User login
Docs prescribe drugs despite possible interaction

Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Photo by Rhoda Baer
Physicians may still prescribe a controversial drug combination despite safety concerns, according to a study published in Pharmacology Research & Perspectives.
Regulatory agencies have warned against prescribing the antiplatelet agent clopidogrel with the proton pump inhibitors (PPIs) omeprazole and esomeprazole.
A PPI may be prescribed with clopidogrel to reduce the risk of gastrointestinal bleeding associated with antiplatelet therapy.
However, concomitant use of clopidogrel and esomeprazole/omeprazole is thought by some to reduce the pharmacological activity of clopidogrel.
In 2009 and 2010, regulatory agencies in Europe and the US published statements advising against concomitant use of clopidogrel and the aforementioned PPIs.
Willemien J. Kruik-Kolloffel, PharmD, of Medisch Spectrum Twente in Enschede, The Netherlands, and his colleagues wanted to determine if this recommendation was followed in The Netherlands.
The researchers studied data spanning the period from 2008 to 2011 and encompassing 39,496 patients. Forty percent of the patients did not use gastroprotective drugs at all during the study period.
Twenty-seven percent of patients were taking gastroprotective drugs before starting clopidogrel, 23% started gastroprotective drugs and clopidogrel concomitantly, and 10% started gastroprotective drugs at least 4 weeks after starting clopidogrel.
Among the patients who started a gastroprotective drug and clopidogrel concomitantly, an average of 40% started on esomeprazole/omeprazole before the first statement from a regulatory agency was released in January 2009.
This percentage decreased to around 20% after the statements were released. The percentage of patients starting on other PPIs rose from 60% to about 80%.
After the last statement was released in February 2010, there was an 11.9% decrease in dispensation of omeprazole and esomeprazole and an increase of 16.0% for other PPIs.
Results were similar among the patients who started taking a gastroprotective drug at least 4 weeks after starting clopidogrel.
These data suggest the regulatory agencies’ advice was followed, though not fully. The researchers said this may be, in part, because physicians doubt the suggested interaction between clopidogrel and esomeprazole/omeprazole.
“Regulatory agencies should base their advice on sound scientific data to convince prescribers,” Dr Kruik-Kolloffel said. “We, the authors, doubt the interaction, as do a lot of professionals all around the world.” ![]()
Overcoming drug resistance in malaria
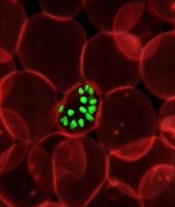
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
infecting a red blood cell
Photo courtesy of St. Jude
Children’s Research Hospital
New research helps explain how one of Plasmodium falciparum’s best weapons against antimalarial drugs can actually be exploited to treat malaria.
Investigators believe the findings, published in PLOS Pathogens, might be used to stop the emergence and spread of drug-resistant malaria.
The team noted that mutations in the P falciparum chloroquine resistance transporter (PfCRT) confer resistance to
chloroquine and related antimalarial drugs by enabling the protein to transport the drugs away from their targets within the parasite’s digestive vacuole.
However, chloroquine resistance-conferring isoforms of PfCRT (PfCRTCQR) also render the parasite hypersensitive to a subset of structurally diverse drugs. And mutations in PfCRTCQR that suppress this hypersensitivity simultaneously reinstate sensitivity to chloroquine and related drugs.
With this study, the investigators uncovered 2 mechanisms by which PfCRT causes P falciparum to become hypersensitive to antimalarial drugs.
First, they found that quinine, which normally exerts its killing effect within the parasite’s digestive vacuole, can bind tightly to certain forms of PfCRT. This blocks the function of the protein, which is essential to the parasite’s survival.
Second, the team found that amantadine, which normally sequesters within the digestive vacuole as well, is leaked back into the cytosol via PfCRT.
The investigators noted that, in both of these cases, mutations that suppress hypersensitivity also revoke PfCRT’s ability to transport chloroquine, which explains why rescue from hypersensitivity restores the parasite’s sensitivity to chloroquine.
“[C]hanges that allow the protein to move chloroquine away from its antimalarial target simultaneously enable the protein to deliver other drugs to their antimalarial targets,” explained study author Rowena Martin, PhD, of Australian National University in Canberra.
“[W]hen the protein adapts itself to fend off one of these drugs, it is no longer able to deal with chloroquine and, hence, the parasite is re-sensitized to chloroquine. Essentially, the parasite can’t have its cake and eat it too. So if chloroquine or a related drug is paired with a drug that is super-active against the modified protein, no matter what the parasite tries to do, it’s ‘checkmate’ for malaria.”
Dr Martin and her colleagues believe their findings provide a foundation for understanding and exploiting the hypersensitivity of chloroquine-resistant parasites to several antimalarial drugs that are currently available.
“Health authorities could use our research to find ways to prolong the lifespan of antimalarial drugs,” said Sashika Richards, a PhD student at Australian National University.
“The current frontline antimalarial drug, artemisinin, is already failing in Asia, and we don’t have anything to replace it. It will be at least 5 years before the next new drug makes it to market. The low-hanging fruit is gone, and it’s now very costly and time-consuming to develop new treatments for malaria.” ![]()
Gene therapy reduces need for FIX prophylaxis

Image courtesy of NIGMS
ORLANDO—The gene therapy AMT-060 can reduce the need for factor IX (FIX) prophylaxis in patients with severe hemophilia B, results of a phase 1/2 study suggest.
All of the patients treated in the low-dose cohort of this study have had sustained improvements in their disease phenotype and continue to maintain durable levels of FIX gene activity for up to 39 weeks post-treatment.
Four of the 5 patients were able to discontinue prophylactic FIX infusions.
In addition, AMT-060 was considered well-tolerated. There were 2 serious adverse events, but both were temporary. And none of the patients developed FIX inhibitors.
These data were presented at the World Federation of Hemophilia 2016 World Congress.* The research is sponsored by uniQure.
“I am very encouraged by the stability of increased FIX activity of AMT-060 and the significant reduction in required infusions of factor replacement,” said study investigator Wolfgang Miesbach, MD, of the University of Frankfurt in Germany.
“This effect is particularly important because it is seen in severe patients with established joint disease who experienced a high frequency of joint bleeds despite intense use of prophylactic FIX prior to study entry.”
Patients and treatment
AMT-060 consists of a codon-optimized wild-type FIX gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform.
In this phase 1/2 trial, Dr Miesbach and his colleagues are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5x1012 gc/kg, and 5 received AMT-060 at 2x1013 gc/kg.
Dr Miesbach presented results observed in the low-dose cohort. Patients in the high-dose cohort are still in the early stages of follow-up.
Most patients in the low-dose cohort were older than 50 years of age (range, 35-72). Four patients had severe hemophilia B, and 4 had advanced joint disease. All of the patients had frequent bleeding episodes, despite receiving once- or twice-weekly FIX prophylaxis.
Efficacy
For all 5 patients in the low-dose cohort, the mean annualized total FIX usage declined 75% after treatment with AMT-060.
“The majority of patients in this low-dose cohort of AMT-060 are showing FIX activity in the range of 5% of normal, and clinical experience has shown that patients in this range generally do not require prophylactic factor replacement and have a very low frequency of spontaneous joint bleeding episodes,” Dr Miesbach said.
Four patients discontinued prophylactic therapy. The 1 patient who remained on prophylactic therapy has sustained an improved disease phenotype and also required materially less FIX concentrate after treatment with AMT-060.
Through up to 9 months of follow-up, the mean steady-state FIX activity for the 4 patients who discontinued prophylactic FIX therapy was 5.4% of normal, with a range from 3.1% to 6.7% of normal. These patients had a mean reduction in annualized total FIX usage of 82%.
Safety and immunogenicity
Two patients experienced serious adverse events. One patient had self-limiting fever in the first 24 hours after receiving AMT-060.
The other patient had a transient elevation of alanine aminotransferase (ALT) that was responsive to tapering prednisolone (60 mg/day start dose) without loss of FIX activity. At baseline, this patient’s ALT was 26 IU/L. It hit a peak of 61 IU/L at week 10, but values returned to baseline levels within 2 weeks of treatment.
As expected, all of the patients developed anti-AAV5 antibodies after week 1. None of the patients developed inhibitory antibodies against FIX.
There was no evidence of sustained AAV5 capsid-specific T-cell activation, although 1 patient had transient T-cell activation slightly above the positive threshold at 1 time point. This patient did not have ALT elevation. ![]()
*Miesbach W et al, Updated results from a dose escalating study in adult patients with haemophilia B treated with AMT-060 (AAV5-hFIX) gene therapy, WFH 2016 World
Congress, July 2016.
Image courtesy of NIGMS
ORLANDO—The gene therapy AMT-060 can reduce the need for factor IX (FIX) prophylaxis in patients with severe hemophilia B, results of a phase 1/2 study suggest.
All of the patients treated in the low-dose cohort of this study have had sustained improvements in their disease phenotype and continue to maintain durable levels of FIX gene activity for up to 39 weeks post-treatment.
Four of the 5 patients were able to discontinue prophylactic FIX infusions.
In addition, AMT-060 was considered well-tolerated. There were 2 serious adverse events, but both were temporary. And none of the patients developed FIX inhibitors.
These data were presented at the World Federation of Hemophilia 2016 World Congress.* The research is sponsored by uniQure.
“I am very encouraged by the stability of increased FIX activity of AMT-060 and the significant reduction in required infusions of factor replacement,” said study investigator Wolfgang Miesbach, MD, of the University of Frankfurt in Germany.
“This effect is particularly important because it is seen in severe patients with established joint disease who experienced a high frequency of joint bleeds despite intense use of prophylactic FIX prior to study entry.”
Patients and treatment
AMT-060 consists of a codon-optimized wild-type FIX gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform.
In this phase 1/2 trial, Dr Miesbach and his colleagues are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5x1012 gc/kg, and 5 received AMT-060 at 2x1013 gc/kg.
Dr Miesbach presented results observed in the low-dose cohort. Patients in the high-dose cohort are still in the early stages of follow-up.
Most patients in the low-dose cohort were older than 50 years of age (range, 35-72). Four patients had severe hemophilia B, and 4 had advanced joint disease. All of the patients had frequent bleeding episodes, despite receiving once- or twice-weekly FIX prophylaxis.
Efficacy
For all 5 patients in the low-dose cohort, the mean annualized total FIX usage declined 75% after treatment with AMT-060.
“The majority of patients in this low-dose cohort of AMT-060 are showing FIX activity in the range of 5% of normal, and clinical experience has shown that patients in this range generally do not require prophylactic factor replacement and have a very low frequency of spontaneous joint bleeding episodes,” Dr Miesbach said.
Four patients discontinued prophylactic therapy. The 1 patient who remained on prophylactic therapy has sustained an improved disease phenotype and also required materially less FIX concentrate after treatment with AMT-060.
Through up to 9 months of follow-up, the mean steady-state FIX activity for the 4 patients who discontinued prophylactic FIX therapy was 5.4% of normal, with a range from 3.1% to 6.7% of normal. These patients had a mean reduction in annualized total FIX usage of 82%.
Safety and immunogenicity
Two patients experienced serious adverse events. One patient had self-limiting fever in the first 24 hours after receiving AMT-060.
The other patient had a transient elevation of alanine aminotransferase (ALT) that was responsive to tapering prednisolone (60 mg/day start dose) without loss of FIX activity. At baseline, this patient’s ALT was 26 IU/L. It hit a peak of 61 IU/L at week 10, but values returned to baseline levels within 2 weeks of treatment.
As expected, all of the patients developed anti-AAV5 antibodies after week 1. None of the patients developed inhibitory antibodies against FIX.
There was no evidence of sustained AAV5 capsid-specific T-cell activation, although 1 patient had transient T-cell activation slightly above the positive threshold at 1 time point. This patient did not have ALT elevation. ![]()
*Miesbach W et al, Updated results from a dose escalating study in adult patients with haemophilia B treated with AMT-060 (AAV5-hFIX) gene therapy, WFH 2016 World
Congress, July 2016.
Image courtesy of NIGMS
ORLANDO—The gene therapy AMT-060 can reduce the need for factor IX (FIX) prophylaxis in patients with severe hemophilia B, results of a phase 1/2 study suggest.
All of the patients treated in the low-dose cohort of this study have had sustained improvements in their disease phenotype and continue to maintain durable levels of FIX gene activity for up to 39 weeks post-treatment.
Four of the 5 patients were able to discontinue prophylactic FIX infusions.
In addition, AMT-060 was considered well-tolerated. There were 2 serious adverse events, but both were temporary. And none of the patients developed FIX inhibitors.
These data were presented at the World Federation of Hemophilia 2016 World Congress.* The research is sponsored by uniQure.
“I am very encouraged by the stability of increased FIX activity of AMT-060 and the significant reduction in required infusions of factor replacement,” said study investigator Wolfgang Miesbach, MD, of the University of Frankfurt in Germany.
“This effect is particularly important because it is seen in severe patients with established joint disease who experienced a high frequency of joint bleeds despite intense use of prophylactic FIX prior to study entry.”
Patients and treatment
AMT-060 consists of a codon-optimized wild-type FIX gene cassette, the LP1 liver promoter, and an AAV5 viral vector manufactured by uniQure using its proprietary insect cell-based technology platform.
In this phase 1/2 trial, Dr Miesbach and his colleagues are testing AMT-060 in 10 patients. All patients had severe or moderately severe hemophilia at baseline, including documented FIX levels less than 1% to 2% of normal, and required chronic infusions of prophylactic or on-demand FIX therapy at the time of enrollment.
Each patient received a 1-time, 30-minute, intravenous dose of AMT-060, without the use of corticosteroids. Five patients received AMT-060 at 5x1012 gc/kg, and 5 received AMT-060 at 2x1013 gc/kg.
Dr Miesbach presented results observed in the low-dose cohort. Patients in the high-dose cohort are still in the early stages of follow-up.
Most patients in the low-dose cohort were older than 50 years of age (range, 35-72). Four patients had severe hemophilia B, and 4 had advanced joint disease. All of the patients had frequent bleeding episodes, despite receiving once- or twice-weekly FIX prophylaxis.
Efficacy
For all 5 patients in the low-dose cohort, the mean annualized total FIX usage declined 75% after treatment with AMT-060.
“The majority of patients in this low-dose cohort of AMT-060 are showing FIX activity in the range of 5% of normal, and clinical experience has shown that patients in this range generally do not require prophylactic factor replacement and have a very low frequency of spontaneous joint bleeding episodes,” Dr Miesbach said.
Four patients discontinued prophylactic therapy. The 1 patient who remained on prophylactic therapy has sustained an improved disease phenotype and also required materially less FIX concentrate after treatment with AMT-060.
Through up to 9 months of follow-up, the mean steady-state FIX activity for the 4 patients who discontinued prophylactic FIX therapy was 5.4% of normal, with a range from 3.1% to 6.7% of normal. These patients had a mean reduction in annualized total FIX usage of 82%.
Safety and immunogenicity
Two patients experienced serious adverse events. One patient had self-limiting fever in the first 24 hours after receiving AMT-060.
The other patient had a transient elevation of alanine aminotransferase (ALT) that was responsive to tapering prednisolone (60 mg/day start dose) without loss of FIX activity. At baseline, this patient’s ALT was 26 IU/L. It hit a peak of 61 IU/L at week 10, but values returned to baseline levels within 2 weeks of treatment.
As expected, all of the patients developed anti-AAV5 antibodies after week 1. None of the patients developed inhibitory antibodies against FIX.
There was no evidence of sustained AAV5 capsid-specific T-cell activation, although 1 patient had transient T-cell activation slightly above the positive threshold at 1 time point. This patient did not have ALT elevation. ![]()
*Miesbach W et al, Updated results from a dose escalating study in adult patients with haemophilia B treated with AMT-060 (AAV5-hFIX) gene therapy, WFH 2016 World
Congress, July 2016.
Microneedle system could replace blood draws, team says

Photo courtesy of
Sahan Ranamukhaarachchi
A new microneedle drug monitoring system could one day replace invasive blood draws, according to researchers.
The system consists of a small, thin patch that is pressed against a patient’s arm during medical treatment and measures drugs in the bloodstream painlessly without drawing any blood.
The tiny projections on this patch resemble hollow cones and don’t pierce the skin like a standard hypodermic needle.
The researchers described this system in Scientific Reports.
“Many groups are researching microneedle technology for painless vaccines and drug delivery,” said study author Sahan Ranamukhaarachchi, a PhD student at the University of British Columbia (UBC) in Vancouver, British Columbia, Canada. “Using them to painlessly monitor drugs is a newer idea.”
The microneedle system Ranamukhaarachchi and his colleagues created was developed to monitor the antibiotic vancomycin. Patients taking vancomycin must be closely monitored because the drug can cause life-threatening side effects, so the patients undergo 3 to 4 blood draws per day.
The researchers discovered they could use fluid found just below the outer layer of skin, instead of blood, to monitor levels of vancomycin in the bloodstream.
The microneedle patch collects a tiny amount of the fluid, less than 1 nL, and a reaction occurs on the inside of the microneedles that can be detected using an optical sensor. This allows the user to quickly determine the concentration of vancomycin.
“This is probably one of the smallest probe volumes ever recorded for a medically relevant analysis,” said study author Urs Häfeli, PhD, of UBC.
This microneedle drug monitoring system was developed out of a research collaboration between Dr Häfeli and Boris Stoeber, PhD, also of UBC. The system is being commercialized by the UBC spin-off Microdermics Inc. ![]()
Photo courtesy of
Sahan Ranamukhaarachchi
A new microneedle drug monitoring system could one day replace invasive blood draws, according to researchers.
The system consists of a small, thin patch that is pressed against a patient’s arm during medical treatment and measures drugs in the bloodstream painlessly without drawing any blood.
The tiny projections on this patch resemble hollow cones and don’t pierce the skin like a standard hypodermic needle.
The researchers described this system in Scientific Reports.
“Many groups are researching microneedle technology for painless vaccines and drug delivery,” said study author Sahan Ranamukhaarachchi, a PhD student at the University of British Columbia (UBC) in Vancouver, British Columbia, Canada. “Using them to painlessly monitor drugs is a newer idea.”
The microneedle system Ranamukhaarachchi and his colleagues created was developed to monitor the antibiotic vancomycin. Patients taking vancomycin must be closely monitored because the drug can cause life-threatening side effects, so the patients undergo 3 to 4 blood draws per day.
The researchers discovered they could use fluid found just below the outer layer of skin, instead of blood, to monitor levels of vancomycin in the bloodstream.
The microneedle patch collects a tiny amount of the fluid, less than 1 nL, and a reaction occurs on the inside of the microneedles that can be detected using an optical sensor. This allows the user to quickly determine the concentration of vancomycin.
“This is probably one of the smallest probe volumes ever recorded for a medically relevant analysis,” said study author Urs Häfeli, PhD, of UBC.
This microneedle drug monitoring system was developed out of a research collaboration between Dr Häfeli and Boris Stoeber, PhD, also of UBC. The system is being commercialized by the UBC spin-off Microdermics Inc. ![]()
Photo courtesy of
Sahan Ranamukhaarachchi
A new microneedle drug monitoring system could one day replace invasive blood draws, according to researchers.
The system consists of a small, thin patch that is pressed against a patient’s arm during medical treatment and measures drugs in the bloodstream painlessly without drawing any blood.
The tiny projections on this patch resemble hollow cones and don’t pierce the skin like a standard hypodermic needle.
The researchers described this system in Scientific Reports.
“Many groups are researching microneedle technology for painless vaccines and drug delivery,” said study author Sahan Ranamukhaarachchi, a PhD student at the University of British Columbia (UBC) in Vancouver, British Columbia, Canada. “Using them to painlessly monitor drugs is a newer idea.”
The microneedle system Ranamukhaarachchi and his colleagues created was developed to monitor the antibiotic vancomycin. Patients taking vancomycin must be closely monitored because the drug can cause life-threatening side effects, so the patients undergo 3 to 4 blood draws per day.
The researchers discovered they could use fluid found just below the outer layer of skin, instead of blood, to monitor levels of vancomycin in the bloodstream.
The microneedle patch collects a tiny amount of the fluid, less than 1 nL, and a reaction occurs on the inside of the microneedles that can be detected using an optical sensor. This allows the user to quickly determine the concentration of vancomycin.
“This is probably one of the smallest probe volumes ever recorded for a medically relevant analysis,” said study author Urs Häfeli, PhD, of UBC.
This microneedle drug monitoring system was developed out of a research collaboration between Dr Häfeli and Boris Stoeber, PhD, also of UBC. The system is being commercialized by the UBC spin-off Microdermics Inc. ![]()
FDA approves reconstitution system for FVIII product
Photo courtesy of Baxalta
The US Food and Drug Administration (FDA) has approved the Baxject III reconstitution system for Adynovate, a pegylated recombinant factor VIII (FVIII) product.
The system is designed to mix a FVIII product with a diluent prior to infusion.
The Baxject III reconstitution system was previously FDA-approved for use with Advate, a recombinant FVIII product.
The latest FDA approval means the system will be available with Adynovate as well.
Adynovate and the diluent will come pre-packaged in the reconstitution system.
The Baxject III reconstitution system with Adynovate will be available to most customers in the fourth quarter of 2016, with a 2 mL diluent for the 250, 500, and 1000 IU potencies; and a 5 mL diluent for the 2000 IU potency.
Adynovate was approved by the FDA in 2015 for use in hemophilia A patients age 12 and older for on-demand treatment and control of bleeding and for prophylaxis to reduce the frequency of bleeding episodes. Full prescribing information is available here.
Advate was first approved by the FDA in 2003. The product is indicated for use in children and adults with hemophilia A for the control and prevention of bleeding episodes, perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes. Full prescribing information is available here.
The Baxject III reconstitution system, Adynovate, and Advate are all products of Baxalta, which is now a part of Shire. ![]()
Photo courtesy of Baxalta
The US Food and Drug Administration (FDA) has approved the Baxject III reconstitution system for Adynovate, a pegylated recombinant factor VIII (FVIII) product.
The system is designed to mix a FVIII product with a diluent prior to infusion.
The Baxject III reconstitution system was previously FDA-approved for use with Advate, a recombinant FVIII product.
The latest FDA approval means the system will be available with Adynovate as well.
Adynovate and the diluent will come pre-packaged in the reconstitution system.
The Baxject III reconstitution system with Adynovate will be available to most customers in the fourth quarter of 2016, with a 2 mL diluent for the 250, 500, and 1000 IU potencies; and a 5 mL diluent for the 2000 IU potency.
Adynovate was approved by the FDA in 2015 for use in hemophilia A patients age 12 and older for on-demand treatment and control of bleeding and for prophylaxis to reduce the frequency of bleeding episodes. Full prescribing information is available here.
Advate was first approved by the FDA in 2003. The product is indicated for use in children and adults with hemophilia A for the control and prevention of bleeding episodes, perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes. Full prescribing information is available here.
The Baxject III reconstitution system, Adynovate, and Advate are all products of Baxalta, which is now a part of Shire. ![]()
Photo courtesy of Baxalta
The US Food and Drug Administration (FDA) has approved the Baxject III reconstitution system for Adynovate, a pegylated recombinant factor VIII (FVIII) product.
The system is designed to mix a FVIII product with a diluent prior to infusion.
The Baxject III reconstitution system was previously FDA-approved for use with Advate, a recombinant FVIII product.
The latest FDA approval means the system will be available with Adynovate as well.
Adynovate and the diluent will come pre-packaged in the reconstitution system.
The Baxject III reconstitution system with Adynovate will be available to most customers in the fourth quarter of 2016, with a 2 mL diluent for the 250, 500, and 1000 IU potencies; and a 5 mL diluent for the 2000 IU potency.
Adynovate was approved by the FDA in 2015 for use in hemophilia A patients age 12 and older for on-demand treatment and control of bleeding and for prophylaxis to reduce the frequency of bleeding episodes. Full prescribing information is available here.
Advate was first approved by the FDA in 2003. The product is indicated for use in children and adults with hemophilia A for the control and prevention of bleeding episodes, perioperative management, and routine prophylaxis to prevent or reduce the frequency of bleeding episodes. Full prescribing information is available here.
The Baxject III reconstitution system, Adynovate, and Advate are all products of Baxalta, which is now a part of Shire. ![]()
HDAC inhibitor granted breakthrough designation
Image by Eric Smith
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the histone deacetylase (HDAC) inhibitor pracinostat to be used in combination with azacitidine to treat newly diagnosed acute myeloid leukemia (AML) patients who are 75 and older or unfit for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough therapy designation for pracinostat is supported by data from a phase 2 study of the HDAC inhibitor in combination with azacitidine in elderly patients with newly diagnosed AML who were not candidates for induction chemotherapy.
Detailed results from this trial were presented at the 20th Congress of the European Hematology Association last year. The research was sponsored by MEI Pharma, the company developing pracinostat.
The study included 50 AML patients who had a median age of 75 (range, 66-84).
The patients received pracinostat at 60 mg orally on days 1, 3, and 5 of each week for 21 days of each 28-day cycle. They received azacitidine subcutaneously or intravenously on days 1-7 or days 1-5 and 8-9 (per site preference) of each 28-day cycle.
According to updated data from MEI Pharma, the complete response rate was 42% (n=21), and the median overall survival was 19.1 months.
The company said these data compare favorably to a phase 3 study of azacitidine (AZA-AML-0011), which showed a median overall survival of 10.4 months with azacitidine alone and a complete response rate of 19.5% in a similar patient population.
The combination of pracinostat and azacitidine was thought to be well tolerated overall, with no unexpected toxicities. The most common grade 3-4 treatment-emergent adverse events included febrile neutropenia, thrombocytopenia, anemia, and fatigue. ![]()
Image by Eric Smith
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the histone deacetylase (HDAC) inhibitor pracinostat to be used in combination with azacitidine to treat newly diagnosed acute myeloid leukemia (AML) patients who are 75 and older or unfit for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough therapy designation for pracinostat is supported by data from a phase 2 study of the HDAC inhibitor in combination with azacitidine in elderly patients with newly diagnosed AML who were not candidates for induction chemotherapy.
Detailed results from this trial were presented at the 20th Congress of the European Hematology Association last year. The research was sponsored by MEI Pharma, the company developing pracinostat.
The study included 50 AML patients who had a median age of 75 (range, 66-84).
The patients received pracinostat at 60 mg orally on days 1, 3, and 5 of each week for 21 days of each 28-day cycle. They received azacitidine subcutaneously or intravenously on days 1-7 or days 1-5 and 8-9 (per site preference) of each 28-day cycle.
According to updated data from MEI Pharma, the complete response rate was 42% (n=21), and the median overall survival was 19.1 months.
The company said these data compare favorably to a phase 3 study of azacitidine (AZA-AML-0011), which showed a median overall survival of 10.4 months with azacitidine alone and a complete response rate of 19.5% in a similar patient population.
The combination of pracinostat and azacitidine was thought to be well tolerated overall, with no unexpected toxicities. The most common grade 3-4 treatment-emergent adverse events included febrile neutropenia, thrombocytopenia, anemia, and fatigue. ![]()
Image by Eric Smith
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for the histone deacetylase (HDAC) inhibitor pracinostat to be used in combination with azacitidine to treat newly diagnosed acute myeloid leukemia (AML) patients who are 75 and older or unfit for intensive chemotherapy.
The FDA’s breakthrough designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
The breakthrough therapy designation for pracinostat is supported by data from a phase 2 study of the HDAC inhibitor in combination with azacitidine in elderly patients with newly diagnosed AML who were not candidates for induction chemotherapy.
Detailed results from this trial were presented at the 20th Congress of the European Hematology Association last year. The research was sponsored by MEI Pharma, the company developing pracinostat.
The study included 50 AML patients who had a median age of 75 (range, 66-84).
The patients received pracinostat at 60 mg orally on days 1, 3, and 5 of each week for 21 days of each 28-day cycle. They received azacitidine subcutaneously or intravenously on days 1-7 or days 1-5 and 8-9 (per site preference) of each 28-day cycle.
According to updated data from MEI Pharma, the complete response rate was 42% (n=21), and the median overall survival was 19.1 months.
The company said these data compare favorably to a phase 3 study of azacitidine (AZA-AML-0011), which showed a median overall survival of 10.4 months with azacitidine alone and a complete response rate of 19.5% in a similar patient population.
The combination of pracinostat and azacitidine was thought to be well tolerated overall, with no unexpected toxicities. The most common grade 3-4 treatment-emergent adverse events included febrile neutropenia, thrombocytopenia, anemia, and fatigue. ![]()
Gene therapy shows promise for severe hemophilia A

Image by Spencer Phillips
ORLANDO—An investigational gene therapy can safely reduce bleeding in patients with severe hemophilia A, a phase 1/2 study suggests.
The therapy is BMN 270, a recombinant adeno-associated virus (AAV) vector coding for human coagulation factor VIII (FVIII).
Six of the 7 patients treated with the highest dose of BMN 270 had FVIII levels above 50%, and the number of bleeding events fell substantially from baseline.
None of the patients developed inhibitors to FVIII, there were no serious adverse events, and none of the patients discontinued the therapy due to safety reasons.
John Pasi, PhD, of Barts and the London School of Medicine and Dentistry in London, UK, presented the results of this study in a late-breaking oral presentation at the World Federation of Hemophilia 2016 World Congress.* The research was funded by BioMarin Pharmaceutical Inc.
This phase 1/2 dose-escalation study was designed to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
The primary endpoints are to assess the safety of a single dose of BMN 270 and the change from baseline of FVIII expression level at 16 weeks after infusion.
Secondary endpoints include assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses. Patients will be monitored for safety and durability of effect for 5 years.
Thus far, 9 patients with severe hemophilia A have received a single dose of BMN 270—1 at 6×1012 vg/kg, 1 at 2×1013 vg/kg, and 7 at 6 x 1013 vg/kg.
As of the July 6 data cutoff, post-treatment follow-up ranges from 12 weeks to 28 weeks.
Safety
The most common adverse events were arthralgia (9 events in 6 subjects), contusion (6 events in 3 subjects), back pain (4 events in 3 subjects), and ALT elevation (6 events in 6 subjects).
No clinically relevant sustained rises in ALT levels or other markers of liver toxicity have been observed.
The maximum ALT levels were between 23 U/L and 82 U/L (less than 2 times the upper limit of normal, which is 43 U/L for the central laboratory in this study) approximately 12 weeks after gene delivery and generally declined over the next few weeks. ALT rises have not been associated with any decrease in FVIII levels.
A steroid regimen administered to all high-dose patients has been well-tolerated. Patients are successfully tapering off of steroids. Two patients have been off steroid therapy for up to 2.5 weeks, with no adverse impact on FVIII expression or ALT levels.
Efficacy
The patient treated at the lowest dose (6×1012 vg/kg) had no change from baseline in FVIII levels. The patient treated at the mid-dose (2×1013 vg/kg) had a stable FVIII activity level greater than 2 IU/dL for more than 28 weeks.
All 7 patients treated at the highest dose (6×1013 vg/kg) had FVIII activity levels greater than 10 IU/dL after week 10.
As of each patient’s most recent reading, 6 of the 7 patients in the high-dose group had FVIII levels above 50%, as a percentage calculated based on the numbers of IU/dL. The seventh patient had levels above 10%.
Four patients who have been followed the longest had a mean FVIII level of 146% at their 20-week visit. Two patients with FVIII levels above 200% had no unexpected events or need for medical intervention.
For the 7 patients treated at the high dose, the median annualized bleeding rate measured from the day of gene transfer to the data cutoff fell from 20 to 5.
After week 7 post-infusion, there were no bleeds in 6 of the 7 patients. There were 10 bleeds from weeks 0 through 2 post-infusion, 7 bleeds from weeks 3 through 8, and 2 bleeds from weeks 9 through 28. From weeks 2 through 28, all but 1 bleed occurred in a single subject who is the lowest responder.
All of the patients in the high-dose cohort have switched to receiving FVIII therapy on-demand. Six of them were previously receiving FVIII therapy as prophylaxis.
“These data provide strong proof-of-concept evidence that restoration of clotting function may be achieved by gene therapy,” Dr Pasi said. “For the first time, patients have reason to hope to avoid bleeding and the opportunity to live a normal life.” ![]()
Image by Spencer Phillips
ORLANDO—An investigational gene therapy can safely reduce bleeding in patients with severe hemophilia A, a phase 1/2 study suggests.
The therapy is BMN 270, a recombinant adeno-associated virus (AAV) vector coding for human coagulation factor VIII (FVIII).
Six of the 7 patients treated with the highest dose of BMN 270 had FVIII levels above 50%, and the number of bleeding events fell substantially from baseline.
None of the patients developed inhibitors to FVIII, there were no serious adverse events, and none of the patients discontinued the therapy due to safety reasons.
John Pasi, PhD, of Barts and the London School of Medicine and Dentistry in London, UK, presented the results of this study in a late-breaking oral presentation at the World Federation of Hemophilia 2016 World Congress.* The research was funded by BioMarin Pharmaceutical Inc.
This phase 1/2 dose-escalation study was designed to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
The primary endpoints are to assess the safety of a single dose of BMN 270 and the change from baseline of FVIII expression level at 16 weeks after infusion.
Secondary endpoints include assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses. Patients will be monitored for safety and durability of effect for 5 years.
Thus far, 9 patients with severe hemophilia A have received a single dose of BMN 270—1 at 6×1012 vg/kg, 1 at 2×1013 vg/kg, and 7 at 6 x 1013 vg/kg.
As of the July 6 data cutoff, post-treatment follow-up ranges from 12 weeks to 28 weeks.
Safety
The most common adverse events were arthralgia (9 events in 6 subjects), contusion (6 events in 3 subjects), back pain (4 events in 3 subjects), and ALT elevation (6 events in 6 subjects).
No clinically relevant sustained rises in ALT levels or other markers of liver toxicity have been observed.
The maximum ALT levels were between 23 U/L and 82 U/L (less than 2 times the upper limit of normal, which is 43 U/L for the central laboratory in this study) approximately 12 weeks after gene delivery and generally declined over the next few weeks. ALT rises have not been associated with any decrease in FVIII levels.
A steroid regimen administered to all high-dose patients has been well-tolerated. Patients are successfully tapering off of steroids. Two patients have been off steroid therapy for up to 2.5 weeks, with no adverse impact on FVIII expression or ALT levels.
Efficacy
The patient treated at the lowest dose (6×1012 vg/kg) had no change from baseline in FVIII levels. The patient treated at the mid-dose (2×1013 vg/kg) had a stable FVIII activity level greater than 2 IU/dL for more than 28 weeks.
All 7 patients treated at the highest dose (6×1013 vg/kg) had FVIII activity levels greater than 10 IU/dL after week 10.
As of each patient’s most recent reading, 6 of the 7 patients in the high-dose group had FVIII levels above 50%, as a percentage calculated based on the numbers of IU/dL. The seventh patient had levels above 10%.
Four patients who have been followed the longest had a mean FVIII level of 146% at their 20-week visit. Two patients with FVIII levels above 200% had no unexpected events or need for medical intervention.
For the 7 patients treated at the high dose, the median annualized bleeding rate measured from the day of gene transfer to the data cutoff fell from 20 to 5.
After week 7 post-infusion, there were no bleeds in 6 of the 7 patients. There were 10 bleeds from weeks 0 through 2 post-infusion, 7 bleeds from weeks 3 through 8, and 2 bleeds from weeks 9 through 28. From weeks 2 through 28, all but 1 bleed occurred in a single subject who is the lowest responder.
All of the patients in the high-dose cohort have switched to receiving FVIII therapy on-demand. Six of them were previously receiving FVIII therapy as prophylaxis.
“These data provide strong proof-of-concept evidence that restoration of clotting function may be achieved by gene therapy,” Dr Pasi said. “For the first time, patients have reason to hope to avoid bleeding and the opportunity to live a normal life.” ![]()
Image by Spencer Phillips
ORLANDO—An investigational gene therapy can safely reduce bleeding in patients with severe hemophilia A, a phase 1/2 study suggests.
The therapy is BMN 270, a recombinant adeno-associated virus (AAV) vector coding for human coagulation factor VIII (FVIII).
Six of the 7 patients treated with the highest dose of BMN 270 had FVIII levels above 50%, and the number of bleeding events fell substantially from baseline.
None of the patients developed inhibitors to FVIII, there were no serious adverse events, and none of the patients discontinued the therapy due to safety reasons.
John Pasi, PhD, of Barts and the London School of Medicine and Dentistry in London, UK, presented the results of this study in a late-breaking oral presentation at the World Federation of Hemophilia 2016 World Congress.* The research was funded by BioMarin Pharmaceutical Inc.
This phase 1/2 dose-escalation study was designed to evaluate the safety and efficacy of BMN 270 in up to 12 patients with severe hemophilia A.
The primary endpoints are to assess the safety of a single dose of BMN 270 and the change from baseline of FVIII expression level at 16 weeks after infusion.
Secondary endpoints include assessing the impact of BMN 270 on the frequency of FVIII replacement therapy, the number of bleeding episodes requiring treatment, and any potential immune responses. Patients will be monitored for safety and durability of effect for 5 years.
Thus far, 9 patients with severe hemophilia A have received a single dose of BMN 270—1 at 6×1012 vg/kg, 1 at 2×1013 vg/kg, and 7 at 6 x 1013 vg/kg.
As of the July 6 data cutoff, post-treatment follow-up ranges from 12 weeks to 28 weeks.
Safety
The most common adverse events were arthralgia (9 events in 6 subjects), contusion (6 events in 3 subjects), back pain (4 events in 3 subjects), and ALT elevation (6 events in 6 subjects).
No clinically relevant sustained rises in ALT levels or other markers of liver toxicity have been observed.
The maximum ALT levels were between 23 U/L and 82 U/L (less than 2 times the upper limit of normal, which is 43 U/L for the central laboratory in this study) approximately 12 weeks after gene delivery and generally declined over the next few weeks. ALT rises have not been associated with any decrease in FVIII levels.
A steroid regimen administered to all high-dose patients has been well-tolerated. Patients are successfully tapering off of steroids. Two patients have been off steroid therapy for up to 2.5 weeks, with no adverse impact on FVIII expression or ALT levels.
Efficacy
The patient treated at the lowest dose (6×1012 vg/kg) had no change from baseline in FVIII levels. The patient treated at the mid-dose (2×1013 vg/kg) had a stable FVIII activity level greater than 2 IU/dL for more than 28 weeks.
All 7 patients treated at the highest dose (6×1013 vg/kg) had FVIII activity levels greater than 10 IU/dL after week 10.
As of each patient’s most recent reading, 6 of the 7 patients in the high-dose group had FVIII levels above 50%, as a percentage calculated based on the numbers of IU/dL. The seventh patient had levels above 10%.
Four patients who have been followed the longest had a mean FVIII level of 146% at their 20-week visit. Two patients with FVIII levels above 200% had no unexpected events or need for medical intervention.
For the 7 patients treated at the high dose, the median annualized bleeding rate measured from the day of gene transfer to the data cutoff fell from 20 to 5.
After week 7 post-infusion, there were no bleeds in 6 of the 7 patients. There were 10 bleeds from weeks 0 through 2 post-infusion, 7 bleeds from weeks 3 through 8, and 2 bleeds from weeks 9 through 28. From weeks 2 through 28, all but 1 bleed occurred in a single subject who is the lowest responder.
All of the patients in the high-dose cohort have switched to receiving FVIII therapy on-demand. Six of them were previously receiving FVIII therapy as prophylaxis.
“These data provide strong proof-of-concept evidence that restoration of clotting function may be achieved by gene therapy,” Dr Pasi said. “For the first time, patients have reason to hope to avoid bleeding and the opportunity to live a normal life.”
Molecule reverses effects of anticoagulants

A genetically engineered coagulation factor can reverse the effects of direct oral anticoagulants in vitro and in vivo, according to research published in Nature Medicine.
Researchers altered the shape of the coagulation factor, factor Xa (FXa), into a variant that appears to be more potent and longer-lasting than wild-type FXa.
The team found this variant, FXaI16L, could counteract the effects of rivaroxaban, apixaban, and dabigatran.
“This molecule holds the potential to fill an important unmet clinical need,” said study author Rodney A. Camire, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“There are limited treatment options to stop uncontrolled bleeding in patients who are using the newer anticoagulant medications.”
Dr Camire and his colleagues first found that FXaI16L could reverse the effects of rivaroxaban and apixaban in vitro, increasing peak thrombin generation to near-normal levels.
The team then showed that FXaI16L restores hemostasis in mice treated with rivaroxaban and significantly decreases blood loss in injured mice treated with the drug.
FXaI16L also significantly decreased blood loss in injured mice treated with dabigatran.
FXaI16L was more than 50 times more potent in the hemostasis models tested than andexanet alfa, a FXa inhibitor antidote currently in clinical development.
The researchers said FXaI16L’s ability to reverse the effects of anticoagulants depends, at least partly, on the ability of the active site inhibitor to hinder antithrombin-dependent FXa inactivation, which allows uninhibited FXa to persist in plasma.
“Our next steps will be to test this approach in large animals to help determine whether this variant is effective and safe and may progress to clinical trials,” Dr Camire said. “If so, we may be able to develop an important treatment to rapidly control bleeding in both children and adults.”
A genetically engineered coagulation factor can reverse the effects of direct oral anticoagulants in vitro and in vivo, according to research published in Nature Medicine.
Researchers altered the shape of the coagulation factor, factor Xa (FXa), into a variant that appears to be more potent and longer-lasting than wild-type FXa.
The team found this variant, FXaI16L, could counteract the effects of rivaroxaban, apixaban, and dabigatran.
“This molecule holds the potential to fill an important unmet clinical need,” said study author Rodney A. Camire, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“There are limited treatment options to stop uncontrolled bleeding in patients who are using the newer anticoagulant medications.”
Dr Camire and his colleagues first found that FXaI16L could reverse the effects of rivaroxaban and apixaban in vitro, increasing peak thrombin generation to near-normal levels.
The team then showed that FXaI16L restores hemostasis in mice treated with rivaroxaban and significantly decreases blood loss in injured mice treated with the drug.
FXaI16L also significantly decreased blood loss in injured mice treated with dabigatran.
FXaI16L was more than 50 times more potent in the hemostasis models tested than andexanet alfa, a FXa inhibitor antidote currently in clinical development.
The researchers said FXaI16L’s ability to reverse the effects of anticoagulants depends, at least partly, on the ability of the active site inhibitor to hinder antithrombin-dependent FXa inactivation, which allows uninhibited FXa to persist in plasma.
“Our next steps will be to test this approach in large animals to help determine whether this variant is effective and safe and may progress to clinical trials,” Dr Camire said. “If so, we may be able to develop an important treatment to rapidly control bleeding in both children and adults.”
A genetically engineered coagulation factor can reverse the effects of direct oral anticoagulants in vitro and in vivo, according to research published in Nature Medicine.
Researchers altered the shape of the coagulation factor, factor Xa (FXa), into a variant that appears to be more potent and longer-lasting than wild-type FXa.
The team found this variant, FXaI16L, could counteract the effects of rivaroxaban, apixaban, and dabigatran.
“This molecule holds the potential to fill an important unmet clinical need,” said study author Rodney A. Camire, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
“There are limited treatment options to stop uncontrolled bleeding in patients who are using the newer anticoagulant medications.”
Dr Camire and his colleagues first found that FXaI16L could reverse the effects of rivaroxaban and apixaban in vitro, increasing peak thrombin generation to near-normal levels.
The team then showed that FXaI16L restores hemostasis in mice treated with rivaroxaban and significantly decreases blood loss in injured mice treated with the drug.
FXaI16L also significantly decreased blood loss in injured mice treated with dabigatran.
FXaI16L was more than 50 times more potent in the hemostasis models tested than andexanet alfa, a FXa inhibitor antidote currently in clinical development.
The researchers said FXaI16L’s ability to reverse the effects of anticoagulants depends, at least partly, on the ability of the active site inhibitor to hinder antithrombin-dependent FXa inactivation, which allows uninhibited FXa to persist in plasma.
“Our next steps will be to test this approach in large animals to help determine whether this variant is effective and safe and may progress to clinical trials,” Dr Camire said. “If so, we may be able to develop an important treatment to rapidly control bleeding in both children and adults.”
Targeted conjugate therapy kills ALL cells

Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals.
Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals.
Photo courtesy of the
University of California Davis
Researchers say they have developed a targeted conjugate therapy that harnesses a monoclonal antibody to deliver antisense DNA to acute lymphoblastic leukemia (ALL) cells.
Once delivered, the therapeutic DNA reduces levels of MXD3, a protein that helps cancer cells survive.
This conjugate therapy proved cytotoxic in ALL cell lines and showed promise in animal models, destroying ALL cells while limiting other damage.
“We’ve shown, for the first time, that anti-CD22 antibody-antisense conjugates are a potential therapeutic agent for ALL,” said Noriko Satake, MD, of the University of California Davis in Sacramento.
“This could be a new type of treatment that kills leukemia cells with few side effects.”
Dr Satake and her colleagues described the treatment in Molecular Medicine.
To create the therapy, the researchers attached antisense DNA that inhibits the MXD3 protein to an antibody that binds to CD22, a protein receptor expressed almost exclusively on ALL cells and normal B cells.
Once the antibody binds to CD22, the conjugate is drawn inside the cell, allowing the antisense molecule to prevent MXD3 production. Without this anti-apoptotic protein, cells are more prone to death.
The conjugate therapy was effective against ALL cell lines and primary ALL cells in a xenograft mouse model. Animals that received the therapy survived significantly longer than those in the control group.
While the conjugate therapy does target healthy B cells along with ALL cells, it is expected to leave hematopoietic stem cells and other tissues unharmed.
“Our novel conjugate is designed so that it does not harm hair, eyes, heart, kidneys, or other types of cells,” Dr Satake said.
She and her colleagues noted that, although this study shows the conjugate therapy can knock down MXD3, it is not clear exactly how this is accomplished. So the researchers plan to investigate the mechanism.
The team also plans to look into combining the conjugate therapy with other treatments. Because it hastens cell death, the conjugate could make traditional chemotherapy drugs more effective, and it might work against other cancers.
“You can see this as proof of principle,” Dr Satake said. “You could switch the target and substitute the antibody, which could be used to treat other cancers or even other diseases.”
This study was not industry-funded, but 4 study authors are employees and stockholders of Isis Pharmaceuticals.
Local Zika transmission and the US blood supply

Photo courtesy of
Muhammad Mahdi Karim
Officials have announced what is likely the first known occurrence of local mosquito-borne Zika virus transmission in the continental US.*
Fourteen cases of Zika virus in 2 Florida counties are believed to have been caused by bites of local Aedes aegypti mosquitoes.
The Florida Department of Health (DOH) believes that active transmission of the Zika virus is only occurring in a small area in Miami-Dade County.
The US Centers for Disease Control and Prevention (CDC) has recommended that women who are pregnant or thinking of becoming pregnant avoid unnecessary travel to the impacted area. The agency has also issued a guidance for people living in or traveling to the area.
“All the evidence we have seen indicates that this is mosquito-borne transmission that occurred several weeks ago in several blocks in Miami,” said Tom Frieden, MD, director of the CDC.
The exact location is within the boundaries of the following area: NW 5th Avenue to the west, US 1 to the east, NW/NE 38th Street to the north and NW/NE 20th Street to the south. This is about one square mile.
Protecting the blood supply
The US Food and Drug Administration (FDA) has requested that all blood centers in Miami-Dade and Broward counties stop collecting blood immediately.
Blood centers in these counties can resume blood collection once they begin testing each unit of blood with an available investigational donor screening test for Zika virus RNA or once they implement the use of an approved or investigational pathogen inactivation technology.
The FDA also recommended that blood centers in nearby counties implement the same precautions as soon as possible to help maintain the safety of the blood supply. The agency has encouraged screening of the blood supply in regions of the US at risk of local mosquito-borne Zika transmission.
The FDA said it is working with companies that are making blood screening tests available under an Investigational New Drug (IND) application to ensure these companies are ready to expand testing as needed. Blood collection centers may choose to participate in testing under an IND even in the absence of local mosquito-borne transmission of Zika virus.
Florida/CDC response
Florida state officials have implemented mosquito control measures and a community-wide search for additional Zika cases. Thus far, Florida’s DOH has conducted testing for the Zika virus in more than 2300 people statewide.
The DOH has activated the Joint Information Center within the State Emergency Operations Center to ensure the area impacted by local transmission of the Zika virus has coordinated access to information and resources.
The DOH has also begun the process of contracting with commercial pest control companies to enhance and expand mosquito mitigation and abatement, including increased spraying, in the impacted area.
Earlier this year, Florida’s governor, Rick Scott, directed the State Surgeon General to activate a Zika Virus Information Hotline for Florida residents and visitors. The number for this hotline is 1-855-622-6735.
The CDC said it is coordinating with Florida officials leading the ongoing investigation into local transmission of the Zika virus. At the state’s request, the CDC sent a medical epidemiologist to provide additional assistance.
Governor Scott has also asked the CDC to activate a CDC Emergency Response Team to assist the DOH and other partners in their investigation, sample collection, and mosquito control efforts.
To date, the CDC has provided Florida with more than $8 million in Zika-specific funding and about $27 million in emergency preparedness funding that can be used for Zika response efforts.
“We have been working with state and local governments to prepare for the likelihood of local mosquito-borne Zika virus transmission in the continental United States and Hawaii,” said Lyle Petersen, MD, incident manager for CDC’s Zika virus response.
“We anticipate that there may be additional cases of ‘homegrown’ Zika in the coming weeks. Our top priority is to protect pregnant women from the potentially devastating harm caused by Zika.”
For more information about the Zika virus, visit http://www.cdc.gov/zika/.
*This story was updated on August 1.
Photo courtesy of
Muhammad Mahdi Karim
Officials have announced what is likely the first known occurrence of local mosquito-borne Zika virus transmission in the continental US.*
Fourteen cases of Zika virus in 2 Florida counties are believed to have been caused by bites of local Aedes aegypti mosquitoes.
The Florida Department of Health (DOH) believes that active transmission of the Zika virus is only occurring in a small area in Miami-Dade County.
The US Centers for Disease Control and Prevention (CDC) has recommended that women who are pregnant or thinking of becoming pregnant avoid unnecessary travel to the impacted area. The agency has also issued a guidance for people living in or traveling to the area.
“All the evidence we have seen indicates that this is mosquito-borne transmission that occurred several weeks ago in several blocks in Miami,” said Tom Frieden, MD, director of the CDC.
The exact location is within the boundaries of the following area: NW 5th Avenue to the west, US 1 to the east, NW/NE 38th Street to the north and NW/NE 20th Street to the south. This is about one square mile.
Protecting the blood supply
The US Food and Drug Administration (FDA) has requested that all blood centers in Miami-Dade and Broward counties stop collecting blood immediately.
Blood centers in these counties can resume blood collection once they begin testing each unit of blood with an available investigational donor screening test for Zika virus RNA or once they implement the use of an approved or investigational pathogen inactivation technology.
The FDA also recommended that blood centers in nearby counties implement the same precautions as soon as possible to help maintain the safety of the blood supply. The agency has encouraged screening of the blood supply in regions of the US at risk of local mosquito-borne Zika transmission.
The FDA said it is working with companies that are making blood screening tests available under an Investigational New Drug (IND) application to ensure these companies are ready to expand testing as needed. Blood collection centers may choose to participate in testing under an IND even in the absence of local mosquito-borne transmission of Zika virus.
Florida/CDC response
Florida state officials have implemented mosquito control measures and a community-wide search for additional Zika cases. Thus far, Florida’s DOH has conducted testing for the Zika virus in more than 2300 people statewide.
The DOH has activated the Joint Information Center within the State Emergency Operations Center to ensure the area impacted by local transmission of the Zika virus has coordinated access to information and resources.
The DOH has also begun the process of contracting with commercial pest control companies to enhance and expand mosquito mitigation and abatement, including increased spraying, in the impacted area.
Earlier this year, Florida’s governor, Rick Scott, directed the State Surgeon General to activate a Zika Virus Information Hotline for Florida residents and visitors. The number for this hotline is 1-855-622-6735.
The CDC said it is coordinating with Florida officials leading the ongoing investigation into local transmission of the Zika virus. At the state’s request, the CDC sent a medical epidemiologist to provide additional assistance.
Governor Scott has also asked the CDC to activate a CDC Emergency Response Team to assist the DOH and other partners in their investigation, sample collection, and mosquito control efforts.
To date, the CDC has provided Florida with more than $8 million in Zika-specific funding and about $27 million in emergency preparedness funding that can be used for Zika response efforts.
“We have been working with state and local governments to prepare for the likelihood of local mosquito-borne Zika virus transmission in the continental United States and Hawaii,” said Lyle Petersen, MD, incident manager for CDC’s Zika virus response.
“We anticipate that there may be additional cases of ‘homegrown’ Zika in the coming weeks. Our top priority is to protect pregnant women from the potentially devastating harm caused by Zika.”
For more information about the Zika virus, visit http://www.cdc.gov/zika/.
*This story was updated on August 1.
Photo courtesy of
Muhammad Mahdi Karim
Officials have announced what is likely the first known occurrence of local mosquito-borne Zika virus transmission in the continental US.*
Fourteen cases of Zika virus in 2 Florida counties are believed to have been caused by bites of local Aedes aegypti mosquitoes.
The Florida Department of Health (DOH) believes that active transmission of the Zika virus is only occurring in a small area in Miami-Dade County.
The US Centers for Disease Control and Prevention (CDC) has recommended that women who are pregnant or thinking of becoming pregnant avoid unnecessary travel to the impacted area. The agency has also issued a guidance for people living in or traveling to the area.
“All the evidence we have seen indicates that this is mosquito-borne transmission that occurred several weeks ago in several blocks in Miami,” said Tom Frieden, MD, director of the CDC.
The exact location is within the boundaries of the following area: NW 5th Avenue to the west, US 1 to the east, NW/NE 38th Street to the north and NW/NE 20th Street to the south. This is about one square mile.
Protecting the blood supply
The US Food and Drug Administration (FDA) has requested that all blood centers in Miami-Dade and Broward counties stop collecting blood immediately.
Blood centers in these counties can resume blood collection once they begin testing each unit of blood with an available investigational donor screening test for Zika virus RNA or once they implement the use of an approved or investigational pathogen inactivation technology.
The FDA also recommended that blood centers in nearby counties implement the same precautions as soon as possible to help maintain the safety of the blood supply. The agency has encouraged screening of the blood supply in regions of the US at risk of local mosquito-borne Zika transmission.
The FDA said it is working with companies that are making blood screening tests available under an Investigational New Drug (IND) application to ensure these companies are ready to expand testing as needed. Blood collection centers may choose to participate in testing under an IND even in the absence of local mosquito-borne transmission of Zika virus.
Florida/CDC response
Florida state officials have implemented mosquito control measures and a community-wide search for additional Zika cases. Thus far, Florida’s DOH has conducted testing for the Zika virus in more than 2300 people statewide.
The DOH has activated the Joint Information Center within the State Emergency Operations Center to ensure the area impacted by local transmission of the Zika virus has coordinated access to information and resources.
The DOH has also begun the process of contracting with commercial pest control companies to enhance and expand mosquito mitigation and abatement, including increased spraying, in the impacted area.
Earlier this year, Florida’s governor, Rick Scott, directed the State Surgeon General to activate a Zika Virus Information Hotline for Florida residents and visitors. The number for this hotline is 1-855-622-6735.
The CDC said it is coordinating with Florida officials leading the ongoing investigation into local transmission of the Zika virus. At the state’s request, the CDC sent a medical epidemiologist to provide additional assistance.
Governor Scott has also asked the CDC to activate a CDC Emergency Response Team to assist the DOH and other partners in their investigation, sample collection, and mosquito control efforts.
To date, the CDC has provided Florida with more than $8 million in Zika-specific funding and about $27 million in emergency preparedness funding that can be used for Zika response efforts.
“We have been working with state and local governments to prepare for the likelihood of local mosquito-borne Zika virus transmission in the continental United States and Hawaii,” said Lyle Petersen, MD, incident manager for CDC’s Zika virus response.
“We anticipate that there may be additional cases of ‘homegrown’ Zika in the coming weeks. Our top priority is to protect pregnant women from the potentially devastating harm caused by Zika.”
For more information about the Zika virus, visit http://www.cdc.gov/zika/.
*This story was updated on August 1.