User login
Team develops multicolor electron microscopy
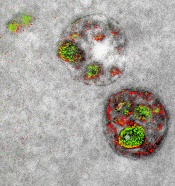
of endosomal uptake
of peptide proteins
Image by Adams et al/
Cell Chemical Biology 2016
Researchers say they have found a way to image specific cellular components using multicolor electron microscopy.
With their method, which the team worked on for nearly 15 years, up to 3 colors at a time (green, red, or yellow) can be used in an image.
A detector on the microscope captures electrons lost from metal ions painted over the specimen and records the metal’s energy loss signature as a color.
A technician must add the ionized metals one at a time and then lay the full color map over the still microscopy image.
The researchers described this method in detail in Cell Chemical Biology.
“This method has many potential applications in biology,” said study author Stephen Adams, PhD, of the University of California, San Diego in La Jolla.
“In the paper, we demonstrate how it can distinguish cellular compartments or track proteins and tag cells.”
For the multicolor effect to work, the researchers needed metal complexes that are stable enough to withstand application (meaning they don’t quickly deteriorate and blur the image) and have a distinct electron energy loss signature.
The researchers used ionized lanthanum (La), cerium (Ce), and praseodymium (Pr)—all metals in the lanthanide family. Each metal complex was laid down sequentially as a precipitate onto the specimen as it sat in the microscope.
“One challenge that kept us from publishing this much earlier, because we had the chemistry and we had an instrument that worked about 4 years ago, was we needed a way to deposit the metal compounds sequentially,” said study author Mark Ellisman, PhD, of the University of California, San Diego.
“We spent an awful lot of time trying to figure out how to deposit one of the lanthanides and then clear it so that it didn’t react when we deposited a second signal on the first site.”
Once the application process had been established, the researchers illustrated the power of multicolor electron microscopy by visualizing 2 brain cells sharing a single synapse. They also showed peptides entering through a cell membrane.
The researchers said there is more chemistry to be done to perfect the metal ion application process as well as produce images with more colors. There may also be ways to increase the amount of metal ions that can be deposited, which could help with resolution.
Many in the biochemical community should be able to begin using this technique right away, as it takes advantage of tools that are already found in laboratories.
This study is one of the last that Roger Tsien, PhD, who won a 2008 Nobel Prize in Chemistry for the discovery and application of green fluorescent protein to biochemical imaging, saw accepted by a journal before his death last August.
He did the first experiments to develop the chemical compounds needed for the multicolor imaging method nearly 15 years ago.
“One theme that has gone through all of Roger’s work is the desire to peer more closely into the workings of the cell,” Dr Adams said.
“With all of the fluorescence techniques that he’s introduced, he was able to do that in live cells and make action movies of them in vivid colors. But he always wanted to look closer, and now he’s left the beginnings for a method where we can add colors to electron microscopy.”
“This is clearly an example of Roger’s brilliance at chemistry and how he saw that, if we could do this, we would be able to enjoy the advantages of electron microscopy,” Dr Ellisman added.
“The biggest advantage of electron microscopy that we saw is that you have weak contrasts by the nature of the way that staining works, so color-specific labels give context to all of the rich information in the scene of which molecules are operating.” ![]()
of endosomal uptake
of peptide proteins
Image by Adams et al/
Cell Chemical Biology 2016
Researchers say they have found a way to image specific cellular components using multicolor electron microscopy.
With their method, which the team worked on for nearly 15 years, up to 3 colors at a time (green, red, or yellow) can be used in an image.
A detector on the microscope captures electrons lost from metal ions painted over the specimen and records the metal’s energy loss signature as a color.
A technician must add the ionized metals one at a time and then lay the full color map over the still microscopy image.
The researchers described this method in detail in Cell Chemical Biology.
“This method has many potential applications in biology,” said study author Stephen Adams, PhD, of the University of California, San Diego in La Jolla.
“In the paper, we demonstrate how it can distinguish cellular compartments or track proteins and tag cells.”
For the multicolor effect to work, the researchers needed metal complexes that are stable enough to withstand application (meaning they don’t quickly deteriorate and blur the image) and have a distinct electron energy loss signature.
The researchers used ionized lanthanum (La), cerium (Ce), and praseodymium (Pr)—all metals in the lanthanide family. Each metal complex was laid down sequentially as a precipitate onto the specimen as it sat in the microscope.
“One challenge that kept us from publishing this much earlier, because we had the chemistry and we had an instrument that worked about 4 years ago, was we needed a way to deposit the metal compounds sequentially,” said study author Mark Ellisman, PhD, of the University of California, San Diego.
“We spent an awful lot of time trying to figure out how to deposit one of the lanthanides and then clear it so that it didn’t react when we deposited a second signal on the first site.”
Once the application process had been established, the researchers illustrated the power of multicolor electron microscopy by visualizing 2 brain cells sharing a single synapse. They also showed peptides entering through a cell membrane.
The researchers said there is more chemistry to be done to perfect the metal ion application process as well as produce images with more colors. There may also be ways to increase the amount of metal ions that can be deposited, which could help with resolution.
Many in the biochemical community should be able to begin using this technique right away, as it takes advantage of tools that are already found in laboratories.
This study is one of the last that Roger Tsien, PhD, who won a 2008 Nobel Prize in Chemistry for the discovery and application of green fluorescent protein to biochemical imaging, saw accepted by a journal before his death last August.
He did the first experiments to develop the chemical compounds needed for the multicolor imaging method nearly 15 years ago.
“One theme that has gone through all of Roger’s work is the desire to peer more closely into the workings of the cell,” Dr Adams said.
“With all of the fluorescence techniques that he’s introduced, he was able to do that in live cells and make action movies of them in vivid colors. But he always wanted to look closer, and now he’s left the beginnings for a method where we can add colors to electron microscopy.”
“This is clearly an example of Roger’s brilliance at chemistry and how he saw that, if we could do this, we would be able to enjoy the advantages of electron microscopy,” Dr Ellisman added.
“The biggest advantage of electron microscopy that we saw is that you have weak contrasts by the nature of the way that staining works, so color-specific labels give context to all of the rich information in the scene of which molecules are operating.” ![]()
of endosomal uptake
of peptide proteins
Image by Adams et al/
Cell Chemical Biology 2016
Researchers say they have found a way to image specific cellular components using multicolor electron microscopy.
With their method, which the team worked on for nearly 15 years, up to 3 colors at a time (green, red, or yellow) can be used in an image.
A detector on the microscope captures electrons lost from metal ions painted over the specimen and records the metal’s energy loss signature as a color.
A technician must add the ionized metals one at a time and then lay the full color map over the still microscopy image.
The researchers described this method in detail in Cell Chemical Biology.
“This method has many potential applications in biology,” said study author Stephen Adams, PhD, of the University of California, San Diego in La Jolla.
“In the paper, we demonstrate how it can distinguish cellular compartments or track proteins and tag cells.”
For the multicolor effect to work, the researchers needed metal complexes that are stable enough to withstand application (meaning they don’t quickly deteriorate and blur the image) and have a distinct electron energy loss signature.
The researchers used ionized lanthanum (La), cerium (Ce), and praseodymium (Pr)—all metals in the lanthanide family. Each metal complex was laid down sequentially as a precipitate onto the specimen as it sat in the microscope.
“One challenge that kept us from publishing this much earlier, because we had the chemistry and we had an instrument that worked about 4 years ago, was we needed a way to deposit the metal compounds sequentially,” said study author Mark Ellisman, PhD, of the University of California, San Diego.
“We spent an awful lot of time trying to figure out how to deposit one of the lanthanides and then clear it so that it didn’t react when we deposited a second signal on the first site.”
Once the application process had been established, the researchers illustrated the power of multicolor electron microscopy by visualizing 2 brain cells sharing a single synapse. They also showed peptides entering through a cell membrane.
The researchers said there is more chemistry to be done to perfect the metal ion application process as well as produce images with more colors. There may also be ways to increase the amount of metal ions that can be deposited, which could help with resolution.
Many in the biochemical community should be able to begin using this technique right away, as it takes advantage of tools that are already found in laboratories.
This study is one of the last that Roger Tsien, PhD, who won a 2008 Nobel Prize in Chemistry for the discovery and application of green fluorescent protein to biochemical imaging, saw accepted by a journal before his death last August.
He did the first experiments to develop the chemical compounds needed for the multicolor imaging method nearly 15 years ago.
“One theme that has gone through all of Roger’s work is the desire to peer more closely into the workings of the cell,” Dr Adams said.
“With all of the fluorescence techniques that he’s introduced, he was able to do that in live cells and make action movies of them in vivid colors. But he always wanted to look closer, and now he’s left the beginnings for a method where we can add colors to electron microscopy.”
“This is clearly an example of Roger’s brilliance at chemistry and how he saw that, if we could do this, we would be able to enjoy the advantages of electron microscopy,” Dr Ellisman added.
“The biggest advantage of electron microscopy that we saw is that you have weak contrasts by the nature of the way that staining works, so color-specific labels give context to all of the rich information in the scene of which molecules are operating.” ![]()
Studies reveal markers of malaria resistance
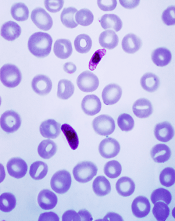
Plasmodium falciparum
Image from CDC/Mae Melvin
Two studies have revealed genetic markers associated with resistance to piperaquine, one of the most commonly used malaria drugs in Southeast Asia.
Investigators found that Plasmodium falciparum parasites were less sensitive to piperaquine if they had the exo-E415G variant or multiple copies of the plasmepsin 2 and plasmepsin 3 genes.
Furthermore, the presence of these markers could identify which malaria patients would fail piperaquine treatment.
However, investigators said more research is needed to establish whether plasmepsin gene amplification and the exo-E415G variant actually cause piperaquine resistance and to explore whether this association extends to other parts of the world.
The 2 groups of investigators reported the findings of their studies in The Lancet Infectious Diseases.
About resistance
The combination of artemisinin and piperaquine (dihydroartemisinin-piperaquine) is a frontline treatment for malaria in Southeast Asia.
However, in recent years, the emergence of resistance to these drugs in Cambodia means that up to 60% of patients fail treatment in several provinces. There are fears that resistance could spread to other regions where malaria is endemic.
Genome-wide association studies have been conducted to identify genetic variations among P falciparum parasites that can be linked to variations in drug resistance.
Recent studies have revealed genetic markers linked to resistance to artemisinin (K13) and mefloquine (Pfmdr1), but, until now, markers for resistance to other combination drugs had not been identified.
Plasmepsin genes
In the first study, Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia, and his colleagues found that amplification of plasmepsin genes in the P falciparum genome was linked to piperaquine resistance.
The investigators first analyzed artemisinin-resistant, Cambodian P falciparum parasite lines, comparing the exomes of lines that were piperaquine-susceptible and those that were piperaquine-resistant.
The team said this revealed an increased copy number of the plasmepsin 2-plasmepsin 3 gene cluster as a putative genetic signature associated with piperaquine resistance.
The investigators then confirmed their findings in a set of 725 P falciparum parasites collected from patients across Cambodia since 2009.
Individuals infected with parasites with multiple copies of the plasmepsin 2 gene were, on average, 20.4 times more likely to experience dihydroartemisinin-piperaquine treatment failure.
Furthermore, the geographical distribution and rise in the proportion of parasites with multiple copies of the plasmepsin 2 and 3 genes in Cambodia corresponded with areas that had reported increases in dihydroartemisinin-piperaquine treatment failures in recent years.
“We now know that amplification of the plasmepsin 2 and plasmepsin 3 genes is an important factor in determining how well piperaquine kills malaria parasites,” Dr Ménard said. “A genetic toolkit combining this new marker with markers of artemisinin and mefloquine resistance could be used to track the spread of resistance and provide timely information for containment policies.”
“Piperaquine resistance, although currently confined to Cambodia, is a major concern, because patients suffering malaria are almost untreatable. At present, alternative treatments are scarce, and the reduced cure rates lead to prolonged parasite carriage, increasing the potential of transmitting the resistant parasites and endangering efforts to control and eliminate malaria.”
Exo-E415G variant
The second study was conducted by Rick Fairhurst, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues.
The investigators analyzed 486 P falciparum parasites collected between July 9, 2010, and December 31, 2013, from 3 different provinces in Cambodia where artemisinin resistance and dihydroartemisinin-piperaquine treatment failure are common (Pursat), emerging (Preah Vihear), or uncommon (Ratanakiri).
The investigators exposed the parasites to therapeutic levels of piperaquine and measured how well each strain grew. At the same time, they sequenced the entire genome of each parasite.
Parasites were more likely to survive exposure to piperaquine if they had 2 or more copies of the plasmepsin 2 and plasmepsin 3 genes, as well as the exo-E415G variant on chromosome 13.
Similarly, in a further analysis of 133 patient samples, the investigators found that individuals infected with parasites carrying multiple copies of the plasmepsin genes were much more likely to fail treatment with dihydroartemisinin-piperaquine, as were individuals who had parasites with the exo-E415G variant.
The team also noted that the prevalence of exo-E415G and plasmepsin 2-3 markers has risen substantially in recent years in Pursat and Preah Vihear, where artemisinin resistance is common and dihydroartemisinin-piperaquine has been the frontline treatment for at least 6 years.
“By surveying the piperaquine resistance marker across Southeast Asia in real-time, we can identify those areas where dihydroartemisinin-piperaquine will not be effective, and this could enable national malaria control programs to immediately recommend alternative therapies, such as artesunate-mefloquine,” Dr Fairhurst said.
“This approach will be crucial to eliminating multidrug-resistant parasites in Southeast Asia before they spread to sub-Saharan Africa, where most of the world’s malaria transmission, illness, and death occur.” ![]()
Plasmodium falciparum
Image from CDC/Mae Melvin
Two studies have revealed genetic markers associated with resistance to piperaquine, one of the most commonly used malaria drugs in Southeast Asia.
Investigators found that Plasmodium falciparum parasites were less sensitive to piperaquine if they had the exo-E415G variant or multiple copies of the plasmepsin 2 and plasmepsin 3 genes.
Furthermore, the presence of these markers could identify which malaria patients would fail piperaquine treatment.
However, investigators said more research is needed to establish whether plasmepsin gene amplification and the exo-E415G variant actually cause piperaquine resistance and to explore whether this association extends to other parts of the world.
The 2 groups of investigators reported the findings of their studies in The Lancet Infectious Diseases.
About resistance
The combination of artemisinin and piperaquine (dihydroartemisinin-piperaquine) is a frontline treatment for malaria in Southeast Asia.
However, in recent years, the emergence of resistance to these drugs in Cambodia means that up to 60% of patients fail treatment in several provinces. There are fears that resistance could spread to other regions where malaria is endemic.
Genome-wide association studies have been conducted to identify genetic variations among P falciparum parasites that can be linked to variations in drug resistance.
Recent studies have revealed genetic markers linked to resistance to artemisinin (K13) and mefloquine (Pfmdr1), but, until now, markers for resistance to other combination drugs had not been identified.
Plasmepsin genes
In the first study, Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia, and his colleagues found that amplification of plasmepsin genes in the P falciparum genome was linked to piperaquine resistance.
The investigators first analyzed artemisinin-resistant, Cambodian P falciparum parasite lines, comparing the exomes of lines that were piperaquine-susceptible and those that were piperaquine-resistant.
The team said this revealed an increased copy number of the plasmepsin 2-plasmepsin 3 gene cluster as a putative genetic signature associated with piperaquine resistance.
The investigators then confirmed their findings in a set of 725 P falciparum parasites collected from patients across Cambodia since 2009.
Individuals infected with parasites with multiple copies of the plasmepsin 2 gene were, on average, 20.4 times more likely to experience dihydroartemisinin-piperaquine treatment failure.
Furthermore, the geographical distribution and rise in the proportion of parasites with multiple copies of the plasmepsin 2 and 3 genes in Cambodia corresponded with areas that had reported increases in dihydroartemisinin-piperaquine treatment failures in recent years.
“We now know that amplification of the plasmepsin 2 and plasmepsin 3 genes is an important factor in determining how well piperaquine kills malaria parasites,” Dr Ménard said. “A genetic toolkit combining this new marker with markers of artemisinin and mefloquine resistance could be used to track the spread of resistance and provide timely information for containment policies.”
“Piperaquine resistance, although currently confined to Cambodia, is a major concern, because patients suffering malaria are almost untreatable. At present, alternative treatments are scarce, and the reduced cure rates lead to prolonged parasite carriage, increasing the potential of transmitting the resistant parasites and endangering efforts to control and eliminate malaria.”
Exo-E415G variant
The second study was conducted by Rick Fairhurst, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues.
The investigators analyzed 486 P falciparum parasites collected between July 9, 2010, and December 31, 2013, from 3 different provinces in Cambodia where artemisinin resistance and dihydroartemisinin-piperaquine treatment failure are common (Pursat), emerging (Preah Vihear), or uncommon (Ratanakiri).
The investigators exposed the parasites to therapeutic levels of piperaquine and measured how well each strain grew. At the same time, they sequenced the entire genome of each parasite.
Parasites were more likely to survive exposure to piperaquine if they had 2 or more copies of the plasmepsin 2 and plasmepsin 3 genes, as well as the exo-E415G variant on chromosome 13.
Similarly, in a further analysis of 133 patient samples, the investigators found that individuals infected with parasites carrying multiple copies of the plasmepsin genes were much more likely to fail treatment with dihydroartemisinin-piperaquine, as were individuals who had parasites with the exo-E415G variant.
The team also noted that the prevalence of exo-E415G and plasmepsin 2-3 markers has risen substantially in recent years in Pursat and Preah Vihear, where artemisinin resistance is common and dihydroartemisinin-piperaquine has been the frontline treatment for at least 6 years.
“By surveying the piperaquine resistance marker across Southeast Asia in real-time, we can identify those areas where dihydroartemisinin-piperaquine will not be effective, and this could enable national malaria control programs to immediately recommend alternative therapies, such as artesunate-mefloquine,” Dr Fairhurst said.
“This approach will be crucial to eliminating multidrug-resistant parasites in Southeast Asia before they spread to sub-Saharan Africa, where most of the world’s malaria transmission, illness, and death occur.” ![]()
Plasmodium falciparum
Image from CDC/Mae Melvin
Two studies have revealed genetic markers associated with resistance to piperaquine, one of the most commonly used malaria drugs in Southeast Asia.
Investigators found that Plasmodium falciparum parasites were less sensitive to piperaquine if they had the exo-E415G variant or multiple copies of the plasmepsin 2 and plasmepsin 3 genes.
Furthermore, the presence of these markers could identify which malaria patients would fail piperaquine treatment.
However, investigators said more research is needed to establish whether plasmepsin gene amplification and the exo-E415G variant actually cause piperaquine resistance and to explore whether this association extends to other parts of the world.
The 2 groups of investigators reported the findings of their studies in The Lancet Infectious Diseases.
About resistance
The combination of artemisinin and piperaquine (dihydroartemisinin-piperaquine) is a frontline treatment for malaria in Southeast Asia.
However, in recent years, the emergence of resistance to these drugs in Cambodia means that up to 60% of patients fail treatment in several provinces. There are fears that resistance could spread to other regions where malaria is endemic.
Genome-wide association studies have been conducted to identify genetic variations among P falciparum parasites that can be linked to variations in drug resistance.
Recent studies have revealed genetic markers linked to resistance to artemisinin (K13) and mefloquine (Pfmdr1), but, until now, markers for resistance to other combination drugs had not been identified.
Plasmepsin genes
In the first study, Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia, and his colleagues found that amplification of plasmepsin genes in the P falciparum genome was linked to piperaquine resistance.
The investigators first analyzed artemisinin-resistant, Cambodian P falciparum parasite lines, comparing the exomes of lines that were piperaquine-susceptible and those that were piperaquine-resistant.
The team said this revealed an increased copy number of the plasmepsin 2-plasmepsin 3 gene cluster as a putative genetic signature associated with piperaquine resistance.
The investigators then confirmed their findings in a set of 725 P falciparum parasites collected from patients across Cambodia since 2009.
Individuals infected with parasites with multiple copies of the plasmepsin 2 gene were, on average, 20.4 times more likely to experience dihydroartemisinin-piperaquine treatment failure.
Furthermore, the geographical distribution and rise in the proportion of parasites with multiple copies of the plasmepsin 2 and 3 genes in Cambodia corresponded with areas that had reported increases in dihydroartemisinin-piperaquine treatment failures in recent years.
“We now know that amplification of the plasmepsin 2 and plasmepsin 3 genes is an important factor in determining how well piperaquine kills malaria parasites,” Dr Ménard said. “A genetic toolkit combining this new marker with markers of artemisinin and mefloquine resistance could be used to track the spread of resistance and provide timely information for containment policies.”
“Piperaquine resistance, although currently confined to Cambodia, is a major concern, because patients suffering malaria are almost untreatable. At present, alternative treatments are scarce, and the reduced cure rates lead to prolonged parasite carriage, increasing the potential of transmitting the resistant parasites and endangering efforts to control and eliminate malaria.”
Exo-E415G variant
The second study was conducted by Rick Fairhurst, MD, of the National Institutes of Health in Bethesda, Maryland, and his colleagues.
The investigators analyzed 486 P falciparum parasites collected between July 9, 2010, and December 31, 2013, from 3 different provinces in Cambodia where artemisinin resistance and dihydroartemisinin-piperaquine treatment failure are common (Pursat), emerging (Preah Vihear), or uncommon (Ratanakiri).
The investigators exposed the parasites to therapeutic levels of piperaquine and measured how well each strain grew. At the same time, they sequenced the entire genome of each parasite.
Parasites were more likely to survive exposure to piperaquine if they had 2 or more copies of the plasmepsin 2 and plasmepsin 3 genes, as well as the exo-E415G variant on chromosome 13.
Similarly, in a further analysis of 133 patient samples, the investigators found that individuals infected with parasites carrying multiple copies of the plasmepsin genes were much more likely to fail treatment with dihydroartemisinin-piperaquine, as were individuals who had parasites with the exo-E415G variant.
The team also noted that the prevalence of exo-E415G and plasmepsin 2-3 markers has risen substantially in recent years in Pursat and Preah Vihear, where artemisinin resistance is common and dihydroartemisinin-piperaquine has been the frontline treatment for at least 6 years.
“By surveying the piperaquine resistance marker across Southeast Asia in real-time, we can identify those areas where dihydroartemisinin-piperaquine will not be effective, and this could enable national malaria control programs to immediately recommend alternative therapies, such as artesunate-mefloquine,” Dr Fairhurst said.
“This approach will be crucial to eliminating multidrug-resistant parasites in Southeast Asia before they spread to sub-Saharan Africa, where most of the world’s malaria transmission, illness, and death occur.” ![]()
Team targets fructose to treat AML

Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Photo from the University
of Hawaii Cancer Center
Researchers say they have discovered a novel treatment strategy for acute myeloid leukemia (AML)—inhibiting fructose utilization.
The team discovered that AML cells are prone to using fructose for energy, and increased fructose utilization predicts poor treatment outcomes in AML patients.
The researchers also found that 2,5-anhydro-D-mannitol (2,5-AM) could inhibit fructose use in AML cells and therefore hinder their growth.
Wei Jia, PhD, of the University of Hawaii Cancer Center in Honolulu, and his colleagues reported these findings in Cancer Cell.
The researchers said their findings highlight the unique ability of AML cells to switch their energy supply from glucose to fructose, when glucose is in short supply.
Fructose is the second most abundant blood sugar in the body and is used as a glucose alternative by AML cells to retain energy. After the switch, AML cells begin to multiply faster.
The study revealed a potential treatment by stopping the glucose transporter GLUT5. This restricts the AML energy supply and effectively slows AML growth.
To target GLUT5, the researchers used 2,5-AM, a fructose analog with high affinity for GLUT5.
The team tested 2,5-AM in AML cells with enhanced fructose utilization and found the drug significantly suppressed fructose-induced proliferation, colony growth, and migration in the absence of glucose or when glucose levels were low.
The researchers tested 2,5-AM in 4 different AML cell lines and found the drug suppressed fructose-induced cell proliferation in a dose-dependent manner in all of the cell lines under glucose-limiting conditions. However, 2,5-AM had little effect on glucose-induced cell proliferation.
The team tested 2,5-AM in normal monocytes as well. They said the drug had a negligible effect on glucose-induced cell growth.
“Our normal cells hardly rely on fructose for growth,” Dr Jia noted. “This makes the fructose transport in cancer cells an attractive drug target.”
Finally, the researchers tested 2,5-AM in combination with ara-C in the 4 AML cell lines. They observed a synergistic effect between the drugs in all cell lines in the absence of glucose or when glucose levels were low.
“We are in the process of developing a GLUT5 inhibitor, thus cutting the cancer cells’ energy source and eventually killing them,” Dr Jia said. “The new GLUT5 inhibitor can potentially be used alone or in addition to the current chemotherapy drugs to enhance anticancer effects.” ![]()
Targeting CD98 to treat AML
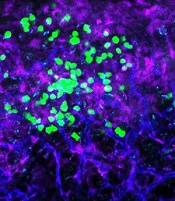
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
with blood vessels (blue).
Image courtesy of
UC San Diego Health
Preclinical research suggests the cell surface molecule CD98 promotes acute myeloid leukemia (AML), and the anti-CD98 antibody IGN523 can inhibit AML growth.
In AML patient cells and mouse models of the disease, IGN523 disrupted the interactions between leukemia cells and the surrounding blood vessels, thereby inhibiting the growth of AML.
Tannishtha Reya, PhD, of the University of California San Diego School of Medicine, and her colleagues reported these findings in Cancer Cell.
The team believes their results suggest IGN523 or other anti-CD98 antibodies might be useful for treating AML, particularly in children.
However, in a phase 1 study presented at the 2015 ASH Annual Meeting, IGN523 demonstrated only modest anti-leukemic activity in adults with AML.
Still, the researchers involved in the phase 1 study said IGN523 may prove effective in combination with other drugs used to treat AML.
Cancer Cell study
“To improve therapeutic strategies for [AML], we need to look not just at the cancer cells themselves but also at their interactions with surrounding cells, tissues, molecules, and blood vessels in the body,” Dr Reya said.
“In this study, we identified CD98 as a critical molecule driving AML growth. We showed that blocking CD98 can effectively reduce leukemia burden and improve survival by preventing cancer cells from receiving support from the surrounding environment.”
Dr Reya’s team engineered mouse models that lacked CD98 and found that loss of this molecule blocked AML growth and improved survival. Furthermore, CD98 loss largely spared normal blood cells, which the researchers said indicates a potential therapeutic window.
Additional experiments revealed that leukemia cells lacking CD98 had fewer stable interactions with the lining of blood vessels—interactions that were needed to fuel AML growth.
So the researchers decided to test the effects of blocking CD98 with a therapeutic inhibitor—IGN523. The team found that IGN523 blocks CD98’s AML-promoting activity in mouse models and human AML cells.
The researchers also transplanted patient-derived AML cells into mice and treated the recipients with either IGN523 or a control antibody. Anti-CD98 treatment effectively eliminated AML cells, while AML in the control mice expanded more than 100-fold.
“This study suggests that human AML can’t get established without CD98 and that blocking the molecule with anti-CD98 antibodies could be beneficial for the treatment of AML in both adults and children,” Dr Reya said.
Moving forward, Dr Reya and her colleagues are working to further define whether CD98 could be used to treat pediatric AML.
“Many of the models we used in this work were based on mutations found in childhood AML,” Dr Reya said. “While many childhood cancers have become very treatable, childhood AML continues to have a high rate of relapse and death.”
“We plan to work with pediatric oncologists to test if anti-CD98 agents can be effective against pediatric AML and whether it can improve responses to current treatments. I think this is particularly important to pursue since the anti-CD98 antibody has already been through phase 1 trials and could be more easily positioned to test in drug-resistant pediatric AML.”
Igenica Biotherapeutics Inc., the company developing IGN523, provided the drug for this study, and one of the study’s authors is an employee of the company. ![]()
Study reveals ‘high-traffic’ routes of malaria importation
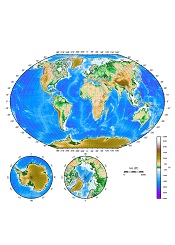
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Results of an international study suggest France and the UK experience the highest number of malaria cases imported from other countries.
Researchers mapped the movement of malaria from endemic countries to 40 countries defined as being malaria-free.
The countries with the highest average number of imported infections per year over the past decade were France (2169), the UK (1898), the US (1511), Italy (637), and Germany (401).
Infection movement was strongly skewed to a small number of “high-traffic” routes, with malaria cases originating from West Africa accounting for 56% (13,947) of all cases detected in non-endemic countries.
These results were published in The Lancet Infectious Diseases.
“This is the first world-wide assessment of imported malaria cases in 20 years, and mapping this data is hugely valuable in helping us understand how we can mitigate against the effects of the global movements of the disease,” said study author Andrew Tatem, PhD, of the University of Southampton in the UK.
“Imported malaria can be expensive to treat, contribute to drug resistance, sometimes cause secondary local transmission, and threaten the long-term goal of eradication. This study forms part of wider efforts to understand patterns of human and malaria parasite movement to help guide elimination strategies.”
For this study, researchers analyzed a database of nationally reported statistics on imported malaria covering more than 50,000 individual cases over 10 years.
Although most incidents of malaria in non-endemic countries originated in West Africa, the study showed that 20% were from India (4988), 13% were from East Africa (3242), and 3% were from Papua New Guinea (748).
And although the routes from West Africa to France and the UK showed the strongest imported malaria link, there were other high-traffic routes. These included India to the US (149 cases on average per year), Papua New Guinea to Australia (97), Pakistan to the UK (69), and Haiti to the US (52).
By mapping this network of malaria movements across continents, the researchers showed that a number of factors, beyond geographic ones, may influence the strength of importation levels.
For example, the researchers believe that historical, economic, language, and cultural ties all play a part. They said population movements with former colonies had particular influence; such as Nigeria, Ghana, and Kenya with the UK, and Mali, Niger, and Chad with France.
The researchers hope to conduct further studies to examine which factors are the drivers behind the patterns of malaria spread between endemic and non-endemic countries. ![]()
Testing could ID cancer patients at high risk of VTE

Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
Image by Andre E.X. Brown
Genetic testing could help identify breast cancer patients with a high risk of developing venous thromboembolism (VTE), according to a study published in Clinical Cancer Research.
The study showed that patients who received chemotherapy, had a higher genetic susceptibility for VTE according to a polygenic risk score (PRS), or had both of these risk factors were more likely to develop VTE than patients without these risk factors.
In addition, researchers found the impact of genetic susceptibility was most pronounced in older patients.
“The risk for [VTE] is increased in cancer patients, particularly in those receiving chemotherapy,” said study author Judith S. Brand, PhD, of Karolinska Institutet in Stockholm, Sweden.
“As one of the most common cancers, breast cancer accounts for a large number of cancer-associated VTE cases.”
Dr Brand and her colleagues sought to identify the individual and joint effects of chemotherapy and genetic susceptibility on VTE risk. They studied 4261 women in the Stockholm region diagnosed with primary invasive breast cancer between 2001 and 2008, and followed until 2012.
Risks were stratified based on chemotherapy status and genetic susceptibility, as determined by a PRS based on 9 established VTE loci, with the top 5% classified as having high genetic susceptibility.
The median follow-up was 7.6 years, and 276 patients experienced a VTE during that time.
The researchers found that receiving chemotherapy and having high genetic susceptibility independently increased the risk of VTE. The hazard ratio was 1.98 for patients receiving chemotherapy and 1.90 for patients in the highest 5% of the PRS.
The 1-year cumulative incidence of VTE was 9.5% among patients who both received chemotherapy and had high genetic susceptibility, compared with 1.3% in the patients who did not have either of these risk factors (P<0.001).
The researchers also found that patient age played a role in VTE risk. The team said the risk-increasing effect of the PRS was stronger in older patients (P interaction = 0.04).
In patients age 60 or older who underwent chemotherapy and had a high genetic susceptibility, the 1-year cumulative incidence of VTE was 25%.
“Breast cancer patients receiving chemotherapy are not routinely being examined for VTE prevention in today’s clinical practice,” Dr Brand said. “Our study demonstrates that information on genetic susceptibility can be used to identify patients at high risk of developing VTE.”
“Combined with other clinical risk factors and biomarkers, these findings will guide future studies evaluating routine VTE risk assessment in chemotherapy outpatients, and prophylaxis for those at highest risk. Because older patients demonstrated a stronger genetic effect and higher VTE incidence, this group requires special attention in future risk stratification efforts.”
Dr Brand added that a limitation of this study is the small number of older patients who had chemotherapy and a high genetic susceptibility. She said larger-scale studies would be necessary to provide more precise risk estimates. And further research is needed to assess the safety and potential benefit of thromboprophylaxis in high-risk cancer patients. ![]()
CTC analysis as good as BM biopsy in MM, team says

Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM. ![]()
Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM. ![]()
Photo by Juan D. Alfonso
Analysis of circulating tumor cells (CTCs) can provide at least as much genetic information about multiple myeloma (MM) as a bone marrow (BM) biopsy, according to a new study.
Researchers found evidence to suggest that analyzing CTCs isolated from the peripheral blood of MM patients could help physicians monitor disease progression over time, track the emergence of drug resistance, and tailor therapies to individual patients.
Jens Lohr, MD, PhD, of the Broad Institute of MIT and Harvard in Cambridge, Massachusetts, and colleagues conducted this research and detailed their findings in Science Translational Medicine.
The researchers said they devised a method that was “highly sensitive” in detecting and sequencing single CTCs, even from patients with low tumor burden.
Specifically, the team found they could isolate CTCs from, and detect mutations in, blood samples from an MM patient who had achieved a very good partial response to treatment and a patient with monoclonal gammopathy of undetermined significance.
Overall, the researchers found that CTCs could be used to identify mutations relevant for prognosis, and some of these mutations were more abundant in CTCs than in BM samples.
For example, the team detected somatic mutations in the KRAS, BRAF, IRF4, and TP53 genes in single CTCs from 3 MM patients. And the same mutations were present in single BM-derived MM cells.
In another 3 patients, the proportion of CTCs harboring TP53 R273C, BRAF G469A, and NRAS G13D mutations was higher than that observed in BM-derived cells. In 2 of the patients, the mutations weren’t detectable in BM cells because of insufficient sample material.
The researchers also found that CTCs could reveal patients with an overabundance of molecules expressed by MM cells—such as CD38 and SLAMF7—that can be targeted by therapies currently approved to treat MM—such as daratumumab and elotuzumab.
The team therefore believes that, with further development, CTC analysis could be a valuable tool for advancing precision medicine in MM.
Drug can fight adenovirus in HSCT recipients
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression.
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression.
Image by Yale Rosen
NEW ORLEANS—Interim results of a phase 3 trial suggest brincidofovir can treat adenovirus (AdV) infection in recipients of allogeneic hematopoietic stem cell transplant (HSCT).
Both pediatric and adult patients experienced a decline in AdV viral load after brincidofovir treatment, but pediatric patients were more likely to respond.
Overall survival rates were better for patients who had a rapid response and were therefore better among pediatric patients than adults.
Investigators said the adverse events (AEs) in this study were consistent with the known safety profile of brincidofovir.
Michael Grimley, MD, of Cincinnati Children’s Hospital in Ohio, and his colleagues presented these results at IDWeek 2016 (abstract 2339). The research was supported by Chimerix, the company developing brincidofovir.
This trial, known as AdVise, was designed to evaluate brincidofovir for the treatment of AdV infection in pediatric and adult patients divided into 3 cohorts:
- Cohort A consists of allogeneic HSCT recipients with asymptomatic or limited AdV infection
- Cohort B consists of allogeneic HSCT recipients with disseminated AdV disease
- Cohort C consists of autologous HSCT recipients, solid organ transplant recipients, and other immunocompromised patients.
All patients were assigned to 12 weeks of oral brincidofovir, administered twice weekly. An additional 12 weeks of treatment was allowed in patients with ongoing or recurrent infection. After completing treatment, all patients were followed until week 36.
Interim analysis
The investigators examined outcomes at 24 weeks after the first brincidofovir dose (12 weeks after prescribed dosing duration) in 158 patients, including:
- Cohort A—23 adults and 43 pediatric patients
- Cohort B—35 adults and 57 pediatric patients.
The investigators noted that many of the patients did not complete the study. The team said this is a reflection of the significant mortality risk of AdV because most of these patients died before they could finish.
Sixty-five percent of adults and 33% of children in Cohort A did not complete the study. The same was true for 71% of adults and 49% of children in Cohort B.
Mortality
The study’s primary efficacy endpoint is all-cause mortality at day 60 after the first brincidofovir dose in allogeneic HSCT recipients with disseminated AdV disease (Cohort B). All-cause mortality at day 60 in this cohort was 19% in pediatric patients and 43% in adults.
In Cohorts A and B, all-cause mortality at 24 weeks was lower in children than adults.
At 24 weeks, pediatric all-cause mortality was 33% in Cohort A and 42% in Cohort B. Adult all-cause mortality was 48% in Cohort A and 71% in Cohort B.
AdV-related mortality at 24 weeks in pediatric patients was 9% in Cohort A and 14% in Cohort B. AdV-related mortality in adults was 4% in Cohort A and 46% in Cohort B.
Declines in viremia
In Cohort A, 61% of patients achieved undetectable viremia at the end of treatment—43% of adults and 70% of children.
In Cohort B, 49% of patients achieved undetectable viremia at the end of treatment—29% of adults and 63% of children.
The median time to undetectable AdV viremia was 43 days (range, 8 to non-estimable) for adults in Cohort A, 14 days (range, 5 to 23) for children in Cohort A, non-estimable (range, 29 days to non-estimable) for adults in Cohort B, and 22 days (range, 15 to 36) for children in Cohort B.
Link between response and survival
The investigators conducted post-hoc analyses to assess the correlation between rapid virologic response to brincidofovir and time to subsequent mortality.
The team compared patients who responded to treatment—defined as achieving a ≥ 2-log10 copies/mL decline, undetectable AdV viremia at week 4, or undetectable AdV viremia at week 6—with non-responders.
Fifty percent of adults and 84% of children who were still alive at week 4 had achieved a ≥ 2 log decline or undetectable AdV viremia at that time.
This type of response was associated with improved survival at week 24. In adults, the mortality rate was 46% in responders and 85% in non-responders (P=0.03). In pediatric patients, the mortality rate was 25% in responders and 71% in non-responders (P=0.01).
In patients who were alive at week 6, 42% of adults and 68% of children achieved undetectable AdV viremia by that time.
This response was associated with improved survival at week 24. In adults, the mortality rate was 30% in responders and 86% in non-responders (P=0.001). In pediatric patients, the mortality rate was 18% in responders and 54% in non-responders (P=0.01).
Safety
All adults had treatment-emergent AEs, as did all pediatric patients in Cohort B and 95% of pediatric patients in Cohort A.
The most common treatment-emergent AEs were gastrointestinal (GI) events, which occurred in 70% of adults and 81% of children in Cohort A, as well as 83% of adults and 74% of children in Cohort B.
Acute graft-versus-host disease (GVHD) was also common, occurring in 22% of adults and 37% of children in Cohort A and 43% of adults and 40% of children in Cohort B. Some patients did have acute GVHD at baseline, however—22%, 26%, 34%, and 19%, respectively.
The percentage of patients with AEs leading to treatment discontinuation was 26% for adults and 28% for children in Cohort A and 31% for adults and 14% for children in Cohort B.
Overall, 20% of pediatric patients and 29% of adults discontinued brincidofovir due to AEs. GI events were cited as the most common reason—5% and 14%, respectively.
The investigators said there were no events reported that were suggestive of drug-related nephrotoxicity or myelosuppression.
Two-drug combination targets LSCs in CML

Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells.
Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells.
Image by Difu Wu
Targeting a pair of transcription factors might improve the treatment of chronic myeloid leukemia (CML), according to researchers.
The team found that p53 and c-MYC have “defining roles” in the survival of leukemia stem cells (LSCs) in CML.
And by targeting these transcription factors with a pair of investigational drugs, the researchers were able to kill LSCs.
The team described this work in Nature.
“This collaborative study combined proteomics, transcriptomics, and systems biology to identify a novel, precision medicine-based approach for eradicating leukemic stem cells,” said study author Tony Whetton, PhD, of the University of Manchester in the UK.
Dr Whetton and his colleagues first discovered that p53 and c-MYC are “central hubs” in a CML network of deregulated proteins. The team also found that CML cells express increased c-MYC and decreased p53 levels.
So the researchers theorized that simultaneously activating p53 and inhibiting c-MYC could be a method for treating CML.
To that end, the team tested 2 drugs—RITA (or NSC652287), which binds p53 and blocks its degradation, and CPI-203, a BET inhibitor that hinders transcription by disrupting chromatin-dependent signal transduction.
The researchers found that CPI-203 successfully downregulated c-MYC but also reduced p53, while RITA increased p53.
Treating CML CD34+ cells with RITA or CPI-203 for 72 hours reduced cell viability and induced significant apoptosis, the team said. Combining the drugs enhanced these effects.
The researchers also found evidence to suggest that c-MYC inhibition induces differentiation of CML CD34+ cells. The team said that labelling with the cell-division tracker carboxyfluorescein succinimidyl ester (CFSE) and CD34 antibody showed that, as CML cells divided in the presence of CPI-203, there was a clear and rapid loss of CD34 expression that was not seen in the presence of RITA.
The researchers did not observe any differences in the effects of RITA and CPI-203 when they were tested in CML CD34+ cells pretreated with imatinib.
Furthermore, RITA and CPI-203, either alone or in combination, had no significant effects on normal CD34+ cells when tested at lower concentrations. However, when CPI-203 was used alone at higher concentrations (2 or 5 μ M) or with RITA at the highest concentrations tested (RITA at 25 nM, CPI-203 at 5 μ M), apoptosis did occur.
In CML cells, the researchers observed “significant apoptosis” with all concentrations of CPI-203 and RITA tested.
The team also exposed CML LSCs, defined as either CFSEmax or CD34+CD38− cells, to CPI-203 and RITA as well as a pair of tyrosine kinase inhibitors.
The CFSEmax population persisted despite 5 days of treatment with dasatinib or nilotinib, but the cells were “significantly reduced” after 5 days of treatment with CPI-203 alone and in combination with RITA.
Similarly, 72 hours of treatment with RITA with CPI-203 eliminated residual CD34+CD38− cells.
The researchers also assessed LSC engraftment after treatment with RITA and/or CPI-203, as well as dasatinib. They exposed CML CD34+ cells to the drugs for 48 hours before transplanting the cells into sublethally irradiated NSG mice.
The team said dasatinib had no significant effect on NSG-repopulating CML LSCs. However, RITA, CPI-203, and the drugs in combination reduced engraftment, as indicated by decreased CD45+, CD34+, CD33+, CD11b+, CD19+ and CD14+ cells.
Tranexamic acid safely reduces need for transfusion, study suggests
Photo by Piotr Bodzek
Results of a large study suggest that tranexamic acid can reduce the need for blood transfusion without increasing the risk of thrombotic complications or death in patients undergoing coronary artery surgery.
Patients who received tranexamic acid had a lower risk of excessive bleeding, required fewer units of blood products, and had a lower risk of emergency reoperation after surgery than patients who received placebo.
In addition, patients who received tranexamic acid had no higher risk of death or thrombotic complications than those who received placebo.
Paul S. Myles, MBBS, MD, of Alfred Hospital in Melbourne, Australia, and his colleagues conducted this study and reported the results in NEJM. The study was also presented at the ANESTHESIOLOGY® 2016 annual meeting.
The study included 4631 patients who underwent surgery and had available outcomes data, 2311 who were assigned to receive tranexamic acid and 2320 who were assigned to receive placebo.
The study’s primary outcome was a composite of death and thrombotic complications (nonfatal myocardial infarction, stroke, pulmonary embolism, renal failure, or bowel infarction) within 30 days after surgery.
There was no significant difference in the primary outcome between the 2 treatment groups. Thrombotic complications/death occurred in 16.7% of patients in the tranexamic acid group and 18.1% in the placebo group (relative risk=0.92; P=0.22).
Patients who received placebo required significantly more units of blood products than patients who received tranexamic acid—7994 and 4331 units, respectively (P<0.001).
And significantly fewer patients in the tranexamic acid group than the placebo group had major hemorrhage or cardiac tamponade leading to emergency reoperations—1.4% and 2.8%, respectively (P=0.001).
However, patients in the tranexamic group had a significantly higher incidence of seizures—0.7% and 0.1%, respectively (P=0.002).
Dr Myles said that although this study was conducted in patients undergoing coronary artery surgery, the results are relevant for patients having many other types of surgery where bleeding and the need for blood transfusion may occur.
Photo by Piotr Bodzek
Results of a large study suggest that tranexamic acid can reduce the need for blood transfusion without increasing the risk of thrombotic complications or death in patients undergoing coronary artery surgery.
Patients who received tranexamic acid had a lower risk of excessive bleeding, required fewer units of blood products, and had a lower risk of emergency reoperation after surgery than patients who received placebo.
In addition, patients who received tranexamic acid had no higher risk of death or thrombotic complications than those who received placebo.
Paul S. Myles, MBBS, MD, of Alfred Hospital in Melbourne, Australia, and his colleagues conducted this study and reported the results in NEJM. The study was also presented at the ANESTHESIOLOGY® 2016 annual meeting.
The study included 4631 patients who underwent surgery and had available outcomes data, 2311 who were assigned to receive tranexamic acid and 2320 who were assigned to receive placebo.
The study’s primary outcome was a composite of death and thrombotic complications (nonfatal myocardial infarction, stroke, pulmonary embolism, renal failure, or bowel infarction) within 30 days after surgery.
There was no significant difference in the primary outcome between the 2 treatment groups. Thrombotic complications/death occurred in 16.7% of patients in the tranexamic acid group and 18.1% in the placebo group (relative risk=0.92; P=0.22).
Patients who received placebo required significantly more units of blood products than patients who received tranexamic acid—7994 and 4331 units, respectively (P<0.001).
And significantly fewer patients in the tranexamic acid group than the placebo group had major hemorrhage or cardiac tamponade leading to emergency reoperations—1.4% and 2.8%, respectively (P=0.001).
However, patients in the tranexamic group had a significantly higher incidence of seizures—0.7% and 0.1%, respectively (P=0.002).
Dr Myles said that although this study was conducted in patients undergoing coronary artery surgery, the results are relevant for patients having many other types of surgery where bleeding and the need for blood transfusion may occur.
Photo by Piotr Bodzek
Results of a large study suggest that tranexamic acid can reduce the need for blood transfusion without increasing the risk of thrombotic complications or death in patients undergoing coronary artery surgery.
Patients who received tranexamic acid had a lower risk of excessive bleeding, required fewer units of blood products, and had a lower risk of emergency reoperation after surgery than patients who received placebo.
In addition, patients who received tranexamic acid had no higher risk of death or thrombotic complications than those who received placebo.
Paul S. Myles, MBBS, MD, of Alfred Hospital in Melbourne, Australia, and his colleagues conducted this study and reported the results in NEJM. The study was also presented at the ANESTHESIOLOGY® 2016 annual meeting.
The study included 4631 patients who underwent surgery and had available outcomes data, 2311 who were assigned to receive tranexamic acid and 2320 who were assigned to receive placebo.
The study’s primary outcome was a composite of death and thrombotic complications (nonfatal myocardial infarction, stroke, pulmonary embolism, renal failure, or bowel infarction) within 30 days after surgery.
There was no significant difference in the primary outcome between the 2 treatment groups. Thrombotic complications/death occurred in 16.7% of patients in the tranexamic acid group and 18.1% in the placebo group (relative risk=0.92; P=0.22).
Patients who received placebo required significantly more units of blood products than patients who received tranexamic acid—7994 and 4331 units, respectively (P<0.001).
And significantly fewer patients in the tranexamic acid group than the placebo group had major hemorrhage or cardiac tamponade leading to emergency reoperations—1.4% and 2.8%, respectively (P=0.001).
However, patients in the tranexamic group had a significantly higher incidence of seizures—0.7% and 0.1%, respectively (P=0.002).
Dr Myles said that although this study was conducted in patients undergoing coronary artery surgery, the results are relevant for patients having many other types of surgery where bleeding and the need for blood transfusion may occur.