User login
Explaining the development of MPNs, leukemia
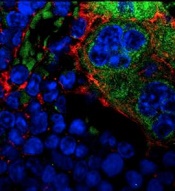
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
MSPCs with mutant PTPN11
(red) and monocytes (green).
Image courtesy of
Dong et al, Nature 2016
New research published in Nature has shown how certain mutations drive the development of myeloproliferative neoplasms (MPNs) and leukemia.
Investigators discovered that PTPN11 activating mutations promote the development and progression of MPNs through “profound detrimental effects” on hematopoietic stem cells (HSCs).
The team also identified a potential method of treating MPNs in patients with Noonan syndrome.
Noonan syndrome can be caused by mutations in several genes, but the most common is PTPN11. Children with Noonan syndrome are known to have an increased risk of developing MPNs/leukemia.
Previous research had established that mutations in PTPN11 have a conventional cell-autonomous effect on HSC growth.
In the current study, investigators showed that PTPN11 mutations also affect mesenchymal stem/progenitor cells (MSPCs) and osteoprogenitors.
The mutations cause over-production of the CC chemokine CCL3, which attracts monocytes into the HSCs’ niches. The monocytes make inflammatory molecules that stimulate the HSCs to differentiate and proliferate, leading to MPNs and leukemia.
“We have identified CCL3 as a potential therapeutic target for controlling leukemic progression in Noonan syndrome and for improving stem cell transplantation therapy in Noonan syndrome-associated leukemias,” said study author Cheng-Kui Qu, MD, PhD, of Emory University School of Medicine in Atlanta, Georgia.
Dr Qu and his colleagues began this research intending to investigate the effects of PTPN11 mutations in the nervous system. The team developed genetically engineered mice that had altered PTPN11 in neural cells.
The mice all developed a condition resembling an MPN at an early age. It turned out that the mice had changes in the PTPN11 gene in their MSPCs and osteoprogenitors (in addition to their neural cells) but not in their HSCs.
The investigators found the MPN in these PTPN11-mutant mice can be treated in the short-term by HSC transplant, but the condition comes back within months.
However, drugs counteracting CCL3 successfully reversed MPN phenotypes. One of the drugs is the CCR5 antagonist maraviroc, which is approved in the US to combat HIV infection, and another is the CCR1 antagonist BX471.
The investigators noted that other Noonan syndrome mutations, in genes besides PTPN11, need to be assessed for their effects on MPN/leukemia formation. ![]()
Gene-editing method cures thalassemia in mice

Photo by Aaron Logan
A new gene-editing strategy may be able to cure thalassemia, according to preclinical research published in Nature Communications.
The technique—which involves a combination of nanoparticles, synthetic pieces of DNA, and an intravenous injection—was able to alleviate symptoms of thalassemia in mice.
The strategy also decreases the risk of off-target mutations, when compared to other gene-editing techniques, according to researchers.
The new technique involves biocompatible nanoparticles containing peptide nucleic acids (PNAs), which are small, nano-sized, synthetic molecules in which a protein-like backbone is combined with the nucleobases found in DNA and RNA.
PNAs are designed to open up double-stranded DNA and bind near the target site in a highly specific manner. The PNAs fit inside a nanoparticle delivery system that is approved by the US Food and Drug Administration (FDA) and has already been used to treat neurodegenerative diseases in humans.
“We have developed a system that uses FDA-approved nanoparticles to deliver our PNA molecule along with a donor DNA to repair a malfunctioning gene in living mice,” said study author Danith Ly, PhD, of Carnegie Mellon University in Pittsburgh, Pennsylvania. “This has not been achieved with CRISPR.”
Dr Ly and his colleagues designed a PNA to target the malfunctioning gene in beta-thalassemia, the hemoglobin subunit beta (HBB) gene.
The researchers then loaded the nanoparticles with the PNAs, a donor strand of DNA encoding the sequence for a functional HBB gene, and a stem cell factor that enhances gene editing.
When the PNA binds to the target site in the DNA, it forms a PNA-DNA-PNA triplex, leaving a displaced DNA strand. Formation of such a complex enables the donor DNA to bind to the faulty DNA site within the vicinity.
Taken together, this altered helix engages the cell’s own DNA repair pathways to correct the malfunctioning HBB gene.
In addition to testing this system on mouse and human hematopoietic stem cells, the researchers used an intravenous injection to deliver the gene-editing package in mouse models of beta-thalassemia.
The results showed up to 6.9% successful gene-editing in hematopoietic stem cells. The mice showed elevated levels of hemoglobin—into the normal range—for several months after treatment, a reduction in reticulocytosis, and reversal of splenomegaly.
The researchers said this represents a striking increase in efficacy over typical gene-editing methods, which produce a 0.1% success rate.
“The effect may only be 7%, but that’s curative,” Dr Ly said. “In the case of this particular disease model, you don’t need a lot of correction. You don’t need 100% to see the phenotype return to normal.”
In addition, this gene-editing strategy had “extremely low off-target effects,” according to study author Peter Glazer, MD, of Yale University in New Haven, Connecticut.
The overall off-target modification frequency was 0.0032%.
If this strategy proves effective in clinical studies, it could lead to the development of gene therapy for patients with thalassemia, sickle cell disease, and other inherited blood disorders, Dr Glazer said.
“We might get enough cells corrected that individuals are not anemic anymore,” he added. “We could achieve a symptomatic cure.” ![]()
Photo by Aaron Logan
A new gene-editing strategy may be able to cure thalassemia, according to preclinical research published in Nature Communications.
The technique—which involves a combination of nanoparticles, synthetic pieces of DNA, and an intravenous injection—was able to alleviate symptoms of thalassemia in mice.
The strategy also decreases the risk of off-target mutations, when compared to other gene-editing techniques, according to researchers.
The new technique involves biocompatible nanoparticles containing peptide nucleic acids (PNAs), which are small, nano-sized, synthetic molecules in which a protein-like backbone is combined with the nucleobases found in DNA and RNA.
PNAs are designed to open up double-stranded DNA and bind near the target site in a highly specific manner. The PNAs fit inside a nanoparticle delivery system that is approved by the US Food and Drug Administration (FDA) and has already been used to treat neurodegenerative diseases in humans.
“We have developed a system that uses FDA-approved nanoparticles to deliver our PNA molecule along with a donor DNA to repair a malfunctioning gene in living mice,” said study author Danith Ly, PhD, of Carnegie Mellon University in Pittsburgh, Pennsylvania. “This has not been achieved with CRISPR.”
Dr Ly and his colleagues designed a PNA to target the malfunctioning gene in beta-thalassemia, the hemoglobin subunit beta (HBB) gene.
The researchers then loaded the nanoparticles with the PNAs, a donor strand of DNA encoding the sequence for a functional HBB gene, and a stem cell factor that enhances gene editing.
When the PNA binds to the target site in the DNA, it forms a PNA-DNA-PNA triplex, leaving a displaced DNA strand. Formation of such a complex enables the donor DNA to bind to the faulty DNA site within the vicinity.
Taken together, this altered helix engages the cell’s own DNA repair pathways to correct the malfunctioning HBB gene.
In addition to testing this system on mouse and human hematopoietic stem cells, the researchers used an intravenous injection to deliver the gene-editing package in mouse models of beta-thalassemia.
The results showed up to 6.9% successful gene-editing in hematopoietic stem cells. The mice showed elevated levels of hemoglobin—into the normal range—for several months after treatment, a reduction in reticulocytosis, and reversal of splenomegaly.
The researchers said this represents a striking increase in efficacy over typical gene-editing methods, which produce a 0.1% success rate.
“The effect may only be 7%, but that’s curative,” Dr Ly said. “In the case of this particular disease model, you don’t need a lot of correction. You don’t need 100% to see the phenotype return to normal.”
In addition, this gene-editing strategy had “extremely low off-target effects,” according to study author Peter Glazer, MD, of Yale University in New Haven, Connecticut.
The overall off-target modification frequency was 0.0032%.
If this strategy proves effective in clinical studies, it could lead to the development of gene therapy for patients with thalassemia, sickle cell disease, and other inherited blood disorders, Dr Glazer said.
“We might get enough cells corrected that individuals are not anemic anymore,” he added. “We could achieve a symptomatic cure.” ![]()
Photo by Aaron Logan
A new gene-editing strategy may be able to cure thalassemia, according to preclinical research published in Nature Communications.
The technique—which involves a combination of nanoparticles, synthetic pieces of DNA, and an intravenous injection—was able to alleviate symptoms of thalassemia in mice.
The strategy also decreases the risk of off-target mutations, when compared to other gene-editing techniques, according to researchers.
The new technique involves biocompatible nanoparticles containing peptide nucleic acids (PNAs), which are small, nano-sized, synthetic molecules in which a protein-like backbone is combined with the nucleobases found in DNA and RNA.
PNAs are designed to open up double-stranded DNA and bind near the target site in a highly specific manner. The PNAs fit inside a nanoparticle delivery system that is approved by the US Food and Drug Administration (FDA) and has already been used to treat neurodegenerative diseases in humans.
“We have developed a system that uses FDA-approved nanoparticles to deliver our PNA molecule along with a donor DNA to repair a malfunctioning gene in living mice,” said study author Danith Ly, PhD, of Carnegie Mellon University in Pittsburgh, Pennsylvania. “This has not been achieved with CRISPR.”
Dr Ly and his colleagues designed a PNA to target the malfunctioning gene in beta-thalassemia, the hemoglobin subunit beta (HBB) gene.
The researchers then loaded the nanoparticles with the PNAs, a donor strand of DNA encoding the sequence for a functional HBB gene, and a stem cell factor that enhances gene editing.
When the PNA binds to the target site in the DNA, it forms a PNA-DNA-PNA triplex, leaving a displaced DNA strand. Formation of such a complex enables the donor DNA to bind to the faulty DNA site within the vicinity.
Taken together, this altered helix engages the cell’s own DNA repair pathways to correct the malfunctioning HBB gene.
In addition to testing this system on mouse and human hematopoietic stem cells, the researchers used an intravenous injection to deliver the gene-editing package in mouse models of beta-thalassemia.
The results showed up to 6.9% successful gene-editing in hematopoietic stem cells. The mice showed elevated levels of hemoglobin—into the normal range—for several months after treatment, a reduction in reticulocytosis, and reversal of splenomegaly.
The researchers said this represents a striking increase in efficacy over typical gene-editing methods, which produce a 0.1% success rate.
“The effect may only be 7%, but that’s curative,” Dr Ly said. “In the case of this particular disease model, you don’t need a lot of correction. You don’t need 100% to see the phenotype return to normal.”
In addition, this gene-editing strategy had “extremely low off-target effects,” according to study author Peter Glazer, MD, of Yale University in New Haven, Connecticut.
The overall off-target modification frequency was 0.0032%.
If this strategy proves effective in clinical studies, it could lead to the development of gene therapy for patients with thalassemia, sickle cell disease, and other inherited blood disorders, Dr Glazer said.
“We might get enough cells corrected that individuals are not anemic anymore,” he added. “We could achieve a symptomatic cure.” ![]()
FDA approves blood screening assay
Photo by Daniel Gay
The US Food and Drug Administration has approved the blood screening assay cobas® MPX for use on the cobas® 6800 and 8800 Systems.
cobas® MPX is a nucleic acid test designed to screen donated blood and plasma for human immunodeficiency virus (HIV),hepatitis B virus (HBV), and hepatitis C virus (HCV).
The test can detect 5 viral targets—HIV-1 Group M, HIV-1 Group O, HIV-2, HBV, and HCV—in a single sample.
cobas® MPX features a dual-target approach with amplification of separate regions of HIV-1 and dual probes for HCV. It eliminates both the need for discriminatory testing between HIV, HBV, and HCV and the potential for discrepant results.
cobas® MPX is a product of Roche Molecular Diagnostics and can be used on Roche’s cobas® 6800 System or cobas® 8800 System.
These systems are used for routine molecular testing in the areas of donor screening, viral load monitoring, women’s health, and microbiology.
Both systems make it possible for labs to perform up to 3 tests in the same run with no pre-sorting required.
In an 8-hour shift, the cobas® 6800 System can provide 384 results, and the cobas® 8800 System can provide 960 results.
The cobas® 6800 system enables up to 8 hours of walk-away time with minimal user interaction, and the cobas® 8800 enables up to 4 hours of walk-away time. ![]()
Photo by Daniel Gay
The US Food and Drug Administration has approved the blood screening assay cobas® MPX for use on the cobas® 6800 and 8800 Systems.
cobas® MPX is a nucleic acid test designed to screen donated blood and plasma for human immunodeficiency virus (HIV),hepatitis B virus (HBV), and hepatitis C virus (HCV).
The test can detect 5 viral targets—HIV-1 Group M, HIV-1 Group O, HIV-2, HBV, and HCV—in a single sample.
cobas® MPX features a dual-target approach with amplification of separate regions of HIV-1 and dual probes for HCV. It eliminates both the need for discriminatory testing between HIV, HBV, and HCV and the potential for discrepant results.
cobas® MPX is a product of Roche Molecular Diagnostics and can be used on Roche’s cobas® 6800 System or cobas® 8800 System.
These systems are used for routine molecular testing in the areas of donor screening, viral load monitoring, women’s health, and microbiology.
Both systems make it possible for labs to perform up to 3 tests in the same run with no pre-sorting required.
In an 8-hour shift, the cobas® 6800 System can provide 384 results, and the cobas® 8800 System can provide 960 results.
The cobas® 6800 system enables up to 8 hours of walk-away time with minimal user interaction, and the cobas® 8800 enables up to 4 hours of walk-away time. ![]()
Photo by Daniel Gay
The US Food and Drug Administration has approved the blood screening assay cobas® MPX for use on the cobas® 6800 and 8800 Systems.
cobas® MPX is a nucleic acid test designed to screen donated blood and plasma for human immunodeficiency virus (HIV),hepatitis B virus (HBV), and hepatitis C virus (HCV).
The test can detect 5 viral targets—HIV-1 Group M, HIV-1 Group O, HIV-2, HBV, and HCV—in a single sample.
cobas® MPX features a dual-target approach with amplification of separate regions of HIV-1 and dual probes for HCV. It eliminates both the need for discriminatory testing between HIV, HBV, and HCV and the potential for discrepant results.
cobas® MPX is a product of Roche Molecular Diagnostics and can be used on Roche’s cobas® 6800 System or cobas® 8800 System.
These systems are used for routine molecular testing in the areas of donor screening, viral load monitoring, women’s health, and microbiology.
Both systems make it possible for labs to perform up to 3 tests in the same run with no pre-sorting required.
In an 8-hour shift, the cobas® 6800 System can provide 384 results, and the cobas® 8800 System can provide 960 results.
The cobas® 6800 system enables up to 8 hours of walk-away time with minimal user interaction, and the cobas® 8800 enables up to 4 hours of walk-away time. ![]()
EMA recommends orphan status for drug in SCD
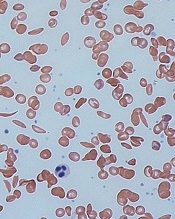
and normal red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending that LJPC-401 receive orphan designation as a treatment for patients with sickle cell disease (SCD).
LJPC-401 is a formulation of synthetic human hepcidin.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload, which can occur in SCD and other diseases.
LJPC-401 already has orphan designation in the European Union as a treatment for patients with beta-thalassemia intermedia and major.
La Jolla recently reported positive results from a phase 1 study of LJPC-401 in patients at risk of iron overload suffering from hereditary hemochromatosis, thalassemia, or SCD. Fifteen patients received LJPC-401 at escalating dose levels ranging from 1 mg to 20 mg.
The researchers observed a dose-dependent, statistically significant reduction in serum iron (P=0.008 for dose response; not adjusted for multiple comparisons).
At the 20 mg dose level, LJPC-401 reduced serum iron by an average of 58.1% from baseline to hour 8 (P=0.001; not adjusted for potential regression to the mean effect), and serum iron had not returned to baseline through day 7 (21.2% reduction from baseline to the end of day 7).
The researchers also said LJPC-401 was well tolerated, with no dose-limiting toxicities. Injection-site reactions were the most commonly reported adverse event. These were all mild or moderate in severity, self-limiting, and fully resolved.
Now, La Jolla is working to initiate a pivotal study of LJPC-401. This will be a randomized, controlled, multicenter study in beta-thalassemia patients suffering from iron overload. La Jolla plans to initiate this study in mid-2017.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure. ![]()
and normal red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending that LJPC-401 receive orphan designation as a treatment for patients with sickle cell disease (SCD).
LJPC-401 is a formulation of synthetic human hepcidin.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload, which can occur in SCD and other diseases.
LJPC-401 already has orphan designation in the European Union as a treatment for patients with beta-thalassemia intermedia and major.
La Jolla recently reported positive results from a phase 1 study of LJPC-401 in patients at risk of iron overload suffering from hereditary hemochromatosis, thalassemia, or SCD. Fifteen patients received LJPC-401 at escalating dose levels ranging from 1 mg to 20 mg.
The researchers observed a dose-dependent, statistically significant reduction in serum iron (P=0.008 for dose response; not adjusted for multiple comparisons).
At the 20 mg dose level, LJPC-401 reduced serum iron by an average of 58.1% from baseline to hour 8 (P=0.001; not adjusted for potential regression to the mean effect), and serum iron had not returned to baseline through day 7 (21.2% reduction from baseline to the end of day 7).
The researchers also said LJPC-401 was well tolerated, with no dose-limiting toxicities. Injection-site reactions were the most commonly reported adverse event. These were all mild or moderate in severity, self-limiting, and fully resolved.
Now, La Jolla is working to initiate a pivotal study of LJPC-401. This will be a randomized, controlled, multicenter study in beta-thalassemia patients suffering from iron overload. La Jolla plans to initiate this study in mid-2017.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure. ![]()
and normal red blood cells
Image by Graham Beards
The European Medicines Agency’s (EMA) Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending that LJPC-401 receive orphan designation as a treatment for patients with sickle cell disease (SCD).
LJPC-401 is a formulation of synthetic human hepcidin.
La Jolla Pharmaceutical Company is developing LJPC-401 for the treatment of iron overload, which can occur in SCD and other diseases.
LJPC-401 already has orphan designation in the European Union as a treatment for patients with beta-thalassemia intermedia and major.
La Jolla recently reported positive results from a phase 1 study of LJPC-401 in patients at risk of iron overload suffering from hereditary hemochromatosis, thalassemia, or SCD. Fifteen patients received LJPC-401 at escalating dose levels ranging from 1 mg to 20 mg.
The researchers observed a dose-dependent, statistically significant reduction in serum iron (P=0.008 for dose response; not adjusted for multiple comparisons).
At the 20 mg dose level, LJPC-401 reduced serum iron by an average of 58.1% from baseline to hour 8 (P=0.001; not adjusted for potential regression to the mean effect), and serum iron had not returned to baseline through day 7 (21.2% reduction from baseline to the end of day 7).
The researchers also said LJPC-401 was well tolerated, with no dose-limiting toxicities. Injection-site reactions were the most commonly reported adverse event. These were all mild or moderate in severity, self-limiting, and fully resolved.
Now, La Jolla is working to initiate a pivotal study of LJPC-401. This will be a randomized, controlled, multicenter study in beta-thalassemia patients suffering from iron overload. La Jolla plans to initiate this study in mid-2017.
About orphan designation
The EMA’s COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat life-threatening or chronically debilitating conditions affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity if the drug receives regulatory approval. The designation also provides incentives for companies seeking protocol assistance from the EMA during the product development phase and direct access to the centralized authorization procedure. ![]()
Test approved to screen donated blood for sickle cell trait
The US Food and Drug Administration (FDA) has approved use of the PreciseType HEA test to screen blood donors for sickle cell trait (SCT).
The test was previously FDA approved for use in determining blood compatibility between donors and transfusion recipients.
The added utility of screening donors for SCT addresses the desire to avoid transfusing red blood cells from SCT donors to neonates or patients with sickle cell disease.
Blood from SCT donors can also present a problem when performing the required filtration of white cells from the blood donation.
The PreciseType HEA test will allow these units to be identified prior to filtration and provide blood center staff with the opportunity to decide how best to utilize the various components of a whole blood donation.
The PreciseType HEA test is manufactured by BioArray Solutions, a wholly owned subsidiary of Immucor, Inc.
“We’ve successfully demonstrated the clinical benefits of our PreciseType HEA test, and this is evident in the FDA broadening its approved use,” said Michael Spigarelli, vice president of medical affairs at Immucor.
“The use of PreciseType HEA to screen donor units for patients with sickle cell disease, neonates, or any individual that may require SCT-negative blood provides a great improvement over previously used methods and offers the first FDA-approved molecular method specifically for screening units.”
SCT screening has traditionally been performed by solubility testing of sickle hemoglobin in buffer, but blood centers have been looking for an alternative due to limitations in this method.
According to Immucor, a molecular approach using PreciseType HEA can overcome the throughput limitations and reduce the false-positive rates observed with the traditional SCT screening method.
“We had already validated the PreciseType HEA test for [SCT screening] in our lab,” said Connie Westhoff, PhD, of the New York Blood Center in New York, New York.
“Our previous screening method required manual testing and interpretation of the results and had high false-positive rates. About 1 in 12 minority donors possess the sickle trait, so accurate results are important to us to avoid unnecessary notifications to donors and deferred blood units. We are now able to identify SCT in our donors utilizing the same PreciseType HEA test we are already running on many of our donors without running additional tests.” ![]()
The US Food and Drug Administration (FDA) has approved use of the PreciseType HEA test to screen blood donors for sickle cell trait (SCT).
The test was previously FDA approved for use in determining blood compatibility between donors and transfusion recipients.
The added utility of screening donors for SCT addresses the desire to avoid transfusing red blood cells from SCT donors to neonates or patients with sickle cell disease.
Blood from SCT donors can also present a problem when performing the required filtration of white cells from the blood donation.
The PreciseType HEA test will allow these units to be identified prior to filtration and provide blood center staff with the opportunity to decide how best to utilize the various components of a whole blood donation.
The PreciseType HEA test is manufactured by BioArray Solutions, a wholly owned subsidiary of Immucor, Inc.
“We’ve successfully demonstrated the clinical benefits of our PreciseType HEA test, and this is evident in the FDA broadening its approved use,” said Michael Spigarelli, vice president of medical affairs at Immucor.
“The use of PreciseType HEA to screen donor units for patients with sickle cell disease, neonates, or any individual that may require SCT-negative blood provides a great improvement over previously used methods and offers the first FDA-approved molecular method specifically for screening units.”
SCT screening has traditionally been performed by solubility testing of sickle hemoglobin in buffer, but blood centers have been looking for an alternative due to limitations in this method.
According to Immucor, a molecular approach using PreciseType HEA can overcome the throughput limitations and reduce the false-positive rates observed with the traditional SCT screening method.
“We had already validated the PreciseType HEA test for [SCT screening] in our lab,” said Connie Westhoff, PhD, of the New York Blood Center in New York, New York.
“Our previous screening method required manual testing and interpretation of the results and had high false-positive rates. About 1 in 12 minority donors possess the sickle trait, so accurate results are important to us to avoid unnecessary notifications to donors and deferred blood units. We are now able to identify SCT in our donors utilizing the same PreciseType HEA test we are already running on many of our donors without running additional tests.” ![]()
The US Food and Drug Administration (FDA) has approved use of the PreciseType HEA test to screen blood donors for sickle cell trait (SCT).
The test was previously FDA approved for use in determining blood compatibility between donors and transfusion recipients.
The added utility of screening donors for SCT addresses the desire to avoid transfusing red blood cells from SCT donors to neonates or patients with sickle cell disease.
Blood from SCT donors can also present a problem when performing the required filtration of white cells from the blood donation.
The PreciseType HEA test will allow these units to be identified prior to filtration and provide blood center staff with the opportunity to decide how best to utilize the various components of a whole blood donation.
The PreciseType HEA test is manufactured by BioArray Solutions, a wholly owned subsidiary of Immucor, Inc.
“We’ve successfully demonstrated the clinical benefits of our PreciseType HEA test, and this is evident in the FDA broadening its approved use,” said Michael Spigarelli, vice president of medical affairs at Immucor.
“The use of PreciseType HEA to screen donor units for patients with sickle cell disease, neonates, or any individual that may require SCT-negative blood provides a great improvement over previously used methods and offers the first FDA-approved molecular method specifically for screening units.”
SCT screening has traditionally been performed by solubility testing of sickle hemoglobin in buffer, but blood centers have been looking for an alternative due to limitations in this method.
According to Immucor, a molecular approach using PreciseType HEA can overcome the throughput limitations and reduce the false-positive rates observed with the traditional SCT screening method.
“We had already validated the PreciseType HEA test for [SCT screening] in our lab,” said Connie Westhoff, PhD, of the New York Blood Center in New York, New York.
“Our previous screening method required manual testing and interpretation of the results and had high false-positive rates. About 1 in 12 minority donors possess the sickle trait, so accurate results are important to us to avoid unnecessary notifications to donors and deferred blood units. We are now able to identify SCT in our donors utilizing the same PreciseType HEA test we are already running on many of our donors without running additional tests.” ![]()
FDA grants drug orphan designation for CTCL
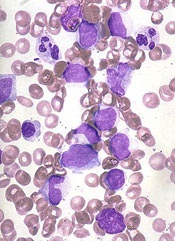
The US Food and Drug Administration (FDA) has granted orphan drug designation to TLC178 for the treatment of cutaneous T-cell lymphoma (CTCL).
TLC178 is a liposomal-encapsulated formulation of the chemotherapy drug vinorelbine, which is FDA approved to treat non-small cell lung cancer.
The goal with TLC178 is to improve the efficacy and decrease the toxicity of vinorelbine to extend the indication beyond solid tumors into lymphoma.
A proprietary technology known as NanoX™ is used to load vinorelbine into liposomes designed to target tumor-specific cell-surface epitopes, extend the circulation time of the drug, increase the concentation of drug delivered to tumor cells, and decrease side effects.
TLC178 is being developed by Taiwan Liposome Company.
The company recently received US FDA approval for its phase 1/2 study (NCT02925000) investigating TLC178 in patients with advanced cancers, including CTCL and other lymphomas.
This trial is planned for sites in Taiwan and the US. The trial will be initiated in Taiwan once approval is granted by the Taiwan FDA.
About orphan designation
The US FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to TLC178 for the treatment of cutaneous T-cell lymphoma (CTCL).
TLC178 is a liposomal-encapsulated formulation of the chemotherapy drug vinorelbine, which is FDA approved to treat non-small cell lung cancer.
The goal with TLC178 is to improve the efficacy and decrease the toxicity of vinorelbine to extend the indication beyond solid tumors into lymphoma.
A proprietary technology known as NanoX™ is used to load vinorelbine into liposomes designed to target tumor-specific cell-surface epitopes, extend the circulation time of the drug, increase the concentation of drug delivered to tumor cells, and decrease side effects.
TLC178 is being developed by Taiwan Liposome Company.
The company recently received US FDA approval for its phase 1/2 study (NCT02925000) investigating TLC178 in patients with advanced cancers, including CTCL and other lymphomas.
This trial is planned for sites in Taiwan and the US. The trial will be initiated in Taiwan once approval is granted by the Taiwan FDA.
About orphan designation
The US FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation to TLC178 for the treatment of cutaneous T-cell lymphoma (CTCL).
TLC178 is a liposomal-encapsulated formulation of the chemotherapy drug vinorelbine, which is FDA approved to treat non-small cell lung cancer.
The goal with TLC178 is to improve the efficacy and decrease the toxicity of vinorelbine to extend the indication beyond solid tumors into lymphoma.
A proprietary technology known as NanoX™ is used to load vinorelbine into liposomes designed to target tumor-specific cell-surface epitopes, extend the circulation time of the drug, increase the concentation of drug delivered to tumor cells, and decrease side effects.
TLC178 is being developed by Taiwan Liposome Company.
The company recently received US FDA approval for its phase 1/2 study (NCT02925000) investigating TLC178 in patients with advanced cancers, including CTCL and other lymphomas.
This trial is planned for sites in Taiwan and the US. The trial will be initiated in Taiwan once approval is granted by the Taiwan FDA.
About orphan designation
The US FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved. ![]()
Resource use lower for patients on dabigatran, data suggest
Photo by ec-jpr
Real-world data suggest that, in the first year of treatment with an anticoagulant, patients with non-valvular atrial fibrillation tend to use fewer healthcare resources if they receive dabigatran rather than warfarin.
Patients treated with dabigatran experienced fewer all-cause hospitalizations, emergency room (ER) visits, and physician office visits than patients treated with warfarin.
These findings were published in The American Journal of Pharmacy Benefits.
The research was supported by Boehringer Ingelheim Pharmaceuticals, Inc., the company that markets dabigatran as Pradaxa.
“While there are many published studies comparing the clinical outcomes of [dabigatran] and warfarin, this is one of the first to compare their respective impact on the use of healthcare resources,” said study author Matthew Sussman, of Boston Health Economics, Inc. in Waltham, Massachusetts.
“Beyond data from clinical studies, it is important for physicians to also understand the experiences patients have in real-world settings, including the economic considerations of their treatment choices.”
Sussman and his colleagues analyzed data on 3890 patients newly diagnosed with non-valvular atrial fibrillation—1945 treated with dabigatran and 1945 treated with warfarin—using de-identified electronic health records from a large, nationwide database of US integrated delivery networks.
Patients in the warfarin cohort were propensity-score matched 1:1 to patients in the dabigatran cohort and were followed up to 1 year after initiating therapy to assess all-cause, stroke-related, and bleed-related healthcare resource use.
The researchers found that dabigatran-treated patients had a significantly lower number of mean per-patient per-year (PPPY) hospitalizations (1.07 vs 1.20, P<0.001), ER visits (0.36 vs 0.51, P<0.001), and physician office visits (10.64 vs 18.13, P<0.001) than patients treated with warfarin.
When it came to stroke-related resource use, dabigatran-treated patients had a significantly lower number of mean PPPY hospitalizations (0.06 vs 0.10, P=0.03) and physician office visits (0.16 vs 0.29, P=0.02). But the difference in ER visits between the dabigatran and warfarin groups was not significant (0 vs 0.01, P=0.65).
As for bleed-related resource use, there was no significant difference between the dabigatran and warfarin groups with regard to the mean number of PPPY hospitalizations (0.05 vs 0. 03, P=0.49) or physician office visits (0.05 vs 0.15, P=0.57). But the difference in ER visits was significant (0.01 vs 0.03, P=0.02). ![]()
Photo by ec-jpr
Real-world data suggest that, in the first year of treatment with an anticoagulant, patients with non-valvular atrial fibrillation tend to use fewer healthcare resources if they receive dabigatran rather than warfarin.
Patients treated with dabigatran experienced fewer all-cause hospitalizations, emergency room (ER) visits, and physician office visits than patients treated with warfarin.
These findings were published in The American Journal of Pharmacy Benefits.
The research was supported by Boehringer Ingelheim Pharmaceuticals, Inc., the company that markets dabigatran as Pradaxa.
“While there are many published studies comparing the clinical outcomes of [dabigatran] and warfarin, this is one of the first to compare their respective impact on the use of healthcare resources,” said study author Matthew Sussman, of Boston Health Economics, Inc. in Waltham, Massachusetts.
“Beyond data from clinical studies, it is important for physicians to also understand the experiences patients have in real-world settings, including the economic considerations of their treatment choices.”
Sussman and his colleagues analyzed data on 3890 patients newly diagnosed with non-valvular atrial fibrillation—1945 treated with dabigatran and 1945 treated with warfarin—using de-identified electronic health records from a large, nationwide database of US integrated delivery networks.
Patients in the warfarin cohort were propensity-score matched 1:1 to patients in the dabigatran cohort and were followed up to 1 year after initiating therapy to assess all-cause, stroke-related, and bleed-related healthcare resource use.
The researchers found that dabigatran-treated patients had a significantly lower number of mean per-patient per-year (PPPY) hospitalizations (1.07 vs 1.20, P<0.001), ER visits (0.36 vs 0.51, P<0.001), and physician office visits (10.64 vs 18.13, P<0.001) than patients treated with warfarin.
When it came to stroke-related resource use, dabigatran-treated patients had a significantly lower number of mean PPPY hospitalizations (0.06 vs 0.10, P=0.03) and physician office visits (0.16 vs 0.29, P=0.02). But the difference in ER visits between the dabigatran and warfarin groups was not significant (0 vs 0.01, P=0.65).
As for bleed-related resource use, there was no significant difference between the dabigatran and warfarin groups with regard to the mean number of PPPY hospitalizations (0.05 vs 0. 03, P=0.49) or physician office visits (0.05 vs 0.15, P=0.57). But the difference in ER visits was significant (0.01 vs 0.03, P=0.02). ![]()
Photo by ec-jpr
Real-world data suggest that, in the first year of treatment with an anticoagulant, patients with non-valvular atrial fibrillation tend to use fewer healthcare resources if they receive dabigatran rather than warfarin.
Patients treated with dabigatran experienced fewer all-cause hospitalizations, emergency room (ER) visits, and physician office visits than patients treated with warfarin.
These findings were published in The American Journal of Pharmacy Benefits.
The research was supported by Boehringer Ingelheim Pharmaceuticals, Inc., the company that markets dabigatran as Pradaxa.
“While there are many published studies comparing the clinical outcomes of [dabigatran] and warfarin, this is one of the first to compare their respective impact on the use of healthcare resources,” said study author Matthew Sussman, of Boston Health Economics, Inc. in Waltham, Massachusetts.
“Beyond data from clinical studies, it is important for physicians to also understand the experiences patients have in real-world settings, including the economic considerations of their treatment choices.”
Sussman and his colleagues analyzed data on 3890 patients newly diagnosed with non-valvular atrial fibrillation—1945 treated with dabigatran and 1945 treated with warfarin—using de-identified electronic health records from a large, nationwide database of US integrated delivery networks.
Patients in the warfarin cohort were propensity-score matched 1:1 to patients in the dabigatran cohort and were followed up to 1 year after initiating therapy to assess all-cause, stroke-related, and bleed-related healthcare resource use.
The researchers found that dabigatran-treated patients had a significantly lower number of mean per-patient per-year (PPPY) hospitalizations (1.07 vs 1.20, P<0.001), ER visits (0.36 vs 0.51, P<0.001), and physician office visits (10.64 vs 18.13, P<0.001) than patients treated with warfarin.
When it came to stroke-related resource use, dabigatran-treated patients had a significantly lower number of mean PPPY hospitalizations (0.06 vs 0.10, P=0.03) and physician office visits (0.16 vs 0.29, P=0.02). But the difference in ER visits between the dabigatran and warfarin groups was not significant (0 vs 0.01, P=0.65).
As for bleed-related resource use, there was no significant difference between the dabigatran and warfarin groups with regard to the mean number of PPPY hospitalizations (0.05 vs 0. 03, P=0.49) or physician office visits (0.05 vs 0.15, P=0.57). But the difference in ER visits was significant (0.01 vs 0.03, P=0.02).
Team identifies genetic hallmarks of B-ALL subtype

Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
Photo courtesy of St. Jude
Children’s Research Hospital
Researchers say they have uncovered a unique paradigm of transcription factor deregulation in B-precursor acute lymphoblastic leukemia (B-ALL).
The team found that deregulation of the homeobox transcription factor gene DUX4 and the ETS transcription factor gene ERG is a hallmark of a subtype of B-ALL that may comprise up to 8% of B-ALL cases.
The researchers reported these findings in Nature Genetics.
“Our work is motivated by a lack of information on the genetic basis of many B-ALL cases,” said study author Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
“We discovered a distinct gene pattern in blood samples from some patients in our study and wanted to determine the underlying molecular events behind this signal.”
The researchers studied a group of 1913 B-ALL patients (including children, adolescents, and young adults) to understand the genetic basis of the disease.
Microarray and transcriptome sequencing revealed that 7.6% of these patients had the distinctive genetic profile the researchers wanted to characterize further.
“Our work revealed that, in this type of B-ALL, there is a sequence of molecular events that involves the interplay of 2 transcription factors,” Dr Mullighan said.
The team observed rearrangement of the gene DUX4 in all cases of this subtype of B-ALL, which resulted in high-level expression of DUX4. DUX4 was shown to bind to the ERG gene, leading to deregulated expression of ERG.
The deregulation of ERG compromised the function of ERG either by deleting part of the gene or by expressing another form of ERG—ERGalt. In both cases, loss of activity was observed for the ERG transcription factor, which led to leukemia.
“These results underscore that there is still more to be learned about the genetic changes in ALL, and that this knowledge can help refine treatment for patients,” said study author Stephen Hunger, MD, of the Children’s Hospital of Philadelphia in Pennsylvania.
The researchers hope identification of the relationships between the 2 transcription factors will lead to new diagnostic tests for patients. DUX4/ERG ALL is linked to favorable outcomes even when other detrimental genetic mutations are present.
Currently, only transcriptome or genome sequencing helps identify the DUX4 rearrangements. The researchers say other detection methods, such as fluorescence hybridization or karyotyping, are not sufficient to recognize genetic changes to DUX4.
FDA clears analyzer for high-volume transfusion labs

Photo courtesy of
PR Newswire and
Ortho Clinical Diagnostics
The US Food and Drug Administration has granted 510(k) clearance for ORTHO VISION® Max, a fully automated blood analyzer for high-volume transfusion medicine laboratories.
Ortho Clinical Diagnostics developed ORTHO VISION Max for labs conducting more than 50 types and screens per day.
ORTHO VISION Max is now commercially available in the US as well as Europe and Japan.
The launch of ORTHO VISION® Max follows the 2015 release of the ORTHO VISION® Analyzer, an instrument designed for small- to mid-sized transfusion labs.
Together, the analyzers form the ORTHO VISION® platform. According to Ortho Clinical Diagnostics, the platform automates more tests than ever before and takes less time to perform those tests.
The platform supports complex immunohematology testing such as serial dilutions for titration studies, reflex tests, and selected cell antibody identification.
The ORTHO VISION platform also has scheduling intelligence, which allows a transfusion medicine department to process routine samples and STAT orders as they are received, rather than waiting for a complete batch before running the instrument.
The platform offers dynamic workflow and lab standardization across instrumentation, technology, procedures, and training. These features are intended to help blood bank labs keep pace with growing industry pressure to increase productivity while remaining operationally efficient.
“Labs are continuously pushed to accomplish more with fewer resources, including staff, and Ortho can now ease those pressures in labs of every size and makeup,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics.
“Whether they perform 15 tests per day or 150, the ORTHO VISION platform helps labs better manage their contribution to the overall critical care path.”
Photo courtesy of
PR Newswire and
Ortho Clinical Diagnostics
The US Food and Drug Administration has granted 510(k) clearance for ORTHO VISION® Max, a fully automated blood analyzer for high-volume transfusion medicine laboratories.
Ortho Clinical Diagnostics developed ORTHO VISION Max for labs conducting more than 50 types and screens per day.
ORTHO VISION Max is now commercially available in the US as well as Europe and Japan.
The launch of ORTHO VISION® Max follows the 2015 release of the ORTHO VISION® Analyzer, an instrument designed for small- to mid-sized transfusion labs.
Together, the analyzers form the ORTHO VISION® platform. According to Ortho Clinical Diagnostics, the platform automates more tests than ever before and takes less time to perform those tests.
The platform supports complex immunohematology testing such as serial dilutions for titration studies, reflex tests, and selected cell antibody identification.
The ORTHO VISION platform also has scheduling intelligence, which allows a transfusion medicine department to process routine samples and STAT orders as they are received, rather than waiting for a complete batch before running the instrument.
The platform offers dynamic workflow and lab standardization across instrumentation, technology, procedures, and training. These features are intended to help blood bank labs keep pace with growing industry pressure to increase productivity while remaining operationally efficient.
“Labs are continuously pushed to accomplish more with fewer resources, including staff, and Ortho can now ease those pressures in labs of every size and makeup,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics.
“Whether they perform 15 tests per day or 150, the ORTHO VISION platform helps labs better manage their contribution to the overall critical care path.”
Photo courtesy of
PR Newswire and
Ortho Clinical Diagnostics
The US Food and Drug Administration has granted 510(k) clearance for ORTHO VISION® Max, a fully automated blood analyzer for high-volume transfusion medicine laboratories.
Ortho Clinical Diagnostics developed ORTHO VISION Max for labs conducting more than 50 types and screens per day.
ORTHO VISION Max is now commercially available in the US as well as Europe and Japan.
The launch of ORTHO VISION® Max follows the 2015 release of the ORTHO VISION® Analyzer, an instrument designed for small- to mid-sized transfusion labs.
Together, the analyzers form the ORTHO VISION® platform. According to Ortho Clinical Diagnostics, the platform automates more tests than ever before and takes less time to perform those tests.
The platform supports complex immunohematology testing such as serial dilutions for titration studies, reflex tests, and selected cell antibody identification.
The ORTHO VISION platform also has scheduling intelligence, which allows a transfusion medicine department to process routine samples and STAT orders as they are received, rather than waiting for a complete batch before running the instrument.
The platform offers dynamic workflow and lab standardization across instrumentation, technology, procedures, and training. These features are intended to help blood bank labs keep pace with growing industry pressure to increase productivity while remaining operationally efficient.
“Labs are continuously pushed to accomplish more with fewer resources, including staff, and Ortho can now ease those pressures in labs of every size and makeup,” said Robert Yates, chief operating officer of Ortho Clinical Diagnostics.
“Whether they perform 15 tests per day or 150, the ORTHO VISION platform helps labs better manage their contribution to the overall critical care path.”
Age of blood doesn’t affect risk of death, study shows

Photo by Elise Amendola
Results of a large, international study suggest the risk of death after transfusion is not significantly affected by the age of blood transfused.
The median storage duration for the fresher blood used in this study was 11 days, and the median storage duration for older blood was 23 days.
The rate of death was 9.1% for the patients who received fresher blood and 8.8% for patients who received older blood (P=0.38).
Researchers reported these and other results from this study in NEJM.
“It’s been a contentious issue, but our study finally puts an end to the question about whether stored blood could be harmful and fresher blood would be better,” said study author Nancy Heddle, of McMaster University in Hamilton, Ontario, Canada.
“Our study provides strong evidence that transfusion of fresh blood does not improve patient outcomes, and this should reassure clinicians that fresher is not better.”
For this study, Heddle and her colleagues enrolled 31,497 adult patients treated at hospitals in Australia, Canada, Israel, and the US. However, 6761 patients did not meet all the enrollment criteria and were excluded after randomization.
A and O blood types
The researchers’ primary analysis included only patients with type A or O blood. Of these 20,858 patients, 6936 were assigned to receive blood stored for a shorter period, and 13,922 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.0 ± 7.6 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-16) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=634) in the short-term storage group and 8.7% (n=1213) in the long-term storage group. The odds ratio was 1.05 (95% CI, 0.95 to 1.16; P=0.34).
All blood types
The researchers also analyzed patients with any blood type. Of the 24,736 patients studied, 8215 were assigned to receive blood stored for a shorter period, and 16,521 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.4 ± 7.7 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-17) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=750) in the short-term storage group and 8.8% (n=1446) in the long-term storage group. The odds ratio was 1.04 (95% CI, 0.95 to 1.14; P=0.38).
The researchers said the overall results were similar to those observed in 3 pre-specified high-risk subgroups—patients undergoing cardiovascular surgery, individuals admitted to intensive care, and patients with cancer.
“Advances in blood storage now allow blood to be stored up to 42 days before transfusion, and the usual practice is to use up the blood that has been in storage the longest,” said study author John Eikelboom, MD, also of McMaster University.
“But because there are biochemical, structural, and functional changes in the blood during storage, there had been concerns about the use of ‘older’ blood. This study reassures us that aging is not bad—even for blood.”
The findings of this study are in line with the recently released AABB recommendations on blood transfusion.
Photo by Elise Amendola
Results of a large, international study suggest the risk of death after transfusion is not significantly affected by the age of blood transfused.
The median storage duration for the fresher blood used in this study was 11 days, and the median storage duration for older blood was 23 days.
The rate of death was 9.1% for the patients who received fresher blood and 8.8% for patients who received older blood (P=0.38).
Researchers reported these and other results from this study in NEJM.
“It’s been a contentious issue, but our study finally puts an end to the question about whether stored blood could be harmful and fresher blood would be better,” said study author Nancy Heddle, of McMaster University in Hamilton, Ontario, Canada.
“Our study provides strong evidence that transfusion of fresh blood does not improve patient outcomes, and this should reassure clinicians that fresher is not better.”
For this study, Heddle and her colleagues enrolled 31,497 adult patients treated at hospitals in Australia, Canada, Israel, and the US. However, 6761 patients did not meet all the enrollment criteria and were excluded after randomization.
A and O blood types
The researchers’ primary analysis included only patients with type A or O blood. Of these 20,858 patients, 6936 were assigned to receive blood stored for a shorter period, and 13,922 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.0 ± 7.6 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-16) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=634) in the short-term storage group and 8.7% (n=1213) in the long-term storage group. The odds ratio was 1.05 (95% CI, 0.95 to 1.16; P=0.34).
All blood types
The researchers also analyzed patients with any blood type. Of the 24,736 patients studied, 8215 were assigned to receive blood stored for a shorter period, and 16,521 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.4 ± 7.7 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-17) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=750) in the short-term storage group and 8.8% (n=1446) in the long-term storage group. The odds ratio was 1.04 (95% CI, 0.95 to 1.14; P=0.38).
The researchers said the overall results were similar to those observed in 3 pre-specified high-risk subgroups—patients undergoing cardiovascular surgery, individuals admitted to intensive care, and patients with cancer.
“Advances in blood storage now allow blood to be stored up to 42 days before transfusion, and the usual practice is to use up the blood that has been in storage the longest,” said study author John Eikelboom, MD, also of McMaster University.
“But because there are biochemical, structural, and functional changes in the blood during storage, there had been concerns about the use of ‘older’ blood. This study reassures us that aging is not bad—even for blood.”
The findings of this study are in line with the recently released AABB recommendations on blood transfusion.
Photo by Elise Amendola
Results of a large, international study suggest the risk of death after transfusion is not significantly affected by the age of blood transfused.
The median storage duration for the fresher blood used in this study was 11 days, and the median storage duration for older blood was 23 days.
The rate of death was 9.1% for the patients who received fresher blood and 8.8% for patients who received older blood (P=0.38).
Researchers reported these and other results from this study in NEJM.
“It’s been a contentious issue, but our study finally puts an end to the question about whether stored blood could be harmful and fresher blood would be better,” said study author Nancy Heddle, of McMaster University in Hamilton, Ontario, Canada.
“Our study provides strong evidence that transfusion of fresh blood does not improve patient outcomes, and this should reassure clinicians that fresher is not better.”
For this study, Heddle and her colleagues enrolled 31,497 adult patients treated at hospitals in Australia, Canada, Israel, and the US. However, 6761 patients did not meet all the enrollment criteria and were excluded after randomization.
A and O blood types
The researchers’ primary analysis included only patients with type A or O blood. Of these 20,858 patients, 6936 were assigned to receive blood stored for a shorter period, and 13,922 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.0 ± 7.6 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-16) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=634) in the short-term storage group and 8.7% (n=1213) in the long-term storage group. The odds ratio was 1.05 (95% CI, 0.95 to 1.16; P=0.34).
All blood types
The researchers also analyzed patients with any blood type. Of the 24,736 patients studied, 8215 were assigned to receive blood stored for a shorter period, and 16,521 were assigned to receive blood stored for a longer period.
The mean storage duration was 13.4 ± 7.7 days in the short-term group and 23.6 ± 8.9 days in the long-term group. The median storage duration was 11 days (range, 8-17) and 23 days (range, 16-31), respectively.
The rate of death was 9.1% (n=750) in the short-term storage group and 8.8% (n=1446) in the long-term storage group. The odds ratio was 1.04 (95% CI, 0.95 to 1.14; P=0.38).
The researchers said the overall results were similar to those observed in 3 pre-specified high-risk subgroups—patients undergoing cardiovascular surgery, individuals admitted to intensive care, and patients with cancer.
“Advances in blood storage now allow blood to be stored up to 42 days before transfusion, and the usual practice is to use up the blood that has been in storage the longest,” said study author John Eikelboom, MD, also of McMaster University.
“But because there are biochemical, structural, and functional changes in the blood during storage, there had been concerns about the use of ‘older’ blood. This study reassures us that aging is not bad—even for blood.”
The findings of this study are in line with the recently released AABB recommendations on blood transfusion.