User login
Study quantifies 5-year survival rates for blood cancers

chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
chemotherapy
Photo by Rhoda Baer
A new study shows that 5-year survival rates for US patients with hematologic malignancies have increased greatly since the 1950s, but there is still room for improvement, particularly for patients with acute myeloid leukemia (AML).
Researchers found the absolute difference in improvement for 5-year survival from 1950-1954 to 2008-2013 ranged from 38.2% for non-Hodgkin lymphoma (NHL) to 56.6% for Hodgkin lymphoma.
And although the 5-year survival rate for Hodgkin lymphoma patients reached 86.6% for 2008-2013, the 5-year survival rate for patients with AML only reached 27.4%.
This study also revealed large disparities in overall cancer mortality rates between different counties across the country.
Ali H. Mokdad, PhD, of the Institute for Health Metrics and Evaluation in Seattle, Washington, and his colleagues reported these findings in JAMA.
Overall cancer deaths
The researchers found there were 19,511,910 cancer deaths recorded in the US between 1980 and 2014. Cancer mortality decreased by 20.1% between 1980 and 2014, from 240.2 deaths per 100,000 people to 192.0 deaths per 100,000 people.
In 1980, cancer mortality ranged from 130.6 per 100,000 in Summit County, Colorado, to 386.9 per 100,000 in North Slope Borough, Alaska.
In 2014, cancer mortality ranged from 70.7 per 100,000 in Summit County, Colorado, to 503.1 per 100,000 in Union County, Florida.
“Such significant disparities among US counties is unacceptable,” Dr Mokdad said. “Every person should have access to early screenings for cancer, as well as adequate treatment.”
Mortality rates for hematologic malignancies
In 2014, the mortality rates, per 100,000 people, for hematologic malignancies were:
- 0.4 for Hodgkin lymphoma (rank out of all cancers, 27)
- 8.3 for NHL (rank, 7)
- 3.9 for multiple myeloma (rank, 16)
- 9.0 for all leukemias (rank, 6)
- 0.7 for acute lymphoid leukemia (ALL)
- 2.6 for chronic lymphoid leukemia (CLL)
- 5.1 for AML
- 0.6 for chronic myeloid leukemia (CML).
The leukemia subtypes were not assigned a rank.
5-year survival rates for hematologic malignancies
Hodgkin lymphoma
- 30% for 1950-54
- 68.6% for 1973-77
- 72.1% for 1978-82
- 86.6% for 2008-2013
- Absolute difference (between the first and latest year of data), 56.6%.
NHL
- 33% for 1950-54
- 45.3% for 1973-77
- 48.7% for 1978-82
- 71.2% for 2008-2013
- Absolute difference, 38.2%.
Multiple myeloma
- 6% for 1950-54
- 23.4% for 1973-77
- 26.6% for 1978-82
- 49.8% for 2008-2013
- Absolute difference, 43.8%.
Leukemia
- 10% for 1950-54
- 34% for 1973-77
- 36.3% for 1978-82
- 60.1% for 2008-2013
- Absolute difference, 50.1%.
ALL
- 39.2% for 1973-77
- 50.5% for 1978-82
- 68.1% for 2008-2013
- Absolute difference, 28.9%.
CLL
- 67% for 1973-77
- 66.3% for 1978-82
- 82.5% for 2008-2013
- Absolute difference, 15.5%.
AML
- 6.2% for 1973-77
- 7.9% for 1978-82
- 27.4% for 2008-2013
- Absolute difference, 21.2%.
CML
- 21.1% for 1973-77
- 25.8% for 1978-82
- 66.4% for 2008-2013
- Absolute difference, 45.3%.
For the leukemia subtypes, there was no data for 1950 to 1954. ![]()
Health Canada expands indication for lenalidomide

Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.” ![]()
Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.” ![]()
Photo courtesy of Celgene
Health Canada has expanded the approved indication for lenalidomide (Revlimid®) to include the treatment of patients with multiple myeloma (MM).
Lenalidomide is now approved for use in combination with dexamethasone to treat patients newly diagnosed with MM who are not eligible for stem cell transplant.
Lenalidomide was previously approved in Canada for the treatment of patients with transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes associated with a deletion 5q cytogenetic abnormality, with or without additional cytogenetic abnormalities.
Lenalidomide is a product of Celgene Corporation.
“The expanded indication of Revlimid® provides [MM] patients with a treatment much earlier in their disease and offers this patient population an all-oral, melphalan-free option for a disease that continues to be difficult to treat,” said Donna Reece, MD, of Princess Margaret Hospital in Toronto, Ontario, Canada.
The expanded approval of lenalidomide is based on safety and efficacy results from the phase 3 FIRST trial. Updated results from this study were published in the Journal of Clinical Oncology last November.
The trial included 1623 patients with newly diagnosed MM who were not eligible for stem cell transplant.
Patients were randomized to receive:
- Lenalidomide and low-dose dexamethasone (Rd) in 28-day cycles until disease progression (n=535)
- 18 cycles of Rd (Rd18) for 72 weeks (n=541)
- Melphalan, prednisone, and thalidomide (MPT) for 72 weeks (n=547).
In the intent-to-treat population, the overall response rate was 81% for the continuous Rd group, 79% for the Rd18 group, and 67% in the MPT group. The complete response rates were 21%, 20%, and 12%, respectively.
The median progression-free survival (PFS) was 26.0 months in the continuous Rd group, 21.0 months in the Rd18 group, and 21.9 months in the MPT group. At 4 years, the PFS rates were 33%, 14%, and 13%, respectively.
The median overall survival (OS) was 58.9 months in the continuous Rd group, 56.7 months in the Rd18 group, and 48.5 months in the MPT group. At 4 years, the OS rates were 60%, 57%, and 51%, respectively.
The most frequent grade 3/4 hematologic treatment-emergent adverse events were neutropenia and anemia. The rate of grade 3/4 neutropenia was higher in the MPT group than the continuous Rd or Rd18 groups.
Infections were the most common grade 3/4 non-hematologic treatment-emergent adverse events. The rate of grade 3/4 infections was higher in the Rd groups than the MPT group.
“With this new clinical evidence, we know that keeping newly diagnosed multiple myeloma patients on Revlimid® may help delay disease progression and reduce the risk of death,” Dr Reece said. “As such, we are looking forward to having Revlimid® as a key option in the first-line setting for the appropriate patients.” ![]()
Improving the efficacy of obinutuzumab

Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
Preclinical research suggests that immune stimulation through Toll-like receptor 7 (TLR7) agonism can enhance the efficacy of obinutuzumab in lymphoma.
Researchers found that combining the anti-CD20 monoclonal antibody obinutuzumab with the TLR7 agonist R848 improved survival in lab mice with lymphoma.
The combination also demonstrated efficacy against chronic lymphocytic leukemia (CLL) cells in vitro.
Tim Illidge, PhD, MBBS, of the University of Manchester in the UK, and his colleagues reported these findings in the journal Leukemia.
The research was funded by the Kay Kendall Leukaemia Fund and Cancer Research UK in collaboration with Roche Pharmaceutical Research and Early Development.
The researchers said they initially found that R848 activates immune cells in vivo and enhances obinutuzumab-mediated antitumor effector mechanisms in vitro.
The team therefore went on to test R848 and obinutuzumab in C57Bl/6 mice bearing human CD20+ lymphoma (EL4). The mice received obinutuzumab modified to express the murine glycoengineered IgG2a Fc region (m2a) starting 1 day after tumor inoculation and systemic R848 once weekly for 4 weeks.
The researchers found that monotherapy with either obinutuzumab or R848 significantly improved survival compared to control (P<0.0001), but only 8% to 15% of mice that received monotherapy were long-term survivors (living more than 90 days).
Mice that received obinutuzumab in combination with R848 had significantly better survival than mice that received either monotherapy (P<0.0001). And about 70% of mice receiving the combination remained tumor-free out to 95 days.
Furthermore, long-term survivors that had received the combination treatment were protected from tumor re-challenge.
The researchers also tested the combination in a second model—human CD20 transgenic mice, which express the human CD20 antigen on normal B cells. The team said this model is more akin to the clinical situation.
The mice received treatment 7 days after the inoculation of EL4hCD20 cells. Mice that received obinutuzumab monotherapy had significantly better survival than control mice (P=0.02), but there were no long-term survivors. For mice that received R848 monotherapy, survival was not significantly different from that of controls.
Mice that received R848 in combination with obinutuzumab had significantly better survival than mice that received obinutuzumab alone (P=0.003).
In fact, 6 of the 12 mice that received the combination were long-term survivors. And 5 of these mice rejected tumor re-challenge.
“We were excited when we discovered that combining obinutuzumab with TLR7 activation significantly enhanced survival of animals with lymphoma by effectively eradicating tumors,” Dr Illidge said. “Clearly, more work needs to be done to assess the impact of this combination on humans, but this study is, nevertheless, very promising.”
The researchers said the primary antitumor activity of the combination is dependent on natural killer cells and CD4 helper T cells but not on CD8 killer T cells.
“While the combination therapy was highly effective, CD8 killer T cells did not play a major role in the therapy,” said Eleanor Cheadle, PhD, also of the University of Manchester.
“Given the important role that killer T cells can play in long-term protection from tumor regrowth, we are looking at ways to enhance activation of these cells after obinutuzumab therapy.”
The researchers also found that, in vitro, R848 significantly enhanced natural killer cell-mediated antibody-dependent cellular cytotoxicity against obinutuzumab-opsonized CLL cells and significantly increased non-specific, antibody-independent killing of CLL cells. ![]()
CMA report reveals successes and shortcomings

The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program. ![]()
The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program. ![]()
The European Medicines Agency (EMA) has released a report showing both successes and room for improvement regarding conditional marketing authorizations (CMAs).
CMA is one of the tools available to regulators to support the development of and early access to drugs that address unmet medical needs of patients in the European Union.
Drugs are granted CMA if the public health benefit of their immediate availability is thought to outweigh the risk of an authorization on the basis of less comprehensive data than normally required.
A CMA is valid for 1 year. As part of the authorization, the drug’s developer is obliged to carry out further studies to obtain complete data.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) assesses the data generated by these specific post-authorization obligations at least annually to ensure the balance of benefits and risks of the drug continues to remain positive.
At the end of its assessment, the CHMP issues a recommendation regarding the renewal of the CMA or its conversion into a standard marketing authorization.
Overview
The EMA’s report summarizes the experience with CMAs from the first use of this authorization type in 2006 until June 30, 2016.
During this time, a total of 30 drugs have received a CMA, including several
hematology drugs—Adcetris (brentuximab vedotin),
Arzerra (ofatumumab), Blincyto (blinatumomab), Bosulif (bosutinib), Darzalex (daratumumab), and Pixuvri (pixantrone).
Eleven CMAs have been converted into standard marketing authorizations (including Arzerra’s CMA), 2 have been withdrawn for commercial reasons, and 17 are still conditional authorizations.
None of the drugs that still have CMAs have been authorized for more than 5 years. And none of the CMAs issued since 2006 have had to be revoked or suspended.
Successes
According to the EMA’s analysis, marketing authorization holders comply with the specific obligations imposed by the agency.
More than 90% of completed specific obligations did not result in major changes of scope, and about 70% of specific obligations did not require an extension to the originally specified timelines.
The report shows that it took an average of 4 years to generate the additional data needed and to convert a CMA into a full marketing authorization.
This suggests patients with life-threatening or seriously debilitating conditions had access to promising drugs much earlier than they would have under standard authorization.
Areas for improvement
The EMA’s analysis also revealed room for improvement.
The report showed that, relatively frequently, CMA was first
considered only during the assessment of the drug application, which meant granting a CMA took longer than intended.
Therefore, the EMA recommends that drug developers engage in early dialogue with the EMA
and prospectively plan to apply for a CMA.
The agency said this should support
prompt assessment of such applications and could also facilitate prompt
completion of additional studies and timely availability of
comprehensive data.
The EMA said another area for improvement is engaging other stakeholders involved in bringing drugs to patients—in particular, Health Technology Assessment bodies—to facilitate the generation of all data needed for decision-making through one development program. ![]()
Iron-fortified nutrition bars combat anemia in India

Consuming an iron-fortified nutrition bar daily for a few months can fight anemia without producing side effects, according to research published in the American Journal of Clinical Nutrition.
Anemic
women in India who consumed an iron-fortified nutrition bar each day for 90 days
were much more likely to experience increases in hemoglobin and
hematocrit and to be cured of their anemia than women who did not consume
such bars.
Rajvi Mehta, a medical student at Duke University in Durham, North Carolina, developed the nutrition bars used in this study, known as GudNesS bars.
The bars are made with iron-rich, natural, local (to India), and culturally accepted ingredients. They contain the World Health Organization’s daily recommended dose of iron.
In 2011, Mehta worked with nutritionists and physicians in India to establish a social venture there called Let’s Be Well Red (LBWR) to begin large-scale production of the bars.
The study, conducted from March to August 2014 in Mumbai and Navi Mumbai, India, involved 179 anemic, non-pregnant participants of reproductive age (18-35) at 10 demographically diverse sites.
The sites were randomly placed in either a control group or an intervention group.
Women in the intervention group received 1 iron-fortified nutrition bar (containing 14 mg Fe) daily for 90 days, and women in the control group received nothing. Baseline characteristics were comparable between the groups.
Each group underwent 3 blood tests during the 90-day follow-up period—at 15 days, 45 days, and 90 days—to measure their hemoglobin and hematocrit.
Seventy-six percent of subjects (n=136) completed all follow-up assessments (65 intervention and 71 control subjects).
The primary outcomes were 90-day changes from baseline in hemoglobin concentrations and hematocrit percentages.
The researchers said the mean hemoglobin and hematocrit increases after 90 days were greater for the intervention group than the control group, at 1.4 g/dL and 2.7%, respectively.
And subjects in the intervention group had a much greater decrease in anemia than those in the control group. At 90 days, 29.2% of subjects in the intervention group still had anemia, compared to 98.6% of those in the control group. The odds ratio was 0.007.
The researchers said no side effects were reported.
“We are encouraged by the results of this study, which show a positive connection between consuming an iron-fortified nutrition bar and a reduction in anemia prevalence,” said study author Elizabeth Turner, PhD, of Duke University.
“It appears to be a practical and well-tolerated solution to a significant health challenge in India.”
Let’s Be Well Red is currently operating in 3 locations in India and produces 100,000 bars each year that it distributes throughout the country.
“Anemia is a debilitating condition that can have severe health consequences,” Mehta said. “I am thrilled that my colleagues and I were able to develop a solution that has proven to be effective among a high-risk population. Making an impact in global health has long been a goal of mine.” ![]()
Consuming an iron-fortified nutrition bar daily for a few months can fight anemia without producing side effects, according to research published in the American Journal of Clinical Nutrition.
Anemic
women in India who consumed an iron-fortified nutrition bar each day for 90 days
were much more likely to experience increases in hemoglobin and
hematocrit and to be cured of their anemia than women who did not consume
such bars.
Rajvi Mehta, a medical student at Duke University in Durham, North Carolina, developed the nutrition bars used in this study, known as GudNesS bars.
The bars are made with iron-rich, natural, local (to India), and culturally accepted ingredients. They contain the World Health Organization’s daily recommended dose of iron.
In 2011, Mehta worked with nutritionists and physicians in India to establish a social venture there called Let’s Be Well Red (LBWR) to begin large-scale production of the bars.
The study, conducted from March to August 2014 in Mumbai and Navi Mumbai, India, involved 179 anemic, non-pregnant participants of reproductive age (18-35) at 10 demographically diverse sites.
The sites were randomly placed in either a control group or an intervention group.
Women in the intervention group received 1 iron-fortified nutrition bar (containing 14 mg Fe) daily for 90 days, and women in the control group received nothing. Baseline characteristics were comparable between the groups.
Each group underwent 3 blood tests during the 90-day follow-up period—at 15 days, 45 days, and 90 days—to measure their hemoglobin and hematocrit.
Seventy-six percent of subjects (n=136) completed all follow-up assessments (65 intervention and 71 control subjects).
The primary outcomes were 90-day changes from baseline in hemoglobin concentrations and hematocrit percentages.
The researchers said the mean hemoglobin and hematocrit increases after 90 days were greater for the intervention group than the control group, at 1.4 g/dL and 2.7%, respectively.
And subjects in the intervention group had a much greater decrease in anemia than those in the control group. At 90 days, 29.2% of subjects in the intervention group still had anemia, compared to 98.6% of those in the control group. The odds ratio was 0.007.
The researchers said no side effects were reported.
“We are encouraged by the results of this study, which show a positive connection between consuming an iron-fortified nutrition bar and a reduction in anemia prevalence,” said study author Elizabeth Turner, PhD, of Duke University.
“It appears to be a practical and well-tolerated solution to a significant health challenge in India.”
Let’s Be Well Red is currently operating in 3 locations in India and produces 100,000 bars each year that it distributes throughout the country.
“Anemia is a debilitating condition that can have severe health consequences,” Mehta said. “I am thrilled that my colleagues and I were able to develop a solution that has proven to be effective among a high-risk population. Making an impact in global health has long been a goal of mine.” ![]()
Consuming an iron-fortified nutrition bar daily for a few months can fight anemia without producing side effects, according to research published in the American Journal of Clinical Nutrition.
Anemic
women in India who consumed an iron-fortified nutrition bar each day for 90 days
were much more likely to experience increases in hemoglobin and
hematocrit and to be cured of their anemia than women who did not consume
such bars.
Rajvi Mehta, a medical student at Duke University in Durham, North Carolina, developed the nutrition bars used in this study, known as GudNesS bars.
The bars are made with iron-rich, natural, local (to India), and culturally accepted ingredients. They contain the World Health Organization’s daily recommended dose of iron.
In 2011, Mehta worked with nutritionists and physicians in India to establish a social venture there called Let’s Be Well Red (LBWR) to begin large-scale production of the bars.
The study, conducted from March to August 2014 in Mumbai and Navi Mumbai, India, involved 179 anemic, non-pregnant participants of reproductive age (18-35) at 10 demographically diverse sites.
The sites were randomly placed in either a control group or an intervention group.
Women in the intervention group received 1 iron-fortified nutrition bar (containing 14 mg Fe) daily for 90 days, and women in the control group received nothing. Baseline characteristics were comparable between the groups.
Each group underwent 3 blood tests during the 90-day follow-up period—at 15 days, 45 days, and 90 days—to measure their hemoglobin and hematocrit.
Seventy-six percent of subjects (n=136) completed all follow-up assessments (65 intervention and 71 control subjects).
The primary outcomes were 90-day changes from baseline in hemoglobin concentrations and hematocrit percentages.
The researchers said the mean hemoglobin and hematocrit increases after 90 days were greater for the intervention group than the control group, at 1.4 g/dL and 2.7%, respectively.
And subjects in the intervention group had a much greater decrease in anemia than those in the control group. At 90 days, 29.2% of subjects in the intervention group still had anemia, compared to 98.6% of those in the control group. The odds ratio was 0.007.
The researchers said no side effects were reported.
“We are encouraged by the results of this study, which show a positive connection between consuming an iron-fortified nutrition bar and a reduction in anemia prevalence,” said study author Elizabeth Turner, PhD, of Duke University.
“It appears to be a practical and well-tolerated solution to a significant health challenge in India.”
Let’s Be Well Red is currently operating in 3 locations in India and produces 100,000 bars each year that it distributes throughout the country.
“Anemia is a debilitating condition that can have severe health consequences,” Mehta said. “I am thrilled that my colleagues and I were able to develop a solution that has proven to be effective among a high-risk population. Making an impact in global health has long been a goal of mine.” ![]()
New recommendations for pediatric AMKL
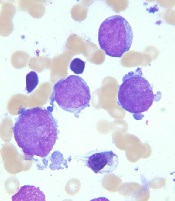
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
Image courtesy of St. Jude
Children’s Research Hospital
and Tina Motroni
Research has revealed genetic alterations that may prove useful for predicting treatment outcomes in pediatric patients with acute megakaryoblastic leukemia (AMKL) who do not have Down syndrome.
The study suggests that 3
genetic alterations can be used to identify high-risk patients who may benefit
from allogeneic stem cell transplant, and 1 alteration may identify
low-risk patients who require less chemotherapy than their peers.
Researchers said these findings, published in Nature Genetics, support revised diagnostic screening and treatment recommendations for pediatric AMKL.
“Because long-term survival for pediatric AMKL patients without Down syndrome is poor, just 14% to 34%, the standard recommendation by many pediatric oncologists has been to treat all patients with allogeneic stem cell transplantation during their first remission,” said study author Tanja Gruber, MD, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“In this study, we identified several genetic alterations that are important predictors of treatment success. All newly identified pediatric AMKL patients without Down syndrome should be screened for these prognostic indicators at diagnosis. The results will help identify which patients need allogeneic stem cell transplants during their first remission and which do not.”
This study involved next-generation sequencing of the whole exome or RNA of 99 AMKL patients without Down syndrome (75 pediatric and 24 adult patients). Along with the sequencing data, researchers analyzed patients’ gene expression and long-term survival.
Results
The results showed that non-Down syndrome pediatric AMKL can be divided into 7 subgroups based on the underlying genetic alteration, pattern of gene expression, and treatment outcome.
The subgroups include the newly identified HOX subgroup. About 15% of the pediatric patients in this study were in the HOX subgroup, which is characterized by several different HOX fusion genes.
The researchers also identified cooperating mutations that help fuel AMKL in different subgroups. The cooperating mutations include changes in the RB1 gene and recurring mutations in the RAS and JAK pathways.
In addition, the study showed that 3 genetic alterations—CBFA2T3-GLIS2, KMT2A rearrangements, and NUP98-KDM5A—are associated with reduced survival in pediatric AMKL subtypes.
The researchers said patients with these alterations may benefit from allogeneic stem cell transplants, so non-Down syndrome pediatric AMKL patients should be screened for these alterations at diagnosis.
The team also recommended testing AMKL patients for mutations in the GATA1 gene. GATA1 mutations are a hallmark of AMKL in children with Down syndrome, who almost always survive the leukemia. In this study, AMKL patients with GATA1 mutations and no fusion gene had the same favorable outcomes.
“The results raise the possibility that pediatric AMKL patients without Down syndrome who have mutations in GATA1 may benefit from the same reduced chemotherapy used to treat the leukemia in patients with Down syndrome,” Dr Gruber said.
These revised diagnostic screening and treatment recommendations are being implemented at St. Jude.
This study also showed that adults with AMKL lacked recurrent fusion genes. The most common mutations found in the adult patients were in TP53 (21%), cohesin genes (17%), splicing factor genes (17%), ASXL genes (17%), and DNMT3A (13%). About 4% had GATA1 mutations. ![]()
Overcoming glucocorticoid resistance in lymphoma

Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas. ![]()
Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas. ![]()
Image by Ed Uthman
Targeting RUNX1 could combat glucocorticoid resistance in patients with lymphoma, according to research published in the Journal of Cellular Biochemistry.
Researchers found an over activity of RUNX1 in lymphoma cells interfered with sphingolipids and caused cells to become resistant to dexamethasone.
Dexamethasone works, in part, through the control of sphingolipid enzymes, which play a role in instructing cells to live or die.
Specifically, the researchers said they found that ectopic expression of RUNX1 in lymphoma cells consistently perturbs the sphingolipid rheostat and confers increased resistance to glucocorticoid-mediated apoptosis.
The team also described the mechanism of cross-talk between glucocorticoid and sphingolipid metabolism through the enzyme Sgpp1.
The researchers said dexamethasone induces expression of Sgpp1 in T-lymphoma cells and drives cell death, which is reduced by partial knockdown of Sgpp1 with short hairpin RNA or direct transcriptional repression of Sgpp1 by ectopic RUNX1.
These findings suggest that drugs targeting RUNX1 may be able to reverse glucocorticoid resistance in lymphoma patients.
“The possibility of making existing therapies more active and specific by combining [them] with drugs that inhibit RUNX is a new and exciting prospect,” said study author James Neil, of The University of Glasgow in Scotland.
“Our collaborators in the US have recently developed drugs that inhibit RUNX, and we plan to test these with existing therapies in blood cancers where MYC and RUNX are both implicated, including multiple myeloma and Burkitt lymphoma.”
An earlier study by Dr Neil and his colleagues suggested that RUNX1 was a potential therapeutic target in MYC-driven lymphomas.
Reducing the risk of device-related thrombosis

“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices.
“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices.
“superomniphobic” surface.
Photo from the Kota lab
at Colorado State University
Researchers say they have engineered “superhemophobic” titanium surfaces that could be used to create implantable medical devices that don’t pose a risk of thrombosis.
The team described this work in Advanced Healthcare Materials.
The undesirable interaction of blood with foreign materials is an ongoing problem in medical research, said study author Ketul Popat, PhD, of Colorado State University in Fort Collins, Colorado.
He and his colleagues noted that, when implanted medical devices come in contact with blood, platelet adhesion and activation occur, which may lead to thrombosis and device failure.
“If we can design materials where blood barely contacts the surface, there is virtually no chance of clotting, which is a coordinated set of events,” Dr Popat said. “Here, we’re targeting the prevention of the first set of events.”
Dr Popat and his colleagues started with sheets of titanium, which are commonly used in medical devices. The team then grew chemically altered surfaces that act as barriers between the titanium and blood.
They analyzed variations of titanium surfaces, including different textures and chemistries, and compared the extent of platelet adhesion and activation.
These experiments revealed that fluorinated nanotubes offered the best protection against clotting.
Having implantable medical devices that repel blood might seem counterintuitive, the researchers noted, as biomedical scientists often use materials with an affinity to blood to make them biologically compatible.
“What we are doing is the exact opposite,” said study author Arun Kota, PhD, of Colorado State University.
“We are taking a material that blood hates to come in contact with, in order to make it compatible with blood.”
In essence, the titanium surface is so repellent that blood is “tricked” into “believing” there’s virtually no foreign material there at all.
Growing a surface and testing it in the lab is only the beginning, the researchers said. They want to continue examining other clotting factors, and eventually, to test real medical devices.
Vaccine candidate can protect humans from malaria
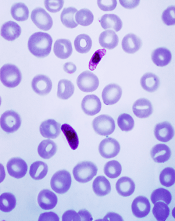
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.”
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.”
Plasmodium falciparum
Photo by Mae Melvin, CDC
In a phase 2 trial, a malaria vaccine candidate was able to prevent volunteers from contracting Plasmodium falciparum malaria.
The vaccine, known as PfSPZ vaccine, is composed of live but weakened P falciparum sporozoites.
Three to 5 doses of PfSPZ vaccine protected subjects against malaria parasites similar to those in the vaccine as well as parasites different from those in the vaccine.
A majority of subjects were protected at 3 weeks after vaccination. For some subjects, this protection was sustained at 24 weeks.
PfSPZ vaccine was considered well-tolerated, as all adverse events (AEs) in this trial were grade 1 or 2.
These results were published in JCI Insight.
The study was funded by the US Department of Defense through the Joint Warfighter Program, the Military Infectious Disease Research Program, and US Navy Advanced Medical Development, with additional support from Sanaria, Inc., the company developing PfSPZ vaccine.
“Our military continues to be at risk from malaria as it deploys worldwide,” said Kenneth A. Bertram, MD, of the US Army Medical Research and Materiel Command in Ft Detrick, Maryland.
“We are excited about the results of this clinical trial and are now investing in the ongoing clinical trial to finalize the vaccination regimen for PfSPZ vaccine.”
The current trial included 67 volunteers with a median age of 29.6 (range, 19 to 45).
They were randomized to 3 treatment groups. Forty-five subjects were set to receive the PfSPZ vaccine, and 22 subjects served as controls.
The volunteers underwent controlled human malaria infection (CHMI) 3 weeks after vaccinated subjects received their final vaccine dose and then again at 24 weeks.
Efficacy at 3 weeks
Group 1
In this group, 13 subjects received 5 doses of the vaccine at 2.7 × 105. They and 6 control subjects underwent CHMI with a homologous strain of P falciparum, Pf3D7.
Twelve of the 13 fully immunized subjects, or 92.3%, did not develop parasitemia. However, all 6 control subjects did, with a median prepatent period of 11.6 days. The prepatent period in the immunized subject who developed parasitemia was 13.9 days.
Group 2
In this group, 5 subjects received 5 doses of the vaccine at 2.7 × 105. They and 4 control subjects underwent CHMI with a heterologous strain of P falciparum, Pf7G8.
Four of the 5 fully immunized subjects, or 80%, did not develop parasitemia. However, all 4 control subjects did, with a median prepatent period of 11.9 days. The prepatent period in the fully immunized subject who developed parasitemia was 11.9 days.
Group 3
In this group, 15 subjects received 3 doses of the vaccine at 4.5 × 105 and underwent homologous Pf3D7 CHMI. The control subjects for this group were the same as those in group 1.
Thirteen of the 15 immunized subjects, or 86.7%, did not develop parasitemia. The prepatent periods in the 2 immunized subjects who did develop parasitemia were 13.9 days and 16.9 days.
Efficacy at 24 weeks
Study subjects underwent a second CHMI at 24 weeks after immunized participants received their final dose of vaccine.
Group 1
Seven of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (70%). However, all 6 control subjects did, with a median prepatent period of 11.6 days. The median prepatent period in the 3 fully immunized subjects who developed parasitemia was 15.4 days.
Group 2
One of the 10 fully immunized subjects who underwent a second CHMI did not develop parasitemia (10%). However, all control subjects did, with a median prepatent period of 10.9 days. The median prepatent period in the 9 fully immunized subjects who developed parasitemia was 11.9 days.
Group 3
Eight of the 14 fully immunized subjects who underwent a second CHMI did not develop parasitemia (57.1%). The median prepatent period in the 6 fully immunized subjects who did develop parasitemia was 14.0 days.
Safety
There were 66 solicited AEs reported within 7 days of immunization that were considered possibly, probably, or definitely related to vaccination. Ninety-two percent of these AEs were grade 1, and 8% were grade 2. All unsolicited AEs reported within 7 days of immunization were grade 1.
The incidence of AEs was not higher in group 3 than in groups 1 or 2, and the incidence of AEs did not increase as subjects received additional doses of the vaccine.
The most common AEs (with an incidence of 10% or higher in at least 1 group) were injection site pain, headache, fatigue, malaise, myalgia, injection site hemorrhage, and cough.
“The results of this clinical trial, along with recent results from other trials of this vaccine in the US and Africa, were critical to our decision to move forward with a trial involving 400 infants in Kenya,” said Tina Oneko, MD, of the Kenya Medical Research Institute, who is the principal investigator of the Kenya trial but was not involved in the current trial.
“This represents significant progress toward the development of a regimen for PfSPZ vaccine that we anticipate will provide a high level [of] efficacy for malaria prevention in all age groups in Africa.”
Meta-analysis supports statins as VTE prophylaxis

Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.”
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.”
Photo courtesy of the CDC
Results of a meta-analysis support the idea that statins can reduce the risk of venous thromboembolism (VTE).
Researchers analyzed data from 36 studies involving more than 3.2 million people and found an “extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE.”
The team believes these findings could potentially lead to new guidelines and an expansion of the use of statins.
“These findings underscore a potential beneficial role of statin therapy on VTE in addition to its established role in cardiovascular disease prevention,” said Kamlesh Khunti, MD, PhD, of the University of Leicester in the UK.
Dr Khunti and his colleagues reported these findings in The Lancet Haematology.
The researchers analyzed data from 36 studies. This included 13 cohort studies with a total of 3,148,259 subjects and 23 randomized, controlled trials (RCTs) of 118,464 subjects in which statins were compared to placebo or no treatment.
In the observational studies, statin use was associated with a significant reduction in the risk of VTE, when compared to no statin use. The pooled relative risk (RR) was 0.75 (P<0.0001).
Statin use was associated with a significant reduction in the risk of deep vein thrombosis (DVT), with an RR of 0.77 (P<0.0001). (There were only 2 observational studies looking specifically at DVT outcomes.)
However, there was no significant reduction in the risk of pulmonary embolism (PE) with statin use. The RR was 1.02 (P=0.90). (There was only 1 observational study reporting specifically on PE outcomes.)
In the RCTs, statin use was associated with a significant reduction in the risk of VTE, with an RR of 0.85 (P=0.038).
Statin use was also associated with a significant reduction in DVT risk, with an RR of 0.45, but not PE risk, with an RR of 0.77 (no P values available). (There was only 1 RCT that reported specifically on DVT and PE outcomes.)
The RCTs also showed significant differences in VTE risk according to type of statin used. Patients receiving rosuvastatin had the lowest risk of VTE when compared to those receiving other statins, with an RR of 0.57 (P=0.015).
“Currently, statins are only approved for lipid lowering in the primary and secondary prevention of cardiovascular disease,” said study author Setor Kunutsor, MBChB, PhD, of the University of Bristol in the UK.
“But they have shown great promise beyond their established lipid-lowering effects, and these include potential beneficial impact on multiple disease conditions. These results provide an extensive body of evidence on the clinical benefit of statin[s] in the occurrence of VTE and may support a true protective effect.”