, the FDA has contracted with the National Academy of Medicine to provide advice on how to incorporate current evidence about the public health impact of opioid use (for patients who are prescribed opioids as well as for nonpatients) into regulatory activities concerning opioids.
The FDA shares the responsibility of keeping patients safe. Working with the health care community and federal and state partners to help reduce opioid misuse and abuse and improve appropriate opioid prescribing while ensuring that patients in pain continue to have appropriate access to opioid analgesics is a top priority for the FDA and part of the targeted approach of the HHS focused on prevention, treatment, and intervention.
Opioid Abuse-Deterrent Formulations
The meaning of the term abuse-deterrent is often misunderstood to mean abuse-proof. The FDA defines abuse-deterrent properties as those properties expected to meaningfully deter abuse even if they do not fully prevent abuse. Abuse-deterrent properties make certain types of abuse, such as crushing in order to snort or dissolving in order to inject, more difficult or less rewarding. However, this does not mean that the product is impossible to abuse or that these properties will necessarily prevent addiction, overdose, or death.
Of note, currently marketed abuse-deterrent formulation technologies do not effectively deter one of the most common forms of opioid abuse—simply swallowing a number of intact tablets or capsules. Abuse-deterrent opioids do not reduce the risk for opioid addiction, and they carry the same warnings about the risk for addiction as do conventional opioids.
Abuse and Misuse Data
The FDA is encouraging pharmaceutical industry efforts to develop pain medicines that are more difficult to abuse and to prioritize the need for data and study methods that will help evaluate the impact of abuse-deterrent opioids on misuse and abuse in the community. To collect this important information, the FDA requires that all companies that have brand-name opioids with labeling describing abuse-deterrent properties conduct postmarketing studies to determine the impact of abuse-deterrent formulation technologies in the real world. Each company is given a time line to which they must adhere. These types of studies take several years to conduct and analyze. Data collected will include the amount prescribed for each product; adverse events related to the use, abuse, and misuse of the products; and epidemiologic data on the rates of abuse and misuse and their consequences (addiction, overdose, and death). These studies should allow the FDA to assess the impact in the community, if any, attributable to the abuse-deterrent properties.
The science of abuse deterrence is relatively new, and both the formulation technologies and the analytical, clinical, and statistical methods for evaluating those technologies ar
Key Points for Practitioners
The FDA’s work to facilitate the safe use of opioids is taking place within a larger policy framework aimed at addressing opioid abuse while ensuring appropriate access to pain treatment. The FDA has undertaken several efforts helpful to clinicians. The FDA’s Extended-Release and Long-Acting Opioid Analgesics Risk Evaluation and Mitigation Strategy (ER/LA REMS) Program is required for all companies who make these products. The program’s goal is to reduce serious adverse outcomes of inappropriate prescribing, misuse, and abuse of ER/LA opioid analgesics while maintaining patient access to pain medications. Adverse outcomes of concern include addiction, unintentional overdose, and death.
As part of the REMS, all ER/LA opioid analgesic pharmaceutical companies must provide education for prescribers of their medications through accredited continuing education activities that are supported by independent educational grants. Companies must also provide information that prescribers can use when counseling patients about the risks and benefits associated with ER/LA opioid analgesic use.
The FDA has developed core messages that are communicated to prescribers in the Blueprint for Prescriber Education. The Blueprint is directed to prescribers of ER/LA opioid analgesics but also may be relevant for other health care professionals (eg, pharmacists). Companies involved in the ER/LA Opioid Analgesics REMS Program have collaborated to implement a single shared REMS. This group provides a list of REMS-compliant continuing education activities, which can be found at http://www.er-la-opioidrems.com.
It is important for practitioners to understand that all currently approved abuse-deterrent opioid products still can be abused, and as scheduled controlled substances, they are addictive. The abuse-deterrent properties are expected to deter but do not wholly prevent abuse. Because in the end opioid medications must be able to deliver the opioid to the patient, there probably always will be potential for abuse of these products. Consequently, practitioners should counsel their patients on the following:
- Keep medicines in a secure location out of the reach and out of sight of children and pets. Put away medicines after every use. Accidental exposure to medicine in the home is a major source of unintentional poisonings in the U.S.
- If medicines are no longer needed, dispose of them properly. Disposing of all unused opioid analgesics reduces access to these medications by family members and household guests seeking opioids for abuse.
- The FDA recommends returning most prescription medications through a local or U.S. Drug Enforcement Administration (DEA)-sponsored take-back program or DEA-authorized collector. For opioid analgesics, the FDA recommends immediate removal from the home by flushing them down the toilet or sink.
Opioids Action Plan
In February 2016, FDA Commissioner Robert Califf (then the deputy commissioner for medical products and tobacco) announced the FDA Opioids Action Plan. The plan focuses on policies aimed at reversing the opioid epidemic while still providing patients in pain access to effective pain relief. The FDA actions include:
- Convening an expert advisory committee before approving any new drug application for an opioid that does not have abuse-deterrent properties;
- Consulting with the Pediatric Advisory Committee about a framework for pediatric opioid labeling before any new labeling is approved;
- Updating the REMS requirements for ER/LA opioid analgesics after considering the advisory committee’s recommendations from a meeting held in May 2016 and reviewing existing requirements;
- Improving access to naloxone (by facilitating the development of an over-the-counter version of naloxone, which is currently available only by prescription, thereby making it more accessible to treat opioid overdose), and medication-assisted treatment options for patients with opioid use disorders; and
- Supporting better pain management options, including alternative, nonaddictive treatments for pain.
The FDA is conducting research on pain measurements for conditions such as chronic low back pain, osteoarthritis, diabetic neuropathy, postherpetic neuralgia, and fibromyalgia. The FDA is also working to support the development of nonopioid options for these patients.
Consistent with the plan, in March 2016, the FDA announced that it was requiring changes to the labeling on immediate-release opioids, including additional warnings and safety information that incorporate elements similar to the ER/LA opioid analgesics labeling. Furthermore, among other steps, the FDA has contracted with the National Academy of Medicine to provide advice on how to incorporate current evidence about the public health impact of opioid use (for patients who are prescribed opioids as well as for nonpatients) into regulatory activities concerning opioids.
The FDA shares the responsibility of keeping patients safe. Working with the health care community and federal and state partners to help reduce opioid misuse and abuse and improve appropriate opioid prescribing while ensuring that patients in pain continue to have appropriate access to opioid analgesics is a top priority for the FDA and part of the targeted approach of the HHS focused on prevention, treatment, and intervention.