User login
COMP recommends orphan status for drug to treat PNH

The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
The European Medicines Agency’s Committee for Orphan Medicinal Products (COMP) has issued a positive opinion recommending orphan designation for Coversin for the treatment of paroxysmal nocturnal hemoglobinuria (PNH).
Coversin is a second-generation complement inhibitor that acts on complement component-C5, preventing release of C5a and formation of C5b-9 (also known as the membrane attack complex).
Coversin is a recombinant small protein (16,740 Da) derived from a native protein found in the saliva of the Ornithodoros moubata tick.
The drug is being developed by Akari Therapeutics.
In vitro experiments have shown that Coversin inhibits red blood cell lysis in PNH, and the drug can achieve full complement inhibition in the blood of PNH patients who are resistant to the drug eculizumab.
In a phase 1a trial of healthy volunteers, Coversin completely inhibited complement C5 activity within 12 hours of administration.
Akari Therapeutics is currently conducting a phase 1b study of Coversin in healthy volunteers and is administering the drug to a patient with eculizumab-resistant PNH. Thus far, Coversin has prevented hemolytic episodes and improved disease symptoms in this patient. And the only drug-related adverse event has been occasional local and transient irritation at the injection site.
Coversin is also being studied in atypical hemolytic uremic syndrome and Guillain Barré syndrome.
About orphan designation
The COMP adopts an opinion on the granting of orphan drug designation, and that opinion is submitted to the European Commission for a final decision.
Orphan designation provides regulatory and financial incentives for companies to develop and market therapies that treat a life-threatening or chronically debilitating condition affecting no more than 5 in 10,000 people in the European Union, and where no satisfactory treatment is available.
Orphan designation provides a 10-year period of marketing exclusivity in the European Union if the drug receives regulatory approval.
The designation also provides incentives for companies seeking protocol assistance from the European Medicines Agency during the product development phase and direct access to the centralized authorization procedure. ![]()
Drug could be disease-modifying for SCD, team says
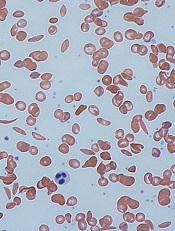
Image by Graham Beards
Researchers say the small molecule GBT440 could be a disease-modifying agent for patients with sickle cell disease (SCD).
Preclinical data showed that GBT440 can reduce sickling, extend the circulating half-life of red blood cells (RBCs), and decrease excessive erythropoiesis in SCD.
GBT440 binds specifically to hemoglobin and is designed to inhibit sickle hemoglobin (HbS) polymer formation.
“One promising strategy for preventing red blood cell sickling and subsequently modifying sickle cell disease over the long term involves inhibiting polymerization of HbS in red blood cells,” said David R. Archer, PhD, of Emory University School of Medicine in Atlanta, Georgia.
“This can be achieved by increasing the proportion of oxygenated HbS in those cells. We believe our preclinical results provide strong evidence that GBT440 inhibits HbS polymerization and red blood cell sickling, which is important because it addresses the underlying pathophysiology of sickle cell disease and has the potential to change its devastating clinical course.”
Dr Archer and his colleagues reported these results in the British Journal of Haematology. The research was supported by Global Blood Therapeutics, Inc., the company developing GBT440.
The researchers reported that, in vitro, GBT440 dose-dependently increased the affinity of HbS for oxygen, delayed polymerization of HbS, and reduced the number of sickled RBCs in whole blood from SCD patients.
In an animal model of SCD, GBT440 inhibited RBC sickling, prolonged the half-life of RBCs, and reduced reticulocyte counts.
The researchers said the drug also exhibited favorable pharmacokinetic properties in various animal species, suggesting the potential for once-daily oral dosing in SCD patients.
“Our preclinical work has developed a foundation of evidence that GBT440 is a potent inhibitor of the polymerization of HbS,” said Ted W. Love, MD, chief executive officer of Global Blood Therapeutics, Inc.
“We continue to build on these data with our ongoing phase 1/2 study, which has shown that GBT440 was well-tolerated over 90 days of dosing and that all SCD patients who received multiple doses of GBT440 exhibited improvements in one or more clinical markers of hemolysis and anemia. Our next step is to initiate a pivotal trial in adults with SCD later this year.” ![]()
Image by Graham Beards
Researchers say the small molecule GBT440 could be a disease-modifying agent for patients with sickle cell disease (SCD).
Preclinical data showed that GBT440 can reduce sickling, extend the circulating half-life of red blood cells (RBCs), and decrease excessive erythropoiesis in SCD.
GBT440 binds specifically to hemoglobin and is designed to inhibit sickle hemoglobin (HbS) polymer formation.
“One promising strategy for preventing red blood cell sickling and subsequently modifying sickle cell disease over the long term involves inhibiting polymerization of HbS in red blood cells,” said David R. Archer, PhD, of Emory University School of Medicine in Atlanta, Georgia.
“This can be achieved by increasing the proportion of oxygenated HbS in those cells. We believe our preclinical results provide strong evidence that GBT440 inhibits HbS polymerization and red blood cell sickling, which is important because it addresses the underlying pathophysiology of sickle cell disease and has the potential to change its devastating clinical course.”
Dr Archer and his colleagues reported these results in the British Journal of Haematology. The research was supported by Global Blood Therapeutics, Inc., the company developing GBT440.
The researchers reported that, in vitro, GBT440 dose-dependently increased the affinity of HbS for oxygen, delayed polymerization of HbS, and reduced the number of sickled RBCs in whole blood from SCD patients.
In an animal model of SCD, GBT440 inhibited RBC sickling, prolonged the half-life of RBCs, and reduced reticulocyte counts.
The researchers said the drug also exhibited favorable pharmacokinetic properties in various animal species, suggesting the potential for once-daily oral dosing in SCD patients.
“Our preclinical work has developed a foundation of evidence that GBT440 is a potent inhibitor of the polymerization of HbS,” said Ted W. Love, MD, chief executive officer of Global Blood Therapeutics, Inc.
“We continue to build on these data with our ongoing phase 1/2 study, which has shown that GBT440 was well-tolerated over 90 days of dosing and that all SCD patients who received multiple doses of GBT440 exhibited improvements in one or more clinical markers of hemolysis and anemia. Our next step is to initiate a pivotal trial in adults with SCD later this year.” ![]()
Image by Graham Beards
Researchers say the small molecule GBT440 could be a disease-modifying agent for patients with sickle cell disease (SCD).
Preclinical data showed that GBT440 can reduce sickling, extend the circulating half-life of red blood cells (RBCs), and decrease excessive erythropoiesis in SCD.
GBT440 binds specifically to hemoglobin and is designed to inhibit sickle hemoglobin (HbS) polymer formation.
“One promising strategy for preventing red blood cell sickling and subsequently modifying sickle cell disease over the long term involves inhibiting polymerization of HbS in red blood cells,” said David R. Archer, PhD, of Emory University School of Medicine in Atlanta, Georgia.
“This can be achieved by increasing the proportion of oxygenated HbS in those cells. We believe our preclinical results provide strong evidence that GBT440 inhibits HbS polymerization and red blood cell sickling, which is important because it addresses the underlying pathophysiology of sickle cell disease and has the potential to change its devastating clinical course.”
Dr Archer and his colleagues reported these results in the British Journal of Haematology. The research was supported by Global Blood Therapeutics, Inc., the company developing GBT440.
The researchers reported that, in vitro, GBT440 dose-dependently increased the affinity of HbS for oxygen, delayed polymerization of HbS, and reduced the number of sickled RBCs in whole blood from SCD patients.
In an animal model of SCD, GBT440 inhibited RBC sickling, prolonged the half-life of RBCs, and reduced reticulocyte counts.
The researchers said the drug also exhibited favorable pharmacokinetic properties in various animal species, suggesting the potential for once-daily oral dosing in SCD patients.
“Our preclinical work has developed a foundation of evidence that GBT440 is a potent inhibitor of the polymerization of HbS,” said Ted W. Love, MD, chief executive officer of Global Blood Therapeutics, Inc.
“We continue to build on these data with our ongoing phase 1/2 study, which has shown that GBT440 was well-tolerated over 90 days of dosing and that all SCD patients who received multiple doses of GBT440 exhibited improvements in one or more clinical markers of hemolysis and anemia. Our next step is to initiate a pivotal trial in adults with SCD later this year.” ![]()
Method reveals cells of origin in AML

in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
in the bone marrow
Whole-genome profiling of open chromatin is a reliable way to identify the cells of origin in acute myeloid leukemia (AML), according to research published in Nature Communications.
“Knowing the cell of origin of cancer cells can provide insight into tumor subtypes and possibly diagnostic and therapeutic benefit,” said study author Jennifer Trowbridge, PhD, of the Jackson Laboratory for Mammalian Genetics in Bar Harbor, Maine.
“But existing methods to identify cell of origin from bulk tumor cell samples have been unsuccessful.”
Dr Trowbridge and her colleagues hypothesized that analyzing open chromatin in bulk tumor cells might provide a better method for identifying cancer cells of origin because of the cell-type specificity of chromatin structure.
The researchers worked with a mouse model of AML driven by expression of MLL-AF9, a fusion oncogene formed by a chromosome translocation between chromosomes 9 and 11.
The team began with 5 distinct, normal cell types found in the bone marrow in both mice and humans: long-term hematopoietic stem cells (HSCs), short-term HSCs, multipotent progenitors, common myeloid progenitors, and granulocyte macrophage progenitors.
The AML that developed from these different cells of origin had different penetrance and aggressiveness when engrafted in mice. The stem cell-derived lines proved the most aggressive and the committed progenitor lines the least aggressive.
These patterns were also reflected in the frequency of leukemia-initiating cells in each cell line, with HSCs having the highest frequency and committed progenitors having the lowest.
The researchers then set out to profile the open chromatin in these distinct AML samples and compare them to open chromatin patterns in normal cells using computational models.
The team identified open chromatin signatures and gene expression patterns in AML samples that may allow stem-cell-derived AML to be distinguished from progenitor-cell-of-origin AML.
These results support findings in human data suggesting the stage of a progenitor cell when it transforms to leukemia impacts clinical progression, with earlier-stage cell-of-origin cancers being more aggressive.
The researchers noted that, with further study of open chromatin in normal human stem and progenitor cell types as well as AML patient cohorts, this profiling approach could reveal precise regions with prognostic significance based on cell of origin; in other words, a valuable cancer biomarker. ![]()
US cancer centers spend millions in advertising

chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
chemotherapy
Photo by Rhoda Baer
US cancer centers have substantially increased spending for consumer-directed advertising in recent years, according to researchers.
The team looked at consumer advertising for 890 cancer centers and found that total advertising spending for these centers increased from $54,229,849 in 2005 to $173,510,900 in 2014.
The researchers reported these findings in a letter to JAMA Internal Medicine.
The team analyzed data from an agency that tracks the content and number of advertisements and calculates expenditures. An advertiser was considered a cancer center if its name contained certain key words.
Advertising expenditure data covered 6 media outlets: television, magazines, radio, newspapers, billboards, and the Internet.
According to these data, from 2005 to 2014, 890 cancer centers advertised to the public. In general, inflation-adjusted spending increased for all of the types of advertising the researchers analyzed.
The team said the greatest relative growth in spending was for Internet display advertisements, which increased from less than 1% of total advertising spending in 2005 ($302,030/$54,229,849) to 5% in 2014 ($8,633,000/$173,510,900).
The researchers also found that, in 2014, 20 cancer centers accounted for 86% of the roughly $174 million total advertising spending.
Cancer Treatment Centers of America spent the most (at $101.7 million), followed by MD Anderson Cancer Center (at $13.9 million) and Memorial Sloan Kettering Cancer Center (at $9.1 million).
Among the 20 centers, 5 were for-profit, 17 were Commission on Cancer-accredited, and 9 were National Cancer Institute-designated.
The researchers said their findings likely underestimate advertising spending because the available data didn’t include all types of advertising, such as ads in cancer-specific magazines.
The team also said the effect of cancer-center advertising on the quality and costs of cancer care should be investigated. ![]()
Finding could help treat and prevent MM, team says

Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()
Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()
Photo by Daniel Sone
Research published in Leukemia suggests bone marrow mesenchymal stem cells (BMMSCs) are altered in monoclonal gammopathy of undetermined significance (MGUS) as well as in multiple myeloma (MM).
This discovery indicates that changes in the bone marrow needed for MM to grow have already taken hold in patients with MGUS, raising the possibility that early medical intervention could prevent the development of MM.
“It is now clear that the bone marrow of patients with MGUS, traditionally thought of as a benign condition, is significantly different to that of healthy individuals,” said study author Daniel Tennant, PhD, of the University of Birmingham in the UK.
“The bone marrow environment in these patients appears capable of supporting cancer growth, even though the majority of patients will not progress to myeloma. While this research is in the early stages, it offers the exciting possibility that early intervention could potentially delay or even prevent cancer development.”
With this study, Dr Tennant and his colleagues found that, early on in MGUS development, BMMSCs change their behavior and become more supportive of cancer growth.
The team found that a key gene, PADI2, becomes particularly overactive in BMMSCs, which leads to overproduction of the signaling molecule interleukin-6 (IL-6).
BMMSCs release IL-6 into the bone marrow, where it binds with receptors on the surface of cancerous plasma cells, instructing them to multiply rapidly and resist cell death signals.
It is already known that high levels of IL-6 in a patient’s bone marrow significantly reduces the effectiveness of the drug bortezomib.
The researchers believe that drugs designed to target the PADI2 gene in MGUS and MM patients could significantly reduce the supportive signaling that MM cells depend on, and such drugs may increase the effectiveness of current treatments.
The team also noted that the PADI2 gene has been linked to other types of cancer, rheumatoid arthritis, Alzheimer’s disease, and autoimmune disease. So any drug developed could have wider applications beyond MM treatment.
“There is an urgent need for new treatments for myeloma, which, as well as being largely incurable, can have a devastating impact on quality of life,” said Alasdair Rankin, director of research at the blood cancer charity Bloodwise, which helped to fund this study.
“With an increasing elderly population, MGUS and myeloma are only going to become more common. Drugs designed to remove the support system myeloma uses to grow could be an effective way of treating the disease, or even preventing it altogether.” ![]()
RBC donor age, gender may affect risk of death

Photo by Elise Amendola
A large study has revealed an unexpected association between blood donor characteristics and transfusion recipients’ outcomes.
It is the first study to suggest that red blood cell (RBC) transfusions from younger donors or female donors may increase the risk of death in recipients.
“We need further research to confirm these findings and to look at possible biological mechanisms,” said Michaël Chassé, MD, PhD, of Université Laval in Quebec, Canada.
“One possibility is that components in the blood of younger donors or female donors may affect the immune system of the transfusion recipient.”
For this study, Dr Chassé and his colleagues linked 30,503 patients who received a transfusion at The Ottawa Hospital between October 2006 and December 2013 with their respective blood donors (80,755 donors in total).
The average age of the recipients was 66.2 years. They were followed for an average of 2.3 years, with a maximum follow-up time of 7.2 years.
The researchers found that recipients of RBCs from female donors had an 8% increased risk of death from any cause per unit transfused, when compared with recipients of RBCs from male donors. The adjusted hazard ratio (aHR) was 1.08 (95% CI, 1.06-1.09; P<0.001).
A similar increased risk of death was observed for recipients of RBCs from younger donors. When compared to recipients of RBCs from donors ages 40 to 49.9, the risk of death was higher for recipients of RBCs from donors ages 17 to 19.9 (aHR=1.08; 95% CI, 1.06-1.10; P<0.001) and recipients of RBCs from donors ages 20 to 29.9 (aHR=1.06; 95% CI, 1.04-1.09; P<0.001).
“Though our research suggests that we should investigate what’s behind the associations that we found, there is no definitive evidence yet that proves that one type of blood is better or worse for patients,” said Jason Acker, PhD, of Canadian Blood Services in Edmonton, Alberta.
“This study opens up new areas of investigation where we can really dig into the biological explanations and understand true cause and effect.”
The study was published in JAMA Internal Medicine. ![]()
Photo by Elise Amendola
A large study has revealed an unexpected association between blood donor characteristics and transfusion recipients’ outcomes.
It is the first study to suggest that red blood cell (RBC) transfusions from younger donors or female donors may increase the risk of death in recipients.
“We need further research to confirm these findings and to look at possible biological mechanisms,” said Michaël Chassé, MD, PhD, of Université Laval in Quebec, Canada.
“One possibility is that components in the blood of younger donors or female donors may affect the immune system of the transfusion recipient.”
For this study, Dr Chassé and his colleagues linked 30,503 patients who received a transfusion at The Ottawa Hospital between October 2006 and December 2013 with their respective blood donors (80,755 donors in total).
The average age of the recipients was 66.2 years. They were followed for an average of 2.3 years, with a maximum follow-up time of 7.2 years.
The researchers found that recipients of RBCs from female donors had an 8% increased risk of death from any cause per unit transfused, when compared with recipients of RBCs from male donors. The adjusted hazard ratio (aHR) was 1.08 (95% CI, 1.06-1.09; P<0.001).
A similar increased risk of death was observed for recipients of RBCs from younger donors. When compared to recipients of RBCs from donors ages 40 to 49.9, the risk of death was higher for recipients of RBCs from donors ages 17 to 19.9 (aHR=1.08; 95% CI, 1.06-1.10; P<0.001) and recipients of RBCs from donors ages 20 to 29.9 (aHR=1.06; 95% CI, 1.04-1.09; P<0.001).
“Though our research suggests that we should investigate what’s behind the associations that we found, there is no definitive evidence yet that proves that one type of blood is better or worse for patients,” said Jason Acker, PhD, of Canadian Blood Services in Edmonton, Alberta.
“This study opens up new areas of investigation where we can really dig into the biological explanations and understand true cause and effect.”
The study was published in JAMA Internal Medicine. ![]()
Photo by Elise Amendola
A large study has revealed an unexpected association between blood donor characteristics and transfusion recipients’ outcomes.
It is the first study to suggest that red blood cell (RBC) transfusions from younger donors or female donors may increase the risk of death in recipients.
“We need further research to confirm these findings and to look at possible biological mechanisms,” said Michaël Chassé, MD, PhD, of Université Laval in Quebec, Canada.
“One possibility is that components in the blood of younger donors or female donors may affect the immune system of the transfusion recipient.”
For this study, Dr Chassé and his colleagues linked 30,503 patients who received a transfusion at The Ottawa Hospital between October 2006 and December 2013 with their respective blood donors (80,755 donors in total).
The average age of the recipients was 66.2 years. They were followed for an average of 2.3 years, with a maximum follow-up time of 7.2 years.
The researchers found that recipients of RBCs from female donors had an 8% increased risk of death from any cause per unit transfused, when compared with recipients of RBCs from male donors. The adjusted hazard ratio (aHR) was 1.08 (95% CI, 1.06-1.09; P<0.001).
A similar increased risk of death was observed for recipients of RBCs from younger donors. When compared to recipients of RBCs from donors ages 40 to 49.9, the risk of death was higher for recipients of RBCs from donors ages 17 to 19.9 (aHR=1.08; 95% CI, 1.06-1.10; P<0.001) and recipients of RBCs from donors ages 20 to 29.9 (aHR=1.06; 95% CI, 1.04-1.09; P<0.001).
“Though our research suggests that we should investigate what’s behind the associations that we found, there is no definitive evidence yet that proves that one type of blood is better or worse for patients,” said Jason Acker, PhD, of Canadian Blood Services in Edmonton, Alberta.
“This study opens up new areas of investigation where we can really dig into the biological explanations and understand true cause and effect.”
The study was published in JAMA Internal Medicine. ![]()
EMA reviewing hemophilia A products

The European Medicines Agency (EMA) has started a review of medicines containing factor VIII (FVIII) to assess the risk of inhibitor development among patients starting treatment for hemophilia A.
The agency is conducting this review because results of the SIPPET study suggested that patients are more likely to develop inhibitors if they receive FVIII products made by DNA recombinant technology rather than FVIII products derived from blood.
The EMA is evaluating data from the SIPPET study as well as all other relevant data on blood-derived and recombinant FVIII products.
The agency said it will consider the implications of these data for previously untreated patients with hemophilia A and whether there is a need for risk minimization measures or other changes to the marketing authorizations of these products.
The review will cover all medicines containing FVIII that are authorized for use within the European Union. For details on the products to be reviewed, including the different product names used in each country, visit the EMA website (Factor VIII Article-31 referral - Annex I).
The EMA’s review has been initiated at the request of the Paul-Ehrlich-Institute, under Article 31 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be sent to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt the EMA’s opinion.
Finally, the European Commission will adopt a legally binding decision applicable in all member states of the European Union. ![]()
The European Medicines Agency (EMA) has started a review of medicines containing factor VIII (FVIII) to assess the risk of inhibitor development among patients starting treatment for hemophilia A.
The agency is conducting this review because results of the SIPPET study suggested that patients are more likely to develop inhibitors if they receive FVIII products made by DNA recombinant technology rather than FVIII products derived from blood.
The EMA is evaluating data from the SIPPET study as well as all other relevant data on blood-derived and recombinant FVIII products.
The agency said it will consider the implications of these data for previously untreated patients with hemophilia A and whether there is a need for risk minimization measures or other changes to the marketing authorizations of these products.
The review will cover all medicines containing FVIII that are authorized for use within the European Union. For details on the products to be reviewed, including the different product names used in each country, visit the EMA website (Factor VIII Article-31 referral - Annex I).
The EMA’s review has been initiated at the request of the Paul-Ehrlich-Institute, under Article 31 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be sent to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt the EMA’s opinion.
Finally, the European Commission will adopt a legally binding decision applicable in all member states of the European Union. ![]()
The European Medicines Agency (EMA) has started a review of medicines containing factor VIII (FVIII) to assess the risk of inhibitor development among patients starting treatment for hemophilia A.
The agency is conducting this review because results of the SIPPET study suggested that patients are more likely to develop inhibitors if they receive FVIII products made by DNA recombinant technology rather than FVIII products derived from blood.
The EMA is evaluating data from the SIPPET study as well as all other relevant data on blood-derived and recombinant FVIII products.
The agency said it will consider the implications of these data for previously untreated patients with hemophilia A and whether there is a need for risk minimization measures or other changes to the marketing authorizations of these products.
The review will cover all medicines containing FVIII that are authorized for use within the European Union. For details on the products to be reviewed, including the different product names used in each country, visit the EMA website (Factor VIII Article-31 referral - Annex I).
The EMA’s review has been initiated at the request of the Paul-Ehrlich-Institute, under Article 31 of Directive 2001/83/EC.
The review is being carried out by the EMA’s Pharmacovigilance Risk Assessment Committee, the committee responsible for the evaluation of safety issues for human medicines, which will make a set of recommendations.
Those recommendations will then be sent to the Committee for Medicinal Products for Human Use, which is responsible for questions concerning medicines for human use and will adopt the EMA’s opinion.
Finally, the European Commission will adopt a legally binding decision applicable in all member states of the European Union.
New insights into infant leukemia
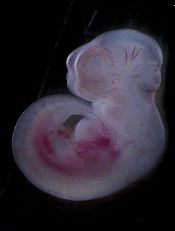
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.”
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.”
Photo by Matthias Zepper
Researchers may have identified the cells responsible for MLL-AF4+ infant B-cell acute lymphoblastic leukemia, according to a paper published in Cell Reports.
The team analyzed mouse embryos and discovered a “window of opportunity” during which a pre-leukemic state can take hold.
They also found evidence to suggest this pre-leukemic state is driven by lymphoid-primed multipotent progenitor cells.
“One of the most common and aggressive types of infant blood cancer is associated with the MLL-AF4 fusion gene, which arises during pregnancy,” said study author Katrin Ottersbach, PhD, of the University of Edinburgh in the UK.
“One of the main impediments to improving the survival rates in infants is the lack of knowledge on where and when during development this mutation arises and how it affects the developing blood system of the baby.”
To gain some insight, Dr Ottersbach and her colleagues bred mice where one parent carries an inactive form of the MLL-AF4 fusion gene and the other parent expresses a gene for an enzyme that activates the fusion gene.
The embryos from this pairing entered a pre-leukemic state between days 12 and 14. The researchers found that MLL-AF4 imparted enhanced B lymphoid potential and increased repopulation and self-renewal capacity during this time.
Further investigation suggested that lymphoid-primed multipotent progenitor cells were the major contributors to the enhanced B lymphoid output and may therefore be the cells of origin.
The researchers noted that the mice did not actually develop infant leukemia, so more research is needed to identify the events required for progression to MLL-AF4+ infant B-cell acute lymphoblastic leukemia.
“Our findings reveal the first changes that take place in blood development caused by the MLL-AF4 mutation during a pre-cancerous state,” Dr Ottersbach said. “This has increased our knowledge on how this aggressive disease develops and will help identify early signs of disease and points for therapeutic intervention.”
Drug’s benefits outweigh risks, PRAC says

Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed.
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed.
Photo courtesy of
Gilead Sciences, Inc.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) has completed its review of the PI3Kδ inhibitor idelalisib (Zyedelig) and concluded that the drug’s benefits outweigh its risks in the treatment of chronic lymphocytic leukemia (CLL) and follicular lymphoma.
However, the PRAC also confirmed that the drug increases the risk of serious infections, including Pneumocystis jirovecii pneumonia.
And the committee updated its previous recommendations to manage this risk.
The PRAC’s recommendations will now be sent to the Committee for Medicinal Products for Human Use, which will adopt the EMA’s final opinion. The final stage of the review procedure is the adoption by the European Commission of a legally binding decision applicable in all member states of the European Union (EU).
About idelalisib
In the EU, idelalisib is approved for use in combination with rituximab to treat adults with CLL who have received at least 1 prior therapy or as first-line treatment in the presence of 17p deletion or TP53 mutation in CLL patients unsuitable for chemo-immunotherapy.
Idelalisib is also approved as monotherapy for adults with follicular lymphoma that is refractory to 2 prior lines of treatment.
About the review
The PRAC’s review of idelalisib began after a higher rate of serious adverse events, including deaths, was seen in 3 clinical trials evaluating the addition of idelalisib to standard therapy in first-line CLL and relapsed indolent non-Hodgkin lymphoma (NHL).
Most of the deaths were related to infections such as Pneumocystis jirovecii pneumonia and cytomegalovirus infection. Other excess deaths were related mainly to respiratory events.
The NHL studies (NCT01732926 and NCT01732913) included patients with disease characteristics different from those covered by the currently approved indications for idelalisib and investigated combinations of drugs that are not currently approved in the EU—idelalisib plus rituximab for NHL and idelalisib plus bendamustine and rituximab for NHL.
The CLL trial (NCT01980888) involved patients who had not received previous treatment, some of whom had the 17p deletion or TP53 mutation. However, the trial also investigated a combination of drugs not currently approved in the EU—idelalisib plus bendamustine and rituximab.
PRAC’s recommendations
The PRAC noted that, although the aforementioned trials did not all use idelalisib as currently authorized, the risk of serious infection is considered relevant to the authorized use.
Therefore, the PRAC recommends that all patients treated with idelalisib receive antibiotics to prevent Pneumocystis jirovecii pneumonia during treatment and for up to 2 to 6 months after treatment has stopped.
Patients should also be monitored for infection and have regular blood tests for white cell counts because low counts can increase their risk of infection.
Furthermore, idelalisib should not be started in patients with a generalized infection.
At the beginning of its review, the PRAC had said idelalisib should not be started in patients with previously untreated CLL and 17p deletion or TP53 mutation.
Now, the PRAC has concluded that idelalisib can be initiated in these patients, provided they cannot take any alternative treatment and that the recommended measures to prevent infection are followed.
Deaths prompt clinical hold for JCAR015 trial

Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.
Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.
Image from UNSW
Update: The hold on this trial has been lifted. Click here for additional details.
A trial of the chimeric antigen receptor (CAR) T-cell therapy JCAR015 has been placed on clinical hold following 3 patient deaths.
The trial, known as ROCKET, is a phase 2 study of adults with relapsed or refractory B-cell acute lymphoblastic leukemia.
The US Food and Drug Administration (FDA) placed a hold on the trial after 3 patients died of cerebral edema.
All 3 patients had received conditioning with fludarabine, and Juno Therapeutics, the company developing JCAR015, believes this may have caused the patients’ deaths.
Patients enrolled on the ROCKET trial previously received conditioning with cyclophosphamide alone, but investigators decided to add fludarabine in hopes of increasing efficacy.
The addition of fludarabine to conditioning had been shown to increase the efficacy of 2 of Juno’s other CAR T-cell therapies, JCAR014 and JCAR017, in phase 1/2 trials.
“However, since adding fludarabine to the preconditioning on the ROCKET trial, we have seen an increase in the incidence of severe neurotoxicity, which has, unfortunately, included 2 patient deaths that occurred last week from cerebral edema that appeared to be treatment-related,” Hans Bishop, Juno’s president and chief executive officer, said in a conference call.
“After the first of these 2 deaths, we immediately paused the trial for an internal review and review with our Data Safety Monitoring Board [DSMB] and the FDA. There was also 1 previous death from cerebral edema on the trial in May. After a review of that event, we, along with the FDA and our DSMB, concluded there were confounding factors, and a change in our plans at the time was not warranted.”
After the more recent deaths, Juno investigated several factors that could have contributed, including the conditioning regimen, patient characteristics, toxicity management, product characteristics, and cell dose.
“Although more than 1 factor may have contributed, based on our review of the data available . . . , we believe the addition of fludarabine, when combined with JCAR015, is the most likely and the most appropriately modifiable factor,” Bishop said.
“Indeed, with cy[clophosphamide] alone, which we have used in the greatest number of patients treated in the ROCKET trial to date, there have not been any treatment-related deaths, and the incidence of severe neurotoxicity is within the range of what we expected in light of the Memorial Sloan-Kettering experience [phase 1 trial of JCAR015].”
Therefore, Juno has proposed continuing the ROCKET trial using conditioning with cyclophosphamide alone.
In response to this request, the FDA has requested that Juno submit:
- A revised patient informed consent form
- A revised investigator brochure
- A revised trial protocol
- A copy of a presentation the company made to the FDA.
The FDA said it will expedite the review of these documents and expects to complete the review within 30 days of receiving them.
If the clinical hold on the ROCKET trial is lifted, Juno plans to continue the trial. However, the hold will likely impact the company’s goal of gaining FDA approval for JCAR015 in 2017.
Juno’s trials and plans for its other CD19-directed CAR-T cell product candidates are not affected by the clinical hold placed on ROCKET.
ROCKET is not the first trial of JCAR015 to be placed on hold. The phase 1 trial of the therapy was placed on clinical hold in 2014, after 2 patients died of cytokine release syndrome.
That hold was lifted following changes to enrollment criteria and dosing. Results from this trial were presented at ASCO 2015 and ASCO 2016.