User login
Are new tools for correcting prolapse and incontinence better just because they’re new?
The author reports no financial relationships relevant to this article.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html.
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling (FIGURE) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
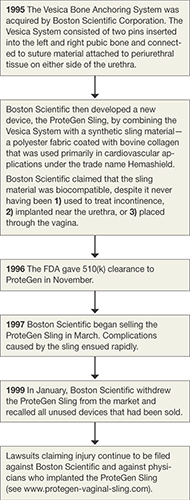
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073) and replaced it with a new guideline (Issue 85, September 20074). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5)
organ prolapse
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
We want to hear from you! Tell us what you think.
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
just because they’re new;Anne M. Weber MD;Commentary;economic factors;urinary incontinence;pelvic organ prolapse;POP;transvaginal mesh;urethral sling;UI;innovation in treatment;FDA;medical device;510(k);ProteGen Sling;ObTape;TVT;Gynecare;TVT Secur;early adopters;ACOG;Clinical Practice Bulletin;informed consent;financial conflict of interest;radiofrequency therapy;SURx Transvaginal system;Renessa;do no harm;FDA Alert;
The author reports no financial relationships relevant to this article.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html.
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling (FIGURE) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073) and replaced it with a new guideline (Issue 85, September 20074). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5)
organ prolapse
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
We want to hear from you! Tell us what you think.
The author reports no financial relationships relevant to this article.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html.
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling (FIGURE) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073) and replaced it with a new guideline (Issue 85, September 20074). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5)
organ prolapse
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
We want to hear from you! Tell us what you think.
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
just because they’re new;Anne M. Weber MD;Commentary;economic factors;urinary incontinence;pelvic organ prolapse;POP;transvaginal mesh;urethral sling;UI;innovation in treatment;FDA;medical device;510(k);ProteGen Sling;ObTape;TVT;Gynecare;TVT Secur;early adopters;ACOG;Clinical Practice Bulletin;informed consent;financial conflict of interest;radiofrequency therapy;SURx Transvaginal system;Renessa;do no harm;FDA Alert;
just because they’re new;Anne M. Weber MD;Commentary;economic factors;urinary incontinence;pelvic organ prolapse;POP;transvaginal mesh;urethral sling;UI;innovation in treatment;FDA;medical device;510(k);ProteGen Sling;ObTape;TVT;Gynecare;TVT Secur;early adopters;ACOG;Clinical Practice Bulletin;informed consent;financial conflict of interest;radiofrequency therapy;SURx Transvaginal system;Renessa;do no harm;FDA Alert;
Are new tools for correcting prolapse and incontinence better just because they’re new?
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html .
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling ( FIGURE ) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
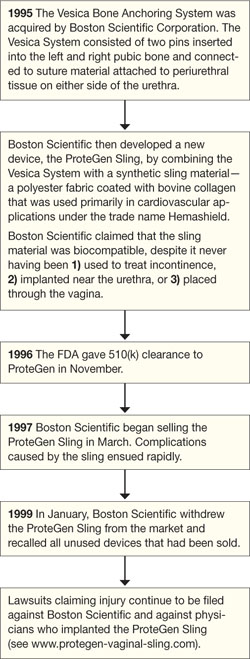
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073 ) and replaced it with a new guideline (Issue 85, September 20074 ). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT ).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5 )
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management, at www.obgmanagement.com.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html .
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling ( FIGURE ) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073 ) and replaced it with a new guideline (Issue 85, September 20074 ). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT ).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5 )
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management, at www.obgmanagement.com.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries1,2 and visit http://www.fda.gov/cdrh/devadvice/314.html .
In a nutshell, the 510(k) process requires only that an applicant demonstrate that a new medical device has “substantial equivalence” to an already legally marketed device, known as the predicate, which may also have been cleared only through the 510(k) process. That means it’s possible to have generations of products cleared on the basis of one predicate device that was itself never studied adequately.
Indeed, this is the case with most medical devices that have been sold for the surgical treatment of POP and UI—from before the ProteGen Sling (Boston Scientific), through Tension-Free Vaginal Tape (TVT) (Gynecare), and continuing with the newest devices.
The story of the ProteGen Sling ( FIGURE ) offers a cautionary tale about what can go wrong when new devices are cleared by the FDA through 510(k), rather than evaluated through rigorous clinical trials, as drugs are. More recently, experience with the ObTape (Mentor Corporation) followed virtually the same trajectory of events; the product was pulled from the market in 2006 and is now the focus of lawsuits nationwide.
Fortunately, for our patients, experience with TVT (Gynecare) has been favorable. Although TVT was also cleared by the FDA through 510(k), clinical research performed after TVT was introduced has demonstrated its effectiveness and relative safety. Indeed, TVT has revolutionized the treatment of stress UI in women—and, even, our understanding of its etiology.
Several companies are capitalizing on the success of TVT by introducing competing products that are designed to be 1) similar enough to ride on the coattails of TVT yet 2) different enough to capture their own share of market—without evidence of safety or effectiveness required. Even Gynecare (part of Ethicon Women’s Health and Urology, a subsidiary of Johnson & Johnson) has introduced TVT SECUR to compete with its own TVT—again, without independent evidence of safety or effectiveness.
The current market in devices for stress UI is a moving target that makes it nearly impossible—even for research organizations, such as the federally funded Pelvic Floor Disorders Network, that are independent of industry—to develop and implement sound clinical trials of those devices. Why do I say “moving target”? First, there are no assurances that any device chosen for study will remain the same for the duration of a trial. Second, there is no way to foresee which products will be abandoned over the time required for a large clinical trial.
FIGURE The saga of the ProteGen Sling
Transparency over what might be considered “experimental”
Until the FDA changes its process—to one in which 1) medical devices are adequately assessed before they reach market and 2) postmarketing surveillance is required—it’s our duty to insist on evidence of safety and effectiveness before adopting the latest and greatest products that companies have to offer.
Of all the questions that a patient might ask before treatment, three of the most important, surely, are:
- “Will this help me?”
- “If it helps me, how long will it help?”
- “Whether or not this treatment helps me, what risks—in the short-term and over the long-term—does it present?”
Until we can provide our patients with answers that are supported by evidence, products that lack such evidence should be considered experimental, and patients should be counseled accordingly.
Some patients may accept what they’ve been advised are new and unproven treatments—in the way that some physicians are early adopters. Nevertheless, I am concerned that some clinicians do not appear to appreciate the true lack of evidence that accompanies most marketed devices for prolapse and incontinence. They may mistake the FDA 510(k) process of clearance for something similar to the agency’s extended and complex drug approval process. They may accept claims made in industry-produced white papers that are often largely promotional materials, and fail to look further into those claims.
Now, more than ever and above all else, we must stand between marketing and our patients’ safety. We are familiar with the toll that prolapse and incontinence, as chronic conditions, take on our patients; yet it’s that very chronic nature that should lead us to adopt patience and caution in accepting new treatments before they have been adequately studied.
If we cannot always rely on industry to provide clear information about the risks and benefits of new devices, neither can we routinely look to our professional organizations for unbiased information. Often, professional organizations accept cash contributions from industry, raising the question of conflict of interest that may undermine their actions when the priorities of industry do not align with the goal of safeguarding patients’ well-being.
In an unprecedented example of how a professional association can interfere with its own, expert-authored clinical practice guidelines, the American College of Obstetricians and Gynecologists (ACOG) more than a year ago rescinded one of its published guidelines on POP (Issue 79, February 20073 ) and replaced it with a new guideline (Issue 85, September 20074 ). The new guideline is nearly identical to the prior one—save for one sentence, in which “experimental” is deleted in a discussion of kits of trocar-based synthetic materials sold for the surgical treatment of pelvic organ prolapse (see the EXCERPT ).
The deletion is crucial because offering informed consent for surgery requires a patient to accept risks in balance with an expectation of benefit. A patient cannot be appropriately informed when no evidence of benefit exists and evidence of postoperative risk is extremely limited.
Now, I am not declaring that ACOG acted with bias because of a financial conflict of interest with industry in this instance; the fact that a financial conflict of interest exists for ACOG, however, cannot be disputed if one examines the College’s Annual Report, where contributors are listed. (For a comprehensive, if disillusioning, treatise on the many effects of financial conflict of interest within medicine, I recommend the book On The Take.5 )
Differences between the two bulletins are marked in boldface
Bulletin #79 (original wording): “Given the limited data and frequent changes in the marketed products (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), if clinicians recommend these procedures before evidence of their risk-benefit is fully understood, the procedures should be considered experimental and patients consented for surgery with that understanding.”
Bulletin #84 (revised wording): “Given the limited data and frequent changes in the marketed products for vaginal surgery for prolapse repair (particularly with regard to type of mesh material itself, which is most closely associated with several of the postoperative risks especially mesh erosion), patients should consent to surgery with an understanding of the post-operative risks and lack of long-term outcomes data.”
Case: Radiofrequency therapy. Even when clinical experience demonstrates lack of effectiveness or an unacceptable rate of complications associated with certain techniques or devices, unequivocal evidence of such problems does not always appear in the literature. One example of this is how a technique was translated into a treatment for incontinence by way of its use in other fields.
Radiofrequency has, among other uses, been used to ablate nerves in intractable chronic pain and to address joint instability in orthopedic surgery. Radiofrequency energy was then, by extension, applied transvaginally to tissue (known as “endopelvic” fascia, of a distinctly different nature than parietal fascia involved in orthopedic procedures) surrounding the urethra. The goal was to coagulate “supporting” tissues and “correct” urethral hypermobility that purportedly causes stress incontinence.
Marketing of the SURx Transvaginal System (CooperSurgical, Inc.) began in 2002, followed by reports of success. One industry-sponsored study, for example, reported a 73% rate of either continence or improvement after 12 months.6
Despite such favorable early results, however, in 2006 CooperSurgical decided to abandon this system, citing “technique-dependent” results of the procedure. Since then, independent research has shown a very low initial success rate that declines rapidly—within weeks or months—of treatment.7,8
A different radiofrequency technique continues to be marketed—the Renessa System™ (NovaSys Medical), which uses a urethral catheter-mounted system to deliver radiofrequency energy through the urethral mucosa to the submucosa and adjacent tissues. Once again, initial reports of industry-sponsored research showed promising results; one study of 110 patients reported 74% achieved continence or improvement after 1 year.9 In a follow-up report of 21 of the original 110 patients, “improvement” was reported in 74% after 3 years.10 Independent research has yet to be reported in the literature.
Of particular concern, no data exist on the long-term effect of denaturing collagen in the urethra and adjacent tissues in relation to UI, other aspects of bladder function, or sexual function. An apparent lack of immediate complications cannot be equated with safety; we need long-term studies to determine whether urethral function is affected adversely compared with that in untreated women and women treated with surgery.
Bring on innovation—in context!
For those who consider my argument anti-innovation, let me repeat: I believe strongly in innovation to improve care for our patients. Am I anti-industry? Only when there is an unbridled race to profit from marketing products without safeguards to ensure, first and foremost, the safety of our patients and, second, their long-term effectiveness. Knowingly or unknowingly, patients then become the guinea pigs on whom these products are tested—just not in the appropriate context of clinical research and informed consent for participation.
Instead (as happened in the US Public Health Service’s Tuskegee syphilis experiments), patients serve as research subjects without their consent when they receive untested products and undergo unproven treatments. And because clinicians are the conduit through which patients receive untested and unproven treatments, who is ultimately responsible for the outcome?
Industry brings innovation to clinical practice. But it is incumbent on clinicians to recognize, with unflinching honesty, the bottom line on which industry operates. Prolapse and incontinence are deeply distressing for our patients, but these chronic conditions are not life-threatening; virtually all women who suffer these conditions have been symptomatic for years before they come for care. I see no need, except to increase that bottom line, to rush products to market before they have been evaluated sufficiently to determine whether “new” is actually “better.”
For clinicians who style themselves as early adopters, remember: It’s not you, but your patient, who is “adopting” a foreign material and having it placed deep in the most intimate area of her body—a foreign material intended to stay for life (except for those unfortunate patients who must have it removed). Above all, we must do no harm—an elusive goal when some of us try to attract patients by being the first to use a product before evidence of its risk and utility have been established in practice.
Does this kind of talk encourage litigation?
Does a commentary like this one provide fodder for plaintiff attorneys who are seeking grounds for product liability lawsuits against manufacturers and malpractice claims against clinicians? Please! Spend a moment on the Web, and you will see that the lawyers are already busy—especially since the FDA’s October 2008 alert about complications with surgical mesh for prolapse and incontinence. [See “FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management, at www.obgmanagement.com.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
1. Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
2. Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
3. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
4. ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
5. Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
6. Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
7. Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
8. Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
9. Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
10. Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
Are new tools for correcting prolapse and incontinence better just because they’re new?
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries“FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
- Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
- Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
- Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
- Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
- Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
- Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
- Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
- Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries“FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
From my vantage point, it appears that economic factors are playing an increasingly important role in how pelvic organ prolapse (POP) and urinary incontinence (UI) are managed—particularly, in regard to the use of surgical devices. As such, the topic of treating POP and UI deserves our attention to ensure that we make the best decisions for our patients.
Now, I’m a staunch supporter of innovation in treatment; certainly, there is room for improvement in current approaches—particularly in surgery—for treating POP and UI. At the same time, I strongly believe that innovation must be demonstrated to be an improvement before it is incorporated into practice. Although innovation is commonly taken on faith, we should know better than to equate “new” with “better” until evidence, gathered through clinical research, has demonstrated this conclusively. A look at the US Food and Drug Administration’s (FDA’s) process for clearing medical devices for clinical use reveals that such a standard often doesn’t apply—and this should matter to us.
The meaning of 510(k)
Most medical devices are evaluated through an FDA clearance mechanism known as the 510(k) process. This is wholly distinct from the agency’s drug approval process with which most of us are familiar. It’s beyond the scope of this commentary for me to go into detail about 510(k); if you are interested, see two recent commentaries“FDA alert: Transvaginal placement of surgical mesh carries serious risks,” in the December 2008 issue of OBG Management.] It’s worth noting how these lawyers see themselves: They would likely tell you that they “provide an important service in protecting patients from unscrupulous manufacturers who profit from the vulnerability of people seeking treatment for distressing conditions.” As clinicians, are we absolutely sure that we can say the same of ourselves?
Is it wrong to harp on what happened in the past?
Why revisit events surrounding, for example, the ProteGen Sling? My reply is another question: Where is the evidence that such sequences of events are in the past? Among clinicians, who knows which is best in a collection of kits that changes from one month to the next, without their promoters skipping a beat in proclaiming theirs as the “best”? It isn’t shameful to admit that one doesn’t know which one is best; but it is a shame to act as if one does know, especially when the risk falls to another. The names change; the events are the same.
What is the solution to this problem?
Businesses succeed only when their products are purchased. If clinicians refused to be participants whenever the device industry introduces unproven treatments into the market, industry would be compelled to test their products beforehand. Patients would benefit—by being able to make truly informed choices, with adequate information about risk and benefit. Clinicians would benefit—by being able to provide the most effective treatment without sacrificing their integrity in the process. Ultimately, industry would benefit, by profiting appropriately from products that truly help our patients. Is that an impossible wish?
- Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
- Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
- Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
- Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
- Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
- Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
- Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
- Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
- Goldman HB. Is new always better? Curr Urol Rep. 2007;8(4):253-254.
- Ostergard DR. Lessons from the past: directions for the future. Do new marketed surgical procedures and grafts produce ethical, personal liability, and legal concerns for physicians? Int Urogynecol J Pelvic Floor Dysfunct. 2007;18:591-598.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 79: Pelvic organ prolapse. Obstet Gynecol. 2007;109(2 Pt 1):461-473.
- ACOG Committee on Practice Bulletins–Gynecology, American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 85: Pelvic organ prolapse. Obstet Gynecol. 2007;110:717-729.
- Kassirer JP. On the Take: How Medicine’s Complicity with Big Business Can Endanger Your Health. New York: Oxford University Press; 2005.
- Dmochowski RR, Avon M, Ross J, et al. Transvaginal radiofrequency treatment of the endopelvic fascia: a prospective evaluation for the treatment of genuine stress urinary incontinence. J Urol. 2003;169:1028-1032.
- Buchsbaum GM, McConville J, Korni R, Duecy EE. Outcome of transvaginal radiofrequency for treatment of women with stress urinary incontinence. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(3):263-265.
- Ismail SI. Radiofrequency remodelling of the endopelvic fascia is not an effective procedure for urodynamic stress incontinence in women. Int Urogynecol J Pelvic Floor Dysfunct. 2008;19:1205-1209.
- Appell RA, Juma S, Wells WG, et al. Transurethral radio-frequency energy collagen micro-remodeling for the treatment of female stress urinary incontinence. Neurourol Urodyn. 2006;25(4):331-336.
- Appell RA, Singh G, Klimberg IW, et al. Nonsurgical radiofrequency collagen denaturation for stress urinary incontinence: retrospective 3-year evaluation. Expert Rev Med Devices. 2007;4:455-461.
URINARY INCONTINENCE
The author reports no financial relationships relevant to this article.
The past year has seen the publication of much useful evidence regarding urinary incontinence, from both epidemiologic studies and clinical trials. Research into the pathophysiology of incontinence continues to move forward, slowly but surely, measured not in breakthroughs but in gradually increasing knowledge of how the urethra and bladder function in the continent person and how that function can break down, leading to incontinence and other urinary symptoms.
Highlighted here are four notable studies from 2007, as well as progress notes on a trial mentioned early this year in Examining the Evidence (January issue).
New data clarify incidence and uncloak the effect of weight gain
Townsend MK, Danforth KN, Liffort KL, et al. Incidence and remission of urinary incontinence in middle-aged women. Am J Obstet Gynecol. 2007;197:167.e1–167.e5.
Townsend MK, Danforth KN, Liffort KL, et al. Body mass index, weight gain, and incident urinary incontinence in middle-aged women. Obstet Gynecol. 2007;110:346–353.
Studies of urinary incontinence in numerous populations have reported its prevalence—i.e., the percentage of people who have the condition at any point in time—but few have attempted to define its incidence—i.e., the rate at which it develops during a defined period.
Incidence is a true rate, described with a unit of time in the denominator. Prevalence is not a rate (although it is commonly referred to as such) and is described as a percentage only, without time in the denominator. With that distinction in mind, it is easy to see why prevalence data greatly outnumber incidence data: Prevalence can be obtained by means of cross-sectional study, with one-time collection of data. In contrast, incidence data require a population that is free of the condition of interest at baseline; that population is then followed to determine how many people who were initially free of the condition go on to develop it.
Lack of a standard definition makes it hard to measure incontinence
Reported prevalence can range from less than 10% to more than 90%, depending on how incontinence is defined:
- Very low prevalence is found when the definition is limited to persons with the greatest severity or frequency of symptoms
- at the other end of the spectrum, very high prevalence—even approaching 100%—can be found using a definition that includes people who have “ever” leaked urine.
The same issues complicate estimates of incidence. Because there is no consensus over what constitutes a clinically significant threshold for incontinence, investigators are forced to develop their own definitions.
In a pair of studies, Townsend and colleagues neatly circumvent this problem. Using data from the Nurses’ Health Study II, they used a series of definitions of incontinence, ranging from less severe to more severe, to describe their findings in ways that are easily transferred to clinical practice. They focused their attention on women aged 36 to 55 years to estimate the incidence of incontinence over a 2-year period. At baseline, women were considered at risk of incident incontinence if they reported never leaking or leaking only a few drops less than once a month. Three categories of incontinence were then defined, based on symptoms 2 years later:
- incident incontinence: any urine loss, defined as leaking 1–3 times a month
- frequent incontinence: urine loss at least once a week
- severe incontinence: urine loss at least once a week of sufficient volume to at least wet underwear.
Incidence rose with BMI, weight gain
In almost 34,000 continent women from 2001 to follow-up in 2003, the overall (average) incidence of urinary incontinence was 6.9 women for every 100 woman-years. Frequent incontinence developed in, on average, 1.8 women for every 100 woman-years; severe incontinence, in 0.6 women for every 100 woman-years.
Using multivariable logistic regression models, the authors analyzed the likelihood of incident incontinence by body mass index (BMI) and estimated weight gain from the age of 18 until 2001. For either variable, odds ratios (OR) showed a highly significant trend (P<.001) for an increased risk of incident incontinence.
For example, at a BMI greater than 35 kg/m2, the likelihood of:
- any incontinence increased by a factor of about 2 (OR, 2.11; 95% confidence interval [CI], 1.84–2.42)
- frequent incontinence increased by a factor of almost 4 (OR, 3.85; 95% CI, 3.05–4.85)
- severe incontinence increased by a factor of more than 5 (OR, 5.52; 95% CI, 3.72–8.18).
The trend for weight gain was similar, with a gain of more than 30 kg showing odds ratios and 95% confidence intervals of similar magnitude to those seen with a BMI greater than 35.
But one third of incontinent women improved after 2 years
Although urinary incontinence is usually understood as a chronic condition, albeit under the influence of other factors, such as weight gain, data on remission are even scarcer than data on incidence. Using the same dataset, the authors determined that almost 31,000 women were incontinent at baseline in 2001, with incontinence occurring at least monthly. Complete remission, defined as no leaking in 2003, occurred in almost 14% of women. One third reported improvement, defined as either complete remission or a decrease in leaking frequency from 2001 to 2003.
It’s interesting that complete remission was more common in younger women. It also was more common in women who experienced frequent incontinence than in those who reported occasional incontinence. The remaining percentage of women—almost 60%—reported a similar or increased frequency of incontinence over the 2 years of follow-up.
The authors did not collect data on treatment. Estimates of persistence, improvement, and remission could be affected, therefore, if women received effective treatment between 2001 and 2003. However, only about one third of women reported mentioning their symptoms to a physician, and only 13% reported receiving treatment for incontinence. The magnitude of the effect of treatment on remission or improvement of urinary incontinence symptoms therefore seems limited.
Women remain reticent about incontinence
Several points underline the clinical importance of these data, including the relatively high incidence of incontinence symptoms and the strong influence of BMI and weight gain on that incidence. Also notable, and described in previous studies, is the vast underreporting and undertreatment of incontinence in women—an observation that should motivate all clinicians to include screening for urinary incontinence as part of regular well-woman care. Clinicians should also be prepared to refer women with incontinence or to initiate evaluation and management.
Some reports have suggested that the stigma of urinary incontinence has diminished slightly in light of widespread direct-to-consumer advertising for products related to the care (e.g., pads) or treatment (e.g., pharmaceuticals) of incontinence. The data from Townsend and colleagues are relatively recent, yet the majority of women failed to report their symptoms, and an even higher percentage received no treatment. The authors recommend that health-care providers initiate a discussion of urinary symptoms even in middle-aged women, who may be targeted for screening less frequently than older women.
In fascial sling vs Burch, sling prevails but is linked to more adverse effects
Albo ME, Richter HE, Brubaker L, et al, for the Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143–2155; comment: 2198–2200.
Eagerly anticipated results of the Urinary Incontinence Treatment Network’s first surgical trial, which compared the fascial sling procedure with Burch colposuspension for stress incontinence, were published in May in the New England Journal of Medicine. The Urinary Incontinence Treatment Network is a multicenter clinical trials group that was established in 2000 and is sponsored by the National Institutes of Health (specifically, by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of Child Health and Human Development).
Women were eligible for the trial if they experienced symptoms of stress incontinence; symptoms of mixed incontinence were allowed as long as stress symptoms predominated. Of 655 women in the trial, 326 were randomly assigned to undergo placement of an autologous rectus fascia pubovaginal sling, and 329 were randomized to Burch colposuspension.
Overall success was defined as:
- negative pad test
- no urinary incontinence reported in a 3-day diary
- negative stress test to cough and Valsalva maneuver
- no self-reported symptoms of stress incontinence
- no retreatment for stress incontinence.
“Stress success,” or stress continence, was defined using the last three criteria.
At 2 years after the index surgery, 520 women (79%) were available for follow-up. Overall success and stress success were slightly higher in women who underwent sling placement than in those treated by Burch: overall success, 47% versus 38%, and stress success, 66% versus 49%, respectively. However, women who had slings experienced more adverse outcomes, including urinary tract infection, difficulty voiding, and postoperative urge incontinence.
Success rates were much lower than previously reported
These findings are particularly striking because the success rates are lower than in previous reports—and lower than the figures commonly used by surgeons to counsel women about likely results. “Success” for either procedure has been commonly quoted in the 80% to 90% range, not the 30% to 40% range found here. The authors are to be commended for the stringent definition of “success,” which included elements that invariably result in a lower success rate. It is these numbers that women are most interested in when they are considering this type of surgery.
The difference between sling and Burch procedures was particularly remarkable in regard to stress success (17 percentage points). The smaller difference seen for overall success (9 percentage points) can be attributed to the increase in postoperative urge incontinence among women undergoing the sling procedure.
If the other adverse events associated with the sling procedure (i.e., urinary tract infection and voiding difficulty) had been included in the composite measure of success, it seems possible, if not likely, that a smaller difference—or no difference at all—would have been seen between the sling and Burch groups.
Additional data still to come
Follow-up of women in this trial has been extended for up to 5 years and should provide much-needed information on longer-term results after these surgeries.
Transobturator mid-urethral sling linked to fewer complications
Sung VW, Schleinitz MD, Rardin CR, Ward RM, Myers DL. Comparison of retropubic versus transobturator approach to mid-urethral slings: a systematic review and meta-analysis. Am J Obstet Gynecol. 2007;197:3–11.
In this Update 1 year ago, I remarked on the need for more comparative information about the various mid-urethral slings currently on the market, particularly in regard to complications—information necessary to make recommendations and guide clinical decision-making.
Originally, the procedure for mid-urethral sling placement was modified from the retropubic approach to the obturator approach with the aim of reducing the risk of major bladder and urethral injury and vascular complications (FIGURE 1). Recent data suggest that that goal has been achieved. In a systematic review and meta-analysis of 17 studies that compared retropubic and transobturator approaches, Sung and colleagues found the transobturator route to be associated with fewer complications.
FIGURE 1 Transobturator approach lives up to promise
The retropubic approach (A) was modified to create the transobturator approach (B), with the aim of protecting the bladder, urethra, and vascular structures. A recent meta-analysis indicates that this goal was achieved.
Subjective and objective outcomes were similar for the two approaches
Overall, 492 women in six trials were randomly assigned to receive either a retropubic or transobturator mid-urethral sling for treatment of stress incontinence. Although some trials specified exactly which device was used, others did not. Follow-up ranged from 1 to 15 months.
Because the studies used different definitions of objective success as outcomes, it was not possible to obtain a pooled estimate for objective outcomes. However, the authors were able to calculate a pooled estimate for subjective outcomes by defining subjective success as a woman reporting either continence or improved status after surgery, and by defining failure as a woman reporting unchanged or deteriorating incontinence status.
The pooled odds ratio for subjective failure after transobturator placement of a mid-urethral sling was 0.85, compared with the retropubic approach (95% CI, 0.38–1.92). Results were relatively stable despite changes in definitions of success and failure and restriction to studies with more than 1 year of follow-up. Sung and colleagues concluded that evidence was insufficient to support one or the other approach in regard to subjective or objective outcomes.
Bladder perforation was most common complication
Findings regarding complications were more conclusive. Again drawing on data from six randomized trials, the authors estimated a pooled odds ratio for complications from transobturator placement of 0.40, compared with the retropubic approach (95% CI, 0.19–0.83). Using data from both randomized trials and cohort studies, the most common complications were:
- bladder perforation: 3.5% for retropubic placement, 0.2% for the transobturator route
- hematoma: 1.5% for retropubic placement, 0.08% for the transobturator route.
More definitive data are in the works
As noted here last year, the Urinary Incontinence Treatment Network is enrolling women with stress or stress-predominant mixed incontinence in a randomized trial to compare the retropubic and transobturator approaches for mid-urethral slings. With a sample size of 655 women and 2-year follow-up planned, this trial should be adequately powered to detect clinically important differences, if they exist, in both continence outcomes and complications. Enrollment is projected to close in 2008, with results to follow 2 years later.
Botox injection for detrusor overactivity is no quick fix after all
Interest continues to rise in treating detrusor overactivity—with or without incontinence—with botulinum toxin A. Only one commercial product is available in the United States, sold by Allergan under the trade name Botox. Last year, the Pelvic Floor Disorders Network, sponsored by the National Institute of Child Health and Human Development and the Office of Research in Women’s Health, began a placebo-controlled trial of cystoscopic detrusor injection of 200 U of Botox versus placebo, randomized in a 2:1 ratio, for women with incontinence caused by refractory idiopathic detrusor overactivity (FIGURE 2).
Although a sample size of 210 subjects was planned, enrollment was halted after 43 women received injections (28 with Botox, 15 with placebo). The reason: A higher-than-expected rate of urinary retention.
The trial had defined urinary retention as:
- use of catheterization for more than 4 weeks after the date of injection, or
- postvoid residual (PVR) urine of 200 mL or more at the 4-week visit. (The protocol mandated that a patient with this degree of retention be catheterized or that catheterization be considered by the clinician.)
FIGURE 2 Botox relieves detrusor overactivity—but only temporarily
A trial intended to encompass 210 women was halted early because the rate of urinary retention was significantly higher than expected. Twenty-eight women underwent injection of 200 U of Botox, and almost half were classified as having urinary retention 4 weeks after the procedure.
Rate of urinary retention proved to be much higher than anticipated
At the time the study protocol was finalized, most existing studies had focused on patients with neurogenic detrusor overactivity incontinence, many of whom already had impaired bladder emptying treated with self-catheterization. Communication with clinicians using Botox off-label for idiopathic detrusor overactivity incontinence suggested that the occurrence of urinary retention requiring intervention was less than 5%. However, of the 28 women who received Botox, 12 experienced urinary retention; most of these women (9 of 12) had elevated PVR at 4 weeks after injection. Although this elevation was temporary, some women required catheterization for months.
Of the 43 women included in the trial, 12 (28%) experienced urinary retention. However, counting only subjects who received Botox, the proportion with retention was 12 of 28 (43%). None of the women who received placebo experienced urinary retention.
There was also a higher incidence of urinary tract infection in women who developed retention after Botox injection and performed self-catheterization.
Follow-up continues for all 43 subjects, as the protocol provided for monitoring up to 1 year after injection. A full report on the safety and effectiveness of Botox for idiopathic detrusor overactivity in this trial is pending (manuscript submitted for publication).
Catheterization may raise risk of infection without providing a benefit
Ideal management of women who experience elevated PVR after Botox injection is unclear. Many clinicians wonder whether treatment—i.e., catheterization—is necessary for the type of impaired bladder emptying that occurs after Botox injection. It is even possible that catheterization increases the risk of urinary tract infection (or colonization) without providing a benefit to balance that risk.
Botox may still be an option, provided the patient is counseled about risks
Our understanding, albeit incomplete, of the mechanism of action when Botox is used to treat detrusor overactivity does not suggest an increased risk of elevated intravesical pressure leading to ureteral reflux and kidney damage; in fact, normal bladder pressure has been observed in the few studies in which it was measured after Botox injection. However, until we have further information about the short- and long-term risks, if any, of elevated PVR after Botox injection, clinicians should counsel patients about this possibility before proceeding with off-label use of Botox for detrusor overactivity.
Patients unlikely to tolerate repeated Botox injection over several years
Whether or not Botox in its current form will prove to be a useful treatment for women who have detrusor overactivity incontinence remains to be proven conclusively. Even if Botox relieves symptoms, especially in women who have not obtained relief from other treatments, current evidence suggests that the effect is time-limited, probably on the order of several months—although occasional patients obtain relief of greater duration, suggesting an effect that lasts beyond direct Botox action.
Given that most women experience these symptoms on a chronic basis—perhaps especially those who are refractory to usual treatment—it seems unlikely that repeated injections at intervals of only several months can be sustained for years. Ideally, development of second-generation products and further research will produce longer-lasting effects without the need for repeated injections at regular intervals.
The author reports no financial relationships relevant to this article.
The past year has seen the publication of much useful evidence regarding urinary incontinence, from both epidemiologic studies and clinical trials. Research into the pathophysiology of incontinence continues to move forward, slowly but surely, measured not in breakthroughs but in gradually increasing knowledge of how the urethra and bladder function in the continent person and how that function can break down, leading to incontinence and other urinary symptoms.
Highlighted here are four notable studies from 2007, as well as progress notes on a trial mentioned early this year in Examining the Evidence (January issue).
New data clarify incidence and uncloak the effect of weight gain
Townsend MK, Danforth KN, Liffort KL, et al. Incidence and remission of urinary incontinence in middle-aged women. Am J Obstet Gynecol. 2007;197:167.e1–167.e5.
Townsend MK, Danforth KN, Liffort KL, et al. Body mass index, weight gain, and incident urinary incontinence in middle-aged women. Obstet Gynecol. 2007;110:346–353.
Studies of urinary incontinence in numerous populations have reported its prevalence—i.e., the percentage of people who have the condition at any point in time—but few have attempted to define its incidence—i.e., the rate at which it develops during a defined period.
Incidence is a true rate, described with a unit of time in the denominator. Prevalence is not a rate (although it is commonly referred to as such) and is described as a percentage only, without time in the denominator. With that distinction in mind, it is easy to see why prevalence data greatly outnumber incidence data: Prevalence can be obtained by means of cross-sectional study, with one-time collection of data. In contrast, incidence data require a population that is free of the condition of interest at baseline; that population is then followed to determine how many people who were initially free of the condition go on to develop it.
Lack of a standard definition makes it hard to measure incontinence
Reported prevalence can range from less than 10% to more than 90%, depending on how incontinence is defined:
- Very low prevalence is found when the definition is limited to persons with the greatest severity or frequency of symptoms
- at the other end of the spectrum, very high prevalence—even approaching 100%—can be found using a definition that includes people who have “ever” leaked urine.
The same issues complicate estimates of incidence. Because there is no consensus over what constitutes a clinically significant threshold for incontinence, investigators are forced to develop their own definitions.
In a pair of studies, Townsend and colleagues neatly circumvent this problem. Using data from the Nurses’ Health Study II, they used a series of definitions of incontinence, ranging from less severe to more severe, to describe their findings in ways that are easily transferred to clinical practice. They focused their attention on women aged 36 to 55 years to estimate the incidence of incontinence over a 2-year period. At baseline, women were considered at risk of incident incontinence if they reported never leaking or leaking only a few drops less than once a month. Three categories of incontinence were then defined, based on symptoms 2 years later:
- incident incontinence: any urine loss, defined as leaking 1–3 times a month
- frequent incontinence: urine loss at least once a week
- severe incontinence: urine loss at least once a week of sufficient volume to at least wet underwear.
Incidence rose with BMI, weight gain
In almost 34,000 continent women from 2001 to follow-up in 2003, the overall (average) incidence of urinary incontinence was 6.9 women for every 100 woman-years. Frequent incontinence developed in, on average, 1.8 women for every 100 woman-years; severe incontinence, in 0.6 women for every 100 woman-years.
Using multivariable logistic regression models, the authors analyzed the likelihood of incident incontinence by body mass index (BMI) and estimated weight gain from the age of 18 until 2001. For either variable, odds ratios (OR) showed a highly significant trend (P<.001) for an increased risk of incident incontinence.
For example, at a BMI greater than 35 kg/m2, the likelihood of:
- any incontinence increased by a factor of about 2 (OR, 2.11; 95% confidence interval [CI], 1.84–2.42)
- frequent incontinence increased by a factor of almost 4 (OR, 3.85; 95% CI, 3.05–4.85)
- severe incontinence increased by a factor of more than 5 (OR, 5.52; 95% CI, 3.72–8.18).
The trend for weight gain was similar, with a gain of more than 30 kg showing odds ratios and 95% confidence intervals of similar magnitude to those seen with a BMI greater than 35.
But one third of incontinent women improved after 2 years
Although urinary incontinence is usually understood as a chronic condition, albeit under the influence of other factors, such as weight gain, data on remission are even scarcer than data on incidence. Using the same dataset, the authors determined that almost 31,000 women were incontinent at baseline in 2001, with incontinence occurring at least monthly. Complete remission, defined as no leaking in 2003, occurred in almost 14% of women. One third reported improvement, defined as either complete remission or a decrease in leaking frequency from 2001 to 2003.
It’s interesting that complete remission was more common in younger women. It also was more common in women who experienced frequent incontinence than in those who reported occasional incontinence. The remaining percentage of women—almost 60%—reported a similar or increased frequency of incontinence over the 2 years of follow-up.
The authors did not collect data on treatment. Estimates of persistence, improvement, and remission could be affected, therefore, if women received effective treatment between 2001 and 2003. However, only about one third of women reported mentioning their symptoms to a physician, and only 13% reported receiving treatment for incontinence. The magnitude of the effect of treatment on remission or improvement of urinary incontinence symptoms therefore seems limited.
Women remain reticent about incontinence
Several points underline the clinical importance of these data, including the relatively high incidence of incontinence symptoms and the strong influence of BMI and weight gain on that incidence. Also notable, and described in previous studies, is the vast underreporting and undertreatment of incontinence in women—an observation that should motivate all clinicians to include screening for urinary incontinence as part of regular well-woman care. Clinicians should also be prepared to refer women with incontinence or to initiate evaluation and management.
Some reports have suggested that the stigma of urinary incontinence has diminished slightly in light of widespread direct-to-consumer advertising for products related to the care (e.g., pads) or treatment (e.g., pharmaceuticals) of incontinence. The data from Townsend and colleagues are relatively recent, yet the majority of women failed to report their symptoms, and an even higher percentage received no treatment. The authors recommend that health-care providers initiate a discussion of urinary symptoms even in middle-aged women, who may be targeted for screening less frequently than older women.
In fascial sling vs Burch, sling prevails but is linked to more adverse effects
Albo ME, Richter HE, Brubaker L, et al, for the Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143–2155; comment: 2198–2200.
Eagerly anticipated results of the Urinary Incontinence Treatment Network’s first surgical trial, which compared the fascial sling procedure with Burch colposuspension for stress incontinence, were published in May in the New England Journal of Medicine. The Urinary Incontinence Treatment Network is a multicenter clinical trials group that was established in 2000 and is sponsored by the National Institutes of Health (specifically, by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of Child Health and Human Development).
Women were eligible for the trial if they experienced symptoms of stress incontinence; symptoms of mixed incontinence were allowed as long as stress symptoms predominated. Of 655 women in the trial, 326 were randomly assigned to undergo placement of an autologous rectus fascia pubovaginal sling, and 329 were randomized to Burch colposuspension.
Overall success was defined as:
- negative pad test
- no urinary incontinence reported in a 3-day diary
- negative stress test to cough and Valsalva maneuver
- no self-reported symptoms of stress incontinence
- no retreatment for stress incontinence.
“Stress success,” or stress continence, was defined using the last three criteria.
At 2 years after the index surgery, 520 women (79%) were available for follow-up. Overall success and stress success were slightly higher in women who underwent sling placement than in those treated by Burch: overall success, 47% versus 38%, and stress success, 66% versus 49%, respectively. However, women who had slings experienced more adverse outcomes, including urinary tract infection, difficulty voiding, and postoperative urge incontinence.
Success rates were much lower than previously reported
These findings are particularly striking because the success rates are lower than in previous reports—and lower than the figures commonly used by surgeons to counsel women about likely results. “Success” for either procedure has been commonly quoted in the 80% to 90% range, not the 30% to 40% range found here. The authors are to be commended for the stringent definition of “success,” which included elements that invariably result in a lower success rate. It is these numbers that women are most interested in when they are considering this type of surgery.
The difference between sling and Burch procedures was particularly remarkable in regard to stress success (17 percentage points). The smaller difference seen for overall success (9 percentage points) can be attributed to the increase in postoperative urge incontinence among women undergoing the sling procedure.
If the other adverse events associated with the sling procedure (i.e., urinary tract infection and voiding difficulty) had been included in the composite measure of success, it seems possible, if not likely, that a smaller difference—or no difference at all—would have been seen between the sling and Burch groups.
Additional data still to come
Follow-up of women in this trial has been extended for up to 5 years and should provide much-needed information on longer-term results after these surgeries.
Transobturator mid-urethral sling linked to fewer complications
Sung VW, Schleinitz MD, Rardin CR, Ward RM, Myers DL. Comparison of retropubic versus transobturator approach to mid-urethral slings: a systematic review and meta-analysis. Am J Obstet Gynecol. 2007;197:3–11.
In this Update 1 year ago, I remarked on the need for more comparative information about the various mid-urethral slings currently on the market, particularly in regard to complications—information necessary to make recommendations and guide clinical decision-making.
Originally, the procedure for mid-urethral sling placement was modified from the retropubic approach to the obturator approach with the aim of reducing the risk of major bladder and urethral injury and vascular complications (FIGURE 1). Recent data suggest that that goal has been achieved. In a systematic review and meta-analysis of 17 studies that compared retropubic and transobturator approaches, Sung and colleagues found the transobturator route to be associated with fewer complications.
FIGURE 1 Transobturator approach lives up to promise
The retropubic approach (A) was modified to create the transobturator approach (B), with the aim of protecting the bladder, urethra, and vascular structures. A recent meta-analysis indicates that this goal was achieved.
Subjective and objective outcomes were similar for the two approaches
Overall, 492 women in six trials were randomly assigned to receive either a retropubic or transobturator mid-urethral sling for treatment of stress incontinence. Although some trials specified exactly which device was used, others did not. Follow-up ranged from 1 to 15 months.
Because the studies used different definitions of objective success as outcomes, it was not possible to obtain a pooled estimate for objective outcomes. However, the authors were able to calculate a pooled estimate for subjective outcomes by defining subjective success as a woman reporting either continence or improved status after surgery, and by defining failure as a woman reporting unchanged or deteriorating incontinence status.
The pooled odds ratio for subjective failure after transobturator placement of a mid-urethral sling was 0.85, compared with the retropubic approach (95% CI, 0.38–1.92). Results were relatively stable despite changes in definitions of success and failure and restriction to studies with more than 1 year of follow-up. Sung and colleagues concluded that evidence was insufficient to support one or the other approach in regard to subjective or objective outcomes.
Bladder perforation was most common complication
Findings regarding complications were more conclusive. Again drawing on data from six randomized trials, the authors estimated a pooled odds ratio for complications from transobturator placement of 0.40, compared with the retropubic approach (95% CI, 0.19–0.83). Using data from both randomized trials and cohort studies, the most common complications were:
- bladder perforation: 3.5% for retropubic placement, 0.2% for the transobturator route
- hematoma: 1.5% for retropubic placement, 0.08% for the transobturator route.
More definitive data are in the works
As noted here last year, the Urinary Incontinence Treatment Network is enrolling women with stress or stress-predominant mixed incontinence in a randomized trial to compare the retropubic and transobturator approaches for mid-urethral slings. With a sample size of 655 women and 2-year follow-up planned, this trial should be adequately powered to detect clinically important differences, if they exist, in both continence outcomes and complications. Enrollment is projected to close in 2008, with results to follow 2 years later.
Botox injection for detrusor overactivity is no quick fix after all
Interest continues to rise in treating detrusor overactivity—with or without incontinence—with botulinum toxin A. Only one commercial product is available in the United States, sold by Allergan under the trade name Botox. Last year, the Pelvic Floor Disorders Network, sponsored by the National Institute of Child Health and Human Development and the Office of Research in Women’s Health, began a placebo-controlled trial of cystoscopic detrusor injection of 200 U of Botox versus placebo, randomized in a 2:1 ratio, for women with incontinence caused by refractory idiopathic detrusor overactivity (FIGURE 2).
Although a sample size of 210 subjects was planned, enrollment was halted after 43 women received injections (28 with Botox, 15 with placebo). The reason: A higher-than-expected rate of urinary retention.
The trial had defined urinary retention as:
- use of catheterization for more than 4 weeks after the date of injection, or
- postvoid residual (PVR) urine of 200 mL or more at the 4-week visit. (The protocol mandated that a patient with this degree of retention be catheterized or that catheterization be considered by the clinician.)
FIGURE 2 Botox relieves detrusor overactivity—but only temporarily
A trial intended to encompass 210 women was halted early because the rate of urinary retention was significantly higher than expected. Twenty-eight women underwent injection of 200 U of Botox, and almost half were classified as having urinary retention 4 weeks after the procedure.
Rate of urinary retention proved to be much higher than anticipated
At the time the study protocol was finalized, most existing studies had focused on patients with neurogenic detrusor overactivity incontinence, many of whom already had impaired bladder emptying treated with self-catheterization. Communication with clinicians using Botox off-label for idiopathic detrusor overactivity incontinence suggested that the occurrence of urinary retention requiring intervention was less than 5%. However, of the 28 women who received Botox, 12 experienced urinary retention; most of these women (9 of 12) had elevated PVR at 4 weeks after injection. Although this elevation was temporary, some women required catheterization for months.
Of the 43 women included in the trial, 12 (28%) experienced urinary retention. However, counting only subjects who received Botox, the proportion with retention was 12 of 28 (43%). None of the women who received placebo experienced urinary retention.
There was also a higher incidence of urinary tract infection in women who developed retention after Botox injection and performed self-catheterization.
Follow-up continues for all 43 subjects, as the protocol provided for monitoring up to 1 year after injection. A full report on the safety and effectiveness of Botox for idiopathic detrusor overactivity in this trial is pending (manuscript submitted for publication).
Catheterization may raise risk of infection without providing a benefit
Ideal management of women who experience elevated PVR after Botox injection is unclear. Many clinicians wonder whether treatment—i.e., catheterization—is necessary for the type of impaired bladder emptying that occurs after Botox injection. It is even possible that catheterization increases the risk of urinary tract infection (or colonization) without providing a benefit to balance that risk.
Botox may still be an option, provided the patient is counseled about risks
Our understanding, albeit incomplete, of the mechanism of action when Botox is used to treat detrusor overactivity does not suggest an increased risk of elevated intravesical pressure leading to ureteral reflux and kidney damage; in fact, normal bladder pressure has been observed in the few studies in which it was measured after Botox injection. However, until we have further information about the short- and long-term risks, if any, of elevated PVR after Botox injection, clinicians should counsel patients about this possibility before proceeding with off-label use of Botox for detrusor overactivity.
Patients unlikely to tolerate repeated Botox injection over several years
Whether or not Botox in its current form will prove to be a useful treatment for women who have detrusor overactivity incontinence remains to be proven conclusively. Even if Botox relieves symptoms, especially in women who have not obtained relief from other treatments, current evidence suggests that the effect is time-limited, probably on the order of several months—although occasional patients obtain relief of greater duration, suggesting an effect that lasts beyond direct Botox action.
Given that most women experience these symptoms on a chronic basis—perhaps especially those who are refractory to usual treatment—it seems unlikely that repeated injections at intervals of only several months can be sustained for years. Ideally, development of second-generation products and further research will produce longer-lasting effects without the need for repeated injections at regular intervals.
The author reports no financial relationships relevant to this article.
The past year has seen the publication of much useful evidence regarding urinary incontinence, from both epidemiologic studies and clinical trials. Research into the pathophysiology of incontinence continues to move forward, slowly but surely, measured not in breakthroughs but in gradually increasing knowledge of how the urethra and bladder function in the continent person and how that function can break down, leading to incontinence and other urinary symptoms.
Highlighted here are four notable studies from 2007, as well as progress notes on a trial mentioned early this year in Examining the Evidence (January issue).
New data clarify incidence and uncloak the effect of weight gain
Townsend MK, Danforth KN, Liffort KL, et al. Incidence and remission of urinary incontinence in middle-aged women. Am J Obstet Gynecol. 2007;197:167.e1–167.e5.
Townsend MK, Danforth KN, Liffort KL, et al. Body mass index, weight gain, and incident urinary incontinence in middle-aged women. Obstet Gynecol. 2007;110:346–353.
Studies of urinary incontinence in numerous populations have reported its prevalence—i.e., the percentage of people who have the condition at any point in time—but few have attempted to define its incidence—i.e., the rate at which it develops during a defined period.
Incidence is a true rate, described with a unit of time in the denominator. Prevalence is not a rate (although it is commonly referred to as such) and is described as a percentage only, without time in the denominator. With that distinction in mind, it is easy to see why prevalence data greatly outnumber incidence data: Prevalence can be obtained by means of cross-sectional study, with one-time collection of data. In contrast, incidence data require a population that is free of the condition of interest at baseline; that population is then followed to determine how many people who were initially free of the condition go on to develop it.
Lack of a standard definition makes it hard to measure incontinence
Reported prevalence can range from less than 10% to more than 90%, depending on how incontinence is defined:
- Very low prevalence is found when the definition is limited to persons with the greatest severity or frequency of symptoms
- at the other end of the spectrum, very high prevalence—even approaching 100%—can be found using a definition that includes people who have “ever” leaked urine.
The same issues complicate estimates of incidence. Because there is no consensus over what constitutes a clinically significant threshold for incontinence, investigators are forced to develop their own definitions.
In a pair of studies, Townsend and colleagues neatly circumvent this problem. Using data from the Nurses’ Health Study II, they used a series of definitions of incontinence, ranging from less severe to more severe, to describe their findings in ways that are easily transferred to clinical practice. They focused their attention on women aged 36 to 55 years to estimate the incidence of incontinence over a 2-year period. At baseline, women were considered at risk of incident incontinence if they reported never leaking or leaking only a few drops less than once a month. Three categories of incontinence were then defined, based on symptoms 2 years later:
- incident incontinence: any urine loss, defined as leaking 1–3 times a month
- frequent incontinence: urine loss at least once a week
- severe incontinence: urine loss at least once a week of sufficient volume to at least wet underwear.
Incidence rose with BMI, weight gain
In almost 34,000 continent women from 2001 to follow-up in 2003, the overall (average) incidence of urinary incontinence was 6.9 women for every 100 woman-years. Frequent incontinence developed in, on average, 1.8 women for every 100 woman-years; severe incontinence, in 0.6 women for every 100 woman-years.
Using multivariable logistic regression models, the authors analyzed the likelihood of incident incontinence by body mass index (BMI) and estimated weight gain from the age of 18 until 2001. For either variable, odds ratios (OR) showed a highly significant trend (P<.001) for an increased risk of incident incontinence.
For example, at a BMI greater than 35 kg/m2, the likelihood of:
- any incontinence increased by a factor of about 2 (OR, 2.11; 95% confidence interval [CI], 1.84–2.42)
- frequent incontinence increased by a factor of almost 4 (OR, 3.85; 95% CI, 3.05–4.85)
- severe incontinence increased by a factor of more than 5 (OR, 5.52; 95% CI, 3.72–8.18).
The trend for weight gain was similar, with a gain of more than 30 kg showing odds ratios and 95% confidence intervals of similar magnitude to those seen with a BMI greater than 35.
But one third of incontinent women improved after 2 years
Although urinary incontinence is usually understood as a chronic condition, albeit under the influence of other factors, such as weight gain, data on remission are even scarcer than data on incidence. Using the same dataset, the authors determined that almost 31,000 women were incontinent at baseline in 2001, with incontinence occurring at least monthly. Complete remission, defined as no leaking in 2003, occurred in almost 14% of women. One third reported improvement, defined as either complete remission or a decrease in leaking frequency from 2001 to 2003.
It’s interesting that complete remission was more common in younger women. It also was more common in women who experienced frequent incontinence than in those who reported occasional incontinence. The remaining percentage of women—almost 60%—reported a similar or increased frequency of incontinence over the 2 years of follow-up.
The authors did not collect data on treatment. Estimates of persistence, improvement, and remission could be affected, therefore, if women received effective treatment between 2001 and 2003. However, only about one third of women reported mentioning their symptoms to a physician, and only 13% reported receiving treatment for incontinence. The magnitude of the effect of treatment on remission or improvement of urinary incontinence symptoms therefore seems limited.
Women remain reticent about incontinence
Several points underline the clinical importance of these data, including the relatively high incidence of incontinence symptoms and the strong influence of BMI and weight gain on that incidence. Also notable, and described in previous studies, is the vast underreporting and undertreatment of incontinence in women—an observation that should motivate all clinicians to include screening for urinary incontinence as part of regular well-woman care. Clinicians should also be prepared to refer women with incontinence or to initiate evaluation and management.
Some reports have suggested that the stigma of urinary incontinence has diminished slightly in light of widespread direct-to-consumer advertising for products related to the care (e.g., pads) or treatment (e.g., pharmaceuticals) of incontinence. The data from Townsend and colleagues are relatively recent, yet the majority of women failed to report their symptoms, and an even higher percentage received no treatment. The authors recommend that health-care providers initiate a discussion of urinary symptoms even in middle-aged women, who may be targeted for screening less frequently than older women.
In fascial sling vs Burch, sling prevails but is linked to more adverse effects
Albo ME, Richter HE, Brubaker L, et al, for the Urinary Incontinence Treatment Network. Burch colposuspension versus fascial sling to reduce urinary stress incontinence. N Engl J Med. 2007;356:2143–2155; comment: 2198–2200.
Eagerly anticipated results of the Urinary Incontinence Treatment Network’s first surgical trial, which compared the fascial sling procedure with Burch colposuspension for stress incontinence, were published in May in the New England Journal of Medicine. The Urinary Incontinence Treatment Network is a multicenter clinical trials group that was established in 2000 and is sponsored by the National Institutes of Health (specifically, by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of Child Health and Human Development).
Women were eligible for the trial if they experienced symptoms of stress incontinence; symptoms of mixed incontinence were allowed as long as stress symptoms predominated. Of 655 women in the trial, 326 were randomly assigned to undergo placement of an autologous rectus fascia pubovaginal sling, and 329 were randomized to Burch colposuspension.
Overall success was defined as:
- negative pad test
- no urinary incontinence reported in a 3-day diary
- negative stress test to cough and Valsalva maneuver
- no self-reported symptoms of stress incontinence
- no retreatment for stress incontinence.
“Stress success,” or stress continence, was defined using the last three criteria.
At 2 years after the index surgery, 520 women (79%) were available for follow-up. Overall success and stress success were slightly higher in women who underwent sling placement than in those treated by Burch: overall success, 47% versus 38%, and stress success, 66% versus 49%, respectively. However, women who had slings experienced more adverse outcomes, including urinary tract infection, difficulty voiding, and postoperative urge incontinence.
Success rates were much lower than previously reported
These findings are particularly striking because the success rates are lower than in previous reports—and lower than the figures commonly used by surgeons to counsel women about likely results. “Success” for either procedure has been commonly quoted in the 80% to 90% range, not the 30% to 40% range found here. The authors are to be commended for the stringent definition of “success,” which included elements that invariably result in a lower success rate. It is these numbers that women are most interested in when they are considering this type of surgery.
The difference between sling and Burch procedures was particularly remarkable in regard to stress success (17 percentage points). The smaller difference seen for overall success (9 percentage points) can be attributed to the increase in postoperative urge incontinence among women undergoing the sling procedure.
If the other adverse events associated with the sling procedure (i.e., urinary tract infection and voiding difficulty) had been included in the composite measure of success, it seems possible, if not likely, that a smaller difference—or no difference at all—would have been seen between the sling and Burch groups.
Additional data still to come
Follow-up of women in this trial has been extended for up to 5 years and should provide much-needed information on longer-term results after these surgeries.
Transobturator mid-urethral sling linked to fewer complications
Sung VW, Schleinitz MD, Rardin CR, Ward RM, Myers DL. Comparison of retropubic versus transobturator approach to mid-urethral slings: a systematic review and meta-analysis. Am J Obstet Gynecol. 2007;197:3–11.
In this Update 1 year ago, I remarked on the need for more comparative information about the various mid-urethral slings currently on the market, particularly in regard to complications—information necessary to make recommendations and guide clinical decision-making.
Originally, the procedure for mid-urethral sling placement was modified from the retropubic approach to the obturator approach with the aim of reducing the risk of major bladder and urethral injury and vascular complications (FIGURE 1). Recent data suggest that that goal has been achieved. In a systematic review and meta-analysis of 17 studies that compared retropubic and transobturator approaches, Sung and colleagues found the transobturator route to be associated with fewer complications.
FIGURE 1 Transobturator approach lives up to promise
The retropubic approach (A) was modified to create the transobturator approach (B), with the aim of protecting the bladder, urethra, and vascular structures. A recent meta-analysis indicates that this goal was achieved.
Subjective and objective outcomes were similar for the two approaches
Overall, 492 women in six trials were randomly assigned to receive either a retropubic or transobturator mid-urethral sling for treatment of stress incontinence. Although some trials specified exactly which device was used, others did not. Follow-up ranged from 1 to 15 months.
Because the studies used different definitions of objective success as outcomes, it was not possible to obtain a pooled estimate for objective outcomes. However, the authors were able to calculate a pooled estimate for subjective outcomes by defining subjective success as a woman reporting either continence or improved status after surgery, and by defining failure as a woman reporting unchanged or deteriorating incontinence status.
The pooled odds ratio for subjective failure after transobturator placement of a mid-urethral sling was 0.85, compared with the retropubic approach (95% CI, 0.38–1.92). Results were relatively stable despite changes in definitions of success and failure and restriction to studies with more than 1 year of follow-up. Sung and colleagues concluded that evidence was insufficient to support one or the other approach in regard to subjective or objective outcomes.
Bladder perforation was most common complication
Findings regarding complications were more conclusive. Again drawing on data from six randomized trials, the authors estimated a pooled odds ratio for complications from transobturator placement of 0.40, compared with the retropubic approach (95% CI, 0.19–0.83). Using data from both randomized trials and cohort studies, the most common complications were:
- bladder perforation: 3.5% for retropubic placement, 0.2% for the transobturator route
- hematoma: 1.5% for retropubic placement, 0.08% for the transobturator route.
More definitive data are in the works
As noted here last year, the Urinary Incontinence Treatment Network is enrolling women with stress or stress-predominant mixed incontinence in a randomized trial to compare the retropubic and transobturator approaches for mid-urethral slings. With a sample size of 655 women and 2-year follow-up planned, this trial should be adequately powered to detect clinically important differences, if they exist, in both continence outcomes and complications. Enrollment is projected to close in 2008, with results to follow 2 years later.
Botox injection for detrusor overactivity is no quick fix after all
Interest continues to rise in treating detrusor overactivity—with or without incontinence—with botulinum toxin A. Only one commercial product is available in the United States, sold by Allergan under the trade name Botox. Last year, the Pelvic Floor Disorders Network, sponsored by the National Institute of Child Health and Human Development and the Office of Research in Women’s Health, began a placebo-controlled trial of cystoscopic detrusor injection of 200 U of Botox versus placebo, randomized in a 2:1 ratio, for women with incontinence caused by refractory idiopathic detrusor overactivity (FIGURE 2).
Although a sample size of 210 subjects was planned, enrollment was halted after 43 women received injections (28 with Botox, 15 with placebo). The reason: A higher-than-expected rate of urinary retention.
The trial had defined urinary retention as:
- use of catheterization for more than 4 weeks after the date of injection, or
- postvoid residual (PVR) urine of 200 mL or more at the 4-week visit. (The protocol mandated that a patient with this degree of retention be catheterized or that catheterization be considered by the clinician.)
FIGURE 2 Botox relieves detrusor overactivity—but only temporarily
A trial intended to encompass 210 women was halted early because the rate of urinary retention was significantly higher than expected. Twenty-eight women underwent injection of 200 U of Botox, and almost half were classified as having urinary retention 4 weeks after the procedure.
Rate of urinary retention proved to be much higher than anticipated
At the time the study protocol was finalized, most existing studies had focused on patients with neurogenic detrusor overactivity incontinence, many of whom already had impaired bladder emptying treated with self-catheterization. Communication with clinicians using Botox off-label for idiopathic detrusor overactivity incontinence suggested that the occurrence of urinary retention requiring intervention was less than 5%. However, of the 28 women who received Botox, 12 experienced urinary retention; most of these women (9 of 12) had elevated PVR at 4 weeks after injection. Although this elevation was temporary, some women required catheterization for months.
Of the 43 women included in the trial, 12 (28%) experienced urinary retention. However, counting only subjects who received Botox, the proportion with retention was 12 of 28 (43%). None of the women who received placebo experienced urinary retention.
There was also a higher incidence of urinary tract infection in women who developed retention after Botox injection and performed self-catheterization.
Follow-up continues for all 43 subjects, as the protocol provided for monitoring up to 1 year after injection. A full report on the safety and effectiveness of Botox for idiopathic detrusor overactivity in this trial is pending (manuscript submitted for publication).
Catheterization may raise risk of infection without providing a benefit
Ideal management of women who experience elevated PVR after Botox injection is unclear. Many clinicians wonder whether treatment—i.e., catheterization—is necessary for the type of impaired bladder emptying that occurs after Botox injection. It is even possible that catheterization increases the risk of urinary tract infection (or colonization) without providing a benefit to balance that risk.
Botox may still be an option, provided the patient is counseled about risks
Our understanding, albeit incomplete, of the mechanism of action when Botox is used to treat detrusor overactivity does not suggest an increased risk of elevated intravesical pressure leading to ureteral reflux and kidney damage; in fact, normal bladder pressure has been observed in the few studies in which it was measured after Botox injection. However, until we have further information about the short- and long-term risks, if any, of elevated PVR after Botox injection, clinicians should counsel patients about this possibility before proceeding with off-label use of Botox for detrusor overactivity.
Patients unlikely to tolerate repeated Botox injection over several years
Whether or not Botox in its current form will prove to be a useful treatment for women who have detrusor overactivity incontinence remains to be proven conclusively. Even if Botox relieves symptoms, especially in women who have not obtained relief from other treatments, current evidence suggests that the effect is time-limited, probably on the order of several months—although occasional patients obtain relief of greater duration, suggesting an effect that lasts beyond direct Botox action.
Given that most women experience these symptoms on a chronic basis—perhaps especially those who are refractory to usual treatment—it seems unlikely that repeated injections at intervals of only several months can be sustained for years. Ideally, development of second-generation products and further research will produce longer-lasting effects without the need for repeated injections at regular intervals.
Q Does Botox relieve urinary urgency and urge incontinence?
<huc>A</huc> Yes, but improvements are time-limited. In this series of 100 cases of idiopathic detrusor overactivity treated with botulinum toxin A (Botox), symptoms resolved in 74% of patients with urge incontinence by 4 weeks after treatment, and in 80% of patients by 12 weeks after treatment. Urgency resolved in 72% at 4 weeks and 66% at 12 weeks.
Expert Commentary
This fairly large case series helps clarify the potential of Botox to ease symptoms in patients who do not respond to current therapies for idiopathic detrusor overactivity.
Among the first authors to report successful use of Botox for neurogenic detrusor overactivity was a research team from Switzerland led by Brigitte Schurch—the same group that published this case series. Participants met detailed criteria for “overactive bladder syndrome,” as defined by the International Continence Society, with either (1) urodynamically demonstrated nonneurogenic (idiopathic) detrusor overactivity with or without incontinence or (2) hypersensitive bladder with premature filling sensation (even when maximum bladder capacity was normal) and more than 8 voids per 24 hours (urgency–frequency syndrome with or without incontinence).
Participants included 77 women and 23 men, each of whom received 100 U of Botox diluted with saline and injected at 30 sites in the bladder under cystoscopic guidance, sparing the trigone.
Risks versus benefits
Adverse events reported in this study included urinary tract infections in 10 patients (10%) and urinary retention requiring intermittent self-catheterization in 4 patients (4%). However, retention was otherwise not well characterized. Mean postvoid residual did not return to baseline values until 9 months after injection.
Why these data are imperfect
Although the study included both women and men (ratio about 3:1), data were not reported by sex. Moreover, according to the International Continence Society definition, patients with overactive bladder syndrome constitute a heterogeneous group based on urinary symptoms (which may or may not include urge incontinence) and urodynamic findings (which may or may not include detrusor overactivity).
A small percentage of patients had apparent detrusor hypocontractility or acontractility with elevated postvoid residual urine volumes—a group clinically distinct from patients with otherwise “normal” bladder emptying despite symptoms attributed to the detrusor muscle.
A robust placebo effect is likely
In this open-label series, as in clinical practice, both patients and clinicians expected treatment to be beneficial—raising the possibility of a placebo effect that may explain part (or most) of the improvement. In many placebo-controlled drug trials involving patients with detrusor overactivity, a relatively large portion of the “treatment effect” may be attributed to the placebo effect; as many as 40% of patients taking placebo report some relief of symptoms.
Beyond beauty: Botox for the bladder
Under cystoscopic guidance, Schmid et al injected 100 U of saline-diluted Botox (tinted with indigo carmine to aid in spacing) at 30 sites in the bladder, excluding the trigone. Most women reported temporary relief of symptoms.
More data are needed
To address these gaps in knowledge, especially as use of Botox is being rapidly incorporated into clinical practice, investigators in the NIH-sponsored Pelvic Floor Disorders Network are conducting a randomized, placebo-controlled trial in women with idiopathic detrusor overactivity incontinence. The aim of the trial: to describe the percentage of women whose symptoms resolve, followed by time to recurrence, after treatment with Botox versus placebo, with 1 year of follow-up.
To obtain focused data, inclusion and exclusion criteria were developed to ensure that the population of this triple-blinded study is relatively homogeneous. As the occurrence and consequences of urinary retention have not been well described in prior studies, subjects in the NIH trial are being carefully assessed for urinary retention. Enrollment should be complete by mid-2007, and follow-up by mid-2008.
Should ObGyns consider Botox for women with intractable symptoms?
For clinicians experienced with cystoscopically guided injections, Botox might be a reasonable option for highly selected women who are truly refractory to all other treatments for detrusor overactivity incontinence. Such women should understand that Botox is not FDA-approved for this indication and that its use is experimental. No evidence identifies the optimal dose of Botox to be injected cystoscopically for incontinence related to detrusor overactivity. However, dosing information may be forthcoming from a Phase II study being performed by the company that markets Botox (Allergan).
Greatest risk is urinary retention
The most important immediate or short-term risk is probably urinary retention. Although experienced clinicians have estimated a low risk of retention, a much higher rate can be found when the postvoid residual is measured routinely after Botox injection. It still seems likely that Botox-associated retention is temporary, but it may last as long as the effect on symptoms, on the order of several months. Women should be counseled carefully to be sure they understand this.
Further, because urinary sensation may be altered after Botox, women may not experience bothersome symptoms of retention. Nevertheless, more data on possible short- and long-term consequences are needed before changing clinical management of this type of retention (ie, I recommend continuing to treat retention rather than watchful waiting).
Until more data are available on risks versus benefits, I recommend against using Botox in women at high risk for complications associated with partial urinary retention. I would be particularly concerned about elderly women at risk of urinary tract infection that may lead to urosepsis.
<huc>A</huc> Yes, but improvements are time-limited. In this series of 100 cases of idiopathic detrusor overactivity treated with botulinum toxin A (Botox), symptoms resolved in 74% of patients with urge incontinence by 4 weeks after treatment, and in 80% of patients by 12 weeks after treatment. Urgency resolved in 72% at 4 weeks and 66% at 12 weeks.
Expert Commentary
This fairly large case series helps clarify the potential of Botox to ease symptoms in patients who do not respond to current therapies for idiopathic detrusor overactivity.
Among the first authors to report successful use of Botox for neurogenic detrusor overactivity was a research team from Switzerland led by Brigitte Schurch—the same group that published this case series. Participants met detailed criteria for “overactive bladder syndrome,” as defined by the International Continence Society, with either (1) urodynamically demonstrated nonneurogenic (idiopathic) detrusor overactivity with or without incontinence or (2) hypersensitive bladder with premature filling sensation (even when maximum bladder capacity was normal) and more than 8 voids per 24 hours (urgency–frequency syndrome with or without incontinence).
Participants included 77 women and 23 men, each of whom received 100 U of Botox diluted with saline and injected at 30 sites in the bladder under cystoscopic guidance, sparing the trigone.
Risks versus benefits
Adverse events reported in this study included urinary tract infections in 10 patients (10%) and urinary retention requiring intermittent self-catheterization in 4 patients (4%). However, retention was otherwise not well characterized. Mean postvoid residual did not return to baseline values until 9 months after injection.
Why these data are imperfect
Although the study included both women and men (ratio about 3:1), data were not reported by sex. Moreover, according to the International Continence Society definition, patients with overactive bladder syndrome constitute a heterogeneous group based on urinary symptoms (which may or may not include urge incontinence) and urodynamic findings (which may or may not include detrusor overactivity).
A small percentage of patients had apparent detrusor hypocontractility or acontractility with elevated postvoid residual urine volumes—a group clinically distinct from patients with otherwise “normal” bladder emptying despite symptoms attributed to the detrusor muscle.
A robust placebo effect is likely
In this open-label series, as in clinical practice, both patients and clinicians expected treatment to be beneficial—raising the possibility of a placebo effect that may explain part (or most) of the improvement. In many placebo-controlled drug trials involving patients with detrusor overactivity, a relatively large portion of the “treatment effect” may be attributed to the placebo effect; as many as 40% of patients taking placebo report some relief of symptoms.
Beyond beauty: Botox for the bladder
Under cystoscopic guidance, Schmid et al injected 100 U of saline-diluted Botox (tinted with indigo carmine to aid in spacing) at 30 sites in the bladder, excluding the trigone. Most women reported temporary relief of symptoms.
More data are needed
To address these gaps in knowledge, especially as use of Botox is being rapidly incorporated into clinical practice, investigators in the NIH-sponsored Pelvic Floor Disorders Network are conducting a randomized, placebo-controlled trial in women with idiopathic detrusor overactivity incontinence. The aim of the trial: to describe the percentage of women whose symptoms resolve, followed by time to recurrence, after treatment with Botox versus placebo, with 1 year of follow-up.
To obtain focused data, inclusion and exclusion criteria were developed to ensure that the population of this triple-blinded study is relatively homogeneous. As the occurrence and consequences of urinary retention have not been well described in prior studies, subjects in the NIH trial are being carefully assessed for urinary retention. Enrollment should be complete by mid-2007, and follow-up by mid-2008.
Should ObGyns consider Botox for women with intractable symptoms?
For clinicians experienced with cystoscopically guided injections, Botox might be a reasonable option for highly selected women who are truly refractory to all other treatments for detrusor overactivity incontinence. Such women should understand that Botox is not FDA-approved for this indication and that its use is experimental. No evidence identifies the optimal dose of Botox to be injected cystoscopically for incontinence related to detrusor overactivity. However, dosing information may be forthcoming from a Phase II study being performed by the company that markets Botox (Allergan).
Greatest risk is urinary retention
The most important immediate or short-term risk is probably urinary retention. Although experienced clinicians have estimated a low risk of retention, a much higher rate can be found when the postvoid residual is measured routinely after Botox injection. It still seems likely that Botox-associated retention is temporary, but it may last as long as the effect on symptoms, on the order of several months. Women should be counseled carefully to be sure they understand this.
Further, because urinary sensation may be altered after Botox, women may not experience bothersome symptoms of retention. Nevertheless, more data on possible short- and long-term consequences are needed before changing clinical management of this type of retention (ie, I recommend continuing to treat retention rather than watchful waiting).
Until more data are available on risks versus benefits, I recommend against using Botox in women at high risk for complications associated with partial urinary retention. I would be particularly concerned about elderly women at risk of urinary tract infection that may lead to urosepsis.
<huc>A</huc> Yes, but improvements are time-limited. In this series of 100 cases of idiopathic detrusor overactivity treated with botulinum toxin A (Botox), symptoms resolved in 74% of patients with urge incontinence by 4 weeks after treatment, and in 80% of patients by 12 weeks after treatment. Urgency resolved in 72% at 4 weeks and 66% at 12 weeks.
Expert Commentary
This fairly large case series helps clarify the potential of Botox to ease symptoms in patients who do not respond to current therapies for idiopathic detrusor overactivity.
Among the first authors to report successful use of Botox for neurogenic detrusor overactivity was a research team from Switzerland led by Brigitte Schurch—the same group that published this case series. Participants met detailed criteria for “overactive bladder syndrome,” as defined by the International Continence Society, with either (1) urodynamically demonstrated nonneurogenic (idiopathic) detrusor overactivity with or without incontinence or (2) hypersensitive bladder with premature filling sensation (even when maximum bladder capacity was normal) and more than 8 voids per 24 hours (urgency–frequency syndrome with or without incontinence).
Participants included 77 women and 23 men, each of whom received 100 U of Botox diluted with saline and injected at 30 sites in the bladder under cystoscopic guidance, sparing the trigone.
Risks versus benefits
Adverse events reported in this study included urinary tract infections in 10 patients (10%) and urinary retention requiring intermittent self-catheterization in 4 patients (4%). However, retention was otherwise not well characterized. Mean postvoid residual did not return to baseline values until 9 months after injection.
Why these data are imperfect
Although the study included both women and men (ratio about 3:1), data were not reported by sex. Moreover, according to the International Continence Society definition, patients with overactive bladder syndrome constitute a heterogeneous group based on urinary symptoms (which may or may not include urge incontinence) and urodynamic findings (which may or may not include detrusor overactivity).
A small percentage of patients had apparent detrusor hypocontractility or acontractility with elevated postvoid residual urine volumes—a group clinically distinct from patients with otherwise “normal” bladder emptying despite symptoms attributed to the detrusor muscle.
A robust placebo effect is likely
In this open-label series, as in clinical practice, both patients and clinicians expected treatment to be beneficial—raising the possibility of a placebo effect that may explain part (or most) of the improvement. In many placebo-controlled drug trials involving patients with detrusor overactivity, a relatively large portion of the “treatment effect” may be attributed to the placebo effect; as many as 40% of patients taking placebo report some relief of symptoms.
Beyond beauty: Botox for the bladder
Under cystoscopic guidance, Schmid et al injected 100 U of saline-diluted Botox (tinted with indigo carmine to aid in spacing) at 30 sites in the bladder, excluding the trigone. Most women reported temporary relief of symptoms.
More data are needed
To address these gaps in knowledge, especially as use of Botox is being rapidly incorporated into clinical practice, investigators in the NIH-sponsored Pelvic Floor Disorders Network are conducting a randomized, placebo-controlled trial in women with idiopathic detrusor overactivity incontinence. The aim of the trial: to describe the percentage of women whose symptoms resolve, followed by time to recurrence, after treatment with Botox versus placebo, with 1 year of follow-up.
To obtain focused data, inclusion and exclusion criteria were developed to ensure that the population of this triple-blinded study is relatively homogeneous. As the occurrence and consequences of urinary retention have not been well described in prior studies, subjects in the NIH trial are being carefully assessed for urinary retention. Enrollment should be complete by mid-2007, and follow-up by mid-2008.
Should ObGyns consider Botox for women with intractable symptoms?
For clinicians experienced with cystoscopically guided injections, Botox might be a reasonable option for highly selected women who are truly refractory to all other treatments for detrusor overactivity incontinence. Such women should understand that Botox is not FDA-approved for this indication and that its use is experimental. No evidence identifies the optimal dose of Botox to be injected cystoscopically for incontinence related to detrusor overactivity. However, dosing information may be forthcoming from a Phase II study being performed by the company that markets Botox (Allergan).
Greatest risk is urinary retention
The most important immediate or short-term risk is probably urinary retention. Although experienced clinicians have estimated a low risk of retention, a much higher rate can be found when the postvoid residual is measured routinely after Botox injection. It still seems likely that Botox-associated retention is temporary, but it may last as long as the effect on symptoms, on the order of several months. Women should be counseled carefully to be sure they understand this.
Further, because urinary sensation may be altered after Botox, women may not experience bothersome symptoms of retention. Nevertheless, more data on possible short- and long-term consequences are needed before changing clinical management of this type of retention (ie, I recommend continuing to treat retention rather than watchful waiting).
Until more data are available on risks versus benefits, I recommend against using Botox in women at high risk for complications associated with partial urinary retention. I would be particularly concerned about elderly women at risk of urinary tract infection that may lead to urosepsis.
URINARY INCONTINENCE
Surgery for stress incontinence
Another year is drawing to a close and, looking back, what have we learned about urinary incontinence? A clear understanding of etiology stubbornly eludes us. How would a clear understanding of etiology affect management? It’s difficult to be specific until we actually do understand it, but generally:
The “multiple-hit” theory probably applies to urinary incontinence, too
The “multiple-hit” theory usually ascribed to cancer probably also fits the development of urinary incontinence, a likewise multifaceted condition. A woman begins life with genetic predisposition at some level that we cannot currently measure, but which is influenced by the environment (eg, nutrition, toxic exposures) and life events (eg, childbirth, aging)—all of which determine her likelihood of developing incontinence.
Until the time when we do have a clear understanding on which to base diagnosis, treatment, and prevention, of course, we must continue to manage incontinence with the tools of today.
A few pieces of the puzzle are slowly coming together.
Surgery for stress incontinence
Even as new surgical techniques or modifications continue to proliferate, evidence to guide clinical practice accumulates belatedly. MEDLINE lists 325 articles since 1996 (combining surgical mesh and urinary incontinence, limited to human females and published in English). Nonetheless, a consensus may be emerging that the safest synthetic material is monofilament polypropylene with pore size larger than 70 μm.
Unfortunately, by the time research reports are published showing higher complications with certain products, countless women have already been treated.
Mesh erosion (or exposure, extrusion), sometimes accompanied by infection, is a common complication when multifilament or small-pore meshes are used. Even worse, companies commonly withdraw products, modify them, and re-introduce them to the market, accompanied by intensive marketing but, as with the original product, without any real evidence of safety and effectiveness.
In an ideal world, clinicians (and patients) would insist on evidence before accepting new products and techniques. Failing that, clinicians (and patients!) should clearly understand that all new products and techniques are experimental until they are proven equal to or better than traditional techniques. As we have learned with the most subtle differences between synthetic materials, “almost the same” or “looks the same” is not the same.
Among the most important evidence on slings this year are reports of investigations that demonstrated what should not be done.
Are monofilament, large-pore mesh products safer?
Abdel-Fattah M, Sivanesan K, Ramsay I, Pringle S, Bjornsson S. How common are tape erosions? A comparison of two versions of the transobturator tension-free vaginal tape procedure. BJU Int. 2006;98:594–598.
Yamada BS, Govier FE, Stefanovic KB, Kobashi KC. High rate of vaginal erosions associated with the Mentor Obtape. J Urol. 2006;176:651–654.
Siegel AL, Kim M, Goldstein M, Levey S, Ilbeigi P. High incidence of vaginal mesh extrusion using the Intravaginal Slingplasty sling. J Urol. 2005;174:1308–1311.
The risk of vaginal erosion is much higher with synthetic meshes used for sling procedures when the mesh is multifilament and/or small-pore (<70 μm). In some cases, companies have replaced products (Mentor Obtape small-pore polypropylene sling product was replaced with macroporous Aris), whereas others continue to market products reported to have unacceptably high rates of vaginal erosion and mesh extrusion (Intravaginal Slingplasty multifilament mesh) (TABLE 1).
TABLE 1
Comparison of selected multifilament and small-pore mesh products
| REFERENCE | SLING PRODUCT | EROSION RATE | MANAGEMENT |
|---|---|---|---|
| Abdel-Fattah et al (2006) | Obtape | 7.3% (14 of 192) | 7: partial excision 7: complete excision with infection |
| TVT-O | 1.8% (2 of 112) | 1: partial excision 1: vaginal closure | |
| Yamada et al (2006) | Obtape | 13.4% (9 of 67) | 9: complete excision (1 with abscess) |
| Monarc | 0 of 56 | — | |
| Siegel et al (2005) | IVS | 17% (6 of 35) | 6: complete excision (1 with pelvic abscess) |
Avoid cadaveric fascia in sling procedures
Howden NS, Zyczynski HM, Moalli PA, Sagan ER, Meyn LA, Weber AM. Comparison of autologous rectus fascia and cadaveric fascia in pubovaginal sling continence outcomes. Am J Obstet Gynecol. 2006;194:1444–1449.
Evidence has accumulated that sling procedures performed with cadaveric fascia have substantially worse continence outcomes, compared with those using autologous fascia. In a retrospective cohort study of 150 women with cadaveric fascial slings and 153 women who had autologous rectus fascial slings, urinary incontinence (16 vs 5 per 100 women-years) and reoperation for stress incontinence (4 vs 1 per 100 women-years) occurred more frequently after cadaveric versus autologous rectus fascial slings.
Should xenograft materials be used in sling procedures?
Giri SK, Hickey JP, Sil D, et al. Long-term results of pubovaginal sling surgery using acellular cross-linked porcine dermis in the treatment of urodynamic stress incontinence. J Urol. 2006; 175:1788–1792.
Several companies market specific products integrated into sling techniques, such as In-First Ultra (porcine dermal matrix secured with bone anchors) and Stratasis (porcine small intestinal submucosa in urethral sling and tension-free versions). Other companies market only the xenograft for application in sling procedures, such as Pelvicol (acellular porcine collagen matrix). However, relatively little information is available to support or discourage use of xenograft materials in sling procedures.
Giri et al found worse outcomes using Pelvicol compared with autologous rectus fascia for pubovaginal slings. With 3-year follow-up, 54% (26 of 48 women) with Pelvicol were considered successfully cured or improved, compared with 80.4% (37 of 46 women) with rectus fascia. Of interest, women continued to report recurrent incontinence with Pelvicol through the 3-year period, whereas women with rectus fascia had recurrence within the first 9 months after surgery.
Midurethral slings: Retropubic or transobturator?
Waltregny D, Reul O, Mathantu B, Gaspar Y, Bonnet P, de Leval J. Inside out transobturator vaginal tape for the treatment of female stress urinary incontinence: interim results of a prospective study after a 1-year minimum followup. J Urol. 2006;175:2191–2195.
Morey AF, Medendorp AR, Noller MW, et al. Transobturator versus transabdominal midurethral slings: a multi-institutional comparison of obstructive voiding complications. J Urol. 2006;175:1014–1017.
Midurethral sling placement was modified from the retropubic to the obturator approach with the objective of reducing the risk of major bladder and urethral injury and vascular complications. Does the obturator approach actually have fewer intraoperative complications compared with the retropubic approach? Unknown. (As noted below, even within retropubic procedures, it is possible—although currently unknown—that vaginal and abdominal approaches have different complication rates.) This is a good news–bad news problem:
Results of the obturator approach are beginning to appear in the literature as case series and uncontrolled comparative studies. Waltregney et al reported cure of stress incontinence in 91% of 99 patients after 1 year of follow-up. Morey et al reported similar continence outcomes: 89% success for 154 patients after the obturator approach compared with 86% success for 350 patients after the abdominal approach, although follow-up in the abdominal group was substantially longer (mean 20 months, range 18–26) than in the obturator group (mean 9 months, range 6–16). Of interest, urethrolysis was performed more frequently in the abdominal group (2.3%) than in the obturator group (0%).
Randomized trials are necessary to obtain unbiased comparisons of techniques. Investigators in the NIH-sponsored Urinary Incontinence Treatment Network are currently performing a randomized trial comparing obturator and abdominal approaches with midurethral slings for women with stress and stress-predominant mixed incontinence. The primary outcome will compare objective and subjective treatment success between the 2 groups at 1 and 2 years after surgery. Enrollment is expected to be complete by early 2008, and 1-year follow-up by early 2009. Stay tuned for the results!
Retropubic midurethral slings: Which brand?
Gandhi S, Abramov Y, Kwon C, et al. TVT versus SPARC: comparison of outcomes for two midurethral tape procedures. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:125–130.
Lord HE, Taylor JD, Finn JC, et al. A randomized controlled equivalence trial of short-term complications and efficacy of tension-free vaginal tape and suprapubic urethral support sling for treating stress incontinence. BJU Int. 2006;98:367–376.
As the first midurethral sling, the tension-free vaginal tape (Gynecare TVT) has the most evidence and longest follow-up available in the literature. It was originally described using the vaginal approach (“bottom-up”); the company now markets all 3 approaches: vaginal, abdominal (“top-down”), and obturator. Other companies market different products along the same lines, but it cannot be assumed that midurethral slings are interchangeable. Studies are starting to appear that compare different retropubic midurethral slings. In a retrospective case series (Gandhi et al) and a randomized trial (Lord et al), Gynecare TVT had better continence outcomes compared with SPARC (TABLE 2).
TABLE 2
Comparison of 2 retropubic midurethral slings
| OUTCOMES | GYNECARE TVT | SPARC | STATISTICAL SIGNIFICANCE |
|---|---|---|---|
| Subjective continence | |||
| Series by Gandhi et al | 86% (61 of 71) | 60% (28 of 47) | 0.001 |
| RCT by Lord et al | 87% (128 of 147) | 76% (117 of 153) | 0.03 |
| Objective stress continence | |||
| Series by Gandhi et al | 95% (58 of 61) | 70% (32 of 46) | <0.001 |
| RCT by Lord et al | 97.3% (143 of 147) | 97.4% (148 of 152) | NS |
| Follow-up | |||
| Series by Gandhi et al (median, range) | 17 weeks (6–197) | 16 weeks (6–129) | — |
| RCT by Lord et al | 6 weeks | 6 weeks | — |
| Retention requiring reoperation | |||
| Series by Gandhi et al | 2.7% (2 of 73) | 2.0% (1 of 49) | NS |
| RCT by Lord et al | 0 of 147 | 6.5% (10 of 154) | 0.002 |
| Mesh erosions* | |||
| RCT by Lord et al | 4.8% (7 of 147) | 10.5% (16 of 152) | 0.08 |
| *Mesh erosions not reported in Gandhi et al. | |||
Particularly for urge incontinence and associated symptoms falling under the heading of “overactive bladder,” new drugs are always being developed and existing drugs can be found to have a potentially new application. Drugs recently added to those FDA-approved for urge incontinence have relied primarily on their anticholinergic effects. In contrast, tramadol, a drug FDA-approved for pain relief (marketed in the United States as Ultram), was tested for this use. Although the mechanism of action is unknown, the authors proposed a possible change in dopamine receptor activation.
Treated group improved, placebo group did not
Safarinejad MR, Hosseini SY. Safety and efficacy of tramadol in the treatment of idiopathic detrusor overactivity: a double-blind, placebo-controlled, randomized study. Br J Clin Pharmacol. 2006;61:456–463.
This randomized, placebo-controlled trial included 76 men and women with detrusor overactivity. The study population was relatively young, with mean ages of 39 and 37 years in the drug and placebo groups, respectively, and included about 2/3 women. At a sustained-release dose of 100 mg twice a day for 12 weeks of study, tramadol was effective for reducing the number of urge incontinence episodes per 24 hours from a baseline mean of 3.2 ±3.3 episodes to a mean of 1.6 ± 2.8 episodes. In addition, frequency of voiding per 24 hours was reduced (baseline mean 9.3 ± 3.2 episodes, to 5.1 ± 2.1) and the mean volume per void increased substantially (158 ± 32 mL, to 198 ± 76 mL) without an increase in postvoid residual urine volume.
In contrast to the results of many placebo-controlled drug trials and even with the use of 24-hour voiding diaries every 2 weeks for the 12-week study, the placebo group showed essentially no change in clinical and urodynamic outcomes. For example, the number of urge incontinence episodes per 24 hours was unchanged, from a baseline mean of 3.3 ± 3.1 episodes, to a mean of 3.1 ± 3.0 after 12 weeks. Nausea was the most commonly reported side effect (18% vs 5% in the drug and placebo groups, respectively); 2 of 35 participants in the tramadol group dropped out of the study due to nausea.
Confirmation of these results and further study may shed light on the complex control of normal voiding and the true etiology behind the symptoms that we call “detrusor overactivity,” and potentially open a new class of drugs for treatment.
Delivery mode and genetic influences on urinary incontinence
Rohr G, Kragstrup J, Gaist D, Christensen K. Genetic and environmental influences on urinary incontinence: a Danish population-based twin study of middle-aged and elderly women. Acta Obstet Gynecol Scand. 2004;83:978–982.
Goldberg RP, Abramov Y, Botros S, et al. Delivery mode is a major environmental determinant of stress urinary incontinence: results of the Evanston–Northwestern Twin Sisters Study. Am J Obstet Gynecol. 2005;193:2149–2153.
The genetic predisposition for urinary incontinence, seen in clinical practice as clustering in families—mothers, daughters, sisters—has been long suspected, and has been supported in recent studies of nature’s gift to genetic research—twins. In a study of more than 1,000 Danish twins in 2 age groups, Rohr et al were able to quantitatively estimate the heritable component for urinary incontinence, which was categorized by questionnaire into urge, mixed, and stress incontinence. The study included 548 monozygotic twin pairs (who share identical genetic material) and 620 dizygotic twin pairs (who, on average, share 50% of their genes like ordinary sisters).
Urge incontinence, in both age groups, had a similar level of heritability: 42% for ages 46–68 and 49% for ages 70–94.
Mixed incontinence had a lower level of heritability: 27% in middle age and 55% in the older group.
Stress incontinence in the older group had a significant heritable component at 39%, but stress incontinence in the middle-aged group was more strongly associated with environmental factors than with heritability.
Another study focused on stress incontinence in a study of 271 monozygotic twin pairs with a mean age of 47 years. Within the 173 parous twin pairs, environmental factors associated with stress incontinence were identified: age, parity, obesity, and mode of delivery.
Childbirth and genetic factors. These data clarify an important area of (apparently) inconsistent epidemiologic literature on the role of childbirth, and particularly mode of delivery, in lifetime risk of urinary incontinence. The inconsistency resolves once age of the study cohort and type of incontinence are considered. Stress incontinence is influenced most strongly by mode of delivery in middle-aged women. Later in life, genetic factors play a more important role in risk of stress incontinence, and mode of delivery becomes less important. Urge incontinence, perhaps developing along a different etiologic path than stress incontinence, is strongly influenced by heritability in both middle-aged and older women; environmental factors influencing the development of urge incontinence are less important through the lifespan.
Nonetheless, indications of a genetic component do not begin to tell us what exactly is affected that increases the likelihood of urinary incontinence. Speculation is easy enough—perhaps the inherent strength, elasticity, or regeneration potential of critically important tissues in the urethra and pelvis is affected—but the details are not yet fully known.
Again, until we have a clearer understanding, we must continue to manage incontinence with the tools of today.
The author reports no financial relationships.

Anne M. Weber, MD, MS
Program Officer, Female Pelvic
Floor Disorders Program,
Contraception and Reproductive
Health Branch, Center for
Population Research, National
Institute of Child Health
and Human Development,
National Institutes of Health,
Bethesda
Anne M. Weber, MD, MS
Program Officer, Female Pelvic
Floor Disorders Program,
Contraception and Reproductive
Health Branch, Center for
Population Research, National
Institute of Child Health
and Human Development,
National Institutes of Health,
Bethesda
Anne M. Weber, MD, MS
Program Officer, Female Pelvic
Floor Disorders Program,
Contraception and Reproductive
Health Branch, Center for
Population Research, National
Institute of Child Health
and Human Development,
National Institutes of Health,
Bethesda
Surgery for stress incontinence
Another year is drawing to a close and, looking back, what have we learned about urinary incontinence? A clear understanding of etiology stubbornly eludes us. How would a clear understanding of etiology affect management? It’s difficult to be specific until we actually do understand it, but generally:
The “multiple-hit” theory probably applies to urinary incontinence, too
The “multiple-hit” theory usually ascribed to cancer probably also fits the development of urinary incontinence, a likewise multifaceted condition. A woman begins life with genetic predisposition at some level that we cannot currently measure, but which is influenced by the environment (eg, nutrition, toxic exposures) and life events (eg, childbirth, aging)—all of which determine her likelihood of developing incontinence.
Until the time when we do have a clear understanding on which to base diagnosis, treatment, and prevention, of course, we must continue to manage incontinence with the tools of today.
A few pieces of the puzzle are slowly coming together.
Surgery for stress incontinence
Even as new surgical techniques or modifications continue to proliferate, evidence to guide clinical practice accumulates belatedly. MEDLINE lists 325 articles since 1996 (combining surgical mesh and urinary incontinence, limited to human females and published in English). Nonetheless, a consensus may be emerging that the safest synthetic material is monofilament polypropylene with pore size larger than 70 μm.
Unfortunately, by the time research reports are published showing higher complications with certain products, countless women have already been treated.
Mesh erosion (or exposure, extrusion), sometimes accompanied by infection, is a common complication when multifilament or small-pore meshes are used. Even worse, companies commonly withdraw products, modify them, and re-introduce them to the market, accompanied by intensive marketing but, as with the original product, without any real evidence of safety and effectiveness.
In an ideal world, clinicians (and patients) would insist on evidence before accepting new products and techniques. Failing that, clinicians (and patients!) should clearly understand that all new products and techniques are experimental until they are proven equal to or better than traditional techniques. As we have learned with the most subtle differences between synthetic materials, “almost the same” or “looks the same” is not the same.
Among the most important evidence on slings this year are reports of investigations that demonstrated what should not be done.
Are monofilament, large-pore mesh products safer?
Abdel-Fattah M, Sivanesan K, Ramsay I, Pringle S, Bjornsson S. How common are tape erosions? A comparison of two versions of the transobturator tension-free vaginal tape procedure. BJU Int. 2006;98:594–598.
Yamada BS, Govier FE, Stefanovic KB, Kobashi KC. High rate of vaginal erosions associated with the Mentor Obtape. J Urol. 2006;176:651–654.
Siegel AL, Kim M, Goldstein M, Levey S, Ilbeigi P. High incidence of vaginal mesh extrusion using the Intravaginal Slingplasty sling. J Urol. 2005;174:1308–1311.
The risk of vaginal erosion is much higher with synthetic meshes used for sling procedures when the mesh is multifilament and/or small-pore (<70 μm). In some cases, companies have replaced products (Mentor Obtape small-pore polypropylene sling product was replaced with macroporous Aris), whereas others continue to market products reported to have unacceptably high rates of vaginal erosion and mesh extrusion (Intravaginal Slingplasty multifilament mesh) (TABLE 1).
TABLE 1
Comparison of selected multifilament and small-pore mesh products
| REFERENCE | SLING PRODUCT | EROSION RATE | MANAGEMENT |
|---|---|---|---|
| Abdel-Fattah et al (2006) | Obtape | 7.3% (14 of 192) | 7: partial excision 7: complete excision with infection |
| TVT-O | 1.8% (2 of 112) | 1: partial excision 1: vaginal closure | |
| Yamada et al (2006) | Obtape | 13.4% (9 of 67) | 9: complete excision (1 with abscess) |
| Monarc | 0 of 56 | — | |
| Siegel et al (2005) | IVS | 17% (6 of 35) | 6: complete excision (1 with pelvic abscess) |
Avoid cadaveric fascia in sling procedures
Howden NS, Zyczynski HM, Moalli PA, Sagan ER, Meyn LA, Weber AM. Comparison of autologous rectus fascia and cadaveric fascia in pubovaginal sling continence outcomes. Am J Obstet Gynecol. 2006;194:1444–1449.
Evidence has accumulated that sling procedures performed with cadaveric fascia have substantially worse continence outcomes, compared with those using autologous fascia. In a retrospective cohort study of 150 women with cadaveric fascial slings and 153 women who had autologous rectus fascial slings, urinary incontinence (16 vs 5 per 100 women-years) and reoperation for stress incontinence (4 vs 1 per 100 women-years) occurred more frequently after cadaveric versus autologous rectus fascial slings.
Should xenograft materials be used in sling procedures?
Giri SK, Hickey JP, Sil D, et al. Long-term results of pubovaginal sling surgery using acellular cross-linked porcine dermis in the treatment of urodynamic stress incontinence. J Urol. 2006; 175:1788–1792.
Several companies market specific products integrated into sling techniques, such as In-First Ultra (porcine dermal matrix secured with bone anchors) and Stratasis (porcine small intestinal submucosa in urethral sling and tension-free versions). Other companies market only the xenograft for application in sling procedures, such as Pelvicol (acellular porcine collagen matrix). However, relatively little information is available to support or discourage use of xenograft materials in sling procedures.
Giri et al found worse outcomes using Pelvicol compared with autologous rectus fascia for pubovaginal slings. With 3-year follow-up, 54% (26 of 48 women) with Pelvicol were considered successfully cured or improved, compared with 80.4% (37 of 46 women) with rectus fascia. Of interest, women continued to report recurrent incontinence with Pelvicol through the 3-year period, whereas women with rectus fascia had recurrence within the first 9 months after surgery.
Midurethral slings: Retropubic or transobturator?
Waltregny D, Reul O, Mathantu B, Gaspar Y, Bonnet P, de Leval J. Inside out transobturator vaginal tape for the treatment of female stress urinary incontinence: interim results of a prospective study after a 1-year minimum followup. J Urol. 2006;175:2191–2195.
Morey AF, Medendorp AR, Noller MW, et al. Transobturator versus transabdominal midurethral slings: a multi-institutional comparison of obstructive voiding complications. J Urol. 2006;175:1014–1017.
Midurethral sling placement was modified from the retropubic to the obturator approach with the objective of reducing the risk of major bladder and urethral injury and vascular complications. Does the obturator approach actually have fewer intraoperative complications compared with the retropubic approach? Unknown. (As noted below, even within retropubic procedures, it is possible—although currently unknown—that vaginal and abdominal approaches have different complication rates.) This is a good news–bad news problem:
Results of the obturator approach are beginning to appear in the literature as case series and uncontrolled comparative studies. Waltregney et al reported cure of stress incontinence in 91% of 99 patients after 1 year of follow-up. Morey et al reported similar continence outcomes: 89% success for 154 patients after the obturator approach compared with 86% success for 350 patients after the abdominal approach, although follow-up in the abdominal group was substantially longer (mean 20 months, range 18–26) than in the obturator group (mean 9 months, range 6–16). Of interest, urethrolysis was performed more frequently in the abdominal group (2.3%) than in the obturator group (0%).
Randomized trials are necessary to obtain unbiased comparisons of techniques. Investigators in the NIH-sponsored Urinary Incontinence Treatment Network are currently performing a randomized trial comparing obturator and abdominal approaches with midurethral slings for women with stress and stress-predominant mixed incontinence. The primary outcome will compare objective and subjective treatment success between the 2 groups at 1 and 2 years after surgery. Enrollment is expected to be complete by early 2008, and 1-year follow-up by early 2009. Stay tuned for the results!
Retropubic midurethral slings: Which brand?
Gandhi S, Abramov Y, Kwon C, et al. TVT versus SPARC: comparison of outcomes for two midurethral tape procedures. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:125–130.
Lord HE, Taylor JD, Finn JC, et al. A randomized controlled equivalence trial of short-term complications and efficacy of tension-free vaginal tape and suprapubic urethral support sling for treating stress incontinence. BJU Int. 2006;98:367–376.
As the first midurethral sling, the tension-free vaginal tape (Gynecare TVT) has the most evidence and longest follow-up available in the literature. It was originally described using the vaginal approach (“bottom-up”); the company now markets all 3 approaches: vaginal, abdominal (“top-down”), and obturator. Other companies market different products along the same lines, but it cannot be assumed that midurethral slings are interchangeable. Studies are starting to appear that compare different retropubic midurethral slings. In a retrospective case series (Gandhi et al) and a randomized trial (Lord et al), Gynecare TVT had better continence outcomes compared with SPARC (TABLE 2).
TABLE 2
Comparison of 2 retropubic midurethral slings
| OUTCOMES | GYNECARE TVT | SPARC | STATISTICAL SIGNIFICANCE |
|---|---|---|---|
| Subjective continence | |||
| Series by Gandhi et al | 86% (61 of 71) | 60% (28 of 47) | 0.001 |
| RCT by Lord et al | 87% (128 of 147) | 76% (117 of 153) | 0.03 |
| Objective stress continence | |||
| Series by Gandhi et al | 95% (58 of 61) | 70% (32 of 46) | <0.001 |
| RCT by Lord et al | 97.3% (143 of 147) | 97.4% (148 of 152) | NS |
| Follow-up | |||
| Series by Gandhi et al (median, range) | 17 weeks (6–197) | 16 weeks (6–129) | — |
| RCT by Lord et al | 6 weeks | 6 weeks | — |
| Retention requiring reoperation | |||
| Series by Gandhi et al | 2.7% (2 of 73) | 2.0% (1 of 49) | NS |
| RCT by Lord et al | 0 of 147 | 6.5% (10 of 154) | 0.002 |
| Mesh erosions* | |||
| RCT by Lord et al | 4.8% (7 of 147) | 10.5% (16 of 152) | 0.08 |
| *Mesh erosions not reported in Gandhi et al. | |||
Particularly for urge incontinence and associated symptoms falling under the heading of “overactive bladder,” new drugs are always being developed and existing drugs can be found to have a potentially new application. Drugs recently added to those FDA-approved for urge incontinence have relied primarily on their anticholinergic effects. In contrast, tramadol, a drug FDA-approved for pain relief (marketed in the United States as Ultram), was tested for this use. Although the mechanism of action is unknown, the authors proposed a possible change in dopamine receptor activation.
Treated group improved, placebo group did not
Safarinejad MR, Hosseini SY. Safety and efficacy of tramadol in the treatment of idiopathic detrusor overactivity: a double-blind, placebo-controlled, randomized study. Br J Clin Pharmacol. 2006;61:456–463.
This randomized, placebo-controlled trial included 76 men and women with detrusor overactivity. The study population was relatively young, with mean ages of 39 and 37 years in the drug and placebo groups, respectively, and included about 2/3 women. At a sustained-release dose of 100 mg twice a day for 12 weeks of study, tramadol was effective for reducing the number of urge incontinence episodes per 24 hours from a baseline mean of 3.2 ±3.3 episodes to a mean of 1.6 ± 2.8 episodes. In addition, frequency of voiding per 24 hours was reduced (baseline mean 9.3 ± 3.2 episodes, to 5.1 ± 2.1) and the mean volume per void increased substantially (158 ± 32 mL, to 198 ± 76 mL) without an increase in postvoid residual urine volume.
In contrast to the results of many placebo-controlled drug trials and even with the use of 24-hour voiding diaries every 2 weeks for the 12-week study, the placebo group showed essentially no change in clinical and urodynamic outcomes. For example, the number of urge incontinence episodes per 24 hours was unchanged, from a baseline mean of 3.3 ± 3.1 episodes, to a mean of 3.1 ± 3.0 after 12 weeks. Nausea was the most commonly reported side effect (18% vs 5% in the drug and placebo groups, respectively); 2 of 35 participants in the tramadol group dropped out of the study due to nausea.
Confirmation of these results and further study may shed light on the complex control of normal voiding and the true etiology behind the symptoms that we call “detrusor overactivity,” and potentially open a new class of drugs for treatment.
Delivery mode and genetic influences on urinary incontinence
Rohr G, Kragstrup J, Gaist D, Christensen K. Genetic and environmental influences on urinary incontinence: a Danish population-based twin study of middle-aged and elderly women. Acta Obstet Gynecol Scand. 2004;83:978–982.
Goldberg RP, Abramov Y, Botros S, et al. Delivery mode is a major environmental determinant of stress urinary incontinence: results of the Evanston–Northwestern Twin Sisters Study. Am J Obstet Gynecol. 2005;193:2149–2153.
The genetic predisposition for urinary incontinence, seen in clinical practice as clustering in families—mothers, daughters, sisters—has been long suspected, and has been supported in recent studies of nature’s gift to genetic research—twins. In a study of more than 1,000 Danish twins in 2 age groups, Rohr et al were able to quantitatively estimate the heritable component for urinary incontinence, which was categorized by questionnaire into urge, mixed, and stress incontinence. The study included 548 monozygotic twin pairs (who share identical genetic material) and 620 dizygotic twin pairs (who, on average, share 50% of their genes like ordinary sisters).
Urge incontinence, in both age groups, had a similar level of heritability: 42% for ages 46–68 and 49% for ages 70–94.
Mixed incontinence had a lower level of heritability: 27% in middle age and 55% in the older group.
Stress incontinence in the older group had a significant heritable component at 39%, but stress incontinence in the middle-aged group was more strongly associated with environmental factors than with heritability.
Another study focused on stress incontinence in a study of 271 monozygotic twin pairs with a mean age of 47 years. Within the 173 parous twin pairs, environmental factors associated with stress incontinence were identified: age, parity, obesity, and mode of delivery.
Childbirth and genetic factors. These data clarify an important area of (apparently) inconsistent epidemiologic literature on the role of childbirth, and particularly mode of delivery, in lifetime risk of urinary incontinence. The inconsistency resolves once age of the study cohort and type of incontinence are considered. Stress incontinence is influenced most strongly by mode of delivery in middle-aged women. Later in life, genetic factors play a more important role in risk of stress incontinence, and mode of delivery becomes less important. Urge incontinence, perhaps developing along a different etiologic path than stress incontinence, is strongly influenced by heritability in both middle-aged and older women; environmental factors influencing the development of urge incontinence are less important through the lifespan.
Nonetheless, indications of a genetic component do not begin to tell us what exactly is affected that increases the likelihood of urinary incontinence. Speculation is easy enough—perhaps the inherent strength, elasticity, or regeneration potential of critically important tissues in the urethra and pelvis is affected—but the details are not yet fully known.
Again, until we have a clearer understanding, we must continue to manage incontinence with the tools of today.
Surgery for stress incontinence
Another year is drawing to a close and, looking back, what have we learned about urinary incontinence? A clear understanding of etiology stubbornly eludes us. How would a clear understanding of etiology affect management? It’s difficult to be specific until we actually do understand it, but generally:
The “multiple-hit” theory probably applies to urinary incontinence, too
The “multiple-hit” theory usually ascribed to cancer probably also fits the development of urinary incontinence, a likewise multifaceted condition. A woman begins life with genetic predisposition at some level that we cannot currently measure, but which is influenced by the environment (eg, nutrition, toxic exposures) and life events (eg, childbirth, aging)—all of which determine her likelihood of developing incontinence.
Until the time when we do have a clear understanding on which to base diagnosis, treatment, and prevention, of course, we must continue to manage incontinence with the tools of today.
A few pieces of the puzzle are slowly coming together.
Surgery for stress incontinence
Even as new surgical techniques or modifications continue to proliferate, evidence to guide clinical practice accumulates belatedly. MEDLINE lists 325 articles since 1996 (combining surgical mesh and urinary incontinence, limited to human females and published in English). Nonetheless, a consensus may be emerging that the safest synthetic material is monofilament polypropylene with pore size larger than 70 μm.
Unfortunately, by the time research reports are published showing higher complications with certain products, countless women have already been treated.
Mesh erosion (or exposure, extrusion), sometimes accompanied by infection, is a common complication when multifilament or small-pore meshes are used. Even worse, companies commonly withdraw products, modify them, and re-introduce them to the market, accompanied by intensive marketing but, as with the original product, without any real evidence of safety and effectiveness.
In an ideal world, clinicians (and patients) would insist on evidence before accepting new products and techniques. Failing that, clinicians (and patients!) should clearly understand that all new products and techniques are experimental until they are proven equal to or better than traditional techniques. As we have learned with the most subtle differences between synthetic materials, “almost the same” or “looks the same” is not the same.
Among the most important evidence on slings this year are reports of investigations that demonstrated what should not be done.
Are monofilament, large-pore mesh products safer?
Abdel-Fattah M, Sivanesan K, Ramsay I, Pringle S, Bjornsson S. How common are tape erosions? A comparison of two versions of the transobturator tension-free vaginal tape procedure. BJU Int. 2006;98:594–598.
Yamada BS, Govier FE, Stefanovic KB, Kobashi KC. High rate of vaginal erosions associated with the Mentor Obtape. J Urol. 2006;176:651–654.
Siegel AL, Kim M, Goldstein M, Levey S, Ilbeigi P. High incidence of vaginal mesh extrusion using the Intravaginal Slingplasty sling. J Urol. 2005;174:1308–1311.
The risk of vaginal erosion is much higher with synthetic meshes used for sling procedures when the mesh is multifilament and/or small-pore (<70 μm). In some cases, companies have replaced products (Mentor Obtape small-pore polypropylene sling product was replaced with macroporous Aris), whereas others continue to market products reported to have unacceptably high rates of vaginal erosion and mesh extrusion (Intravaginal Slingplasty multifilament mesh) (TABLE 1).
TABLE 1
Comparison of selected multifilament and small-pore mesh products
| REFERENCE | SLING PRODUCT | EROSION RATE | MANAGEMENT |
|---|---|---|---|
| Abdel-Fattah et al (2006) | Obtape | 7.3% (14 of 192) | 7: partial excision 7: complete excision with infection |
| TVT-O | 1.8% (2 of 112) | 1: partial excision 1: vaginal closure | |
| Yamada et al (2006) | Obtape | 13.4% (9 of 67) | 9: complete excision (1 with abscess) |
| Monarc | 0 of 56 | — | |
| Siegel et al (2005) | IVS | 17% (6 of 35) | 6: complete excision (1 with pelvic abscess) |
Avoid cadaveric fascia in sling procedures
Howden NS, Zyczynski HM, Moalli PA, Sagan ER, Meyn LA, Weber AM. Comparison of autologous rectus fascia and cadaveric fascia in pubovaginal sling continence outcomes. Am J Obstet Gynecol. 2006;194:1444–1449.
Evidence has accumulated that sling procedures performed with cadaveric fascia have substantially worse continence outcomes, compared with those using autologous fascia. In a retrospective cohort study of 150 women with cadaveric fascial slings and 153 women who had autologous rectus fascial slings, urinary incontinence (16 vs 5 per 100 women-years) and reoperation for stress incontinence (4 vs 1 per 100 women-years) occurred more frequently after cadaveric versus autologous rectus fascial slings.
Should xenograft materials be used in sling procedures?
Giri SK, Hickey JP, Sil D, et al. Long-term results of pubovaginal sling surgery using acellular cross-linked porcine dermis in the treatment of urodynamic stress incontinence. J Urol. 2006; 175:1788–1792.
Several companies market specific products integrated into sling techniques, such as In-First Ultra (porcine dermal matrix secured with bone anchors) and Stratasis (porcine small intestinal submucosa in urethral sling and tension-free versions). Other companies market only the xenograft for application in sling procedures, such as Pelvicol (acellular porcine collagen matrix). However, relatively little information is available to support or discourage use of xenograft materials in sling procedures.
Giri et al found worse outcomes using Pelvicol compared with autologous rectus fascia for pubovaginal slings. With 3-year follow-up, 54% (26 of 48 women) with Pelvicol were considered successfully cured or improved, compared with 80.4% (37 of 46 women) with rectus fascia. Of interest, women continued to report recurrent incontinence with Pelvicol through the 3-year period, whereas women with rectus fascia had recurrence within the first 9 months after surgery.
Midurethral slings: Retropubic or transobturator?
Waltregny D, Reul O, Mathantu B, Gaspar Y, Bonnet P, de Leval J. Inside out transobturator vaginal tape for the treatment of female stress urinary incontinence: interim results of a prospective study after a 1-year minimum followup. J Urol. 2006;175:2191–2195.
Morey AF, Medendorp AR, Noller MW, et al. Transobturator versus transabdominal midurethral slings: a multi-institutional comparison of obstructive voiding complications. J Urol. 2006;175:1014–1017.
Midurethral sling placement was modified from the retropubic to the obturator approach with the objective of reducing the risk of major bladder and urethral injury and vascular complications. Does the obturator approach actually have fewer intraoperative complications compared with the retropubic approach? Unknown. (As noted below, even within retropubic procedures, it is possible—although currently unknown—that vaginal and abdominal approaches have different complication rates.) This is a good news–bad news problem:
Results of the obturator approach are beginning to appear in the literature as case series and uncontrolled comparative studies. Waltregney et al reported cure of stress incontinence in 91% of 99 patients after 1 year of follow-up. Morey et al reported similar continence outcomes: 89% success for 154 patients after the obturator approach compared with 86% success for 350 patients after the abdominal approach, although follow-up in the abdominal group was substantially longer (mean 20 months, range 18–26) than in the obturator group (mean 9 months, range 6–16). Of interest, urethrolysis was performed more frequently in the abdominal group (2.3%) than in the obturator group (0%).
Randomized trials are necessary to obtain unbiased comparisons of techniques. Investigators in the NIH-sponsored Urinary Incontinence Treatment Network are currently performing a randomized trial comparing obturator and abdominal approaches with midurethral slings for women with stress and stress-predominant mixed incontinence. The primary outcome will compare objective and subjective treatment success between the 2 groups at 1 and 2 years after surgery. Enrollment is expected to be complete by early 2008, and 1-year follow-up by early 2009. Stay tuned for the results!
Retropubic midurethral slings: Which brand?
Gandhi S, Abramov Y, Kwon C, et al. TVT versus SPARC: comparison of outcomes for two midurethral tape procedures. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17:125–130.
Lord HE, Taylor JD, Finn JC, et al. A randomized controlled equivalence trial of short-term complications and efficacy of tension-free vaginal tape and suprapubic urethral support sling for treating stress incontinence. BJU Int. 2006;98:367–376.
As the first midurethral sling, the tension-free vaginal tape (Gynecare TVT) has the most evidence and longest follow-up available in the literature. It was originally described using the vaginal approach (“bottom-up”); the company now markets all 3 approaches: vaginal, abdominal (“top-down”), and obturator. Other companies market different products along the same lines, but it cannot be assumed that midurethral slings are interchangeable. Studies are starting to appear that compare different retropubic midurethral slings. In a retrospective case series (Gandhi et al) and a randomized trial (Lord et al), Gynecare TVT had better continence outcomes compared with SPARC (TABLE 2).
TABLE 2
Comparison of 2 retropubic midurethral slings
| OUTCOMES | GYNECARE TVT | SPARC | STATISTICAL SIGNIFICANCE |
|---|---|---|---|
| Subjective continence | |||
| Series by Gandhi et al | 86% (61 of 71) | 60% (28 of 47) | 0.001 |
| RCT by Lord et al | 87% (128 of 147) | 76% (117 of 153) | 0.03 |
| Objective stress continence | |||
| Series by Gandhi et al | 95% (58 of 61) | 70% (32 of 46) | <0.001 |
| RCT by Lord et al | 97.3% (143 of 147) | 97.4% (148 of 152) | NS |
| Follow-up | |||
| Series by Gandhi et al (median, range) | 17 weeks (6–197) | 16 weeks (6–129) | — |
| RCT by Lord et al | 6 weeks | 6 weeks | — |
| Retention requiring reoperation | |||
| Series by Gandhi et al | 2.7% (2 of 73) | 2.0% (1 of 49) | NS |
| RCT by Lord et al | 0 of 147 | 6.5% (10 of 154) | 0.002 |
| Mesh erosions* | |||
| RCT by Lord et al | 4.8% (7 of 147) | 10.5% (16 of 152) | 0.08 |
| *Mesh erosions not reported in Gandhi et al. | |||
Particularly for urge incontinence and associated symptoms falling under the heading of “overactive bladder,” new drugs are always being developed and existing drugs can be found to have a potentially new application. Drugs recently added to those FDA-approved for urge incontinence have relied primarily on their anticholinergic effects. In contrast, tramadol, a drug FDA-approved for pain relief (marketed in the United States as Ultram), was tested for this use. Although the mechanism of action is unknown, the authors proposed a possible change in dopamine receptor activation.
Treated group improved, placebo group did not
Safarinejad MR, Hosseini SY. Safety and efficacy of tramadol in the treatment of idiopathic detrusor overactivity: a double-blind, placebo-controlled, randomized study. Br J Clin Pharmacol. 2006;61:456–463.
This randomized, placebo-controlled trial included 76 men and women with detrusor overactivity. The study population was relatively young, with mean ages of 39 and 37 years in the drug and placebo groups, respectively, and included about 2/3 women. At a sustained-release dose of 100 mg twice a day for 12 weeks of study, tramadol was effective for reducing the number of urge incontinence episodes per 24 hours from a baseline mean of 3.2 ±3.3 episodes to a mean of 1.6 ± 2.8 episodes. In addition, frequency of voiding per 24 hours was reduced (baseline mean 9.3 ± 3.2 episodes, to 5.1 ± 2.1) and the mean volume per void increased substantially (158 ± 32 mL, to 198 ± 76 mL) without an increase in postvoid residual urine volume.
In contrast to the results of many placebo-controlled drug trials and even with the use of 24-hour voiding diaries every 2 weeks for the 12-week study, the placebo group showed essentially no change in clinical and urodynamic outcomes. For example, the number of urge incontinence episodes per 24 hours was unchanged, from a baseline mean of 3.3 ± 3.1 episodes, to a mean of 3.1 ± 3.0 after 12 weeks. Nausea was the most commonly reported side effect (18% vs 5% in the drug and placebo groups, respectively); 2 of 35 participants in the tramadol group dropped out of the study due to nausea.
Confirmation of these results and further study may shed light on the complex control of normal voiding and the true etiology behind the symptoms that we call “detrusor overactivity,” and potentially open a new class of drugs for treatment.
Delivery mode and genetic influences on urinary incontinence
Rohr G, Kragstrup J, Gaist D, Christensen K. Genetic and environmental influences on urinary incontinence: a Danish population-based twin study of middle-aged and elderly women. Acta Obstet Gynecol Scand. 2004;83:978–982.
Goldberg RP, Abramov Y, Botros S, et al. Delivery mode is a major environmental determinant of stress urinary incontinence: results of the Evanston–Northwestern Twin Sisters Study. Am J Obstet Gynecol. 2005;193:2149–2153.
The genetic predisposition for urinary incontinence, seen in clinical practice as clustering in families—mothers, daughters, sisters—has been long suspected, and has been supported in recent studies of nature’s gift to genetic research—twins. In a study of more than 1,000 Danish twins in 2 age groups, Rohr et al were able to quantitatively estimate the heritable component for urinary incontinence, which was categorized by questionnaire into urge, mixed, and stress incontinence. The study included 548 monozygotic twin pairs (who share identical genetic material) and 620 dizygotic twin pairs (who, on average, share 50% of their genes like ordinary sisters).
Urge incontinence, in both age groups, had a similar level of heritability: 42% for ages 46–68 and 49% for ages 70–94.
Mixed incontinence had a lower level of heritability: 27% in middle age and 55% in the older group.
Stress incontinence in the older group had a significant heritable component at 39%, but stress incontinence in the middle-aged group was more strongly associated with environmental factors than with heritability.
Another study focused on stress incontinence in a study of 271 monozygotic twin pairs with a mean age of 47 years. Within the 173 parous twin pairs, environmental factors associated with stress incontinence were identified: age, parity, obesity, and mode of delivery.
Childbirth and genetic factors. These data clarify an important area of (apparently) inconsistent epidemiologic literature on the role of childbirth, and particularly mode of delivery, in lifetime risk of urinary incontinence. The inconsistency resolves once age of the study cohort and type of incontinence are considered. Stress incontinence is influenced most strongly by mode of delivery in middle-aged women. Later in life, genetic factors play a more important role in risk of stress incontinence, and mode of delivery becomes less important. Urge incontinence, perhaps developing along a different etiologic path than stress incontinence, is strongly influenced by heritability in both middle-aged and older women; environmental factors influencing the development of urge incontinence are less important through the lifespan.
Nonetheless, indications of a genetic component do not begin to tell us what exactly is affected that increases the likelihood of urinary incontinence. Speculation is easy enough—perhaps the inherent strength, elasticity, or regeneration potential of critically important tissues in the urethra and pelvis is affected—but the details are not yet fully known.
Again, until we have a clearer understanding, we must continue to manage incontinence with the tools of today.
The author reports no financial relationships.
The author reports no financial relationships.