User login
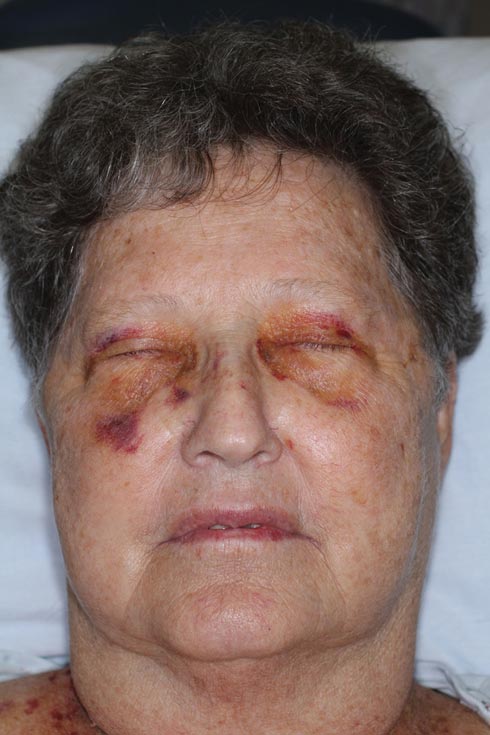
Nonblanchable Violaceous Macules of the Periorbital Skin
The Diagnosis: Primary AL Amyloidosis
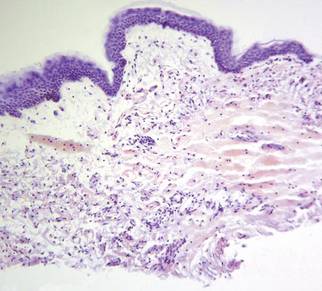
The patient initially presented with bruising around the eyes. She noted characteristic “easy bruising” after minor trauma. Serum protein electrophoresis demonstrated an elevated IgG κ level of 1.4 g/dL (reference range, 0.61–1.04 g/dL) with normal IgA and IgM. Skin biopsy revealed focal amyloid deposition in the dermis (Figures 1 and 2). Liquid chromatography tandem-mass spectrometry performed on peptides extracted from Congo red–positive areas of paraffin-embedded specimen identified peptides representing immunoglobulin κ light chain variable region 1, favoring AL κ-type amyloid deposition.
| 
|
A bone marrow biopsy revealed plasma cell dyscrasia with 15% plasma cells but was negative for amyloid. A fine-needle fat-pad biopsy also was negative for amyloid. Systemic amyloid involvement was evaluated with a 24-hour urine collection but was negative for light chain proteinuria and albuminuria. A complete osseous survey was negative for focal lytic or sclerotic lesions, ruling out multiple myeloma. Echocardiogram and liver function tests were normal. We concluded that this patient exhibited a rare case of primary AL amyloidosis due to plasma cell dyscrasia with only cutaneous involvement. The patient was not a candidate for stem cell therapy because she was older than 70 years. She was initiated on several cycles of melphalan with dexamethasone by a collaborating oncology team.
The amyloidoses are a group of diseases that result from the extracellular deposition of insoluble fibrils in various organs. Amyloidosis can occur primarily from a plasma cell proliferative process or secondarily from a chronic inflammatory process. Light chain (AL) amyloidosis is the most commonform of primary systemic amyloidosis. In AL amyloidosis, an immunoglobulin light chain or a fragment of a light chain is produced by a clonal proliferation of plasma cells, with plasma cell dyscrasia typically ranging from 5% to 10%.1 Rarely, amyloidoses may present primarily as cutaneous lesions, as in this patient, which would warrant an evaluation for systemic involvement.
Skin biopsy was the key to diagnosis, as prior biopsy of bone marrow and fat-pad failed to demonstrate amyloid protein. Further analysis with mass spectrometry was used in conjunction with Congo red staining to increase the sensitivity and specificity of detecting overexpressed light chains. Recognition of the limited differential diagnosis of pinch purpura and appropriate processing of the biopsy specimen allowed diagnosis. The patient improved with cycles of combination melphalan and dexamethasone, which was shown to have similar outcome to those treated with melphalan and autologous stem cell rescue.2 Overall, this case highlights the extensive search for systemic involvement that should be undertaken with cutaneous presentations of amyloidosis and the importance of an interdisciplinary approach to treatment.
1. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-49.
2. Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357:11.
The Diagnosis: Primary AL Amyloidosis
The patient initially presented with bruising around the eyes. She noted characteristic “easy bruising” after minor trauma. Serum protein electrophoresis demonstrated an elevated IgG κ level of 1.4 g/dL (reference range, 0.61–1.04 g/dL) with normal IgA and IgM. Skin biopsy revealed focal amyloid deposition in the dermis (Figures 1 and 2). Liquid chromatography tandem-mass spectrometry performed on peptides extracted from Congo red–positive areas of paraffin-embedded specimen identified peptides representing immunoglobulin κ light chain variable region 1, favoring AL κ-type amyloid deposition.
|
|
|
A bone marrow biopsy revealed plasma cell dyscrasia with 15% plasma cells but was negative for amyloid. A fine-needle fat-pad biopsy also was negative for amyloid. Systemic amyloid involvement was evaluated with a 24-hour urine collection but was negative for light chain proteinuria and albuminuria. A complete osseous survey was negative for focal lytic or sclerotic lesions, ruling out multiple myeloma. Echocardiogram and liver function tests were normal. We concluded that this patient exhibited a rare case of primary AL amyloidosis due to plasma cell dyscrasia with only cutaneous involvement. The patient was not a candidate for stem cell therapy because she was older than 70 years. She was initiated on several cycles of melphalan with dexamethasone by a collaborating oncology team.
The amyloidoses are a group of diseases that result from the extracellular deposition of insoluble fibrils in various organs. Amyloidosis can occur primarily from a plasma cell proliferative process or secondarily from a chronic inflammatory process. Light chain (AL) amyloidosis is the most commonform of primary systemic amyloidosis. In AL amyloidosis, an immunoglobulin light chain or a fragment of a light chain is produced by a clonal proliferation of plasma cells, with plasma cell dyscrasia typically ranging from 5% to 10%.1 Rarely, amyloidoses may present primarily as cutaneous lesions, as in this patient, which would warrant an evaluation for systemic involvement.
Skin biopsy was the key to diagnosis, as prior biopsy of bone marrow and fat-pad failed to demonstrate amyloid protein. Further analysis with mass spectrometry was used in conjunction with Congo red staining to increase the sensitivity and specificity of detecting overexpressed light chains. Recognition of the limited differential diagnosis of pinch purpura and appropriate processing of the biopsy specimen allowed diagnosis. The patient improved with cycles of combination melphalan and dexamethasone, which was shown to have similar outcome to those treated with melphalan and autologous stem cell rescue.2 Overall, this case highlights the extensive search for systemic involvement that should be undertaken with cutaneous presentations of amyloidosis and the importance of an interdisciplinary approach to treatment.
The Diagnosis: Primary AL Amyloidosis
The patient initially presented with bruising around the eyes. She noted characteristic “easy bruising” after minor trauma. Serum protein electrophoresis demonstrated an elevated IgG κ level of 1.4 g/dL (reference range, 0.61–1.04 g/dL) with normal IgA and IgM. Skin biopsy revealed focal amyloid deposition in the dermis (Figures 1 and 2). Liquid chromatography tandem-mass spectrometry performed on peptides extracted from Congo red–positive areas of paraffin-embedded specimen identified peptides representing immunoglobulin κ light chain variable region 1, favoring AL κ-type amyloid deposition.
|
|
|
A bone marrow biopsy revealed plasma cell dyscrasia with 15% plasma cells but was negative for amyloid. A fine-needle fat-pad biopsy also was negative for amyloid. Systemic amyloid involvement was evaluated with a 24-hour urine collection but was negative for light chain proteinuria and albuminuria. A complete osseous survey was negative for focal lytic or sclerotic lesions, ruling out multiple myeloma. Echocardiogram and liver function tests were normal. We concluded that this patient exhibited a rare case of primary AL amyloidosis due to plasma cell dyscrasia with only cutaneous involvement. The patient was not a candidate for stem cell therapy because she was older than 70 years. She was initiated on several cycles of melphalan with dexamethasone by a collaborating oncology team.
The amyloidoses are a group of diseases that result from the extracellular deposition of insoluble fibrils in various organs. Amyloidosis can occur primarily from a plasma cell proliferative process or secondarily from a chronic inflammatory process. Light chain (AL) amyloidosis is the most commonform of primary systemic amyloidosis. In AL amyloidosis, an immunoglobulin light chain or a fragment of a light chain is produced by a clonal proliferation of plasma cells, with plasma cell dyscrasia typically ranging from 5% to 10%.1 Rarely, amyloidoses may present primarily as cutaneous lesions, as in this patient, which would warrant an evaluation for systemic involvement.
Skin biopsy was the key to diagnosis, as prior biopsy of bone marrow and fat-pad failed to demonstrate amyloid protein. Further analysis with mass spectrometry was used in conjunction with Congo red staining to increase the sensitivity and specificity of detecting overexpressed light chains. Recognition of the limited differential diagnosis of pinch purpura and appropriate processing of the biopsy specimen allowed diagnosis. The patient improved with cycles of combination melphalan and dexamethasone, which was shown to have similar outcome to those treated with melphalan and autologous stem cell rescue.2 Overall, this case highlights the extensive search for systemic involvement that should be undertaken with cutaneous presentations of amyloidosis and the importance of an interdisciplinary approach to treatment.
1. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-49.
2. Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357:11.
1. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-49.
2. Jaccard A, Moreau P, Leblond V, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357:11.
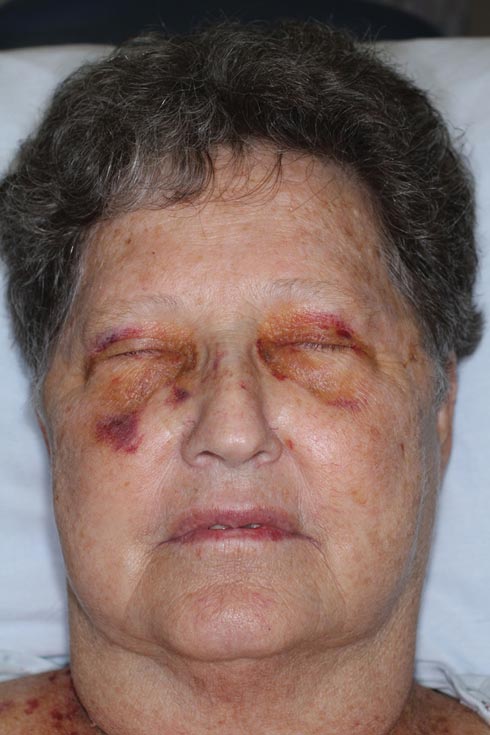
A 71-year-old white woman presented with nonblanchable, violaceous, monomorphic macules involving the bilateral periorbital skin, upper chest, upper arms, and dorsal forearms of 1 year’s duration. Skin and bone marrow biopsies were performed.