User login
P vivax evolving differently in different regions
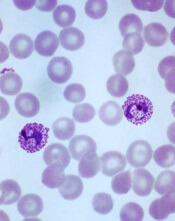
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.” ![]()
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.” ![]()
Plasmodium vivax
Image by Mae Melvin
Genomic research suggests the malaria parasite Plasmodium vivax is evolving rapidly to adapt to conditions in different geographic locations.
Researchers studied more than 200 parasite samples from across the Asia-Pacific region and found that P vivax has evolved differently in different areas.
The team identified substantial differences in the frequency of copy number variations (CNVs) in samples from western Thailand, western Cambodia, and Papua Indonesia.
They believe this is a result of the different antimalarial drugs used in these regions.
The researchers described this work in Nature Genetics.
“For so long, it’s not been possible to study P vivax genomes in detail, on a large-scale, but now we can, and we’re seeing the effect that drug use has on how parasites are evolving,” said study author Dominic Kwiatkowski, of the Wellcome Trust Sanger Institute in the UK.
He and his colleagues studied the genomes of 228 parasite samples, identifying the strains carried by each patient and revealing their infection history. Most samples came from Southeast Asia (Thailand, Cambodia, Vietnam, Laos, Myanmar, and Malaysia) and Oceania (Papua Indonesia and Papua New Guinea), but the team also studied samples from China, India, Sri Lanka, Brazil, and Madagascar.
The researchers performed detailed population genetic analyses using 148 samples from western Thailand, western Cambodia, and Papua Indonesia. This revealed CNVs in 9 regions of the core genome, and the frequency of the 4 most common CNVs varied greatly according to geographical location.
The first common CNV was a 9-kb deletion on chromosome 8 that includes the first 3 exons of a gene encoding a cytoadherence-linked asexual protein. The CNV was present in 73% of Papua Indonesia samples, 6% of western Cambodia samples, and 3% of western Thailand samples.
The second common CNV was a 7-kb duplication on chromosome 6 that encompasses pvdbp, the gene that encodes the Duffy-binding protein, which mediates P vivax’s invasion of erythrocytes. It was present in 5% of Papua Indonesia samples, 35% of western Cambodia samples, and 25% of western Thailand samples.
The third common CNV was a 37-kb duplication on chromosome 10 that includes pvmdr1, which has been associated with resistance to mefloquine and is homologous to the pfmdr1 amplification responsible for mefloquine resistance in P falciparum. This CNV was only present in samples from western Thailand.
The fourth common CNV was a 3-kb duplication on chromosome 14 that includes the gene PVX_101445. It was found only in Papua Indonesia samples.
“Our study shows that the strongest evidence of evolution is in Papua, Indonesia, where resistance of P vivax to chloroquine is now rampant,” said Ric Price, MD, of the University of Oxford in the UK.
“These data provide crucial information from which we can start to identify the mechanisms of drug resistance in P vivax.”
“We can see in the genome that drug resistance is a huge driver for evolution,” added Richard Pearson, PhD, of the Wellcome Trust Sanger Institute.
“Intriguingly, in some places, this process appears to be happening in response to drugs used primarily to treat a different malaria parasite, P falciparum. Although the exact cause isn’t known, this is a worrying sign that drug resistance is becoming deeply entrenched in the parasite population.”
The researchers said there are a few possible reasons why P vivax may be evolving to evade drugs used against P falciparum.
Many people carry mixed infections of both species of parasite, so, in treating one species, the other automatically gets exposed to the drug. Another culprit may be unsupervised drug use—where many people take the most readily available, rather than the most suitable, antimalarial drug.
Another finding from this study was that, when the researchers identified patients who were carrying multiple strains of parasite, the genomic data made it possible to determine how closely the different strains were related to one another.
“This means that we can now start to pull apart the genetic complexity of individual Plasmodium vivax infections and work out whether the parasites came from one or more mosquito bites,” Kwiatkowski said. “It provides a way of addressing fundamental questions about how P vivax is transmitted and how it persists within a community and, in particular, about the biology of relapsing infections.” ![]()
Immunotherapy drugs linked to rheumatic diseases

Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications. ![]()
Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications. ![]()
Photo by Bill Branson
Several case reports have suggested that cancer patients taking the immunotherapy drugs nivolumab and ipilimumab may have a higher-than-normal risk of developing rheumatic diseases.
Between 2012 and 2016, 13 patients at the Johns Hopkins Kimmel Cancer Center who were taking one or both drugs developed inflammatory arthritis or sicca syndrome, a set of autoimmune conditions causing dry eyes and mouth.
The cases were described in Annals of Rheumatic Diseases.
Nivolumab and ipilimumab are both designed to turn off the molecular “checkpoints” some cancers—including lymphoma—use to evade the immune system. When the drugs work, they allow the immune system to detect and attack tumor cells. However, they also turn up the activity of the immune system as a whole and can therefore trigger immune-related side effects.
Clinical trials of ipilimumab and nivolumab have indicated that the drugs confer an increased risk of inflammatory bowel diseases, lung inflammation, autoimmune thyroid disease, and pituitary gland inflammation.
However, those trials were designed primarily to determine efficacy against cancer and not to fully examine all features of rheumatologic side effects, said Laura C. Cappelli, MD, of the Johns Hopkins University School of Medicine in Baltimore, Maryland.
With this in mind, she and her colleagues decided to take a closer look at 13 adults (older than 18) who were treated at the Johns Hopkins Kimmel Cancer Center and reported rheumatologic symptoms after their treatment with nivolumab and/or ipilimumab.
Eight patients were taking both ipilimumab and nivolumab, and 5 were taking 1 of the 2 drugs. They were receiving the drugs to treat melanoma (n=6), non-small-cell lung cancer (n=5), small-cell lung cancer (n=1), and renal cell carcinoma (n=1).
Nine of the patients developed inflammatory arthritis—4 with synovitis confirmed via imaging and 4 with inflammatory synovial fluid—and the remaining 4 patients were diagnosed with sicca syndrome. Other immune-related adverse events included pneumonitis, colitis, interstitial nephritis, and thyroiditis.
The researchers said this is the largest published case series showing a link between checkpoint inhibitors and rheumatic diseases.
The patients described in this case report make up about 1.3% of all patients treated with the drugs—singly or in combination—at The Johns Hopkins Hospital from 2012 to 2016. However, the researchers believe that rate is likely an underestimation of how common rheumatic diseases are in patients taking immune checkpoint inhibitors.
“We keep having referrals coming in from our oncologists as more patients are treated with these drugs,” said Clifton Bingham, MD, of the Johns Hopkins University School of Medicine.
“In particular, as more patients are treated with combinations of multiple immunotherapies, we expect the rate to go up.”
Dr Cappelli said she wants the case report to raise awareness among patients and clinicians that rheumatologic side effects may occur with checkpoint inhibitors.
“It is important when weighing the risk-benefit ratio of prescribing these drugs,” she said. “And it’s important for people to be on the lookout for symptoms so they can see a rheumatologist early in an effort to prevent or limit joint damage.”
Drs Cappelli and Bingham and their colleagues are planning further collaboration with Johns Hopkins oncologists to better track the incidence of rheumatic disease in patients taking immunotherapy drugs and determine whether any particular characteristics put cancer patients at higher risk of such complications. ![]()
Study explains how a mutation spurs AML development
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
A set of faulty genetic instructions keeps hematopoietic stem/progenitor cells (HSPCs) from maturing and contributes to the development of acute myeloid leukemia (AML), according to research published in Cancer Cell.
Researchers found that a mutation in the gene DNMT3A removes a “brake” on the activity of stemness genes, which leads to the creation of immature precursor cells that can become AML cells.
Specifically, the DNMT3A mutational hotspot at Arg882 (DNMT3AR882H) cooperates with an NRAS mutation (NRASG12D) to transform HSPCs and induce AML development.
“Due to a large-scale cancer sequencing project, the DNMT3A gene is now appreciated to be one of the top 3 most frequently mutated genes in human acute myeloid leukemia, and yet the role of its mutation in the disease has remained far from clear,” said G. Greg Wang, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center in Chapel Hill.
“Our findings not only provide a deeper understanding of how this prevalent mutation contributes to the development of AML, but it also offers useful information on how to develop new strategies to treat AML patients.”
In an attempt to understand how the DNMT3A mutation helps drive AML, Dr Wang and his colleagues created one of the first laboratory AML models for studying somatic mutations in DNMT3A.
The DNMT3A gene codes for a protein that binds to specific sections of DNA with a chemical tag that can influence the activity and expression of the underlying genes in cells.
The researchers found that DNMT3AR882H caused AML cells to have a different pattern of chemical tags that affect how the genetic code is interpreted and how the cell develops.
In cancerous cells with DNMT3AR882H, a set of gene enhancers for several stemness genes—including Meis1, Mn1, and the Hoxa gene cluster—were left unchecked. Therefore, HSPCs were left with a constant “on” switch, allowing the cells to “forget” to mature.
“In acute myeloid leukemia, the expression of these stemness genes are aberrantly maintained at a higher level,” Dr Wang said. “As a result, cells ‘forget’ to proceed to normal differentiation and maturation, generating immature precursor blood cells and a prelude to full-blown cancer.”
The researchers also found that, while the DNMT3A mutation is required for AML development, the mutation itself is not sufficient to cause cancer alone. DNMT3AR882H cooperates with another mutation, NRASG12D.
“We found the RAS mutation stimulates these immature blood cells to be hyper-proliferate,” said study author Rui Lu, PhD, of the University of North Carolina Lineberger Comprehensive Cancer Center.
“However, these cells cannot maintain their stem cell properties. While the DNMT3A mutation itself does not have hyper-proliferative effects, [it] does promote stemness properties and generates leukemia stem/initiating cells together with the RAS mutation.”
The researchers also reported testing a potential treatment in cells with the DNMT3A mutation. They found that AML cells with DNMT3AR882H were sensitive to inhibitors of DOT1L, a cellular enzyme involved in modulation of gene expression activities.
As DOT1L inhibitors are currently under clinical investigation, this finding suggests a potential strategy for treating DNMT3A-mutated AML. ![]()
EMA recommends authorization of T-cell product

Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
Image courtesy of NIAID
The European Medicines Agency (EMA) has recommended granting conditional marketing authorization for a T-cell product known as Zalmoxis.
Zalmoxis is intended for use as an adjunctive therapy to aid immune reconstitution and help treat graft-versus-host disease (GVHD) in adults receiving a haploidentical hematopoietic stem cell transplant to treat hematologic malignancy.
Zalmoxis consists of allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (ΔLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2).
This modification makes the T cells susceptible to treatment with the drug ganciclovir. So if a patient develops GVHD, he can be treated with ganciclovir, which should kill the modified T cells and prevent further development of the disease.
Zalmoxis is being developed by MolMed S.p.A.
The EMA’s Committee for Medicinal Products for Human Use (CHMP) recommended conditional approval for Zalmoxis. Conditional approval is one of the agency’s main mechanisms to facilitate earlier access to medicines that fulfill unmet medical needs.
Conditional approval allows the EMA to recommend a medicine for marketing authorization before the availability of confirmatory clinical trial data, if the benefits of making this medicine available to patients immediately outweigh the risks inherent in the lack of comprehensive data.
Zalmoxis was also assessed by the Committee on Advanced Therapies (CAT), the EMA’s specialized scientific committee for advanced therapy medicinal products, such as gene or cell therapies.
At its June 2016 meeting, the CAT recommended a conditional marketing authorization for Zalmoxis. The CHMP then considered the CAT’s recommendation and agreed with it.
The recommendation has been sent to the European Commission, which will adopt a decision on marketing authorization that will apply to the European Economic Area.
If Zalmoxis is granted conditional marketing authorization, MolMed S.p.A. must provide the EMA with results from an ongoing phase 3 trial (TK008; NCT00914628).
Until the complete data from this trial are available, the CAT and the CHMP will review the benefits and risks of Zalmoxis annually to determine whether the conditional marketing authorization can be maintained.
Zalmoxis was designated as an orphan medicinal product in 2003. Orphan designation gives drug developers access to incentives such as fee reductions for scientific advice and the opportunity to obtain 10 years of market exclusivity for an authorized orphan-designated medicine. ![]()
CHMP rejects ofatumumab as maintenance

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended against expanding the approved indication for ofatumumab (Arzerra).
Novartis, which is developing ofatumumab in cooperation with Genmab, had submitted an application requesting that ofatumumab be authorized as maintenance therapy for patients with relapsed chronic lymphocytic leukemia (CLL).
But the CHMP has advised the European Commission (EC) not to grant this authorization.
The CHMP noted that, in the phase 3 PROLONG trial, ofatumumab maintenance improved progression-free survival (PFS) in CLL patients.
However, the committee said the importance of this improvement is not clear because the PFS results were not supported by other measures, such as overall survival or a significant improvement in patients’ quality of life.
The CHMP also said the use of ofatumumab for maintenance treatment should be seen in the context of its side effects. Common side effects of ofatumumab in the PROLONG trial were infusion reactions, neutropenia, and upper respiratory tract infections.
In the end, the CHMP decided that the PROLONG data were not sufficient to conclude that maintenance treatment with ofatumumab is of more benefit than no treatment. So the committee recommended against expanding the drug’s marketing authorization.
This decision does not have any impact on ongoing clinical trials with ofatumumab.
About ofatumumab
Ofatumumab has been authorized for use in the European Union since April 2010.
The EC first granted ofatumumab conditional approval to treat CLL patients who are refractory to fludarabine and alemtuzumab.
Then, in 2014, the EC granted ofatumumab conditional approval for use in combination with chlorambucil or bendamustine in CLL patients who have not received prior therapy and are not eligible for fludarabine-based therapy.
Ofatumumab received conditional approval because the drug’s benefits appear to outweigh the risks it poses in the aforementioned indications. Ofatumumab will not receive full approval until the drug’s developers submit results of additional research to the EC.
About the PROLONG trial
The PROLONG trial was designed to compare ofatumumab maintenance to no further treatment in patients with a complete or partial response after second- or third-line treatment for CLL. Interim results of the study were presented at ASH 2014.
These results—in 474 patients—suggested that ofatumumab can significantly improve PFS. The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance therapy (P<0.0001).
There was no significant difference in the median overall survival, which was not reached in either treatment arm.
The researchers said there were no unexpected safety findings. The most common adverse events (≥10%) were infusion reactions, neutropenia, and upper respiratory tract infection. ![]()
HCPs may underestimate cancer risk from CT scans
Photo by Angela Mary Butler
Healthcare professionals (HCPs) may not be fully aware of a CT scan’s effect on lifetime malignancy risk, according to a study published in the Journal of Medical Imaging and Radiation Sciences.
Researchers surveyed a group of HCPs on radiation exposure from CT.
And although most of the respondents recognized that CT scans confer an increased risk of cancer, many underestimated the actual dose of radiation a person receives from a CT scan.
The survey was given to 308 HCPs—including physicians, radiologists, and technologists—in Saskatchewan, Canada.
Seventy-three percent of physicians, 97% of radiologists, and 76% of technologists correctly reported that there is an increased cancer risk from one abdominal-pelvic CT.
However, only 18% of physicians, 28% of radiologists, and 22% of technologists were able to correctly identify the dose in relation to chest X-rays.
In fact, 14% of physicians and 12% of technologists (but 0% of radiologists) “vastly” underestimated the dose as less than 10 chest X-ray equivalents, according to researchers.
The average radiation dose from an abdominal-pelvic CT is 10 millisieverts (mSv), compared to 0.02 mSv to 0.2 mSv from one chest X-ray, meaning that a radiation dose from a CT scan is equivalent to the dose from 100 to 250 chest radiographs.
“Underestimating radiation dose from a CT scan is more concerning than knowing the exact dose level, particularly when it is a vast underestimation, as this may lead to minimization of the risk estimate when considering a test,” said study author David Leswick, MD, of the University of Saskatchewan in Saskatoon.
“Although [cancer] risk from radiation dose levels in the range of medical imaging procedures is small, it is real, as evidenced from atomic bomb survivors and nuclear industry workers showing significantly increased risk of malignancy after exposure to doses in the range of diagnostic CT.”
“The risk of fatal malignancy may be as high as 1 in 1000 for a 10-mSv exposure. This risk is significant on a population basis, with up to 2% of cancers in the United States population possibly attributable to CT.”
Another aspect highlighted by the survey was some confusion regarding radiation exposure from MRIs and ultrasounds.
MRIs and ultrasounds do not employ ionizing radiation. However, 20% of physicians, 6% of radiologists, and 7% of technologists attributed radiation exposure to MRIs. Eleven percent of physicians, 0% of radiologists, and 7% of technologists believed an ultrasound used radiation.
“Belief that ionizing radiation is utilized by ultrasound and MRI is troubling, as it may result in underutilization of these imaging modalities because of unfounded radiation concerns,” Dr Leswick said.
“It is important for healthcare professionals (including referring physicians, radiologists, and technologists) to be aware of radiation dose levels and risks from imaging tests for several reasons, including the ability to weigh the risks and benefits of tests, counsel patients on relevant risks, optimize protocols to minimize radiation dose, and select appropriate protocols to minimize radiation dose.” ![]()
Photo by Angela Mary Butler
Healthcare professionals (HCPs) may not be fully aware of a CT scan’s effect on lifetime malignancy risk, according to a study published in the Journal of Medical Imaging and Radiation Sciences.
Researchers surveyed a group of HCPs on radiation exposure from CT.
And although most of the respondents recognized that CT scans confer an increased risk of cancer, many underestimated the actual dose of radiation a person receives from a CT scan.
The survey was given to 308 HCPs—including physicians, radiologists, and technologists—in Saskatchewan, Canada.
Seventy-three percent of physicians, 97% of radiologists, and 76% of technologists correctly reported that there is an increased cancer risk from one abdominal-pelvic CT.
However, only 18% of physicians, 28% of radiologists, and 22% of technologists were able to correctly identify the dose in relation to chest X-rays.
In fact, 14% of physicians and 12% of technologists (but 0% of radiologists) “vastly” underestimated the dose as less than 10 chest X-ray equivalents, according to researchers.
The average radiation dose from an abdominal-pelvic CT is 10 millisieverts (mSv), compared to 0.02 mSv to 0.2 mSv from one chest X-ray, meaning that a radiation dose from a CT scan is equivalent to the dose from 100 to 250 chest radiographs.
“Underestimating radiation dose from a CT scan is more concerning than knowing the exact dose level, particularly when it is a vast underestimation, as this may lead to minimization of the risk estimate when considering a test,” said study author David Leswick, MD, of the University of Saskatchewan in Saskatoon.
“Although [cancer] risk from radiation dose levels in the range of medical imaging procedures is small, it is real, as evidenced from atomic bomb survivors and nuclear industry workers showing significantly increased risk of malignancy after exposure to doses in the range of diagnostic CT.”
“The risk of fatal malignancy may be as high as 1 in 1000 for a 10-mSv exposure. This risk is significant on a population basis, with up to 2% of cancers in the United States population possibly attributable to CT.”
Another aspect highlighted by the survey was some confusion regarding radiation exposure from MRIs and ultrasounds.
MRIs and ultrasounds do not employ ionizing radiation. However, 20% of physicians, 6% of radiologists, and 7% of technologists attributed radiation exposure to MRIs. Eleven percent of physicians, 0% of radiologists, and 7% of technologists believed an ultrasound used radiation.
“Belief that ionizing radiation is utilized by ultrasound and MRI is troubling, as it may result in underutilization of these imaging modalities because of unfounded radiation concerns,” Dr Leswick said.
“It is important for healthcare professionals (including referring physicians, radiologists, and technologists) to be aware of radiation dose levels and risks from imaging tests for several reasons, including the ability to weigh the risks and benefits of tests, counsel patients on relevant risks, optimize protocols to minimize radiation dose, and select appropriate protocols to minimize radiation dose.” ![]()
Photo by Angela Mary Butler
Healthcare professionals (HCPs) may not be fully aware of a CT scan’s effect on lifetime malignancy risk, according to a study published in the Journal of Medical Imaging and Radiation Sciences.
Researchers surveyed a group of HCPs on radiation exposure from CT.
And although most of the respondents recognized that CT scans confer an increased risk of cancer, many underestimated the actual dose of radiation a person receives from a CT scan.
The survey was given to 308 HCPs—including physicians, radiologists, and technologists—in Saskatchewan, Canada.
Seventy-three percent of physicians, 97% of radiologists, and 76% of technologists correctly reported that there is an increased cancer risk from one abdominal-pelvic CT.
However, only 18% of physicians, 28% of radiologists, and 22% of technologists were able to correctly identify the dose in relation to chest X-rays.
In fact, 14% of physicians and 12% of technologists (but 0% of radiologists) “vastly” underestimated the dose as less than 10 chest X-ray equivalents, according to researchers.
The average radiation dose from an abdominal-pelvic CT is 10 millisieverts (mSv), compared to 0.02 mSv to 0.2 mSv from one chest X-ray, meaning that a radiation dose from a CT scan is equivalent to the dose from 100 to 250 chest radiographs.
“Underestimating radiation dose from a CT scan is more concerning than knowing the exact dose level, particularly when it is a vast underestimation, as this may lead to minimization of the risk estimate when considering a test,” said study author David Leswick, MD, of the University of Saskatchewan in Saskatoon.
“Although [cancer] risk from radiation dose levels in the range of medical imaging procedures is small, it is real, as evidenced from atomic bomb survivors and nuclear industry workers showing significantly increased risk of malignancy after exposure to doses in the range of diagnostic CT.”
“The risk of fatal malignancy may be as high as 1 in 1000 for a 10-mSv exposure. This risk is significant on a population basis, with up to 2% of cancers in the United States population possibly attributable to CT.”
Another aspect highlighted by the survey was some confusion regarding radiation exposure from MRIs and ultrasounds.
MRIs and ultrasounds do not employ ionizing radiation. However, 20% of physicians, 6% of radiologists, and 7% of technologists attributed radiation exposure to MRIs. Eleven percent of physicians, 0% of radiologists, and 7% of technologists believed an ultrasound used radiation.
“Belief that ionizing radiation is utilized by ultrasound and MRI is troubling, as it may result in underutilization of these imaging modalities because of unfounded radiation concerns,” Dr Leswick said.
“It is important for healthcare professionals (including referring physicians, radiologists, and technologists) to be aware of radiation dose levels and risks from imaging tests for several reasons, including the ability to weigh the risks and benefits of tests, counsel patients on relevant risks, optimize protocols to minimize radiation dose, and select appropriate protocols to minimize radiation dose.” ![]()
Team describes method of targeting LSCs in BC-CML

Photo courtesy of
UC San Diego Health
New research has revealed a method of targeting leukemia stem cells (LSCs) in blast crisis chronic myeloid leukemia (BC-CML).
For this study, investigators used human cells and mouse models to define the role of ADAR1, an RNA editing enzyme, in BC-CML.
The team discovered how ADAR1 promotes LSC generation and identified a small molecule that can disrupt this process to fight BC-CML.
Catriona Jamieson, MD, PhD, of the University of California, San Diego, and her colleagues described this work in Cell Stem Cell.
“In this study, we showed that cancer stem cells co-opt an RNA editing system to clone themselves,” Dr Jamieson said. “What’s more, we found a method to dial it down.”
The investigators knew that ADAR1 can edit the sequence of microRNAs. By swapping out just one microRNA building block for another, ADAR1 alters the carefully orchestrated system cells use to control which genes are turned on or off at which times.
ADAR1 is also known to promote cancer progression and resistance to therapy. But Dr Jamieson’s team wanted to determine ADAR1’s role in governing LSCs.
The investigators conducted experiments with human BC-CML cells and mouse models of BC-CML. And they found that increased JAK2 signaling and BCR-ABL1 amplification activate ADAR1 in BC-CML cells. Then, hyper-ADAR1 editing slows down microRNAs known as let-7.
Ultimately, this activity increases cellular regeneration, turning white blood cell precursors into LSCs. And LSCs promote BC-CML.
After learning how the ADAR1 system works, Dr Jamieson and her colleagues looked for a way to stop it.
By inhibiting ADAR1 with a small-molecule compound known as 8-Aza, the investigators were able to counter ADAR1’s effect on LSC self-renewal and restore let-7.
Treatment with 8-Aza reduced self-renewal of BC-CML cells by approximately 40%, when compared to untreated cells.
“Based on this research, we believe that detecting ADAR1 activity will be important for predicting cancer progression,” Dr Jamieson said.
“In addition, inhibiting this enzyme represents a unique therapeutic vulnerability in cancer stem cells with active inflammatory signaling that may respond to pharmacologic inhibitors of inflammation sensitivity or selective ADAR1 inhibitors that are currently being developed.” ![]()
Photo courtesy of
UC San Diego Health
New research has revealed a method of targeting leukemia stem cells (LSCs) in blast crisis chronic myeloid leukemia (BC-CML).
For this study, investigators used human cells and mouse models to define the role of ADAR1, an RNA editing enzyme, in BC-CML.
The team discovered how ADAR1 promotes LSC generation and identified a small molecule that can disrupt this process to fight BC-CML.
Catriona Jamieson, MD, PhD, of the University of California, San Diego, and her colleagues described this work in Cell Stem Cell.
“In this study, we showed that cancer stem cells co-opt an RNA editing system to clone themselves,” Dr Jamieson said. “What’s more, we found a method to dial it down.”
The investigators knew that ADAR1 can edit the sequence of microRNAs. By swapping out just one microRNA building block for another, ADAR1 alters the carefully orchestrated system cells use to control which genes are turned on or off at which times.
ADAR1 is also known to promote cancer progression and resistance to therapy. But Dr Jamieson’s team wanted to determine ADAR1’s role in governing LSCs.
The investigators conducted experiments with human BC-CML cells and mouse models of BC-CML. And they found that increased JAK2 signaling and BCR-ABL1 amplification activate ADAR1 in BC-CML cells. Then, hyper-ADAR1 editing slows down microRNAs known as let-7.
Ultimately, this activity increases cellular regeneration, turning white blood cell precursors into LSCs. And LSCs promote BC-CML.
After learning how the ADAR1 system works, Dr Jamieson and her colleagues looked for a way to stop it.
By inhibiting ADAR1 with a small-molecule compound known as 8-Aza, the investigators were able to counter ADAR1’s effect on LSC self-renewal and restore let-7.
Treatment with 8-Aza reduced self-renewal of BC-CML cells by approximately 40%, when compared to untreated cells.
“Based on this research, we believe that detecting ADAR1 activity will be important for predicting cancer progression,” Dr Jamieson said.
“In addition, inhibiting this enzyme represents a unique therapeutic vulnerability in cancer stem cells with active inflammatory signaling that may respond to pharmacologic inhibitors of inflammation sensitivity or selective ADAR1 inhibitors that are currently being developed.” ![]()
Photo courtesy of
UC San Diego Health
New research has revealed a method of targeting leukemia stem cells (LSCs) in blast crisis chronic myeloid leukemia (BC-CML).
For this study, investigators used human cells and mouse models to define the role of ADAR1, an RNA editing enzyme, in BC-CML.
The team discovered how ADAR1 promotes LSC generation and identified a small molecule that can disrupt this process to fight BC-CML.
Catriona Jamieson, MD, PhD, of the University of California, San Diego, and her colleagues described this work in Cell Stem Cell.
“In this study, we showed that cancer stem cells co-opt an RNA editing system to clone themselves,” Dr Jamieson said. “What’s more, we found a method to dial it down.”
The investigators knew that ADAR1 can edit the sequence of microRNAs. By swapping out just one microRNA building block for another, ADAR1 alters the carefully orchestrated system cells use to control which genes are turned on or off at which times.
ADAR1 is also known to promote cancer progression and resistance to therapy. But Dr Jamieson’s team wanted to determine ADAR1’s role in governing LSCs.
The investigators conducted experiments with human BC-CML cells and mouse models of BC-CML. And they found that increased JAK2 signaling and BCR-ABL1 amplification activate ADAR1 in BC-CML cells. Then, hyper-ADAR1 editing slows down microRNAs known as let-7.
Ultimately, this activity increases cellular regeneration, turning white blood cell precursors into LSCs. And LSCs promote BC-CML.
After learning how the ADAR1 system works, Dr Jamieson and her colleagues looked for a way to stop it.
By inhibiting ADAR1 with a small-molecule compound known as 8-Aza, the investigators were able to counter ADAR1’s effect on LSC self-renewal and restore let-7.
Treatment with 8-Aza reduced self-renewal of BC-CML cells by approximately 40%, when compared to untreated cells.
“Based on this research, we believe that detecting ADAR1 activity will be important for predicting cancer progression,” Dr Jamieson said.
“In addition, inhibiting this enzyme represents a unique therapeutic vulnerability in cancer stem cells with active inflammatory signaling that may respond to pharmacologic inhibitors of inflammation sensitivity or selective ADAR1 inhibitors that are currently being developed.”
FDA grants drug breakthrough designation to treat GVHD

Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Photo from Business Wire
The US Food and Drug Administration (FDA) has granted breakthrough therapy designation for ruxolitinib (Jakafi) for the treatment of patients with acute graft-versus-host disease (GVHD).
Ruxolitinib is a JAK1/2 inhibitor that is already FDA-approved to treat patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
The drug is also approved to treat patients with intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF, and post-essential thrombocythemia MF.
Ruxolitinib is marketed as Jakafi by Incyte in the US and as Jakavi by Novartis outside the US.
“Receiving breakthrough therapy designation from the FDA recognizes the severe nature of acute GVHD, the clear unmet medical need of these patients, and the potential, based on clinical evidence to date, for ruxolitinib to address the urgent needs of patients with this life-threatening disease,” said Steven Stein, MD, Incyte’s chief medical officer.
The FDA’s breakthrough therapy designation is intended to expedite the development and review of new therapies for serious or life-threatening conditions.
To earn the designation, a treatment must show encouraging early clinical results demonstrating substantial improvement over available therapies with regard to a clinically significant endpoint, or it must fulfill an unmet need.
Company recalls blood collection system

Photo by Charles Haymond
Haemonetics Corporation is recalling all lots of the Leukotrap RC System with RC2D Filter (re-order numbers 129-62 and 129-63) distributed since April 14, 2016.
The company says using these lots of the blood collection system may result in a higher-than-expected level of leukocytes in transfused blood.
Earlier this month, Haemonetics recalled 3 lots of the Leukotrap RC System with RC2D Filter due to reports of higher-than-expected residual white blood cells.
Since then, the company has received additional reports related to other lots.
Blood products that have been processed with the lots distributed since April 14, 2016, should not be re-filtered and should be labeled as non-leukoreduced, unless individual units have been tested and found suitable to be labeled as leukoreduced.
Continued use of these lots will require customers to verify that each unit labeled as leukoreduced was tested and meets standards for residual white blood cells acceptable to the US Food and Drug Administration.
Customers who want to return unused product to Haemonetics should contact their local customer service representative.
Photo by Charles Haymond
Haemonetics Corporation is recalling all lots of the Leukotrap RC System with RC2D Filter (re-order numbers 129-62 and 129-63) distributed since April 14, 2016.
The company says using these lots of the blood collection system may result in a higher-than-expected level of leukocytes in transfused blood.
Earlier this month, Haemonetics recalled 3 lots of the Leukotrap RC System with RC2D Filter due to reports of higher-than-expected residual white blood cells.
Since then, the company has received additional reports related to other lots.
Blood products that have been processed with the lots distributed since April 14, 2016, should not be re-filtered and should be labeled as non-leukoreduced, unless individual units have been tested and found suitable to be labeled as leukoreduced.
Continued use of these lots will require customers to verify that each unit labeled as leukoreduced was tested and meets standards for residual white blood cells acceptable to the US Food and Drug Administration.
Customers who want to return unused product to Haemonetics should contact their local customer service representative.
Photo by Charles Haymond
Haemonetics Corporation is recalling all lots of the Leukotrap RC System with RC2D Filter (re-order numbers 129-62 and 129-63) distributed since April 14, 2016.
The company says using these lots of the blood collection system may result in a higher-than-expected level of leukocytes in transfused blood.
Earlier this month, Haemonetics recalled 3 lots of the Leukotrap RC System with RC2D Filter due to reports of higher-than-expected residual white blood cells.
Since then, the company has received additional reports related to other lots.
Blood products that have been processed with the lots distributed since April 14, 2016, should not be re-filtered and should be labeled as non-leukoreduced, unless individual units have been tested and found suitable to be labeled as leukoreduced.
Continued use of these lots will require customers to verify that each unit labeled as leukoreduced was tested and meets standards for residual white blood cells acceptable to the US Food and Drug Administration.
Customers who want to return unused product to Haemonetics should contact their local customer service representative.
Interim PET-CT can spare HL patients intensive chemo
The use of interim PET-CT scans can spare some advanced Hodgkin lymphoma (HL) patients the toxicity associated with bleomycin, according to researchers.
The team found that patients with negative PET-CT scans after 2 cycles of ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) could go on to receive AVD (doxorubicin, vinblastine, and dacarbazine) without experiencing a significant decrease in progression-free survival (PFS) or overall survival (OS).
Peter Johnson, MD, of the University of Southampton in the UK, and his colleagues reported these findings in NEJM.
“The good news is that the majority of people diagnosed with Hodgkin lymphoma can be cured,” Dr Johnson said. “In this trial, more than 95% of patients are alive after 3 years.”
“But we worry about the long-term side effects from the treatments we use. As we’ve done in this trial, personalizing treatment based on how well it works is a major development for patients with Hodgkin lymphoma and sets a new standard of care.”
Patients and treatment
For this study, Dr Johnson and his colleagues enrolled 1214 patients with newly diagnosed, advanced, classic HL. The patients’ median age was 33 (range, 18 to 79), and 54.5% were male. More patients had stage II disease (41.6%) than stage III (30.2%) or IV (28.3%).
A total of 1119 patients underwent a baseline PET-CT scan, received 2 cycles of ABVD, and underwent an interim PET-CT scan.
The patients with negative interim scans were then randomized to continue treatment with ABVD (n=470) or with AVD (n=465) in cycles 3 through 6.
Patients with positive interim scans (n=182) went on to receive BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone, n=172), salvage treatments (n=6), or ABVD (n=4).
Results
The study’s primary outcome was the difference in 3-year PFS between the randomized groups of PET-CT-negative patients.
With a median follow-up of 41 months, the 3-year PFS was 85.7% in the ABVD group and 84.4% in the AVD group. The hazard ratio was 1.13 (95% CI, 0.81 to 1.57; P=0.48) in the intention-to-treat analysis and 1.10 (95% CI, 0.79 to 1.53; P=0.58) in the per-protocol analysis.
The absolute difference in 3-year PFS (ABVD minus AVD) was 1.6 percentage points (95% CI, −3.2 to 5.3).
The OS rates were 97.2% in the ABVD group and 97.6% in the AVD group. The hazard ratio in the intention-to-treat analysis was 0.90 (95% CI, 0.47 to 1.74; P=0.76).
Patients in the ABVD group had a significantly higher rate of clinical adverse events than patients in the AVD group—31% and 21%, respectively (P<0.005).
Patients in the ABVD group also had significantly (P<0.05) higher rates of fatigue (3% vs 1%), febrile neutropenia (5% vs 2%), pulmonary/upper respiratory events (3% vs 1%), and dyspnea (2% vs <0.5%). But patients in the AVD group had a significantly higher rate of thrombocytopenia (3% vs 1%).
For patients who had positive interim PET-CT scans, the 3-year PFS was 67.5%, and the OS was 87.8%. Among the 172 patients who went on to receive BEACOPP, 74.4% had negative findings on a third PET-CT scan.
Overall, 62 patients died during the trial—24 from HL. So, for the entire study cohort, the 3-year PFS was 82.6%, and the OS was 95.8%.
The use of interim PET-CT scans can spare some advanced Hodgkin lymphoma (HL) patients the toxicity associated with bleomycin, according to researchers.
The team found that patients with negative PET-CT scans after 2 cycles of ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) could go on to receive AVD (doxorubicin, vinblastine, and dacarbazine) without experiencing a significant decrease in progression-free survival (PFS) or overall survival (OS).
Peter Johnson, MD, of the University of Southampton in the UK, and his colleagues reported these findings in NEJM.
“The good news is that the majority of people diagnosed with Hodgkin lymphoma can be cured,” Dr Johnson said. “In this trial, more than 95% of patients are alive after 3 years.”
“But we worry about the long-term side effects from the treatments we use. As we’ve done in this trial, personalizing treatment based on how well it works is a major development for patients with Hodgkin lymphoma and sets a new standard of care.”
Patients and treatment
For this study, Dr Johnson and his colleagues enrolled 1214 patients with newly diagnosed, advanced, classic HL. The patients’ median age was 33 (range, 18 to 79), and 54.5% were male. More patients had stage II disease (41.6%) than stage III (30.2%) or IV (28.3%).
A total of 1119 patients underwent a baseline PET-CT scan, received 2 cycles of ABVD, and underwent an interim PET-CT scan.
The patients with negative interim scans were then randomized to continue treatment with ABVD (n=470) or with AVD (n=465) in cycles 3 through 6.
Patients with positive interim scans (n=182) went on to receive BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone, n=172), salvage treatments (n=6), or ABVD (n=4).
Results
The study’s primary outcome was the difference in 3-year PFS between the randomized groups of PET-CT-negative patients.
With a median follow-up of 41 months, the 3-year PFS was 85.7% in the ABVD group and 84.4% in the AVD group. The hazard ratio was 1.13 (95% CI, 0.81 to 1.57; P=0.48) in the intention-to-treat analysis and 1.10 (95% CI, 0.79 to 1.53; P=0.58) in the per-protocol analysis.
The absolute difference in 3-year PFS (ABVD minus AVD) was 1.6 percentage points (95% CI, −3.2 to 5.3).
The OS rates were 97.2% in the ABVD group and 97.6% in the AVD group. The hazard ratio in the intention-to-treat analysis was 0.90 (95% CI, 0.47 to 1.74; P=0.76).
Patients in the ABVD group had a significantly higher rate of clinical adverse events than patients in the AVD group—31% and 21%, respectively (P<0.005).
Patients in the ABVD group also had significantly (P<0.05) higher rates of fatigue (3% vs 1%), febrile neutropenia (5% vs 2%), pulmonary/upper respiratory events (3% vs 1%), and dyspnea (2% vs <0.5%). But patients in the AVD group had a significantly higher rate of thrombocytopenia (3% vs 1%).
For patients who had positive interim PET-CT scans, the 3-year PFS was 67.5%, and the OS was 87.8%. Among the 172 patients who went on to receive BEACOPP, 74.4% had negative findings on a third PET-CT scan.
Overall, 62 patients died during the trial—24 from HL. So, for the entire study cohort, the 3-year PFS was 82.6%, and the OS was 95.8%.
The use of interim PET-CT scans can spare some advanced Hodgkin lymphoma (HL) patients the toxicity associated with bleomycin, according to researchers.
The team found that patients with negative PET-CT scans after 2 cycles of ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine) could go on to receive AVD (doxorubicin, vinblastine, and dacarbazine) without experiencing a significant decrease in progression-free survival (PFS) or overall survival (OS).
Peter Johnson, MD, of the University of Southampton in the UK, and his colleagues reported these findings in NEJM.
“The good news is that the majority of people diagnosed with Hodgkin lymphoma can be cured,” Dr Johnson said. “In this trial, more than 95% of patients are alive after 3 years.”
“But we worry about the long-term side effects from the treatments we use. As we’ve done in this trial, personalizing treatment based on how well it works is a major development for patients with Hodgkin lymphoma and sets a new standard of care.”
Patients and treatment
For this study, Dr Johnson and his colleagues enrolled 1214 patients with newly diagnosed, advanced, classic HL. The patients’ median age was 33 (range, 18 to 79), and 54.5% were male. More patients had stage II disease (41.6%) than stage III (30.2%) or IV (28.3%).
A total of 1119 patients underwent a baseline PET-CT scan, received 2 cycles of ABVD, and underwent an interim PET-CT scan.
The patients with negative interim scans were then randomized to continue treatment with ABVD (n=470) or with AVD (n=465) in cycles 3 through 6.
Patients with positive interim scans (n=182) went on to receive BEACOPP (bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone, n=172), salvage treatments (n=6), or ABVD (n=4).
Results
The study’s primary outcome was the difference in 3-year PFS between the randomized groups of PET-CT-negative patients.
With a median follow-up of 41 months, the 3-year PFS was 85.7% in the ABVD group and 84.4% in the AVD group. The hazard ratio was 1.13 (95% CI, 0.81 to 1.57; P=0.48) in the intention-to-treat analysis and 1.10 (95% CI, 0.79 to 1.53; P=0.58) in the per-protocol analysis.
The absolute difference in 3-year PFS (ABVD minus AVD) was 1.6 percentage points (95% CI, −3.2 to 5.3).
The OS rates were 97.2% in the ABVD group and 97.6% in the AVD group. The hazard ratio in the intention-to-treat analysis was 0.90 (95% CI, 0.47 to 1.74; P=0.76).
Patients in the ABVD group had a significantly higher rate of clinical adverse events than patients in the AVD group—31% and 21%, respectively (P<0.005).
Patients in the ABVD group also had significantly (P<0.05) higher rates of fatigue (3% vs 1%), febrile neutropenia (5% vs 2%), pulmonary/upper respiratory events (3% vs 1%), and dyspnea (2% vs <0.5%). But patients in the AVD group had a significantly higher rate of thrombocytopenia (3% vs 1%).
For patients who had positive interim PET-CT scans, the 3-year PFS was 67.5%, and the OS was 87.8%. Among the 172 patients who went on to receive BEACOPP, 74.4% had negative findings on a third PET-CT scan.
Overall, 62 patients died during the trial—24 from HL. So, for the entire study cohort, the 3-year PFS was 82.6%, and the OS was 95.8%.