User login
Hemophilia B drug available in larger vial

CSL Behring has announced that Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) is now available in a 3500 IU vial size.
Idelvion is also available in 250 IU, 500 IU, 1000 IU, and 2000 IU vial sizes.
For some patients requiring high doses of Idelvion, the new 3500 IU vial size will reduce the reconstitution time needed to prepare multiple vials for a similar dose.
Idelvion is a fusion protein linking recombinant coagulation factor IX with recombinant albumin, and it is approved by the U.S. Food and Drug Administration to treat children and adults with hemophilia B.
Idelvion can be used as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Idelvion is approved for up to 14-day dosing in appropriate patients.
For more details on Idelvion, see the prescribing information.
CSL Behring has announced that Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) is now available in a 3500 IU vial size.
Idelvion is also available in 250 IU, 500 IU, 1000 IU, and 2000 IU vial sizes.
For some patients requiring high doses of Idelvion, the new 3500 IU vial size will reduce the reconstitution time needed to prepare multiple vials for a similar dose.
Idelvion is a fusion protein linking recombinant coagulation factor IX with recombinant albumin, and it is approved by the U.S. Food and Drug Administration to treat children and adults with hemophilia B.
Idelvion can be used as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Idelvion is approved for up to 14-day dosing in appropriate patients.
For more details on Idelvion, see the prescribing information.
CSL Behring has announced that Idelvion (Coagulation Factor IX [Recombinant], Albumin Fusion Protein) is now available in a 3500 IU vial size.
Idelvion is also available in 250 IU, 500 IU, 1000 IU, and 2000 IU vial sizes.
For some patients requiring high doses of Idelvion, the new 3500 IU vial size will reduce the reconstitution time needed to prepare multiple vials for a similar dose.
Idelvion is a fusion protein linking recombinant coagulation factor IX with recombinant albumin, and it is approved by the U.S. Food and Drug Administration to treat children and adults with hemophilia B.
Idelvion can be used as routine prophylaxis to prevent or reduce the frequency of bleeding episodes, for on-demand control and prevention of bleeding episodes, and for the perioperative management of bleeding.
Idelvion is approved for up to 14-day dosing in appropriate patients.
For more details on Idelvion, see the prescribing information.
Team identifies potential immunotherapy target for AML

New research could aid the development of immunotherapies tailored to patients with acute myeloid leukemia (AML) who are undergoing stem cell transplant (SCT).
Researchers found they could use genetic sequencing and computer software to identify minor histocompatibility antigens (mHAs) known to occur in AML.
The team used this method to predict novel graft-versus-leukemia (GVL) mHAs and demonstrated that one of these mHAs could be a “potentially useful” therapeutic target.
Ben Vincent, MD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill, and his colleagues reported these findings in Blood Advances.
In their retrospective study, the researchers tested whether their software could predict antigenic targets in 101 SCT donor-recipient pairs.
The researchers found they could correctly identify 14 of 18 mHAs known to occur in AML, but they were also able to predict 102 new GVL mHAs.
The researchers then confirmed one of these GVL mHAs, called UNC-GRK4-V, as a potential target for immunotherapy. The team observed immune responses to UNC-GRK4-V in four of nine AML patients who had undergone SCT.
Looking ahead, the researchers want to optimize their software to predict the most common AML-associated mHAs present in the U.S. population and confirm these predicted antigens as valid immunotherapy targets.
The team believes they could potentially use their predictions to engineer donor immune cells to specifically target the cancer cell antigens while preventing graft-versus-host disease.
“We’ve developed a software package that predicts leukemia-specific immune targets in any leukemia patient undergoing a stem cell transplant based on DNA and RNA sequencing and demonstrated that these data can lead to actual targets expressed on leukemia cells,” Dr. Vincent said.
“The next step of our work is to use that information for patient-specific therapies to try to improve cure rates without making graft-versus-host disease worse.”
The current research was supported by a National Cancer Institute grant, an ASCO Young Investigator Award, the North Carolina University Cancer Research Fund, and the Scott Neil Schwirck Fellowship.
New research could aid the development of immunotherapies tailored to patients with acute myeloid leukemia (AML) who are undergoing stem cell transplant (SCT).
Researchers found they could use genetic sequencing and computer software to identify minor histocompatibility antigens (mHAs) known to occur in AML.
The team used this method to predict novel graft-versus-leukemia (GVL) mHAs and demonstrated that one of these mHAs could be a “potentially useful” therapeutic target.
Ben Vincent, MD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill, and his colleagues reported these findings in Blood Advances.
In their retrospective study, the researchers tested whether their software could predict antigenic targets in 101 SCT donor-recipient pairs.
The researchers found they could correctly identify 14 of 18 mHAs known to occur in AML, but they were also able to predict 102 new GVL mHAs.
The researchers then confirmed one of these GVL mHAs, called UNC-GRK4-V, as a potential target for immunotherapy. The team observed immune responses to UNC-GRK4-V in four of nine AML patients who had undergone SCT.
Looking ahead, the researchers want to optimize their software to predict the most common AML-associated mHAs present in the U.S. population and confirm these predicted antigens as valid immunotherapy targets.
The team believes they could potentially use their predictions to engineer donor immune cells to specifically target the cancer cell antigens while preventing graft-versus-host disease.
“We’ve developed a software package that predicts leukemia-specific immune targets in any leukemia patient undergoing a stem cell transplant based on DNA and RNA sequencing and demonstrated that these data can lead to actual targets expressed on leukemia cells,” Dr. Vincent said.
“The next step of our work is to use that information for patient-specific therapies to try to improve cure rates without making graft-versus-host disease worse.”
The current research was supported by a National Cancer Institute grant, an ASCO Young Investigator Award, the North Carolina University Cancer Research Fund, and the Scott Neil Schwirck Fellowship.
New research could aid the development of immunotherapies tailored to patients with acute myeloid leukemia (AML) who are undergoing stem cell transplant (SCT).
Researchers found they could use genetic sequencing and computer software to identify minor histocompatibility antigens (mHAs) known to occur in AML.
The team used this method to predict novel graft-versus-leukemia (GVL) mHAs and demonstrated that one of these mHAs could be a “potentially useful” therapeutic target.
Ben Vincent, MD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill, and his colleagues reported these findings in Blood Advances.
In their retrospective study, the researchers tested whether their software could predict antigenic targets in 101 SCT donor-recipient pairs.
The researchers found they could correctly identify 14 of 18 mHAs known to occur in AML, but they were also able to predict 102 new GVL mHAs.
The researchers then confirmed one of these GVL mHAs, called UNC-GRK4-V, as a potential target for immunotherapy. The team observed immune responses to UNC-GRK4-V in four of nine AML patients who had undergone SCT.
Looking ahead, the researchers want to optimize their software to predict the most common AML-associated mHAs present in the U.S. population and confirm these predicted antigens as valid immunotherapy targets.
The team believes they could potentially use their predictions to engineer donor immune cells to specifically target the cancer cell antigens while preventing graft-versus-host disease.
“We’ve developed a software package that predicts leukemia-specific immune targets in any leukemia patient undergoing a stem cell transplant based on DNA and RNA sequencing and demonstrated that these data can lead to actual targets expressed on leukemia cells,” Dr. Vincent said.
“The next step of our work is to use that information for patient-specific therapies to try to improve cure rates without making graft-versus-host disease worse.”
The current research was supported by a National Cancer Institute grant, an ASCO Young Investigator Award, the North Carolina University Cancer Research Fund, and the Scott Neil Schwirck Fellowship.
Mechanism of cGVHD response to ECP still unclear

A prospective study did not reveal the mechanism driving response to extracorporeal photopheresis (ECP) in patients with chronic graft-versus-host disease (cGVHD).
However, researchers did find that responses occurred independent of risk factors, and results suggested that regulatory T cells (Tregs) are not the dominant mechanism of response to ECP.
Madan Jagasia, MBBS, of Vanderbilt University School of Medicine in Nashville, Tennessee, and his colleagues conducted this research and detailed the findings in Biology of Blood and Marrow Transplantation.
The study was funded by Therakos, Inc., a Mallinckrodt Pharmaceuticals Company.
The study included 77 patients with cGVHD. The median age at transplant was 49, 88% of patients were white, and 62% were male.
Patients had moderate (48%) or severe (52%) cGVHD. Sites of involvement included skin (86%), mouth (52%), gastrointestinal tract (29%), eye (62%), joint and fascia (51%), genital tract (11%), and lung (28%).
Patients had received a median of 2 (range, 0 to 7) prior lines of cGVHD therapy.
For this study, patients received 1689 ECP treatments, an average of 21.9 treatments per patient. The most common regimen was ECP twice a week for 4 weeks, then twice a week every 2 weeks for 8 weeks, then further tapering at the treating physician’s discretion.
Forty-eight patients (62.3%) completed all 6 months of ECP treatment. Reasons for early discontinuation included cGVHD progression (n=6), infection (n=4), cGVHD improvement (n=2), death from cGVHD-related cause (n=2), logistical issues (n=2), loss to follow-up (n=2), unknown reasons (n=4), and other various reasons (n=7) such as a finance issue and non-adherence.
Response
Provider-assessed response rates differed from response rates according to 2005 National Institutes of Health (NIH) consensus criteria.
According to providers, the response rate was 62.3% (48/77), with 14% of patients (n=11) achieving a complete response and 48% (n=37) attaining a partial response. Nineteen percent of patients (n=15) had stable disease, 14% (n=11) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Eight patients did not have enough data to assess NIH response. For the 69 evaluable patients, the NIH response rate was 43.5% (n=30), with 6% of patients (n=4) achieving a complete response and 38% (n=26) attaining a partial response. Fifteen percent of patients (n=10) had stable disease, 38% (n=26) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Risk factors
The researchers said there was no significant difference between responders and nonresponders when it came to age at treatment, platelet count, cGVHD severity, donor gender, and donor type.
For provider-assessed response, the median Karnofsky performance score at study entry was significantly higher in responders than nonresponders (90 vs 80; P=0.001). Responders also had a significantly shorter time from transplant to study entry compared to nonresponders (1.43 vs 2.06 years; P=0.051).
Similarly, according to NIH response, time from transplant to study entry was significantly shorter for responders than nonresponders (1.23 vs 1.92 years; P=0.04).
However, in a logistic regression model, time from transplant to study entry was not associated with provider-assessed or NIH response.
Tregs
The researchers found no significant difference in Treg percentages between responders and nonresponders.
For provider-assessed response, the baseline Treg frequency was 4.4% in responders and 4.8% in nonresponders (P=0.4). Treg percentages at the end of study were 4.2% and 5.5%, respectively (P=0.2). And the change in Treg frequency was 0.3% and 1.3%, respectively (P=0.3).
For NIH response, the baseline Treg frequency was 4.7% in responders and 4.4% in nonresponders (P=0.3). Treg percentages at the end of study were 4.4% and 4.7%, respectively (P=0.6). And the change in Treg percentages was 0.3% and 0.7%, respectively (P=0.4).
These findings run contrary to the researchers’ hypothesis that response to ECP would be associated with an increase in the percentage of Tregs.
The researchers did note that the number of Tregs varied between patients, and the team raised the possibility that the mechanism of Tregs in ECP is not visible by measuring cell abundance.
However, the researchers also said future studies should explore additional mechanisms of action for ECP and look particularly at other T-cell populations, dendritic cells, inhibitory cytokines, and proinflammatory cytokines.
A prospective study did not reveal the mechanism driving response to extracorporeal photopheresis (ECP) in patients with chronic graft-versus-host disease (cGVHD).
However, researchers did find that responses occurred independent of risk factors, and results suggested that regulatory T cells (Tregs) are not the dominant mechanism of response to ECP.
Madan Jagasia, MBBS, of Vanderbilt University School of Medicine in Nashville, Tennessee, and his colleagues conducted this research and detailed the findings in Biology of Blood and Marrow Transplantation.
The study was funded by Therakos, Inc., a Mallinckrodt Pharmaceuticals Company.
The study included 77 patients with cGVHD. The median age at transplant was 49, 88% of patients were white, and 62% were male.
Patients had moderate (48%) or severe (52%) cGVHD. Sites of involvement included skin (86%), mouth (52%), gastrointestinal tract (29%), eye (62%), joint and fascia (51%), genital tract (11%), and lung (28%).
Patients had received a median of 2 (range, 0 to 7) prior lines of cGVHD therapy.
For this study, patients received 1689 ECP treatments, an average of 21.9 treatments per patient. The most common regimen was ECP twice a week for 4 weeks, then twice a week every 2 weeks for 8 weeks, then further tapering at the treating physician’s discretion.
Forty-eight patients (62.3%) completed all 6 months of ECP treatment. Reasons for early discontinuation included cGVHD progression (n=6), infection (n=4), cGVHD improvement (n=2), death from cGVHD-related cause (n=2), logistical issues (n=2), loss to follow-up (n=2), unknown reasons (n=4), and other various reasons (n=7) such as a finance issue and non-adherence.
Response
Provider-assessed response rates differed from response rates according to 2005 National Institutes of Health (NIH) consensus criteria.
According to providers, the response rate was 62.3% (48/77), with 14% of patients (n=11) achieving a complete response and 48% (n=37) attaining a partial response. Nineteen percent of patients (n=15) had stable disease, 14% (n=11) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Eight patients did not have enough data to assess NIH response. For the 69 evaluable patients, the NIH response rate was 43.5% (n=30), with 6% of patients (n=4) achieving a complete response and 38% (n=26) attaining a partial response. Fifteen percent of patients (n=10) had stable disease, 38% (n=26) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Risk factors
The researchers said there was no significant difference between responders and nonresponders when it came to age at treatment, platelet count, cGVHD severity, donor gender, and donor type.
For provider-assessed response, the median Karnofsky performance score at study entry was significantly higher in responders than nonresponders (90 vs 80; P=0.001). Responders also had a significantly shorter time from transplant to study entry compared to nonresponders (1.43 vs 2.06 years; P=0.051).
Similarly, according to NIH response, time from transplant to study entry was significantly shorter for responders than nonresponders (1.23 vs 1.92 years; P=0.04).
However, in a logistic regression model, time from transplant to study entry was not associated with provider-assessed or NIH response.
Tregs
The researchers found no significant difference in Treg percentages between responders and nonresponders.
For provider-assessed response, the baseline Treg frequency was 4.4% in responders and 4.8% in nonresponders (P=0.4). Treg percentages at the end of study were 4.2% and 5.5%, respectively (P=0.2). And the change in Treg frequency was 0.3% and 1.3%, respectively (P=0.3).
For NIH response, the baseline Treg frequency was 4.7% in responders and 4.4% in nonresponders (P=0.3). Treg percentages at the end of study were 4.4% and 4.7%, respectively (P=0.6). And the change in Treg percentages was 0.3% and 0.7%, respectively (P=0.4).
These findings run contrary to the researchers’ hypothesis that response to ECP would be associated with an increase in the percentage of Tregs.
The researchers did note that the number of Tregs varied between patients, and the team raised the possibility that the mechanism of Tregs in ECP is not visible by measuring cell abundance.
However, the researchers also said future studies should explore additional mechanisms of action for ECP and look particularly at other T-cell populations, dendritic cells, inhibitory cytokines, and proinflammatory cytokines.
A prospective study did not reveal the mechanism driving response to extracorporeal photopheresis (ECP) in patients with chronic graft-versus-host disease (cGVHD).
However, researchers did find that responses occurred independent of risk factors, and results suggested that regulatory T cells (Tregs) are not the dominant mechanism of response to ECP.
Madan Jagasia, MBBS, of Vanderbilt University School of Medicine in Nashville, Tennessee, and his colleagues conducted this research and detailed the findings in Biology of Blood and Marrow Transplantation.
The study was funded by Therakos, Inc., a Mallinckrodt Pharmaceuticals Company.
The study included 77 patients with cGVHD. The median age at transplant was 49, 88% of patients were white, and 62% were male.
Patients had moderate (48%) or severe (52%) cGVHD. Sites of involvement included skin (86%), mouth (52%), gastrointestinal tract (29%), eye (62%), joint and fascia (51%), genital tract (11%), and lung (28%).
Patients had received a median of 2 (range, 0 to 7) prior lines of cGVHD therapy.
For this study, patients received 1689 ECP treatments, an average of 21.9 treatments per patient. The most common regimen was ECP twice a week for 4 weeks, then twice a week every 2 weeks for 8 weeks, then further tapering at the treating physician’s discretion.
Forty-eight patients (62.3%) completed all 6 months of ECP treatment. Reasons for early discontinuation included cGVHD progression (n=6), infection (n=4), cGVHD improvement (n=2), death from cGVHD-related cause (n=2), logistical issues (n=2), loss to follow-up (n=2), unknown reasons (n=4), and other various reasons (n=7) such as a finance issue and non-adherence.
Response
Provider-assessed response rates differed from response rates according to 2005 National Institutes of Health (NIH) consensus criteria.
According to providers, the response rate was 62.3% (48/77), with 14% of patients (n=11) achieving a complete response and 48% (n=37) attaining a partial response. Nineteen percent of patients (n=15) had stable disease, 14% (n=11) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Eight patients did not have enough data to assess NIH response. For the 69 evaluable patients, the NIH response rate was 43.5% (n=30), with 6% of patients (n=4) achieving a complete response and 38% (n=26) attaining a partial response. Fifteen percent of patients (n=10) had stable disease, 38% (n=26) progressed, and 4% (n=3) did not have follow-up for cGVHD-related reasons.
Risk factors
The researchers said there was no significant difference between responders and nonresponders when it came to age at treatment, platelet count, cGVHD severity, donor gender, and donor type.
For provider-assessed response, the median Karnofsky performance score at study entry was significantly higher in responders than nonresponders (90 vs 80; P=0.001). Responders also had a significantly shorter time from transplant to study entry compared to nonresponders (1.43 vs 2.06 years; P=0.051).
Similarly, according to NIH response, time from transplant to study entry was significantly shorter for responders than nonresponders (1.23 vs 1.92 years; P=0.04).
However, in a logistic regression model, time from transplant to study entry was not associated with provider-assessed or NIH response.
Tregs
The researchers found no significant difference in Treg percentages between responders and nonresponders.
For provider-assessed response, the baseline Treg frequency was 4.4% in responders and 4.8% in nonresponders (P=0.4). Treg percentages at the end of study were 4.2% and 5.5%, respectively (P=0.2). And the change in Treg frequency was 0.3% and 1.3%, respectively (P=0.3).
For NIH response, the baseline Treg frequency was 4.7% in responders and 4.4% in nonresponders (P=0.3). Treg percentages at the end of study were 4.4% and 4.7%, respectively (P=0.6). And the change in Treg percentages was 0.3% and 0.7%, respectively (P=0.4).
These findings run contrary to the researchers’ hypothesis that response to ECP would be associated with an increase in the percentage of Tregs.
The researchers did note that the number of Tregs varied between patients, and the team raised the possibility that the mechanism of Tregs in ECP is not visible by measuring cell abundance.
However, the researchers also said future studies should explore additional mechanisms of action for ECP and look particularly at other T-cell populations, dendritic cells, inhibitory cytokines, and proinflammatory cytokines.
First CAR T-cell therapy approved in Canada

Health Canada has authorized use of tisagenlecleucel (Kymriah™), making it the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel (formerly CTL019) is approved to treat patients ages 3 to 25 with B-cell acute lymphoblastic leukemia (ALL) who have relapsed after allogeneic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers, and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
JULIET enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
These results were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
ELIANA trial
ELIANA included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi. All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
Ninety-five percent of patients had AEs thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
These results were published in The New England Journal of Medicine in February.
Health Canada has authorized use of tisagenlecleucel (Kymriah™), making it the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel (formerly CTL019) is approved to treat patients ages 3 to 25 with B-cell acute lymphoblastic leukemia (ALL) who have relapsed after allogeneic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers, and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
JULIET enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
These results were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
ELIANA trial
ELIANA included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi. All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
Ninety-five percent of patients had AEs thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
These results were published in The New England Journal of Medicine in February.
Health Canada has authorized use of tisagenlecleucel (Kymriah™), making it the first chimeric antigen receptor (CAR) T-cell therapy to receive regulatory approval in Canada.
Tisagenlecleucel (formerly CTL019) is approved to treat patients ages 3 to 25 with B-cell acute lymphoblastic leukemia (ALL) who have relapsed after allogeneic stem cell transplant (SCT) or are otherwise ineligible for SCT, have experienced second or later relapse, or have refractory disease.
Tisagenlecleucel is also approved in Canada to treat adults who have received two or more lines of systemic therapy and have relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high grade B-cell lymphoma, or DLBCL arising from follicular lymphoma.
Novartis, the company marketing tisagenlecleucel, said it is working with qualified treatment centers in Canada to prepare for the delivery of tisagenlecleucel. Certification and training are underway at these centers, and Novartis is enhancing manufacturing capacity to meet patient needs.
Tisagenlecleucel has been studied in a pair of phase 2 trials—ELIANA and JULIET.
JULIET trial
JULIET enrolled 165 adults with relapsed/refractory DLBCL, and 111 of them received a single infusion of tisagenlecleucel. Ninety-two percent of patients received bridging therapy, and 93% received lymphodepleting chemotherapy prior to tisagenlecleucel.
The overall response rate was 52%, and the complete response (CR) rate was 40%. The median duration of response was not reached with a median follow-up of 13.9 months. At last follow-up, none of the responders had gone on to SCT.
The 12-month overall survival (OS) rate was 49%, and the median OS was 11.7 months. The median OS was not reached for patients in CR.
Within 8 weeks of tisagenlecleucel infusion, 22% of patients had developed grade 3/4 cytokine release syndrome (CRS). Other adverse events (AEs) of interest included grade 3/4 neurologic events (12%), grade 3/4 cytopenias lasting more than 28 days (32%), grade 3/4 infections (20%), and grade 3/4 febrile neutropenia (15%).
These results were presented at the 23rd Annual Congress of the European Hematology Association in June (abstract S799).
ELIANA trial
ELIANA included 75 children and young adults with relapsed/refractory ALL. The patients’ median age was 11 (range, 3 to 23).
All patients received a single infusion of tisagenlecleucel, and 72 received lymphodepleting chemotherapy.
The median duration of follow-up was 13.1 months. The study’s primary endpoint was overall remission rate, which was defined as the rate of a best overall response of either CR or CR with incomplete hematologic recovery (CRi) within 3 months.
The overall remission rate was 81% (61/75), with 60% of patients (n=45) achieving a CR and 21% (n=16) achieving a CRi. All patients whose best response was CR/CRi were negative for minimal residual disease. The median duration of response was not met.
Eight patients proceeded to SCT while in remission. At last follow-up, four were still in remission, and four had unknown disease status.
At 6 months, the event-free survival rate was 73%, and the OS rate was 90%. At 12 months, the rates were 50% and 76%, respectively.
Ninety-five percent of patients had AEs thought to be related to tisagenlecleucel. The incidence of treatment-related grade 3/4 AEs was 73%.
AEs of special interest included CRS (77%), neurologic events (40%), infections (43%), febrile neutropenia (35%), cytopenias not resolved by day 28 (37%), and tumor lysis syndrome (4%).
These results were published in The New England Journal of Medicine in February.
CAR T-cell therapy will soon be available in England, NHS says

The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
Tisagenlecleucel will be made available through the Cancer Drugs Fund, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will only be available for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta®), has not fared as well as tisagenlecleucel. The National Institute for Health and Care Excellence (NICE) recently issued a draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
Tisagenlecleucel will be made available through the Cancer Drugs Fund, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will only be available for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta®), has not fared as well as tisagenlecleucel. The National Institute for Health and Care Excellence (NICE) recently issued a draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund.
The National Health Service (NHS) of England has announced that tisagenlecleucel (Kymriah®), a chimeric antigen receptor (CAR) T-cell therapy, will soon be available for certain leukemia patients.
Tisagenlecleucel will be made available through the Cancer Drugs Fund, and patients could potentially begin receiving the treatment within weeks.
NHS England struck a deal with Novartis to lower the price of tisagenlecleucel, which costs around £282,000 per patient at its full list price. The discount offered to the NHS is confidential.
Tisagenlecleucel was recently approved by the European Commission (EC) to treat patients up to 25 years of age who have B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant, or in second or later relapse.
The EC also approved tisagenlecleucel to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have received two or more lines of systemic therapy.
However, tisagenlecleucel will only be available for ALL patients in England, at least initially. A decision has not been made regarding funding for tisagenlecleucel in DLBCL, and Novartis previously decided to launch tisagenlecleucel in ALL first.
“It’s fantastic news for children and young people with this form of leukemia that CAR T-cell therapy will be made available on the NHS, making them the first in Europe to have routine access to this exciting new type of immunotherapy,” said Charles Swanton, Cancer Research UK’s chief clinician.
The first three NHS hospitals to go through the international accreditation process for the provision of tisagenlecleucel are in London, Manchester, and Newcastle. Subject to passing accreditation requirements, the first treatments could begin in a matter of weeks.
Another CAR T-cell therapy, axicabtagene ciloleucel (Yescarta®), has not fared as well as tisagenlecleucel. The National Institute for Health and Care Excellence (NICE) recently issued a draft guidance recommending against the use of axicabtagene ciloleucel in England.
Axicabtagene ciloleucel was approved by the EC to treat patients with relapsed/refractory DLBCL or primary mediastinal B-cell lymphoma who have received two or more lines of systemic therapy.
However, NICE said it isn’t clear how much of a benefit axicabtagene ciloleucel may provide over salvage chemotherapy. NICE also said the price of axicabtagene ciloleucel is too high for the therapy to be considered a cost-effective use of NHS resources, and axicabtagene ciloleucel does not meet the criteria for inclusion in the Cancer Drugs Fund.
Humans may have more HSCs than we thought

Humans may have ten times more hematopoietic stem cells (HSCs) than previously thought, according to research published in Nature.
Researchers developed a new approach for studying HSCs and found evidence suggesting that HSC numbers increase rapidly through childhood, reach a plateau by adolescence, and remain relatively constant throughout adulthood.
“We discovered that healthy adults have between 50,000 and 200,000 blood stem cells, which is about ten times more than previously thought,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Whereas previous estimates of blood stem cell numbers were extrapolated from studies in mice, cats, or monkeys, this is the first time stem cell numbers have been directly quantified in humans. This new approach opens up avenues into studying stem cells in other human organs and how they change between health and disease, and as we age.”
For this study, Dr Campbell and his colleagues conducted whole-genome sequencing on hematopoietic stem and progenitor colonies from a healthy 59 year-old man. The team adapted a capture-recapture* method to “tag” stem cells and compare them to the population of blood cells.
Specifically, in the “capture” phase, the researchers isolated individual hematopoietic stem and progenitor cells from a bone marrow aspirate and peripheral blood draw from the male subject. The cells were expanded, and the researchers performed whole-genome sequencing on 198 colonies to identify somatic mutations.
In the “recapture” phase, the researchers isolated bulk populations of mature peripheral blood cells from the subject—granulocytes at three time points and B and T lymphocytes at one time point. The team then performed deep, targeted sequencing on these bulk populations.
“The mutations act like barcodes, each of which uniquely tags a stem cell and its descendants,” said study author Henry Lee-Six, of the Wellcome Trust Sanger Institute.
“We then looked for these mutations in the rest of the blood to see what fraction of blood cells carry the same barcodes, and, from this, we could estimate how many stem cells there were in total.”
The researchers’ results suggested the number of HSCs actively contributing to circulating granulocytes at any one time was in the range of 44,000 to 215,000.
The team also estimated that the time between successive self-renewal HSC divisions is likely in the range of 2 to 20 months.
Finally, the researchers observed signs of “rapid” HSC population expansion during early life, which reaches a relatively stable plateau by late childhood or early adolescence that continues into adulthood.
The team said this stability suggests that symmetric self-renewal divisions (when one HSC divides into two) are balanced by HSC death, senescence, and symmetric divisions into committed progenitors.
*Capture-recapture is commonly used in ecology to estimate a species’ population size. A portion of the population is captured, tagged, and released. Later, another portion is captured, and the number of tagged individuals within the sample is counted. Since the number of marked individuals within the second sample should be proportional to the number of marked individuals in the whole population, an estimate of the total population size can be obtained by dividing the number of marked individuals by the proportion of marked individuals in the second sample.
Humans may have ten times more hematopoietic stem cells (HSCs) than previously thought, according to research published in Nature.
Researchers developed a new approach for studying HSCs and found evidence suggesting that HSC numbers increase rapidly through childhood, reach a plateau by adolescence, and remain relatively constant throughout adulthood.
“We discovered that healthy adults have between 50,000 and 200,000 blood stem cells, which is about ten times more than previously thought,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Whereas previous estimates of blood stem cell numbers were extrapolated from studies in mice, cats, or monkeys, this is the first time stem cell numbers have been directly quantified in humans. This new approach opens up avenues into studying stem cells in other human organs and how they change between health and disease, and as we age.”
For this study, Dr Campbell and his colleagues conducted whole-genome sequencing on hematopoietic stem and progenitor colonies from a healthy 59 year-old man. The team adapted a capture-recapture* method to “tag” stem cells and compare them to the population of blood cells.
Specifically, in the “capture” phase, the researchers isolated individual hematopoietic stem and progenitor cells from a bone marrow aspirate and peripheral blood draw from the male subject. The cells were expanded, and the researchers performed whole-genome sequencing on 198 colonies to identify somatic mutations.
In the “recapture” phase, the researchers isolated bulk populations of mature peripheral blood cells from the subject—granulocytes at three time points and B and T lymphocytes at one time point. The team then performed deep, targeted sequencing on these bulk populations.
“The mutations act like barcodes, each of which uniquely tags a stem cell and its descendants,” said study author Henry Lee-Six, of the Wellcome Trust Sanger Institute.
“We then looked for these mutations in the rest of the blood to see what fraction of blood cells carry the same barcodes, and, from this, we could estimate how many stem cells there were in total.”
The researchers’ results suggested the number of HSCs actively contributing to circulating granulocytes at any one time was in the range of 44,000 to 215,000.
The team also estimated that the time between successive self-renewal HSC divisions is likely in the range of 2 to 20 months.
Finally, the researchers observed signs of “rapid” HSC population expansion during early life, which reaches a relatively stable plateau by late childhood or early adolescence that continues into adulthood.
The team said this stability suggests that symmetric self-renewal divisions (when one HSC divides into two) are balanced by HSC death, senescence, and symmetric divisions into committed progenitors.
*Capture-recapture is commonly used in ecology to estimate a species’ population size. A portion of the population is captured, tagged, and released. Later, another portion is captured, and the number of tagged individuals within the sample is counted. Since the number of marked individuals within the second sample should be proportional to the number of marked individuals in the whole population, an estimate of the total population size can be obtained by dividing the number of marked individuals by the proportion of marked individuals in the second sample.
Humans may have ten times more hematopoietic stem cells (HSCs) than previously thought, according to research published in Nature.
Researchers developed a new approach for studying HSCs and found evidence suggesting that HSC numbers increase rapidly through childhood, reach a plateau by adolescence, and remain relatively constant throughout adulthood.
“We discovered that healthy adults have between 50,000 and 200,000 blood stem cells, which is about ten times more than previously thought,” said study author Peter Campbell, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“Whereas previous estimates of blood stem cell numbers were extrapolated from studies in mice, cats, or monkeys, this is the first time stem cell numbers have been directly quantified in humans. This new approach opens up avenues into studying stem cells in other human organs and how they change between health and disease, and as we age.”
For this study, Dr Campbell and his colleagues conducted whole-genome sequencing on hematopoietic stem and progenitor colonies from a healthy 59 year-old man. The team adapted a capture-recapture* method to “tag” stem cells and compare them to the population of blood cells.
Specifically, in the “capture” phase, the researchers isolated individual hematopoietic stem and progenitor cells from a bone marrow aspirate and peripheral blood draw from the male subject. The cells were expanded, and the researchers performed whole-genome sequencing on 198 colonies to identify somatic mutations.
In the “recapture” phase, the researchers isolated bulk populations of mature peripheral blood cells from the subject—granulocytes at three time points and B and T lymphocytes at one time point. The team then performed deep, targeted sequencing on these bulk populations.
“The mutations act like barcodes, each of which uniquely tags a stem cell and its descendants,” said study author Henry Lee-Six, of the Wellcome Trust Sanger Institute.
“We then looked for these mutations in the rest of the blood to see what fraction of blood cells carry the same barcodes, and, from this, we could estimate how many stem cells there were in total.”
The researchers’ results suggested the number of HSCs actively contributing to circulating granulocytes at any one time was in the range of 44,000 to 215,000.
The team also estimated that the time between successive self-renewal HSC divisions is likely in the range of 2 to 20 months.
Finally, the researchers observed signs of “rapid” HSC population expansion during early life, which reaches a relatively stable plateau by late childhood or early adolescence that continues into adulthood.
The team said this stability suggests that symmetric self-renewal divisions (when one HSC divides into two) are balanced by HSC death, senescence, and symmetric divisions into committed progenitors.
*Capture-recapture is commonly used in ecology to estimate a species’ population size. A portion of the population is captured, tagged, and released. Later, another portion is captured, and the number of tagged individuals within the sample is counted. Since the number of marked individuals within the second sample should be proportional to the number of marked individuals in the whole population, an estimate of the total population size can be obtained by dividing the number of marked individuals by the proportion of marked individuals in the second sample.
TYK2 inhibitors could treat ALCL, team says
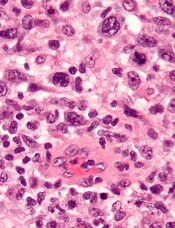
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).
Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in ALCL cells, and it delays tumor onset and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said.
He and his colleagues found that disrupting TYK2—either via gene knockdown or with small-molecule TYK2 inhibitors—induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P<0.0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available . . . ,” said study author Lukas Kenner, MD, of the Medical University of Vienna.
“[I]n the more rare lymphomas, we urgently need better therapies.”
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).
Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in ALCL cells, and it delays tumor onset and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said.
He and his colleagues found that disrupting TYK2—either via gene knockdown or with small-molecule TYK2 inhibitors—induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P<0.0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available . . . ,” said study author Lukas Kenner, MD, of the Medical University of Vienna.
“[I]n the more rare lymphomas, we urgently need better therapies.”
Preclinical research indicates that TYK2 inhibitors could be effective in treating anaplastic large-cell lymphoma (ALCL).
Researchers found evidence to suggest that TYK2 “is highly expressed in all cases of human ALCL.”
The team also discovered that TYK2 inhibition induces apoptosis in ALCL cells, and it delays tumor onset and prolongs survival in a mouse model of ALCL.
Olaf Merkel, PhD, of the Medical University of Vienna in Austria, and his colleagues detailed these findings in Leukemia.
The researchers said their analyses suggest TYK2 is expressed in all types of ALCL, regardless of ALK status, and TYK2 mediates the same anti-apoptotic response across ALCLs.
“Therefore, we could consider TYK2 signaling as the Achilles’ heel of ALCL, as, in all patients we have analyzed, the tumor cells relied on this activity to support the essential survival signal,” Dr. Merkel said.
He and his colleagues found that disrupting TYK2—either via gene knockdown or with small-molecule TYK2 inhibitors—induced apoptosis in human ALCL cells in vitro.
In a mouse model of NPM-ALK-induced lymphoma, Tyk2 deletion slowed the rate of tumor growth and significantly prolonged survival. The median survival was 53.3 weeks in mice with Tyk2 deletion and 16.0 weeks in control mice (P<0.0001).
Additional experiments in human ALCL cell lines showed that “TYK2 is activated by autocrine production of IL-10 and IL-22 and by interaction with specific receptors expressed by the cells,” the researchers said.
They also found that “activated TYK2 leads to STAT1 and STAT3 phosphorylation, activated expression of MCL1, and aberrant ALCL cell survival.”
Taking these findings together, the researchers concluded that TYK2 inhibitors could be effective for treating ALCL.
“We are looking forward to TYK2 inhibitors becoming available . . . ,” said study author Lukas Kenner, MD, of the Medical University of Vienna.
“[I]n the more rare lymphomas, we urgently need better therapies.”
Research may help explain how VOCs occur

Researchers say they have gained new insight that may help explain how vaso-occlusive crises (VOCs) occur in patients with sickle cell disease (SCD).
The team assessed how red blood cell (RBC) adhesion and polymerization of deoxygenated sickle hemoglobin affect the mechanisms underlying VOCs.
Experiments showed that hypoxia enhances sickle RBC adherence, and hemoglobin S polymerization enhances adherence for sickle reticulocytes and mature erythrocytes.
However, sickle reticulocytes have “unique adhesion dynamics” and therefore appear more likely to cause VOCs.
The researchers described these discoveries in an article set to be published this week in the Proceedings of the National Academy of Sciences.
To investigate how RBCs interact with blood vessels to set off a VOC, the researchers built a microfluidic system that mimics post-capillary vessels. These vessels, which carry deoxygenated blood away from the capillaries, are where vaso-occlusions are most likely to occur.
The microfluidic system is designed to allow the researchers to control the oxygen level. The team used the system to test blood from eight SCD patients.
The researchers found that, under hypoxic conditions, sickle RBCs are two to four times more likely to adhere to the blood vessel walls than they are when oxygen levels are normal.
The team also found that hemoglobin S polymerization enhances the adherence of sickle reticulocytes and sickle mature erythrocytes. The hemoglobin S forms stiff fibers that grow and push the cell membrane outward, and these fibers help the cells adhere more firmly to the lining of the blood vessel.
“There has been little understanding of why, under hypoxia, there is much more adhesion,” said study author Subra Suresh, DSc, of Nanyang Technological University in Singapore.
“The experiments of this study provide some key insights into the processes and mechanisms responsible for increased adhesion.”
The researchers also found that, in SCD patients, reticulocytes are more likely than mature erythrocytes to adhere to blood vessels.
“We observed the growth of sickle hemoglobin fibers stretching reticulocytes within minutes,” said study author Dimitrios Papageorgiou, PhD, of the Massachusetts Institute of Technology in Cambridge.
“It looks like they’re trying to grab more of the surface and adhere more strongly.”
The researchers said these and other findings suggest polymerization and adhesion stimulate each other.
The team now hopes to devise a more complete model of vaso-occlusion that combines their new findings with previous work. The previous work involved measuring how long it takes SCD patients’ blood cells to stiffen, making them more likely to block blood flow in tiny blood vessels.
The researchers also hope their findings might help them devise a way to predict VOCs in individual SCD patients.
Researchers say they have gained new insight that may help explain how vaso-occlusive crises (VOCs) occur in patients with sickle cell disease (SCD).
The team assessed how red blood cell (RBC) adhesion and polymerization of deoxygenated sickle hemoglobin affect the mechanisms underlying VOCs.
Experiments showed that hypoxia enhances sickle RBC adherence, and hemoglobin S polymerization enhances adherence for sickle reticulocytes and mature erythrocytes.
However, sickle reticulocytes have “unique adhesion dynamics” and therefore appear more likely to cause VOCs.
The researchers described these discoveries in an article set to be published this week in the Proceedings of the National Academy of Sciences.
To investigate how RBCs interact with blood vessels to set off a VOC, the researchers built a microfluidic system that mimics post-capillary vessels. These vessels, which carry deoxygenated blood away from the capillaries, are where vaso-occlusions are most likely to occur.
The microfluidic system is designed to allow the researchers to control the oxygen level. The team used the system to test blood from eight SCD patients.
The researchers found that, under hypoxic conditions, sickle RBCs are two to four times more likely to adhere to the blood vessel walls than they are when oxygen levels are normal.
The team also found that hemoglobin S polymerization enhances the adherence of sickle reticulocytes and sickle mature erythrocytes. The hemoglobin S forms stiff fibers that grow and push the cell membrane outward, and these fibers help the cells adhere more firmly to the lining of the blood vessel.
“There has been little understanding of why, under hypoxia, there is much more adhesion,” said study author Subra Suresh, DSc, of Nanyang Technological University in Singapore.
“The experiments of this study provide some key insights into the processes and mechanisms responsible for increased adhesion.”
The researchers also found that, in SCD patients, reticulocytes are more likely than mature erythrocytes to adhere to blood vessels.
“We observed the growth of sickle hemoglobin fibers stretching reticulocytes within minutes,” said study author Dimitrios Papageorgiou, PhD, of the Massachusetts Institute of Technology in Cambridge.
“It looks like they’re trying to grab more of the surface and adhere more strongly.”
The researchers said these and other findings suggest polymerization and adhesion stimulate each other.
The team now hopes to devise a more complete model of vaso-occlusion that combines their new findings with previous work. The previous work involved measuring how long it takes SCD patients’ blood cells to stiffen, making them more likely to block blood flow in tiny blood vessels.
The researchers also hope their findings might help them devise a way to predict VOCs in individual SCD patients.
Researchers say they have gained new insight that may help explain how vaso-occlusive crises (VOCs) occur in patients with sickle cell disease (SCD).
The team assessed how red blood cell (RBC) adhesion and polymerization of deoxygenated sickle hemoglobin affect the mechanisms underlying VOCs.
Experiments showed that hypoxia enhances sickle RBC adherence, and hemoglobin S polymerization enhances adherence for sickle reticulocytes and mature erythrocytes.
However, sickle reticulocytes have “unique adhesion dynamics” and therefore appear more likely to cause VOCs.
The researchers described these discoveries in an article set to be published this week in the Proceedings of the National Academy of Sciences.
To investigate how RBCs interact with blood vessels to set off a VOC, the researchers built a microfluidic system that mimics post-capillary vessels. These vessels, which carry deoxygenated blood away from the capillaries, are where vaso-occlusions are most likely to occur.
The microfluidic system is designed to allow the researchers to control the oxygen level. The team used the system to test blood from eight SCD patients.
The researchers found that, under hypoxic conditions, sickle RBCs are two to four times more likely to adhere to the blood vessel walls than they are when oxygen levels are normal.
The team also found that hemoglobin S polymerization enhances the adherence of sickle reticulocytes and sickle mature erythrocytes. The hemoglobin S forms stiff fibers that grow and push the cell membrane outward, and these fibers help the cells adhere more firmly to the lining of the blood vessel.
“There has been little understanding of why, under hypoxia, there is much more adhesion,” said study author Subra Suresh, DSc, of Nanyang Technological University in Singapore.
“The experiments of this study provide some key insights into the processes and mechanisms responsible for increased adhesion.”
The researchers also found that, in SCD patients, reticulocytes are more likely than mature erythrocytes to adhere to blood vessels.
“We observed the growth of sickle hemoglobin fibers stretching reticulocytes within minutes,” said study author Dimitrios Papageorgiou, PhD, of the Massachusetts Institute of Technology in Cambridge.
“It looks like they’re trying to grab more of the surface and adhere more strongly.”
The researchers said these and other findings suggest polymerization and adhesion stimulate each other.
The team now hopes to devise a more complete model of vaso-occlusion that combines their new findings with previous work. The previous work involved measuring how long it takes SCD patients’ blood cells to stiffen, making them more likely to block blood flow in tiny blood vessels.
The researchers also hope their findings might help them devise a way to predict VOCs in individual SCD patients.
Study authors fail to disclose industry payments

New research suggests investigators involved in oncology trials sometimes fail to disclose payments from the pharmaceutical industry.
Researchers looked at clinical trials associated with cancer drugs recently approved in the United States and assessed whether funding was properly disclosed when the trial results were published in scientific journals.
The data showed that roughly a third of investigators failed to completely disclose payments from trial sponsors.
“We know that pharmaceutical companies sponsor trials of their own drugs. That’s not a surprise,” said Cole Wayant, a DO/PhD student at Oklahoma State University in Tulsa.
“But what is a surprise, and what warrants concern, is that this funding is often not disclosed in the publication of clinical trials that form the basis of FDA [U.S. Food and Drug Administration] approvals and clinical practice guidelines.”
Wayant and his colleagues conducted this research and reported the findings in a letter to JAMA Oncology.
The researchers began by searching the FDA Hematology/Oncology (Cancer) Approvals & Safety Notifications website for oncology drugs approved from Jan. 1, 2016, to Aug. 31, 2017.
The team then identified the published trials supporting these drug approvals and searched the Open Payments website for industry payment data for each U.S.-based oncologist involved in the trials.
Finally, the researchers compared the Open Payments data to the disclosure statements from the publications.
There were 344 authors of clinical trials associated with oncology drugs approved during the period studied. Most authors (76.5%) received at least one industry payment, and the total amount they received exceeded $216 million.
Nearly a third of the authors (32%, n=110) did not fully disclose payments from a trial sponsor.
In all, the authors received about $6.3 million in general payments (e.g., speaking fees), and $1.7 million of that was undisclosed.
They received more than $500,000 in research payments (e.g., fees for study coordination), and more than $200,000 of that was undisclosed.
The authors received close to $210 million in associated research payments (e.g., grants), and about $78 million of that was undisclosed.
Wayant and his colleagues said these results suggest financial relationships between the pharmaceutical industry and oncology trial investigators “may be common, expensive, and frequently undisclosed.” However, the research also suggests Open Payments data could be used to ensure complete disclosure of industry payments.
New research suggests investigators involved in oncology trials sometimes fail to disclose payments from the pharmaceutical industry.
Researchers looked at clinical trials associated with cancer drugs recently approved in the United States and assessed whether funding was properly disclosed when the trial results were published in scientific journals.
The data showed that roughly a third of investigators failed to completely disclose payments from trial sponsors.
“We know that pharmaceutical companies sponsor trials of their own drugs. That’s not a surprise,” said Cole Wayant, a DO/PhD student at Oklahoma State University in Tulsa.
“But what is a surprise, and what warrants concern, is that this funding is often not disclosed in the publication of clinical trials that form the basis of FDA [U.S. Food and Drug Administration] approvals and clinical practice guidelines.”
Wayant and his colleagues conducted this research and reported the findings in a letter to JAMA Oncology.
The researchers began by searching the FDA Hematology/Oncology (Cancer) Approvals & Safety Notifications website for oncology drugs approved from Jan. 1, 2016, to Aug. 31, 2017.
The team then identified the published trials supporting these drug approvals and searched the Open Payments website for industry payment data for each U.S.-based oncologist involved in the trials.
Finally, the researchers compared the Open Payments data to the disclosure statements from the publications.
There were 344 authors of clinical trials associated with oncology drugs approved during the period studied. Most authors (76.5%) received at least one industry payment, and the total amount they received exceeded $216 million.
Nearly a third of the authors (32%, n=110) did not fully disclose payments from a trial sponsor.
In all, the authors received about $6.3 million in general payments (e.g., speaking fees), and $1.7 million of that was undisclosed.
They received more than $500,000 in research payments (e.g., fees for study coordination), and more than $200,000 of that was undisclosed.
The authors received close to $210 million in associated research payments (e.g., grants), and about $78 million of that was undisclosed.
Wayant and his colleagues said these results suggest financial relationships between the pharmaceutical industry and oncology trial investigators “may be common, expensive, and frequently undisclosed.” However, the research also suggests Open Payments data could be used to ensure complete disclosure of industry payments.
New research suggests investigators involved in oncology trials sometimes fail to disclose payments from the pharmaceutical industry.
Researchers looked at clinical trials associated with cancer drugs recently approved in the United States and assessed whether funding was properly disclosed when the trial results were published in scientific journals.
The data showed that roughly a third of investigators failed to completely disclose payments from trial sponsors.
“We know that pharmaceutical companies sponsor trials of their own drugs. That’s not a surprise,” said Cole Wayant, a DO/PhD student at Oklahoma State University in Tulsa.
“But what is a surprise, and what warrants concern, is that this funding is often not disclosed in the publication of clinical trials that form the basis of FDA [U.S. Food and Drug Administration] approvals and clinical practice guidelines.”
Wayant and his colleagues conducted this research and reported the findings in a letter to JAMA Oncology.
The researchers began by searching the FDA Hematology/Oncology (Cancer) Approvals & Safety Notifications website for oncology drugs approved from Jan. 1, 2016, to Aug. 31, 2017.
The team then identified the published trials supporting these drug approvals and searched the Open Payments website for industry payment data for each U.S.-based oncologist involved in the trials.
Finally, the researchers compared the Open Payments data to the disclosure statements from the publications.
There were 344 authors of clinical trials associated with oncology drugs approved during the period studied. Most authors (76.5%) received at least one industry payment, and the total amount they received exceeded $216 million.
Nearly a third of the authors (32%, n=110) did not fully disclose payments from a trial sponsor.
In all, the authors received about $6.3 million in general payments (e.g., speaking fees), and $1.7 million of that was undisclosed.
They received more than $500,000 in research payments (e.g., fees for study coordination), and more than $200,000 of that was undisclosed.
The authors received close to $210 million in associated research payments (e.g., grants), and about $78 million of that was undisclosed.
Wayant and his colleagues said these results suggest financial relationships between the pharmaceutical industry and oncology trial investigators “may be common, expensive, and frequently undisclosed.” However, the research also suggests Open Payments data could be used to ensure complete disclosure of industry payments.
Caplacizumab approved to treat aTTP

The European Commission has granted marketing authorization for caplacizumab (Cablivi™), a humanized bivalent nanobody that inhibits the interaction between von Willebrand factor and platelets.
Caplacizumab is now approved to treat adults with acquired thrombotic thrombocytopenic purpura (aTTP) in all member countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Sanofi Genzyme said it will work with relevant local authorities to make caplacizumab available in countries across Europe.
“The approval of Cablivi provides an important addition to the standard-of-care treatment for patients with aTTP in Europe because it can significantly reduce time to platelet count normalization and induce a clinically meaningful reduction in recurrences,” said Marie Scully, MD, of University College Hospital in London, UK.
The European Commission’s approval of caplacizumab is supported by data from the phase 2 TITAN study and the phase 3 HERCULES study.
TITAN
Results from the TITAN trial were published in The New England Journal of Medicine in 2016.
The study included 75 aTTP patients who were randomized to caplacizumab (n=36) or placebo (n=39), with all patients receiving the current standard of care—daily plasma exchange and immunosuppressive therapy.
The study’s primary endpoint was time to response, which was defined as platelet count normalization (150,000/mm3 or higher).
Patients in the caplacizumab arm had a 39% reduction in the median time to response compared to patients in the placebo arm (P=0.005).
Among the 69 patients who had not undergone a plasma exchange session before enrollment, the median time to response was 3.0 days in the caplacizumab arm and 4.9 days in the placebo arm.
Among the 6 patients who did undergo a plasma exchange session before enrollment, the median time to a response was 2.4 days in the caplacizumab arm and 4.3 days in the placebo arm.
The rate of confirmed response was 86.1% (n=31) in the caplacizumab arm and 71.8% (n=28) in the placebo arm.
There were 541 adverse events (AEs) in 34 of the 35 evaluable patients receiving caplacizumab (97%) and 522 AEs in all 37 evaluable patients receiving placebo (100%). TTP exacerbations and relapses were not included as AEs.
The rate of AEs thought to be related to the study drug was 17% in the caplacizumab arm and 11% in the placebo arm. The rate of AEs that were possibly related was 54% and 8%, respectively. The rate of serious AEs was 37% and 32%, respectively.
There were no deaths in the caplacizumab arm and two in the placebo arm. One death was due to severe, refractory TTP, and the other was due to cerebral hemorrhage.
HERCULES
Results from the HERCULES trial were presented at the 2017 ASH Annual Meeting.
The study enrolled patients with an acute episode of aTTP. They were randomized to receive caplacizumab (n=72) or placebo (n=73) in addition to standard care—plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as an initial platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. The incidence of at least one major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
The proportion of patients with at least one study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The proportion of patients with at least one study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively. The rate of discontinuation due to at least one AE was 7.0% and 12.3%, respectively.
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.
The European Commission has granted marketing authorization for caplacizumab (Cablivi™), a humanized bivalent nanobody that inhibits the interaction between von Willebrand factor and platelets.
Caplacizumab is now approved to treat adults with acquired thrombotic thrombocytopenic purpura (aTTP) in all member countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Sanofi Genzyme said it will work with relevant local authorities to make caplacizumab available in countries across Europe.
“The approval of Cablivi provides an important addition to the standard-of-care treatment for patients with aTTP in Europe because it can significantly reduce time to platelet count normalization and induce a clinically meaningful reduction in recurrences,” said Marie Scully, MD, of University College Hospital in London, UK.
The European Commission’s approval of caplacizumab is supported by data from the phase 2 TITAN study and the phase 3 HERCULES study.
TITAN
Results from the TITAN trial were published in The New England Journal of Medicine in 2016.
The study included 75 aTTP patients who were randomized to caplacizumab (n=36) or placebo (n=39), with all patients receiving the current standard of care—daily plasma exchange and immunosuppressive therapy.
The study’s primary endpoint was time to response, which was defined as platelet count normalization (150,000/mm3 or higher).
Patients in the caplacizumab arm had a 39% reduction in the median time to response compared to patients in the placebo arm (P=0.005).
Among the 69 patients who had not undergone a plasma exchange session before enrollment, the median time to response was 3.0 days in the caplacizumab arm and 4.9 days in the placebo arm.
Among the 6 patients who did undergo a plasma exchange session before enrollment, the median time to a response was 2.4 days in the caplacizumab arm and 4.3 days in the placebo arm.
The rate of confirmed response was 86.1% (n=31) in the caplacizumab arm and 71.8% (n=28) in the placebo arm.
There were 541 adverse events (AEs) in 34 of the 35 evaluable patients receiving caplacizumab (97%) and 522 AEs in all 37 evaluable patients receiving placebo (100%). TTP exacerbations and relapses were not included as AEs.
The rate of AEs thought to be related to the study drug was 17% in the caplacizumab arm and 11% in the placebo arm. The rate of AEs that were possibly related was 54% and 8%, respectively. The rate of serious AEs was 37% and 32%, respectively.
There were no deaths in the caplacizumab arm and two in the placebo arm. One death was due to severe, refractory TTP, and the other was due to cerebral hemorrhage.
HERCULES
Results from the HERCULES trial were presented at the 2017 ASH Annual Meeting.
The study enrolled patients with an acute episode of aTTP. They were randomized to receive caplacizumab (n=72) or placebo (n=73) in addition to standard care—plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as an initial platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. The incidence of at least one major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
The proportion of patients with at least one study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The proportion of patients with at least one study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively. The rate of discontinuation due to at least one AE was 7.0% and 12.3%, respectively.
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.
The European Commission has granted marketing authorization for caplacizumab (Cablivi™), a humanized bivalent nanobody that inhibits the interaction between von Willebrand factor and platelets.
Caplacizumab is now approved to treat adults with acquired thrombotic thrombocytopenic purpura (aTTP) in all member countries of the European Union as well as Norway, Iceland, and Liechtenstein.
Sanofi Genzyme said it will work with relevant local authorities to make caplacizumab available in countries across Europe.
“The approval of Cablivi provides an important addition to the standard-of-care treatment for patients with aTTP in Europe because it can significantly reduce time to platelet count normalization and induce a clinically meaningful reduction in recurrences,” said Marie Scully, MD, of University College Hospital in London, UK.
The European Commission’s approval of caplacizumab is supported by data from the phase 2 TITAN study and the phase 3 HERCULES study.
TITAN
Results from the TITAN trial were published in The New England Journal of Medicine in 2016.
The study included 75 aTTP patients who were randomized to caplacizumab (n=36) or placebo (n=39), with all patients receiving the current standard of care—daily plasma exchange and immunosuppressive therapy.
The study’s primary endpoint was time to response, which was defined as platelet count normalization (150,000/mm3 or higher).
Patients in the caplacizumab arm had a 39% reduction in the median time to response compared to patients in the placebo arm (P=0.005).
Among the 69 patients who had not undergone a plasma exchange session before enrollment, the median time to response was 3.0 days in the caplacizumab arm and 4.9 days in the placebo arm.
Among the 6 patients who did undergo a plasma exchange session before enrollment, the median time to a response was 2.4 days in the caplacizumab arm and 4.3 days in the placebo arm.
The rate of confirmed response was 86.1% (n=31) in the caplacizumab arm and 71.8% (n=28) in the placebo arm.
There were 541 adverse events (AEs) in 34 of the 35 evaluable patients receiving caplacizumab (97%) and 522 AEs in all 37 evaluable patients receiving placebo (100%). TTP exacerbations and relapses were not included as AEs.
The rate of AEs thought to be related to the study drug was 17% in the caplacizumab arm and 11% in the placebo arm. The rate of AEs that were possibly related was 54% and 8%, respectively. The rate of serious AEs was 37% and 32%, respectively.
There were no deaths in the caplacizumab arm and two in the placebo arm. One death was due to severe, refractory TTP, and the other was due to cerebral hemorrhage.
HERCULES
Results from the HERCULES trial were presented at the 2017 ASH Annual Meeting.
The study enrolled patients with an acute episode of aTTP. They were randomized to receive caplacizumab (n=72) or placebo (n=73) in addition to standard care—plasma exchange and immunosuppression.
The study’s primary endpoint was the time to platelet count response (normalization), which was defined as an initial platelet count of at least 150 x 109/L with subsequent stop of daily plasma exchange within 5 days.
There was a significant reduction in time to platelet count response in the caplacizumab arm compared to the placebo arm. The platelet normalization rate ratio was 1.55 (P<0.01).
A secondary endpoint was the combination of aTTP-related death, aTTP recurrence, and at least one major thromboembolic event during study treatment. The incidence of this combined endpoint was 12.7% (n=9) in the caplacizumab arm and 49.3% (n=36) in the placebo arm (P<0.0001).
The incidence of aTTP-related death was 0% (n=0) in the caplacizumab arm and 4.1% (n=3) in the placebo arm. The incidence of aTTP recurrence was 4.2% (n=3) and 38.4% (n=28), respectively. The incidence of at least one major thromboembolic event was 8.5% (n=6) and 8.2% (n=6), respectively.
The proportion of patients with at least one study-drug-related AE was 57.7% in the caplacizumab arm and 43.8% in the placebo arm. The proportion of patients with at least one study-drug-related serious AE was 14.1% (n=10) and 5.5% (n=4), respectively. The rate of discontinuation due to at least one AE was 7.0% and 12.3%, respectively.
During the treatment period, there were no deaths in the caplacizumab arm and three deaths in the placebo arm. There was one death in the caplacizumab arm during the follow-up period, but it was considered unrelated to caplacizumab.