User login
Inhibitor produces responses in advanced SM
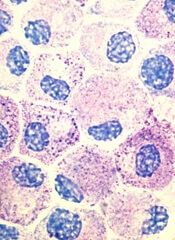
Results of a phase 2 trial suggest the multikinase inhibitor midostaurin can repair organ damage in patients with advanced systemic mastocytosis (SM).
The drug produced a 60% response rate among patients with mastocytosis-related organ damage, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose.
Jason Gotlib, MD, of the Stanford University School of Medicine in California, and his colleagues conducted this study and reported the results in NEJM.
The study was funded by Novartis Inc., which manufactures midostaurin, also known as PKC412.
The researchers noted that roughly 90% of patients with advanced SM have a mutation known as D816V in the gene that encodes the protein KIT, which controls the growth of mast cells.
Unfortunately, the only drug approved to treat advanced SM in the US is the tyrosine kinase inhibitor imatinib, and this drug is not active against the mutated KIT D816V protein. Midostaurin, on the other hand, does inhibit KIT D816V.
With this in mind, Dr Gotlib and his colleagues set out to test midostaurin (given at 100 mg twice daily until disease progression or unacceptable toxicity) in 116 patients.
Eighty-nine of the patients had mastocytosis-related organ damage, 16 had aggressive SM, 57 had SM with an associated hematologic neoplasm, and 16 had mast cell leukemia.
Response
The median follow-up was 26 months (range, 12 to 54), and the study’s primary outcome was the best overall response.
The overall response rate for the primary efficacy population (the 89 patients with mastocytosis-related organ damage) was 60%. Forty-five percent of the patients had a major response, which was defined as complete resolution of at least one type of mastocytosis-related organ damage.
The overall response rate was 75% for patients with aggressive SM, 58% for SM patients with an associated hematologic neoplasm, and 50% for patients with mast-cell leukemia.
Responses occurred in all subgroups, which included patients who were positive for KIT D816V.
The researchers noted that responding patients were less likely to need red blood cell or platelet transfusions, experienced improvements in liver function, and had fewer signs of malabsorption such as weight loss.
The median duration of response for all responders in the primary efficacy population (n=89) was 24.1 months.
Survival
The median overall survival was 28.7 months in the primary efficacy population (n=89) and 33.9 months in the intention-to-treat population (n=116). The median progression-free survival was 14.1 months in the primary efficacy population.
The survival benefit among patients with mast cell leukemia was particularly striking, according to Dr Gotlib. Although most people succumb to this form of the disease within 6 months of diagnosis, the median overall survival of midostaurin-treated patients with mast cell leukemia was 9.4 months.
The median overall survival was not reached among patients with aggressive SM and was 20.7 months among patients who had SM with an associated hematologic neoplasm.
Safety
The most common nonhematologic adverse events were nausea (79%), vomiting (66%), and diarrhea (54%). The most common grade 3/4 nonhematologic adverse events were fatigue (9%) and diarrhea (8%).
Grade 3/4 hematologic adverse events included new or worsening neutropenia (24%), anemia (41%), and thrombocytopenia (29%).
Researchers reduced the dose of midostaurin in 65 patients (56%), mostly due to adverse events (n=48). Twenty-one of these patients (32%) were later able to return to the initial dose.
Eighty-four patients (72%) discontinued treatment, and 32 (28%) were receiving ongoing treatment at the time of data cutoff. The most frequent reasons for treatment discontinuation were disease progression (33%) and adverse events (22%).
Compassionate use
The phase 2 study results are reinforced by a letter published in the same issue of NEJM, which describes a compassionate use program for midostaurin in advanced SM.
The letter includes results in 28 patients. After a median follow-up of 18.5 months, the overall response rate was 71%. The overall survival rate was 42.7%.
The most frequent adverse events were nausea/vomiting in 89% of patients (leading to failure/discontinuation in 18%), lymphocytopenia in 61% (without opportunistic infection), and photosensitivity in 25%. ![]()
Results of a phase 2 trial suggest the multikinase inhibitor midostaurin can repair organ damage in patients with advanced systemic mastocytosis (SM).
The drug produced a 60% response rate among patients with mastocytosis-related organ damage, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose.
Jason Gotlib, MD, of the Stanford University School of Medicine in California, and his colleagues conducted this study and reported the results in NEJM.
The study was funded by Novartis Inc., which manufactures midostaurin, also known as PKC412.
The researchers noted that roughly 90% of patients with advanced SM have a mutation known as D816V in the gene that encodes the protein KIT, which controls the growth of mast cells.
Unfortunately, the only drug approved to treat advanced SM in the US is the tyrosine kinase inhibitor imatinib, and this drug is not active against the mutated KIT D816V protein. Midostaurin, on the other hand, does inhibit KIT D816V.
With this in mind, Dr Gotlib and his colleagues set out to test midostaurin (given at 100 mg twice daily until disease progression or unacceptable toxicity) in 116 patients.
Eighty-nine of the patients had mastocytosis-related organ damage, 16 had aggressive SM, 57 had SM with an associated hematologic neoplasm, and 16 had mast cell leukemia.
Response
The median follow-up was 26 months (range, 12 to 54), and the study’s primary outcome was the best overall response.
The overall response rate for the primary efficacy population (the 89 patients with mastocytosis-related organ damage) was 60%. Forty-five percent of the patients had a major response, which was defined as complete resolution of at least one type of mastocytosis-related organ damage.
The overall response rate was 75% for patients with aggressive SM, 58% for SM patients with an associated hematologic neoplasm, and 50% for patients with mast-cell leukemia.
Responses occurred in all subgroups, which included patients who were positive for KIT D816V.
The researchers noted that responding patients were less likely to need red blood cell or platelet transfusions, experienced improvements in liver function, and had fewer signs of malabsorption such as weight loss.
The median duration of response for all responders in the primary efficacy population (n=89) was 24.1 months.
Survival
The median overall survival was 28.7 months in the primary efficacy population (n=89) and 33.9 months in the intention-to-treat population (n=116). The median progression-free survival was 14.1 months in the primary efficacy population.
The survival benefit among patients with mast cell leukemia was particularly striking, according to Dr Gotlib. Although most people succumb to this form of the disease within 6 months of diagnosis, the median overall survival of midostaurin-treated patients with mast cell leukemia was 9.4 months.
The median overall survival was not reached among patients with aggressive SM and was 20.7 months among patients who had SM with an associated hematologic neoplasm.
Safety
The most common nonhematologic adverse events were nausea (79%), vomiting (66%), and diarrhea (54%). The most common grade 3/4 nonhematologic adverse events were fatigue (9%) and diarrhea (8%).
Grade 3/4 hematologic adverse events included new or worsening neutropenia (24%), anemia (41%), and thrombocytopenia (29%).
Researchers reduced the dose of midostaurin in 65 patients (56%), mostly due to adverse events (n=48). Twenty-one of these patients (32%) were later able to return to the initial dose.
Eighty-four patients (72%) discontinued treatment, and 32 (28%) were receiving ongoing treatment at the time of data cutoff. The most frequent reasons for treatment discontinuation were disease progression (33%) and adverse events (22%).
Compassionate use
The phase 2 study results are reinforced by a letter published in the same issue of NEJM, which describes a compassionate use program for midostaurin in advanced SM.
The letter includes results in 28 patients. After a median follow-up of 18.5 months, the overall response rate was 71%. The overall survival rate was 42.7%.
The most frequent adverse events were nausea/vomiting in 89% of patients (leading to failure/discontinuation in 18%), lymphocytopenia in 61% (without opportunistic infection), and photosensitivity in 25%. ![]()
Results of a phase 2 trial suggest the multikinase inhibitor midostaurin can repair organ damage in patients with advanced systemic mastocytosis (SM).
The drug produced a 60% response rate among patients with mastocytosis-related organ damage, and the median duration of response was 24.1 months.
Fifty-six percent of patients required dose reductions due to toxic effects, but 32% of these patients were able to return to the starting dose.
Jason Gotlib, MD, of the Stanford University School of Medicine in California, and his colleagues conducted this study and reported the results in NEJM.
The study was funded by Novartis Inc., which manufactures midostaurin, also known as PKC412.
The researchers noted that roughly 90% of patients with advanced SM have a mutation known as D816V in the gene that encodes the protein KIT, which controls the growth of mast cells.
Unfortunately, the only drug approved to treat advanced SM in the US is the tyrosine kinase inhibitor imatinib, and this drug is not active against the mutated KIT D816V protein. Midostaurin, on the other hand, does inhibit KIT D816V.
With this in mind, Dr Gotlib and his colleagues set out to test midostaurin (given at 100 mg twice daily until disease progression or unacceptable toxicity) in 116 patients.
Eighty-nine of the patients had mastocytosis-related organ damage, 16 had aggressive SM, 57 had SM with an associated hematologic neoplasm, and 16 had mast cell leukemia.
Response
The median follow-up was 26 months (range, 12 to 54), and the study’s primary outcome was the best overall response.
The overall response rate for the primary efficacy population (the 89 patients with mastocytosis-related organ damage) was 60%. Forty-five percent of the patients had a major response, which was defined as complete resolution of at least one type of mastocytosis-related organ damage.
The overall response rate was 75% for patients with aggressive SM, 58% for SM patients with an associated hematologic neoplasm, and 50% for patients with mast-cell leukemia.
Responses occurred in all subgroups, which included patients who were positive for KIT D816V.
The researchers noted that responding patients were less likely to need red blood cell or platelet transfusions, experienced improvements in liver function, and had fewer signs of malabsorption such as weight loss.
The median duration of response for all responders in the primary efficacy population (n=89) was 24.1 months.
Survival
The median overall survival was 28.7 months in the primary efficacy population (n=89) and 33.9 months in the intention-to-treat population (n=116). The median progression-free survival was 14.1 months in the primary efficacy population.
The survival benefit among patients with mast cell leukemia was particularly striking, according to Dr Gotlib. Although most people succumb to this form of the disease within 6 months of diagnosis, the median overall survival of midostaurin-treated patients with mast cell leukemia was 9.4 months.
The median overall survival was not reached among patients with aggressive SM and was 20.7 months among patients who had SM with an associated hematologic neoplasm.
Safety
The most common nonhematologic adverse events were nausea (79%), vomiting (66%), and diarrhea (54%). The most common grade 3/4 nonhematologic adverse events were fatigue (9%) and diarrhea (8%).
Grade 3/4 hematologic adverse events included new or worsening neutropenia (24%), anemia (41%), and thrombocytopenia (29%).
Researchers reduced the dose of midostaurin in 65 patients (56%), mostly due to adverse events (n=48). Twenty-one of these patients (32%) were later able to return to the initial dose.
Eighty-four patients (72%) discontinued treatment, and 32 (28%) were receiving ongoing treatment at the time of data cutoff. The most frequent reasons for treatment discontinuation were disease progression (33%) and adverse events (22%).
Compassionate use
The phase 2 study results are reinforced by a letter published in the same issue of NEJM, which describes a compassionate use program for midostaurin in advanced SM.
The letter includes results in 28 patients. After a median follow-up of 18.5 months, the overall response rate was 71%. The overall survival rate was 42.7%.
The most frequent adverse events were nausea/vomiting in 89% of patients (leading to failure/discontinuation in 18%), lymphocytopenia in 61% (without opportunistic infection), and photosensitivity in 25%. ![]()
Risk of AML death varies by region

Photo by Rhoda Baer
The risk of death from acute myeloid leukemia (AML) may be influenced by where a patient lives, according to a study published in Cancer.
Three regions in North Carolina were found to be associated with a higher risk of death, when compared to the rest of the state.
Patients had a significantly higher risk of death if they lived in northeastern North Carolina (from Wilson to Roanoke Rapids), in a region around Greenville, and a region around Wake County, including Durham County.
The increased risk remained even when the researchers controlled for other factors.
“The geographic survival disparities we found could not be explained by other sociodemographic variables or proximity to experienced treating facilities,” said study author Ashley Freeman, MD, of the University of North Carolina (UNC) in Chapel Hill.
“This raises the possibility that more complex features of the local healthcare infrastructure, including provider referral and practice patterns, are affecting patient outcomes.”
To study death rates from AML across North Carolina, Dr Freeman and her colleagues analyzed data on 553 adults who were diagnosed with AML between 2003 and 2009 and received inpatient chemotherapy within 30 days of diagnosis.
The team used the UNC Lineberger Integrated Cancer Information and Surveillance System, a database that links insurance claims information to a state information database called the NC Cancer Registry.
The researchers assessed the risk of death in 9 regions defined by the North Carolina Area Health Education Centers (AHEC) Program, a program established in 1972 to address physician shortages and the uneven distribution of healthcare services in North Carolina.
“We looked at geographic disparities because we are trying to improve outcomes for all citizens in North Carolina, consistent with the mission of our cancer center,” said William A. Wood, MD, of UNC.
“We are also trying to find situations in which disparities shouldn’t exist but do for arbitrary reasons—such as where a patient happens to live—so that we can figure out how to improve equity across the state.”
The researchers determined that a region around Greensboro had the lowest risk of death for AML.
Compared to the Greensboro region, the risk of death was 4 times higher in an area of northeastern North Carolina that included Roanoke Rapids, Rocky Mount, and Wilson—the highest in the state.
Compared to the Greensboro region, the risk of death was more than 2 times greater in the eastern region of the state around Greenville, and it was nearly twice as high in the region around Wake County.
“There are areas of the state where there is an elevated mortality, and we need to better understand the factors that are driving that—whether they’re environmental, patient, or provider-related,” said Anne-Marie Meyer, PhD, of UNC.
Nearly half of patients in the study received their care at hospitals not affiliated with one of the state’s 3 National Cancer Institute (NCI) comprehensive cancer centers.
The researchers did not find a significant link between the risk of death and the distance from patients’ homes to their treating facility or the nearest NCI-designated center.
And there was no significant difference in the risk of death at 1 year between patients who received treatment at an NCI-designated cancer center and those who did not. However, patients with a more serious prognosis were more likely to be treated at an NCI-designated cancer center.
The researchers did identify regional differences in healthcare resources. “Area L,” which is the name for the region in northeastern North Carolina that spans from Wilson to Roanoke Rapids, for example, has some of the lowest proportion of general practitioner physicians and radiation oncologists, as well as the highest burden of disease.
However, it is not clear why the disparities continued even after the researchers controlled for regional factors like poverty and education. The team believes other factors could be involved, such as the providers’ experience with treating rare or complex diseases or how supportive care is delivered.
Although there was not a significant association between survival and treatment at an NCI-designated center, there may be other features of treatment facilities, such as patient volume and academic affiliation, that are important for patient outcomes.
The researchers said their next step is to identify those factors and develop programs to try to close the gaps.
“The message here is that acute myeloid leukemia is representative of diseases that are uncommon, involve high-complexity care, and have high risk for morbidity and mortality,” Dr Wood said.
“If we can figure out how to coordinate and improve delivery of effective interventions for this disease throughout the state of North Carolina, then we may be able to develop a model for improving outcomes in many other diseases throughout the state as well.” ![]()
Photo by Rhoda Baer
The risk of death from acute myeloid leukemia (AML) may be influenced by where a patient lives, according to a study published in Cancer.
Three regions in North Carolina were found to be associated with a higher risk of death, when compared to the rest of the state.
Patients had a significantly higher risk of death if they lived in northeastern North Carolina (from Wilson to Roanoke Rapids), in a region around Greenville, and a region around Wake County, including Durham County.
The increased risk remained even when the researchers controlled for other factors.
“The geographic survival disparities we found could not be explained by other sociodemographic variables or proximity to experienced treating facilities,” said study author Ashley Freeman, MD, of the University of North Carolina (UNC) in Chapel Hill.
“This raises the possibility that more complex features of the local healthcare infrastructure, including provider referral and practice patterns, are affecting patient outcomes.”
To study death rates from AML across North Carolina, Dr Freeman and her colleagues analyzed data on 553 adults who were diagnosed with AML between 2003 and 2009 and received inpatient chemotherapy within 30 days of diagnosis.
The team used the UNC Lineberger Integrated Cancer Information and Surveillance System, a database that links insurance claims information to a state information database called the NC Cancer Registry.
The researchers assessed the risk of death in 9 regions defined by the North Carolina Area Health Education Centers (AHEC) Program, a program established in 1972 to address physician shortages and the uneven distribution of healthcare services in North Carolina.
“We looked at geographic disparities because we are trying to improve outcomes for all citizens in North Carolina, consistent with the mission of our cancer center,” said William A. Wood, MD, of UNC.
“We are also trying to find situations in which disparities shouldn’t exist but do for arbitrary reasons—such as where a patient happens to live—so that we can figure out how to improve equity across the state.”
The researchers determined that a region around Greensboro had the lowest risk of death for AML.
Compared to the Greensboro region, the risk of death was 4 times higher in an area of northeastern North Carolina that included Roanoke Rapids, Rocky Mount, and Wilson—the highest in the state.
Compared to the Greensboro region, the risk of death was more than 2 times greater in the eastern region of the state around Greenville, and it was nearly twice as high in the region around Wake County.
“There are areas of the state where there is an elevated mortality, and we need to better understand the factors that are driving that—whether they’re environmental, patient, or provider-related,” said Anne-Marie Meyer, PhD, of UNC.
Nearly half of patients in the study received their care at hospitals not affiliated with one of the state’s 3 National Cancer Institute (NCI) comprehensive cancer centers.
The researchers did not find a significant link between the risk of death and the distance from patients’ homes to their treating facility or the nearest NCI-designated center.
And there was no significant difference in the risk of death at 1 year between patients who received treatment at an NCI-designated cancer center and those who did not. However, patients with a more serious prognosis were more likely to be treated at an NCI-designated cancer center.
The researchers did identify regional differences in healthcare resources. “Area L,” which is the name for the region in northeastern North Carolina that spans from Wilson to Roanoke Rapids, for example, has some of the lowest proportion of general practitioner physicians and radiation oncologists, as well as the highest burden of disease.
However, it is not clear why the disparities continued even after the researchers controlled for regional factors like poverty and education. The team believes other factors could be involved, such as the providers’ experience with treating rare or complex diseases or how supportive care is delivered.
Although there was not a significant association between survival and treatment at an NCI-designated center, there may be other features of treatment facilities, such as patient volume and academic affiliation, that are important for patient outcomes.
The researchers said their next step is to identify those factors and develop programs to try to close the gaps.
“The message here is that acute myeloid leukemia is representative of diseases that are uncommon, involve high-complexity care, and have high risk for morbidity and mortality,” Dr Wood said.
“If we can figure out how to coordinate and improve delivery of effective interventions for this disease throughout the state of North Carolina, then we may be able to develop a model for improving outcomes in many other diseases throughout the state as well.” ![]()
Photo by Rhoda Baer
The risk of death from acute myeloid leukemia (AML) may be influenced by where a patient lives, according to a study published in Cancer.
Three regions in North Carolina were found to be associated with a higher risk of death, when compared to the rest of the state.
Patients had a significantly higher risk of death if they lived in northeastern North Carolina (from Wilson to Roanoke Rapids), in a region around Greenville, and a region around Wake County, including Durham County.
The increased risk remained even when the researchers controlled for other factors.
“The geographic survival disparities we found could not be explained by other sociodemographic variables or proximity to experienced treating facilities,” said study author Ashley Freeman, MD, of the University of North Carolina (UNC) in Chapel Hill.
“This raises the possibility that more complex features of the local healthcare infrastructure, including provider referral and practice patterns, are affecting patient outcomes.”
To study death rates from AML across North Carolina, Dr Freeman and her colleagues analyzed data on 553 adults who were diagnosed with AML between 2003 and 2009 and received inpatient chemotherapy within 30 days of diagnosis.
The team used the UNC Lineberger Integrated Cancer Information and Surveillance System, a database that links insurance claims information to a state information database called the NC Cancer Registry.
The researchers assessed the risk of death in 9 regions defined by the North Carolina Area Health Education Centers (AHEC) Program, a program established in 1972 to address physician shortages and the uneven distribution of healthcare services in North Carolina.
“We looked at geographic disparities because we are trying to improve outcomes for all citizens in North Carolina, consistent with the mission of our cancer center,” said William A. Wood, MD, of UNC.
“We are also trying to find situations in which disparities shouldn’t exist but do for arbitrary reasons—such as where a patient happens to live—so that we can figure out how to improve equity across the state.”
The researchers determined that a region around Greensboro had the lowest risk of death for AML.
Compared to the Greensboro region, the risk of death was 4 times higher in an area of northeastern North Carolina that included Roanoke Rapids, Rocky Mount, and Wilson—the highest in the state.
Compared to the Greensboro region, the risk of death was more than 2 times greater in the eastern region of the state around Greenville, and it was nearly twice as high in the region around Wake County.
“There are areas of the state where there is an elevated mortality, and we need to better understand the factors that are driving that—whether they’re environmental, patient, or provider-related,” said Anne-Marie Meyer, PhD, of UNC.
Nearly half of patients in the study received their care at hospitals not affiliated with one of the state’s 3 National Cancer Institute (NCI) comprehensive cancer centers.
The researchers did not find a significant link between the risk of death and the distance from patients’ homes to their treating facility or the nearest NCI-designated center.
And there was no significant difference in the risk of death at 1 year between patients who received treatment at an NCI-designated cancer center and those who did not. However, patients with a more serious prognosis were more likely to be treated at an NCI-designated cancer center.
The researchers did identify regional differences in healthcare resources. “Area L,” which is the name for the region in northeastern North Carolina that spans from Wilson to Roanoke Rapids, for example, has some of the lowest proportion of general practitioner physicians and radiation oncologists, as well as the highest burden of disease.
However, it is not clear why the disparities continued even after the researchers controlled for regional factors like poverty and education. The team believes other factors could be involved, such as the providers’ experience with treating rare or complex diseases or how supportive care is delivered.
Although there was not a significant association between survival and treatment at an NCI-designated center, there may be other features of treatment facilities, such as patient volume and academic affiliation, that are important for patient outcomes.
The researchers said their next step is to identify those factors and develop programs to try to close the gaps.
“The message here is that acute myeloid leukemia is representative of diseases that are uncommon, involve high-complexity care, and have high risk for morbidity and mortality,” Dr Wood said.
“If we can figure out how to coordinate and improve delivery of effective interventions for this disease throughout the state of North Carolina, then we may be able to develop a model for improving outcomes in many other diseases throughout the state as well.” ![]()
Drug granted breakthrough, orphan designation for cGVHD

Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Photo courtesy of Janssen
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ibrutinib (Imbruvica), a Bruton’s tyrosine kinase inhibitor, as a potential treatment for chronic graft-versus-host-disease (cGVHD) in patients who have failed 1 or more lines of systemic therapy.
The FDA has also granted ibrutinib orphan drug designation for this indication.
The request for breakthrough therapy designation and orphan designation for ibrutinib in patients with cGVHD was based on preliminary data from a phase 1b/2 study of patients with steroid-dependent or refractory cGVHD.
Results from this trial were presented at the 2015 ASCO Annual Meeting (abstract 7024) and the 2016 EBMT meeting (abstract P124).
About ibrutinib
Ibrutinib is an oral, once-daily therapy that inhibits Bruton’s tyrosine kinase, a signaling molecule in the B-cell receptor signaling complex that plays an important role in the survival and spread of malignant B cells.
Ibrutinib is FDA-approved to treat patients with chronic lymphocytic leukemia (CLL) and small lymphocytic lymphoma (SLL), including those with 17p deletion, patients with mantle cell lymphoma (MCL) who have received at least 1 prior therapy, and patients with Waldenström’s macroglobulinemia.
Accelerated approval was granted for the MCL indication based on overall response rate. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted ibrutinib breakthrough designation for the treatment of relapsed or refractory MCL, Waldenström’s macroglobulinemia, and CLL/SLL patients with 17p deletion. The FDA also granted ibrutinib orphan designation for all 3 indications.
Ibrutinib is jointly developed and commercialized by Pharmacyclics LLC, an AbbVie company, and Janssen Biotech, Inc.
About breakthrough designation
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
About orphan designation
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the drug is approved. ![]()
Combo shows promise for treating DLBCL

The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
The mTOR inhibitor everolimus may provide an additional benefit when combined with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) to treat patients with newly diagnosed diffuse large B-cell lymphoma (DLBCL), according to researchers.
The combination was considered well-tolerated in a phase 1 trial, and 96% of patients responded to the treatment.
“There is an unmet need to develop new therapies based on R-CHOP to try to increase the cure rate for diffuse large B-cell lymphoma,” said Patrick Johnston, MD, PhD, of the Mayo Clinic in Rochester, Minnesota.
“This pilot study suggests that adding mTOR inhibitors to standard therapy could improve outcomes, though it needs to be validated in a larger clinical trial.”
Results from this study were published in The Lancet Haematology.
Patients and treatment
Dr Johnston and his colleagues conducted this study in 24 previously untreated DLBCL patients. Their median age was 58.5 (range, 49.5-71.5), and 58% were male. Most patients had stage IV disease (54%), followed by stage II (25%), and stage III (21%). Five patients (21%) had bulky disease.
The patients received standard R-CHOP-21 (rituximab at 375 mg/m2, cyclophosphamide at 750 mg/m2, doxorubicin at 50 mg/m2, and vincristine at 1.4 mg/m2—all on day 1 of the 21-day cycle—as well as oral prednisone at 100 mg/m2 each day on days 1–5 of the cycle) for 6 cycles, with scheduled pegfilgrastim at 6 mg on day 2 of each cycle.
They also received everolimus at 10 mg/day on 2 different schedules. Nine patients were enrolled initially—3 given everolimus on days 1–10 and 6 receiving it on days 1–14. As there were no dose-limiting toxicities in these patients, another 15 patients went on to receive everolimus on days 1–14.
Results
The median follow-up was 21.5 months. Twenty-three patients (96%) achieved an overall response and a complete metabolic response as assessed by PET. The remaining patient withdrew consent during cycle 1 and achieved a complete response with R-CHOP alone.
The 12-month event-free survival rate was 100%. Nine patients had sufficient follow-up and were event-free at 24 months. At last follow-up (March 30, 2016), no deaths or relapses had occurred.
The most common adverse events were hematologic, such as grade 4 neutropenia (75%) and grade 3 febrile neutropenia (21%).
Three patients experienced “significant” toxicity, according to the researchers. One patient had a treatment delay of 12 days due to grade 3 hypokalemia, which was considered possibly related to everolimus.
A second patient had grade 4 sepsis that was possibly related to treatment, and a third patient had a treatment delay of 10 days due to grade 3 infection that was possibly related to everolimus.
Ten patients (42%) had their dose of everolimus reduced, 2 patients permanently discontinued the drug after cycles 3 and 4, respectively, and 2 patients omitted everolimus for 1 and 2 cycles, respectively, then resumed everolimus for subsequent cycles.
“This study is the first to integrate a P13K-mTOR agent with standard R-CHOP,” Dr Johnston said.
“The encouraging outcome results and toxicity profile of this new regimen, along with the worldwide availability of everolimus, make it potentially applicable to the large population of DLBCL patients.” ![]()
Study: CMV doesn’t lower risk of relapse, death
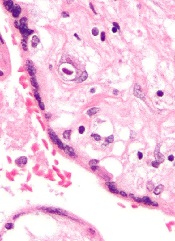
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Small studies have suggested that early cytomegalovirus (CMV) reactivation may protect against leukemia relapse and even death after hematopoietic stem cell transplant.
However, a new study, based on data from about 9500 patients, suggests otherwise.
Results showed no association between CMV reactivation and relapse but suggested CMV reactivation increases the risk of non-relapse mortality.
Researchers reported these findings in Blood.
“The original purpose of the study was to confirm that CMV infection may prevent leukemia relapse, prevent death, and become a major therapeutic tool for improving patient survival rates,” said study author Pierre Teira, MD, of the University of Montreal in Quebec, Canada.
“However, we found the exact opposite. Our results clearly show that . . . the virus not only does not prevent leukemia relapse [it] also remains a major factor associated with the risk of death. Monitoring of CMV after transplantation remains a priority for patients.”
For this study, Dr Teira and his colleagues analyzed data from 9469 patients who received a transplant between 2003 and 2010.
The patients had acute myeloid leukemia (AML, n=5310), acute lymphoblastic leukemia (ALL, n=1883), chronic myeloid leukemia (CML, n=1079), or myelodysplastic syndromes (MDS, n=1197).
The median time to initial CMV reactivation was 41 days (range, 1-362 days).
The researchers found no significant association between CMV reactivation and disease relapse for AML (P=0.60), ALL (P=0.08), CML (P=0.94), or MDS (P=0.58).
However, CMV reactivation was associated with a significantly higher risk of nonrelapse mortality for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0004), and MDS (P=0.0002).
Therefore, CMV reactivation was associated with significantly lower overall survival for AML (P<0.0001), ALL (P<0.0001), CML (P=0.0005), and MDS (P=0.003).
“Deaths due to uncontrolled CMV reactivation are virtually zero in this study, so uncontrolled CMV reactivation is not what reduces survival rates after transplantation,” Dr Teira noted. “The link between this common virus and increased risk of death remains a biological mystery.”
One possible explanation is that CMV decreases the ability of the patient’s immune system to fight against other types of infection. This is supported by the fact that death rates from infections other than CMV are higher in patients infected with CMV or patients whose donors were.
For researchers, the next step is therefore to verify whether the latest generation of anti-CMV treatments can prevent both reactivation of the virus and weakening of the patient’s immune system against other types of infection in the presence of CMV infection.
“CMV has a complex impact on the outcomes for transplant patients, and, each year, more than 30,000 patients around the world receive bone marrow transplants from donors,” Dr Teira said.
“It is therefore essential for future research to better understand the role played by CMV after bone marrow transplantation and improve the chances of success of the transplant. This will help to better choose the right donor for the right patient.” ![]()
Dementia isn’t passed on via transfusion, team says

Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Photo by Elise Amendola
Results of a large, retrospective study suggest that neurological diseases are not transmitted via blood transfusion.
Previous studies have shown that such diseases can be induced in healthy laboratory animals through the injection of diseased brain tissue from humans.
This has caused concern that neurological diseases might be transmitted from human to human via blood transfusions.
However, a study published in Annals of Internal Medicine suggests such transmission does not occur.
“The results are unusually clear for such a complicated subject as this,” said study author Gustaf Edgren, PhD, of Karolinska Institutet in Stockholm, Sweden.
“We’ve been working with this question for a long time now and have found no indication that these diseases can be transmitted via transfusions.”
Dr Edgren and his colleagues conducted this study by analyzing data from 1,465,845 patients who received blood transfusions in Sweden or Denmark between 1968 and 2012.
The team used multivariable Cox regression models (taking into account sex, age, place of residence, blood group, number of transfusions, and time since first transfusion) to estimate hazard ratios for dementia of any type, Alzheimer’s disease, and Parkinson’s disease in patients who received transfusions from donors who were later diagnosed with any of these diseases, compared to patients who received blood from healthy donors.
In all, 2.9% of patients received a transfusion from a donor diagnosed with one of the aforementioned neurological diseases. And there was no evidence of disease transmission via transfusion.
The hazard ratio for dementia in transfusion recipients whose donors were diagnosed with dementia, compared to recipients of blood from healthy donors, was 1.04 (95% CI, 0.99 to 1.09).
The hazard ratios for Alzheimer’s disease and Parkinson’s disease were 0.99 (95% CI, 0.85 to 1.15) and 0.94 (95% CI, 0.78 to 1.14), respectively.
“Blood transfusions are extremely safe in the Western world today, but, even so, we are working continuously and proactively on identifying any overlooked risks,” Dr Edgren said.
“The Swedish-Danish database that we have built up and used in many similar studies clearly demonstrates the value of our vast health registries. This kind of study would have simply been extremely difficult anywhere else in the world.” ![]()
Artemisinin resistance confined to Asia, study shows
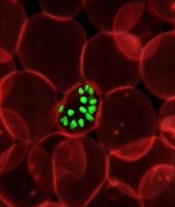
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.” ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.” ![]()
infecting a red blood cell
Image courtesy of St. Jude
Children’s Research Hospital
The first global mapping of artemisinin resistance indicates that resistance to the drug, which is used to treat Plasmodium falciparum malaria, is confined to Southeast Asia and has not yet spread to sub-Saharan Africa.
Results of the effort, known as the KARMA study, were published in NEJM.
The study builds on the 2014 discovery that the K13 gene is the major determinant of P falciparum’s resistance to artemisinin.
Researchers studied the diversity of the K13 gene in 14,037 blood samples taken from P falciparum-infected patients in 59 malaria-endemic countries—72% in Africa, 19% in Asia, 8% in Latin America, and 1% in Oceania. All samples were collected after 2012.
The researchers identified 108 nonsynonymous K13 mutations. In Asia, 36.5% of the mutations were distributed within 2 areas—Cambodia-Vietnam-Laos and western Thailand-Myanmar-China—with no overlap.
In samples from Africa, the researchers identified nonsynonymous K13 mutations that were not associated with artemisinin resistance, including the most frequent mutation found in Africa, A578S.
“We suspect that only a small number of mutations appear to be associated with resistance, which should facilitate global monitoring of resistance to artemisinin,” said study author Odile Mercereau-Puijalon, PhD, of the Institut Pasteur in Paris, France.
“Until now, scientists have not had the tools to be properly informed about the nature of resistance to antimalarial drugs in key affected regions such as sub-Saharan Africa,” added Didier Ménard, PhD, of the Institut Pasteur in Phnom Penh, Cambodia.
“We now have the capacity, thanks to molecular markers, to be able to trace—at a global level and virtually in real-time—resistance to antimalarial drugs. We must ensure that we use this technology to keep us a step ahead of the parasite.”
Agreements may constrain publication of trial results

for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted.
for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted.
for a clinical trial
Photo by Esther Dyson
Publication agreements between industry and academic investigators involved in clinical trials are not often reported in the publications themselves, according to a study published in PLOS Medicine.
In most of the agreements studied, industry had the right to reject or review manuscripts before publication.
Therefore, according to researchers, publication agreements may compromise the scientific evidence base established by randomized clinical trials.
Matthias Briel, MD, of the University Hospital Basel in Switzerland, and his colleagues sought to understand how publication agreements might constrain the publication of trial results.
The researchers examined publication agreements in 647 randomized trial protocols approved from 2000 to 2003 by 6 research ethics committees in Switzerland, Canada, and Germany, as well as the 388 corresponding journal publications.
The team found that 71% of protocols mentioned an agreement on publication rights between industry and academic investigators.
In 86% of those agreements, industry retained the right to disapprove or at least review manuscripts before publication.
And 74% of the agreements documented in protocols were not mentioned in corresponding journal articles.
The researchers noted that half of the included journal articles were published before 2008, leaving open the possibility that these findings do not reflect current practice.
Nonetheless, the team said the findings suggest that more transparency on publication constraints is warranted.
Team maps chromatin landscape in CLL

Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.”
Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.”
Researchers say they have performed the first large-scale analysis of the chromatin landscape in chronic lymphocytic leukemia (CLL).
And, in doing so, they have identified shared gene regulatory networks as well as heterogeneity between patients and CLL subtypes.
The group says this work should enable deeper investigation into chromatin regulation in CLL and the identification of therapeutically relevant mechanisms of disease.
The work has been published in Nature Communications.
The researchers performed chromatin accessibility mapping—via the assay for transposase-accessible chromatin using sequencing (ATAC-seq)—on 88 CLL samples from 55 patients.
For 10 of the samples, the team also established histone profiles using ChIPmentation for 3 histone marks (H3K4me1, H3K27ac, and H3K27me3) and transcriptome profiles using RNA sequencing.
The researchers then developed a bioinformatic method for linking the chromatin profiles to clinical annotations and molecular diagnostics data, and they analyzed gene regulatory networks that underlie the major disease subtypes of CLL.
The work revealed a “shared core” of regulatory regions in CLL patients as well as variations between the samples.
Furthermore, the chromatin profiles and gene regulatory networks accurately predicted IGHV mutation status and pinpointed differences between IGVH-mutated and IGVH-unmutated CLL.
“Our study has been able to dissect the variability that exists in the epigenome of CLL patients and helped to identify disease-specific changes, which will hopefully be informative for distinguishing disease subtypes or identifying suitable treatments,” said study author Jonathan Strefford, PhD, of the University of Southampton in the UK.
“Epigenetics can offer a useful doorway into ways of improving disease diagnosis and more personalized treatment choices for patients.”
EBV-CTL product classified as ATMP
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US.
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US.
among uninfected cells (blue)
Image courtesy of Benjamin
Chaigne-Delalande
A cytotoxic T-lymphocyte product that targets Epstein-Barr virus (EBV-CTLs) has been classified as an advanced therapy medicinal product (ATMP) by the European Medicines Agency (EMA).
The EBV-CTLs are being developed by Atara Biotherapeutics, Inc., to treat patients with EBV post-transplant lymphoproliferative disorder (EBV-PTLD).
ATMP classification was established to regulate cell and gene therapy and tissue-engineered medicinal products, support the development of these products, and provide a benchmark for the level of quality compliance for pharmaceutical practices.
ATMP classification can provide developers with scientific regulatory guidance, help clarify the applicable regulatory framework and development path, and provide access to all relevant services and incentives offered by the EMA. It can also be advantageous when submitting clinical trial dossiers to national regulatory authorities within the European Union.
About EBV-CTLs
Atara Bio’s EBV-CTL product utilizes a technology in which T cells are collected from the blood of third-party donors and then exposed to EBV antigens. The activated T cells are then expanded, characterized, and stored for future use in a partially HLA-matched patient.
In the context of EBV-PTLD, the EBV-CTLs find the cancer cells expressing EBV and kill them.
Atara Bio’s EBV-CTL product is currently being studied in phase 2 trials of patients with EBV-associated cancers, including PTLD and nasopharyngeal carcinoma.
Results of a phase 1/2 study of EBV-CTLs were presented at the APHON 37th Annual Conference and Exhibit and the 2015 ASCO Annual Meeting.
Atara Bio’s EBV-CTL product has orphan designation in the European Union and the US, as well as breakthrough designation in the US.