User login
MicroRNA could be used to treat DLBCL, team says

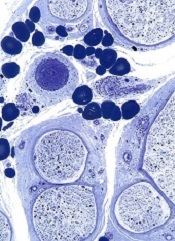
A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
A microRNA known as miR-181a dampens signals from the NF-κB pathway and affects the pathogenesis of diffuse large B-cell lymphoma (DLBCL), according to research published in Blood.
The study showed that, by reducing NF-κB signaling, miR-181a hinders tumor cell proliferation and survival.
And the effect is more pronounced in activated B-cell-like (ABC) DLBCL than in germinal center B-cell-like (GCB) DLBCL.
The researchers therefore believe miR-181a could be used to treat ABC DLBCL.
“The miR-181a microRNA is one of the first examples of a pathway that deactivates NF-κB at multiple levels, functioning as a master regulator,” said study author Izidore S. Lossos, MD, of the University of Miami Miller School of Medicine in Florida.
“In certain tumors, there is no expression of this microRNA, which allows cells to propagate. We believe miR-181a could eventually be used
therapeutically.”
To understand the role of miR-181a in the different types of DLBCL, Dr Lossos and his colleaguese studied both cell lines and mouse models.
The team found that miR-181a levels were significantly lower in ABC DLBCL than in GCB DLBCL.
When they increased miR-181a expression in DLBCL cell lines, the researchers observed a reduction in NF-κB activity and a decrease in cell proliferation and survival. These effects were more potent in ABC DLBCL than in GCB DLBCL.
When the researchers increased miR-181a expression in the mouse models, they observed a significant reduction in tumor growth and a significant increase in animal survival, but only in ABC DLBCL. In GCB DLBCL, there were no significant changes compared to controls.
The researchers said the ability of miR-181a to reduce NF-κB levels may be why the presence of miR-181a has been linked to better outcomes for certain DLBCL patients. Previous studies by Dr Lossos’s group have shown that DLBCL patients whose tumors contain more miR-181a have better prognoses.
With the current study, the team found that miR-181a is a master regulator, turning off a number of genes in the NF-κB pathway, including CARD11, a known DLBCL oncogene, and a number of transcription factors that drive NF-κB signaling.
“We knew that miR181a was biomarker for survival,” Dr Lossos said. “This explains the mechanisms behind it.”
In addition to providing a better understanding of the NF-κB pathway, these results provide hope that miR-181a could be used to improve treatment for patients with ABC DLBCL.
“We are trying to develop miR-181a as a potential therapy,” Dr Lossos said. “But we are only at the beginning. Much more work needs to be done. It will not be a simple journey, but we are sure it can be done and tested in humans eventually to see that it indeed will improve patients’ outcomes.” ![]()
Study provides new insight into blood vessel formation
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
A study published in Nature Cell Biology helps explain how hemodynamic forces contribute to the formation of new vascular lumens during blood vessel morphogenesis.
Investigators found that blood flow drives lumen expansion during sprouting angiogenesis in vivo by inducing spherical deformations of the apical
membrane of endothelial cells, in a process dubbed “inverse blebbing.”
“This work, combined with previous studies, highlights the importance of balanced endothelial cell contractility in allowing the expansion and maintenance of endothelial lumens during blood vessel development,” said study author Holger Gerhardt, PhD, of the Max Delbrück Center for Molecular Medicine in Berlin, Germany.
These results challenge the previous idea that sprouting cells expand lumens independently of blood flow during angiogenesis through the generation and fusion of intracellular vacuoles.
The investigators showed that hemodynamic forces dynamically shape the apical membrane of single or groups of endothelial cells during angiogenesis to form and expand new lumenized vascular tubes.
“We find that this process relies on a tight balance between the forces applied on the membrane and the local contractile responses from the endothelial cells, as impairing this balance either way leads to lumen defects,” Dr Gerhardt said.
These findings suggest the process of blebbing does not require a specific polarity but is likely to be generally applicable to situations in which external versus internal pressure differences challenge the stability and elasticity of the actin cortex.
It more generally raises the question of the role of apical membrane contractility in the adaptation to varying hemodynamic environments, both during blood vessel morphogenesis, as connections form or remodel, and in pathological settings.
“Understanding whether and how this plasticity of the apical membrane and its underlying cortex is challenged in pathological conditions, where vessels exhibit altered perfusion and lack organized structure, has the potential to provide deeper insight into mechanisms of vascular adaptation and maladaptation,” Dr Gerhardt said. “We will definitely further investigate this.” ![]()
Gene discovery could help fight malaria

Photo by James Gathany
Researchers believe they may have discovered a male-determining gene in the malaria-carrying mosquito species Anopheles gambiae.
The discovery of this gene provides scientists with a foundation for studying male mosquito biology.
And this is significant because male mosquitoes offer the potential to develop novel vector control strategies to combat malaria because males do not feed on blood or transmit disease.
One vector control method under development involves genetic modification of the mosquito to bias the population sex ratio toward males.
Modeling has shown the most efficient means for genetic modification of mosquitoes is engineering a driving Y chromosome.
A molecular-level understanding of the Y chromosome of the malaria-carrying mosquito is important to inform and optimize this type of a strategy.
So Omar Akbari, PhD, of the University of California, Riverside, and his colleagues set out to gain such an understanding.
The team used multiple genome sequencing techniques to identify an extensive dataset of Y chromosome sequences and explore their organization and evolution in the Anopheles gambiae complex, a group of at least 7 morphologically indistinguishable species of mosquitoes in the genus Anopheles.
This revealed that only 1 gene, YG2, is exclusive to the Y chromosome across the species complex and may therefore be a male-determining gene.
Dr Akbari and his colleagues described this discovery in PNAS.
Photo by James Gathany
Researchers believe they may have discovered a male-determining gene in the malaria-carrying mosquito species Anopheles gambiae.
The discovery of this gene provides scientists with a foundation for studying male mosquito biology.
And this is significant because male mosquitoes offer the potential to develop novel vector control strategies to combat malaria because males do not feed on blood or transmit disease.
One vector control method under development involves genetic modification of the mosquito to bias the population sex ratio toward males.
Modeling has shown the most efficient means for genetic modification of mosquitoes is engineering a driving Y chromosome.
A molecular-level understanding of the Y chromosome of the malaria-carrying mosquito is important to inform and optimize this type of a strategy.
So Omar Akbari, PhD, of the University of California, Riverside, and his colleagues set out to gain such an understanding.
The team used multiple genome sequencing techniques to identify an extensive dataset of Y chromosome sequences and explore their organization and evolution in the Anopheles gambiae complex, a group of at least 7 morphologically indistinguishable species of mosquitoes in the genus Anopheles.
This revealed that only 1 gene, YG2, is exclusive to the Y chromosome across the species complex and may therefore be a male-determining gene.
Dr Akbari and his colleagues described this discovery in PNAS.
Photo by James Gathany
Researchers believe they may have discovered a male-determining gene in the malaria-carrying mosquito species Anopheles gambiae.
The discovery of this gene provides scientists with a foundation for studying male mosquito biology.
And this is significant because male mosquitoes offer the potential to develop novel vector control strategies to combat malaria because males do not feed on blood or transmit disease.
One vector control method under development involves genetic modification of the mosquito to bias the population sex ratio toward males.
Modeling has shown the most efficient means for genetic modification of mosquitoes is engineering a driving Y chromosome.
A molecular-level understanding of the Y chromosome of the malaria-carrying mosquito is important to inform and optimize this type of a strategy.
So Omar Akbari, PhD, of the University of California, Riverside, and his colleagues set out to gain such an understanding.
The team used multiple genome sequencing techniques to identify an extensive dataset of Y chromosome sequences and explore their organization and evolution in the Anopheles gambiae complex, a group of at least 7 morphologically indistinguishable species of mosquitoes in the genus Anopheles.
This revealed that only 1 gene, YG2, is exclusive to the Y chromosome across the species complex and may therefore be a male-determining gene.
Dr Akbari and his colleagues described this discovery in PNAS.
CHMP recommends daratumumab for MM

Photo courtesy of Janssen
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended conditional marketing authorization for daratumumab (Darzalex), a first-in-class monoclonal antibody targeting CD38.
The recommended indication for daratumumab is as monotherapy for adults with relapsed and refractory multiple myeloma (MM).
The patients must have progressed on their last therapy and have received treatment with both a proteasome inhibitor and an immunomodulatory agent.
The CHMP’s positive opinion will now be reviewed by the European Commission, which has the authority to grant marketing authorization for medicines in the European Economic Area.
The European Commission’s final decision on daratumumab is anticipated in the coming months.
About conditional authorization
A product may receive conditional marketing authorization if the CHMP finds that, although comprehensive clinical data on the safety and efficacy of the product are not available, all of the following requirements are met:
- The risk-benefit balance of the product is positive
- The company developing the product will likely be in a position to provide comprehensive clinical data in the future
- Unmet medical needs will be fulfilled
- The benefit to public health of the immediate availability of the product outweighs the risk inherent in the fact that additional data are still required.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder will be required to complete ongoing studies or to conduct new studies with a view to confirming that the benefit-risk balance of a product is positive. In addition, specific obligations may be imposed in relation to the collection of pharmacovigilance data.
About daratumumab
Daratumumab is the first CD38-directed monoclonal antibody recommended for approval in Europe. It works by binding to CD38, a signaling molecule highly expressed on the surface of MM cells regardless of stage of disease.
In binding to CD38, daratumumab triggers the patient’s own immune system to attack the cancer cells, resulting in rapid tumor cell death through multiple, immune-mediated mechanisms of action and through immunomodulatory effects, in addition to direct tumor cell death via apoptosis.
The CHMP’s positive opinion of daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and 3 additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the drug’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. Janssen licensed daratumumab from Genmab A/S in August 2012. ![]()
Photo courtesy of Janssen
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended conditional marketing authorization for daratumumab (Darzalex), a first-in-class monoclonal antibody targeting CD38.
The recommended indication for daratumumab is as monotherapy for adults with relapsed and refractory multiple myeloma (MM).
The patients must have progressed on their last therapy and have received treatment with both a proteasome inhibitor and an immunomodulatory agent.
The CHMP’s positive opinion will now be reviewed by the European Commission, which has the authority to grant marketing authorization for medicines in the European Economic Area.
The European Commission’s final decision on daratumumab is anticipated in the coming months.
About conditional authorization
A product may receive conditional marketing authorization if the CHMP finds that, although comprehensive clinical data on the safety and efficacy of the product are not available, all of the following requirements are met:
- The risk-benefit balance of the product is positive
- The company developing the product will likely be in a position to provide comprehensive clinical data in the future
- Unmet medical needs will be fulfilled
- The benefit to public health of the immediate availability of the product outweighs the risk inherent in the fact that additional data are still required.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder will be required to complete ongoing studies or to conduct new studies with a view to confirming that the benefit-risk balance of a product is positive. In addition, specific obligations may be imposed in relation to the collection of pharmacovigilance data.
About daratumumab
Daratumumab is the first CD38-directed monoclonal antibody recommended for approval in Europe. It works by binding to CD38, a signaling molecule highly expressed on the surface of MM cells regardless of stage of disease.
In binding to CD38, daratumumab triggers the patient’s own immune system to attack the cancer cells, resulting in rapid tumor cell death through multiple, immune-mediated mechanisms of action and through immunomodulatory effects, in addition to direct tumor cell death via apoptosis.
The CHMP’s positive opinion of daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and 3 additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the drug’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. Janssen licensed daratumumab from Genmab A/S in August 2012. ![]()
Photo courtesy of Janssen
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended conditional marketing authorization for daratumumab (Darzalex), a first-in-class monoclonal antibody targeting CD38.
The recommended indication for daratumumab is as monotherapy for adults with relapsed and refractory multiple myeloma (MM).
The patients must have progressed on their last therapy and have received treatment with both a proteasome inhibitor and an immunomodulatory agent.
The CHMP’s positive opinion will now be reviewed by the European Commission, which has the authority to grant marketing authorization for medicines in the European Economic Area.
The European Commission’s final decision on daratumumab is anticipated in the coming months.
About conditional authorization
A product may receive conditional marketing authorization if the CHMP finds that, although comprehensive clinical data on the safety and efficacy of the product are not available, all of the following requirements are met:
- The risk-benefit balance of the product is positive
- The company developing the product will likely be in a position to provide comprehensive clinical data in the future
- Unmet medical needs will be fulfilled
- The benefit to public health of the immediate availability of the product outweighs the risk inherent in the fact that additional data are still required.
Conditional marketing authorizations are valid for 1 year, on a renewable basis. The holder will be required to complete ongoing studies or to conduct new studies with a view to confirming that the benefit-risk balance of a product is positive. In addition, specific obligations may be imposed in relation to the collection of pharmacovigilance data.
About daratumumab
Daratumumab is the first CD38-directed monoclonal antibody recommended for approval in Europe. It works by binding to CD38, a signaling molecule highly expressed on the surface of MM cells regardless of stage of disease.
In binding to CD38, daratumumab triggers the patient’s own immune system to attack the cancer cells, resulting in rapid tumor cell death through multiple, immune-mediated mechanisms of action and through immunomodulatory effects, in addition to direct tumor cell death via apoptosis.
The CHMP’s positive opinion of daratumumab was based on a review of data from the phase 2 SIRIUS study, the phase 1/2 GEN501 study, and 3 additional supportive studies.
The GEN501 study enrolled 102 patients with relapsed MM or relapsed MM that was refractory to 2 or more prior lines of therapy. The patients received daratumumab at a range of doses and on a number of different schedules.
The results suggested daratumumab is most effective at a dose of 16 mg/kg. At this dose, the overall response rate was 36%. Most adverse events in this study were grade 1 or 2, although serious events did occur.
The SIRIUS study enrolled 124 MM patients who had received 3 or more prior lines of therapy. They received daratumumab at different doses and on different schedules, but 106 patients received the drug at 16 mg/kg.
Twenty-nine percent of the 106 patients responded to treatment, and the median duration of response was 7 months. Thirty percent of patients experienced serious adverse events.
Findings from a combined efficacy analysis of the GEN501 and SIRIUS trials demonstrated that, after a mean follow-up of 14.8 months, the estimated median overall survival for patients who received single-agent daratumumab at 16 mg/kg was 20 months.
Five phase 3 clinical studies with daratumumab in MM patients—in relapsed and frontline settings—are ongoing. Additional studies are ongoing or planned to assess the drug’s potential in other malignant and pre-malignant diseases in which CD38 is expressed.
Janssen has exclusive worldwide rights to the development, manufacturing, and commercialization of daratumumab for all potential indications. Janssen licensed daratumumab from Genmab A/S in August 2012. ![]()
VTE risk varies with hormone therapy

Image by Andre E.X. Brown
Results of a case-control study indicate that estrogen-only hormone therapy carries a lower risk of venous thromboembolism (VTE) than combined estrogen-progestogen therapy.
The study also suggests the type of progestogen a patient receives does not significantly impact the risk of VTE, but the route of administration for estrogen does.
Annica Bergendal, MD, PhD, of Karolinska Institutet in Stockholm, Sweden, and her colleagues reported these findings in Menopause.
The study was conducted in Sweden between 2003 and 2009. It included 838 women with VTE and 891 age-matched control subjects.
Analyses suggested the risk of VTE was almost 2-fold higher in women currently on hormone therapy than in those not taking hormones. The odds ratio (OR)—which was adjusted for smoking, body mass index, and immobilization—was 1.72 (95% CI 1.34-2.20).
Women who took combined estrogen-progestogen therapy had nearly 3 times the VTE risk of those who took no hormones (OR 2.85, 95% CI 2.08-3.90), but the risk was much lower for women who took estrogen alone (OR 1.31, 95% CI 0.78-2.21).
The risk of VTE with combined estrogen-progestogen treatment was about double that of estrogen alone (OR 2.18, 95% CI 1.21-3.92).
Researchers have wondered whether the type of progestogen used makes a difference in the risk of VTE, but this study didn’t show any significant difference in risk between 2 commonly used progestogens.
When oral estrogen was combined with progestogen, the risk of VTE was somewhat, but not significantly, higher among users of medroxyprogesterone acetate (OR 2.94, 95% CI 1.67-5.36) than among users of norethisterone acetate (OR 2.29, 95% CI 1.50-3.40).
On the other hand, the way estrogen was delivered appeared to impact the risk of VTE, with oral estrogen conferring the highest risk.
When the researchers used transdermal estrogen—given alone—as reference, they observed an increased risk of VTE associated with oral estrogen alone (OR 1.84, 95% CI 0.62-5.52).
And when the researchers used locally (vaginally) administered estrogen alone as reference, they saw an increased risk of VTE associated with oral estrogen alone (OR 2.64, 95% CI 1.30-5.38).
Among women using combined estrogen-progestogen treatment, with transdermal estrogen as a reference, there was an increase in VTE risk associated with oral estrogen (OR 2.21, 95% CI 0.88-5.60).
“This study adds to our knowledge that transdermal estrogen therapies are safer than oral, and that different estrogen or progestogen combinations may have different risks,” said JoAnn V. Pinkerton, MD, executive director of the North American Menopause Society, who was not involved in this study.
“The lack of blood clots with transdermal estrogen and with vaginal estrogen is very reassuring for women who need to continue taking hormones as they age, when risk of blood clots increases.” ![]()
Image by Andre E.X. Brown
Results of a case-control study indicate that estrogen-only hormone therapy carries a lower risk of venous thromboembolism (VTE) than combined estrogen-progestogen therapy.
The study also suggests the type of progestogen a patient receives does not significantly impact the risk of VTE, but the route of administration for estrogen does.
Annica Bergendal, MD, PhD, of Karolinska Institutet in Stockholm, Sweden, and her colleagues reported these findings in Menopause.
The study was conducted in Sweden between 2003 and 2009. It included 838 women with VTE and 891 age-matched control subjects.
Analyses suggested the risk of VTE was almost 2-fold higher in women currently on hormone therapy than in those not taking hormones. The odds ratio (OR)—which was adjusted for smoking, body mass index, and immobilization—was 1.72 (95% CI 1.34-2.20).
Women who took combined estrogen-progestogen therapy had nearly 3 times the VTE risk of those who took no hormones (OR 2.85, 95% CI 2.08-3.90), but the risk was much lower for women who took estrogen alone (OR 1.31, 95% CI 0.78-2.21).
The risk of VTE with combined estrogen-progestogen treatment was about double that of estrogen alone (OR 2.18, 95% CI 1.21-3.92).
Researchers have wondered whether the type of progestogen used makes a difference in the risk of VTE, but this study didn’t show any significant difference in risk between 2 commonly used progestogens.
When oral estrogen was combined with progestogen, the risk of VTE was somewhat, but not significantly, higher among users of medroxyprogesterone acetate (OR 2.94, 95% CI 1.67-5.36) than among users of norethisterone acetate (OR 2.29, 95% CI 1.50-3.40).
On the other hand, the way estrogen was delivered appeared to impact the risk of VTE, with oral estrogen conferring the highest risk.
When the researchers used transdermal estrogen—given alone—as reference, they observed an increased risk of VTE associated with oral estrogen alone (OR 1.84, 95% CI 0.62-5.52).
And when the researchers used locally (vaginally) administered estrogen alone as reference, they saw an increased risk of VTE associated with oral estrogen alone (OR 2.64, 95% CI 1.30-5.38).
Among women using combined estrogen-progestogen treatment, with transdermal estrogen as a reference, there was an increase in VTE risk associated with oral estrogen (OR 2.21, 95% CI 0.88-5.60).
“This study adds to our knowledge that transdermal estrogen therapies are safer than oral, and that different estrogen or progestogen combinations may have different risks,” said JoAnn V. Pinkerton, MD, executive director of the North American Menopause Society, who was not involved in this study.
“The lack of blood clots with transdermal estrogen and with vaginal estrogen is very reassuring for women who need to continue taking hormones as they age, when risk of blood clots increases.” ![]()
Image by Andre E.X. Brown
Results of a case-control study indicate that estrogen-only hormone therapy carries a lower risk of venous thromboembolism (VTE) than combined estrogen-progestogen therapy.
The study also suggests the type of progestogen a patient receives does not significantly impact the risk of VTE, but the route of administration for estrogen does.
Annica Bergendal, MD, PhD, of Karolinska Institutet in Stockholm, Sweden, and her colleagues reported these findings in Menopause.
The study was conducted in Sweden between 2003 and 2009. It included 838 women with VTE and 891 age-matched control subjects.
Analyses suggested the risk of VTE was almost 2-fold higher in women currently on hormone therapy than in those not taking hormones. The odds ratio (OR)—which was adjusted for smoking, body mass index, and immobilization—was 1.72 (95% CI 1.34-2.20).
Women who took combined estrogen-progestogen therapy had nearly 3 times the VTE risk of those who took no hormones (OR 2.85, 95% CI 2.08-3.90), but the risk was much lower for women who took estrogen alone (OR 1.31, 95% CI 0.78-2.21).
The risk of VTE with combined estrogen-progestogen treatment was about double that of estrogen alone (OR 2.18, 95% CI 1.21-3.92).
Researchers have wondered whether the type of progestogen used makes a difference in the risk of VTE, but this study didn’t show any significant difference in risk between 2 commonly used progestogens.
When oral estrogen was combined with progestogen, the risk of VTE was somewhat, but not significantly, higher among users of medroxyprogesterone acetate (OR 2.94, 95% CI 1.67-5.36) than among users of norethisterone acetate (OR 2.29, 95% CI 1.50-3.40).
On the other hand, the way estrogen was delivered appeared to impact the risk of VTE, with oral estrogen conferring the highest risk.
When the researchers used transdermal estrogen—given alone—as reference, they observed an increased risk of VTE associated with oral estrogen alone (OR 1.84, 95% CI 0.62-5.52).
And when the researchers used locally (vaginally) administered estrogen alone as reference, they saw an increased risk of VTE associated with oral estrogen alone (OR 2.64, 95% CI 1.30-5.38).
Among women using combined estrogen-progestogen treatment, with transdermal estrogen as a reference, there was an increase in VTE risk associated with oral estrogen (OR 2.21, 95% CI 0.88-5.60).
“This study adds to our knowledge that transdermal estrogen therapies are safer than oral, and that different estrogen or progestogen combinations may have different risks,” said JoAnn V. Pinkerton, MD, executive director of the North American Menopause Society, who was not involved in this study.
“The lack of blood clots with transdermal estrogen and with vaginal estrogen is very reassuring for women who need to continue taking hormones as they age, when risk of blood clots increases.” ![]()
Study implicates circular RNAs in leukemia, other cancers

Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
Image courtesy of The Armed
Forces Institute of Pathology
A class of circular RNAs may play a key role in the development and progression of certain leukemias and other cancers, according to research published in Cell.
Investigators found that cancer-associated chromosomal translocations give rise to fusion circular RNAs (f-circRNAs).
And these f-circRNAs aid cellular transformation, promote cell viability, confer treatment resistance, and exhibit tumor-promoting properties in vivo.
“Cancer is essentially a disease of mutated or broken genes, so that motivated us to examine whether circular RNAs, like proteins, can be affected by these chromosomal breaks,” said study author Pier Paolo Pandolfi, MD, PhD, of Beth Israel Deaconess Medical Center in Boston, Massachusetts.
“Our work paves the way to discovering many more of these unusual RNAs and how they contribute to cancer, which could reveal new mechanisms and druggable pathways involved in tumor progression.”
Curious about the possibility of circular RNAs contributing to cancer, Dr Pandolfi and his colleagues set out to see if they could detect relevant changes in tumors known to harbor distinct fusion proteins.
The team examined acute promyelocytic leukemia, which often carries a translocation between the PML and RARα genes, and acute myeloid leukemia, which can harbor a translocation between the MLL and AF9 genes.
The investigators found f-circRNAs corresponding to different exons associated with the PML-RARα gene fusion and the MLL-AF9 gene fusion. Normally, multiple circular RNAs can be generated from a single gene, so the team was not surprised to find different f-circRNAs emerging from the same fusion gene.
Dr Pandolfi and his colleagues also uncovered f-circRNAs in solid tumors—in Ewing sarcoma and lung cancer.
The team identified the f-circRNAs using 2 distinct methods—PCR-based amplification and sequencing-based approaches. They said this suggests f-circRNAs are bona fide biological entities, rather than experimental artifacts.
“Our ability to readily detect these fusion-circular RNAs—and their normal, non-fused counterparts—will be enhanced by advances in sequencing technology and analytic methods,” said study author Jlenia Guarnerio, PhD, also of Beth Israel Deaconess Medical Center.
“Indeed, as we look ahead to cataloguing them comprehensively across all cancers and to deeply understanding their mechanisms of action, we will need to propel these new methodologies even further.”
To determine whether f-circRNAs play a functional role in cancer, the investigators introduced the RNAs into cells. This caused the cells to increase their proliferation and tendency to overgrow—features shared by tumor cells.
On the other hand, when the team blocked f-circRNA activity, the cells’ normal behaviors were restored.
Dr Pandolfi and his colleagues also conducted experiments using a mouse model of leukemia. They focused on a specific f-circRNA associated with the MLL-AF9 fusion gene, called f-circM9.
Although f-circM9 could not trigger leukemia on its own, it appeared to work with other cancer-promoting signals—such as the MLL-AF9 fusion protein—to cause leukemia.
Additional experiments suggested that f-circM9 may also help tumor cells persist despite treatment with anticancer drugs.
“These results are particularly exciting because they suggest that drugs directed at fusion-circular RNAs could be a powerful strategy to pursue for future therapeutic development in cancer,” Dr Pandolfi said.
“[However,] our knowledge of circular RNAs is really in its infancy. We know that, normally, they can bind proteins as well as DNA and microRNAs, but much more needs to be done to understand how fusion-circular RNAs work. We have only scratched the surface of these RNAs and their roles in cancer and other diseases.” ![]()
FDA grants product orphan designation for AML

Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Image by Lance Liotta
The US Food and Drug Administration (FDA) has granted orphan designation for the radioimmunoconjugate Iomab-B to be used as a conditioning agent for patients with relapsed or refractory acute myeloid leukemia (AML) who are undergoing hematopoietic stem cell transplant (HSCT).
Iomab-B is a radioimmunoconjugate consisting of BC8, a novel murine monoclonal antibody, and the radioisotope iodine-131.
BC8 targets CD45, a pan-leukocytic antigen widely expressed on white blood cells. This makes BC8 potentially useful in targeting white blood cells in preparation for HSCT.
When labeled with radioactive isotopes, BC8 carries radioactivity directly to the site of cancerous growth and bone marrow, while avoiding the effects of radiation on most healthy tissues, according to Actinium Pharmaceuticals, Inc., the company developing Iomab-B.
Actinium said Iomab-B has been tested as a myeloconditioning/myeloablative agent in more than 250 patients with incurable hematologic malignancies.
The company has released data from a phase 1/2 trial of Iomab-B in patients with relapsed/refractory AML who are older than 50.
The data show that patients who received Iomab-B before HSCT (n=27) had higher rates of survival at 1 and 2 years than patients who underwent HSCT with conventional myeloablative conditioning (n=10) or chemotherapy (n=61).
One-year survival rates were 30% in the Iomab-B arm and 10% each in the conventional conditioning and chemotherapy arms. Two-year survival rates were 19%, 0%, and 0%, respectively.
Now, Actinium is planning a phase 3 trial of Iomab-B in relapsed/refractory AML patients over the age of 55.
About orphan designation
The FDA grants orphan designation to drugs intended to treat diseases or conditions affecting fewer than 200,000 patients in the US.
The designation provides the drug’s sponsor with various development incentives, including opportunities to apply for research-related tax credits and grant funding, assistance in designing clinical trials, and 7 years of US market exclusivity if the drug is approved. ![]()
Drug bests placebo in iron deficiency anemia trial

Top-line results from a phase 3 trial suggest the oral, iron-based drug ferric citrate is more effective than placebo for treating iron deficiency anemia in adults with stage 3-5, non-dialysis-dependent chronic kidney disease.
Fifty-two percent of patients who received ferric citrate achieved at least a 1 g/dL increase in hemoglobin over a 16-week period, compared to 19% of patients who received placebo.
Researchers said the safety profile of ferric citrate in this trial was consistent with that in previous studies.
Keryx Biopharmaceuticals, Inc., the company developing ferric citrate, recently announced these results.
Patients and treatment
In this phase 3 study, researchers compared treatment with ferric citrate to placebo in 234 patients who previously had not adequately responded to or tolerated current oral iron therapies. The patients were not allowed to receive any iron (intravenous or oral) or erythropoiesis-stimulating agents during this study.
The patients were randomized 1:1 to receive ferric citrate (n=117) or placebo (n=115). Two patients in the placebo arm discontinued the study and were not included in the efficacy analysis. One discontinued after randomization prior to receiving placebo, and the other discontinued after taking a dose of placebo but before having laboratory values drawn.
The study had a 16-week, randomized, double-blind, placebo-controlled efficacy period, followed by an 8-week, open-label safety extension period. During the extension period, all patients remaining in the study, including the placebo arm, received ferric citrate.
During the efficacy period, ferric citrate was administered at a starting dose of 3 tablets per day, with food, and could be titrated every 4 weeks by an additional 3 tablets, for up to 12 tablets per day. The mean dose of ferric citrate was 5 tablets per day.
Baseline laboratory values were similar between the treatment arms. The mean hemoglobin was 10.4 g/dL in both arms.
The mean transferrin saturation was 20.2% in the ferric citrate arm and 19.6% in the placebo arm. The mean ferritin was 85.9 ng/mL and 81.7 ng/mL, respectively. And the mean serum phosphate was 4.2 mg/dL and 4.1 mg/dL, respectively.
Efficacy results
The study achieved its primary endpoint, with 52.1% (61/117) of patients who received ferric citrate achieving a 1g/dL or greater rise in hemoglobin at any time point during the 16-week efficacy period, compared to 19.1% (22/115) of patients in the placebo arm (P<0.001).
The researchers also observed significant differences in all pre-specified secondary efficacy endpoints.
The mean change in hemoglobin was 0.75 g/dL in the ferric citrate arm and -0.08 g/dL in the placebo arm (P<0.001). The mean change in transferrin saturation was 17.8% and -0.6%, respectively (P<0.001).
The mean change in ferritin was 162.5 ng/mL and -7.7 ng/mL, respectively (P<0.001). And the mean change in serum phosphate was -0.43 mg/dL and -0.22 mg/dL, respectively (P=0.02).
The proportion of patients with a durable response during the efficacy period was 48.7% in the ferric citrate arm and 14.8% in the placebo arm (P<0.001).
A durable response was defined as a mean change in hemoglobin from baseline of at least 0.75 g/dL over any 4-week time period during the efficacy period, provided that an increase of at least 1.0 g/dL had occurred during that 4-week period.
Safety results
During the efficacy period, the majority of adverse events (AEs) were mild to moderate. The most common AEs—in the ferric citrate and placebo arms, respectively—were diarrhea (20.5% vs 16.4%), constipation (18.8% vs 12.9%), discolored feces (14.5% vs 0%), and nausea (11.1% vs 2.6%).
Hypophosphatemia was reported in 4 patients—1 in the ferric citrate arm and 3 in the placebo arm.
Twenty-six percent (31/117) of ferric citrate-treated patients and 30% (35/116) of patients receiving placebo discontinued treatment during the efficacy period. Twelve patients treated with ferric citrate discontinued due to an AE, as did 10 patients who received placebo.
During the efficacy period, the rate of serious AEs was balanced between the ferric citrate and placebo arms, at 12% and 10%, respectively. None of the serious AEs were deemed drug-related.
Over the course of the study, there were 2 deaths reported. Both occurred in patients receiving ferric citrate, but neither were considered drug-related. ![]()
Top-line results from a phase 3 trial suggest the oral, iron-based drug ferric citrate is more effective than placebo for treating iron deficiency anemia in adults with stage 3-5, non-dialysis-dependent chronic kidney disease.
Fifty-two percent of patients who received ferric citrate achieved at least a 1 g/dL increase in hemoglobin over a 16-week period, compared to 19% of patients who received placebo.
Researchers said the safety profile of ferric citrate in this trial was consistent with that in previous studies.
Keryx Biopharmaceuticals, Inc., the company developing ferric citrate, recently announced these results.
Patients and treatment
In this phase 3 study, researchers compared treatment with ferric citrate to placebo in 234 patients who previously had not adequately responded to or tolerated current oral iron therapies. The patients were not allowed to receive any iron (intravenous or oral) or erythropoiesis-stimulating agents during this study.
The patients were randomized 1:1 to receive ferric citrate (n=117) or placebo (n=115). Two patients in the placebo arm discontinued the study and were not included in the efficacy analysis. One discontinued after randomization prior to receiving placebo, and the other discontinued after taking a dose of placebo but before having laboratory values drawn.
The study had a 16-week, randomized, double-blind, placebo-controlled efficacy period, followed by an 8-week, open-label safety extension period. During the extension period, all patients remaining in the study, including the placebo arm, received ferric citrate.
During the efficacy period, ferric citrate was administered at a starting dose of 3 tablets per day, with food, and could be titrated every 4 weeks by an additional 3 tablets, for up to 12 tablets per day. The mean dose of ferric citrate was 5 tablets per day.
Baseline laboratory values were similar between the treatment arms. The mean hemoglobin was 10.4 g/dL in both arms.
The mean transferrin saturation was 20.2% in the ferric citrate arm and 19.6% in the placebo arm. The mean ferritin was 85.9 ng/mL and 81.7 ng/mL, respectively. And the mean serum phosphate was 4.2 mg/dL and 4.1 mg/dL, respectively.
Efficacy results
The study achieved its primary endpoint, with 52.1% (61/117) of patients who received ferric citrate achieving a 1g/dL or greater rise in hemoglobin at any time point during the 16-week efficacy period, compared to 19.1% (22/115) of patients in the placebo arm (P<0.001).
The researchers also observed significant differences in all pre-specified secondary efficacy endpoints.
The mean change in hemoglobin was 0.75 g/dL in the ferric citrate arm and -0.08 g/dL in the placebo arm (P<0.001). The mean change in transferrin saturation was 17.8% and -0.6%, respectively (P<0.001).
The mean change in ferritin was 162.5 ng/mL and -7.7 ng/mL, respectively (P<0.001). And the mean change in serum phosphate was -0.43 mg/dL and -0.22 mg/dL, respectively (P=0.02).
The proportion of patients with a durable response during the efficacy period was 48.7% in the ferric citrate arm and 14.8% in the placebo arm (P<0.001).
A durable response was defined as a mean change in hemoglobin from baseline of at least 0.75 g/dL over any 4-week time period during the efficacy period, provided that an increase of at least 1.0 g/dL had occurred during that 4-week period.
Safety results
During the efficacy period, the majority of adverse events (AEs) were mild to moderate. The most common AEs—in the ferric citrate and placebo arms, respectively—were diarrhea (20.5% vs 16.4%), constipation (18.8% vs 12.9%), discolored feces (14.5% vs 0%), and nausea (11.1% vs 2.6%).
Hypophosphatemia was reported in 4 patients—1 in the ferric citrate arm and 3 in the placebo arm.
Twenty-six percent (31/117) of ferric citrate-treated patients and 30% (35/116) of patients receiving placebo discontinued treatment during the efficacy period. Twelve patients treated with ferric citrate discontinued due to an AE, as did 10 patients who received placebo.
During the efficacy period, the rate of serious AEs was balanced between the ferric citrate and placebo arms, at 12% and 10%, respectively. None of the serious AEs were deemed drug-related.
Over the course of the study, there were 2 deaths reported. Both occurred in patients receiving ferric citrate, but neither were considered drug-related. ![]()
Top-line results from a phase 3 trial suggest the oral, iron-based drug ferric citrate is more effective than placebo for treating iron deficiency anemia in adults with stage 3-5, non-dialysis-dependent chronic kidney disease.
Fifty-two percent of patients who received ferric citrate achieved at least a 1 g/dL increase in hemoglobin over a 16-week period, compared to 19% of patients who received placebo.
Researchers said the safety profile of ferric citrate in this trial was consistent with that in previous studies.
Keryx Biopharmaceuticals, Inc., the company developing ferric citrate, recently announced these results.
Patients and treatment
In this phase 3 study, researchers compared treatment with ferric citrate to placebo in 234 patients who previously had not adequately responded to or tolerated current oral iron therapies. The patients were not allowed to receive any iron (intravenous or oral) or erythropoiesis-stimulating agents during this study.
The patients were randomized 1:1 to receive ferric citrate (n=117) or placebo (n=115). Two patients in the placebo arm discontinued the study and were not included in the efficacy analysis. One discontinued after randomization prior to receiving placebo, and the other discontinued after taking a dose of placebo but before having laboratory values drawn.
The study had a 16-week, randomized, double-blind, placebo-controlled efficacy period, followed by an 8-week, open-label safety extension period. During the extension period, all patients remaining in the study, including the placebo arm, received ferric citrate.
During the efficacy period, ferric citrate was administered at a starting dose of 3 tablets per day, with food, and could be titrated every 4 weeks by an additional 3 tablets, for up to 12 tablets per day. The mean dose of ferric citrate was 5 tablets per day.
Baseline laboratory values were similar between the treatment arms. The mean hemoglobin was 10.4 g/dL in both arms.
The mean transferrin saturation was 20.2% in the ferric citrate arm and 19.6% in the placebo arm. The mean ferritin was 85.9 ng/mL and 81.7 ng/mL, respectively. And the mean serum phosphate was 4.2 mg/dL and 4.1 mg/dL, respectively.
Efficacy results
The study achieved its primary endpoint, with 52.1% (61/117) of patients who received ferric citrate achieving a 1g/dL or greater rise in hemoglobin at any time point during the 16-week efficacy period, compared to 19.1% (22/115) of patients in the placebo arm (P<0.001).
The researchers also observed significant differences in all pre-specified secondary efficacy endpoints.
The mean change in hemoglobin was 0.75 g/dL in the ferric citrate arm and -0.08 g/dL in the placebo arm (P<0.001). The mean change in transferrin saturation was 17.8% and -0.6%, respectively (P<0.001).
The mean change in ferritin was 162.5 ng/mL and -7.7 ng/mL, respectively (P<0.001). And the mean change in serum phosphate was -0.43 mg/dL and -0.22 mg/dL, respectively (P=0.02).
The proportion of patients with a durable response during the efficacy period was 48.7% in the ferric citrate arm and 14.8% in the placebo arm (P<0.001).
A durable response was defined as a mean change in hemoglobin from baseline of at least 0.75 g/dL over any 4-week time period during the efficacy period, provided that an increase of at least 1.0 g/dL had occurred during that 4-week period.
Safety results
During the efficacy period, the majority of adverse events (AEs) were mild to moderate. The most common AEs—in the ferric citrate and placebo arms, respectively—were diarrhea (20.5% vs 16.4%), constipation (18.8% vs 12.9%), discolored feces (14.5% vs 0%), and nausea (11.1% vs 2.6%).
Hypophosphatemia was reported in 4 patients—1 in the ferric citrate arm and 3 in the placebo arm.
Twenty-six percent (31/117) of ferric citrate-treated patients and 30% (35/116) of patients receiving placebo discontinued treatment during the efficacy period. Twelve patients treated with ferric citrate discontinued due to an AE, as did 10 patients who received placebo.
During the efficacy period, the rate of serious AEs was balanced between the ferric citrate and placebo arms, at 12% and 10%, respectively. None of the serious AEs were deemed drug-related.
Over the course of the study, there were 2 deaths reported. Both occurred in patients receiving ferric citrate, but neither were considered drug-related.
AHA and ACC update guidelines for DAPT

Photo courtesy of AstraZeneca
The American College of Cardiology (ACC) and American Heart Association (AHA) have released updated guidelines for the use of dual antiplatelet therapy (DAPT) in patients with coronary artery disease.
DAPT, the combination of aspirin and a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), is used to reduce the risks of future heart attack and coronary stent thrombosis in this patient population.
Overall, the new guidelines, which update recommendations from 6 previous guidelines, recommend an individualized approach to DAPT.
The guidelines were published in the Journal of the American College of Cardiology.
The new recommendations are based on the current use of coronary stents that present a lower risk of thrombosis than some older stents.
The recommendations are also based on the findings of recent studies investigating the duration of DAPT in patients with coronary artery disease, specifically those with myocardial infarction and those undergoing coronary stent implantation.
Studies examining shorter duration (3 to 6 months) of DAPT compared with a standard 12 months of DAPT in select, generally lower-risk patients did not show an increased risk of stent thrombosis. And, in some cases, a shorter treatment duration was associated with less bleeding.
Other studies investigating extending DAPT for an additional 18 or 36 months (beyond a year) showed a decrease in the risk of heart attack and stent thrombosis at the expense of an increase in bleeding risk.
Overview of recommendations
In general, the recommendations regarding DAPT duration consist of a Class I recommendation of “should be given” for a minimum period of time (usually 6 to 12 months), and a Class IIb recommendation of “may be considered” for continuation beyond that time.
Shorter duration of DAPT is recommended for patients at lower ischemic risk with high bleeding risk, whereas longer duration of DAPT may be reasonable for patients at higher ischemic risk with lower bleeding risk.
These recommendations for duration of DAPT apply to newer-generation stents and, in general, only to those not treated with oral anticoagulant therapy.
An aspirin dose of 81 mg daily (range, 75-100 mg) is now recommended in patients treated with DAPT. Regardless of the duration of DAPT, aspirin is almost always continued indefinitely in patients with coronary artery disease.
The updated guidelines also address DAPT after coronary artery bypass grafting and issues regarding the timing of non-cardiac surgery in patients treated with coronary stent implantation and DAPT.
Decisions about the timing of surgery and whether to discontinue DAPT after coronary stent implantation involve weighing the particular surgical procedure and the risks of delaying the procedure, the risks of ischemia and stent thrombosis, and the risk and consequences of bleeding, and are therefore best individualized, according to the guidelines.
The new guidelines update recommendations on the duration of DAPT across 6 previously published guidelines:
- The 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention
- The 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery
- The 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease
- The 2013 ACC/AHA Guideline for the Management of ST-Elevation Myocardial Infarction
- The 2014 ACC/AHA Guideline for Non-ST-Elevation Acute Coronary Syndromes
- The 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery.
Photo courtesy of AstraZeneca
The American College of Cardiology (ACC) and American Heart Association (AHA) have released updated guidelines for the use of dual antiplatelet therapy (DAPT) in patients with coronary artery disease.
DAPT, the combination of aspirin and a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), is used to reduce the risks of future heart attack and coronary stent thrombosis in this patient population.
Overall, the new guidelines, which update recommendations from 6 previous guidelines, recommend an individualized approach to DAPT.
The guidelines were published in the Journal of the American College of Cardiology.
The new recommendations are based on the current use of coronary stents that present a lower risk of thrombosis than some older stents.
The recommendations are also based on the findings of recent studies investigating the duration of DAPT in patients with coronary artery disease, specifically those with myocardial infarction and those undergoing coronary stent implantation.
Studies examining shorter duration (3 to 6 months) of DAPT compared with a standard 12 months of DAPT in select, generally lower-risk patients did not show an increased risk of stent thrombosis. And, in some cases, a shorter treatment duration was associated with less bleeding.
Other studies investigating extending DAPT for an additional 18 or 36 months (beyond a year) showed a decrease in the risk of heart attack and stent thrombosis at the expense of an increase in bleeding risk.
Overview of recommendations
In general, the recommendations regarding DAPT duration consist of a Class I recommendation of “should be given” for a minimum period of time (usually 6 to 12 months), and a Class IIb recommendation of “may be considered” for continuation beyond that time.
Shorter duration of DAPT is recommended for patients at lower ischemic risk with high bleeding risk, whereas longer duration of DAPT may be reasonable for patients at higher ischemic risk with lower bleeding risk.
These recommendations for duration of DAPT apply to newer-generation stents and, in general, only to those not treated with oral anticoagulant therapy.
An aspirin dose of 81 mg daily (range, 75-100 mg) is now recommended in patients treated with DAPT. Regardless of the duration of DAPT, aspirin is almost always continued indefinitely in patients with coronary artery disease.
The updated guidelines also address DAPT after coronary artery bypass grafting and issues regarding the timing of non-cardiac surgery in patients treated with coronary stent implantation and DAPT.
Decisions about the timing of surgery and whether to discontinue DAPT after coronary stent implantation involve weighing the particular surgical procedure and the risks of delaying the procedure, the risks of ischemia and stent thrombosis, and the risk and consequences of bleeding, and are therefore best individualized, according to the guidelines.
The new guidelines update recommendations on the duration of DAPT across 6 previously published guidelines:
- The 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention
- The 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery
- The 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease
- The 2013 ACC/AHA Guideline for the Management of ST-Elevation Myocardial Infarction
- The 2014 ACC/AHA Guideline for Non-ST-Elevation Acute Coronary Syndromes
- The 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery.
Photo courtesy of AstraZeneca
The American College of Cardiology (ACC) and American Heart Association (AHA) have released updated guidelines for the use of dual antiplatelet therapy (DAPT) in patients with coronary artery disease.
DAPT, the combination of aspirin and a P2Y12 inhibitor (clopidogrel, prasugrel, or ticagrelor), is used to reduce the risks of future heart attack and coronary stent thrombosis in this patient population.
Overall, the new guidelines, which update recommendations from 6 previous guidelines, recommend an individualized approach to DAPT.
The guidelines were published in the Journal of the American College of Cardiology.
The new recommendations are based on the current use of coronary stents that present a lower risk of thrombosis than some older stents.
The recommendations are also based on the findings of recent studies investigating the duration of DAPT in patients with coronary artery disease, specifically those with myocardial infarction and those undergoing coronary stent implantation.
Studies examining shorter duration (3 to 6 months) of DAPT compared with a standard 12 months of DAPT in select, generally lower-risk patients did not show an increased risk of stent thrombosis. And, in some cases, a shorter treatment duration was associated with less bleeding.
Other studies investigating extending DAPT for an additional 18 or 36 months (beyond a year) showed a decrease in the risk of heart attack and stent thrombosis at the expense of an increase in bleeding risk.
Overview of recommendations
In general, the recommendations regarding DAPT duration consist of a Class I recommendation of “should be given” for a minimum period of time (usually 6 to 12 months), and a Class IIb recommendation of “may be considered” for continuation beyond that time.
Shorter duration of DAPT is recommended for patients at lower ischemic risk with high bleeding risk, whereas longer duration of DAPT may be reasonable for patients at higher ischemic risk with lower bleeding risk.
These recommendations for duration of DAPT apply to newer-generation stents and, in general, only to those not treated with oral anticoagulant therapy.
An aspirin dose of 81 mg daily (range, 75-100 mg) is now recommended in patients treated with DAPT. Regardless of the duration of DAPT, aspirin is almost always continued indefinitely in patients with coronary artery disease.
The updated guidelines also address DAPT after coronary artery bypass grafting and issues regarding the timing of non-cardiac surgery in patients treated with coronary stent implantation and DAPT.
Decisions about the timing of surgery and whether to discontinue DAPT after coronary stent implantation involve weighing the particular surgical procedure and the risks of delaying the procedure, the risks of ischemia and stent thrombosis, and the risk and consequences of bleeding, and are therefore best individualized, according to the guidelines.
The new guidelines update recommendations on the duration of DAPT across 6 previously published guidelines:
- The 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention
- The 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery
- The 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients With Stable Ischemic Heart Disease
- The 2013 ACC/AHA Guideline for the Management of ST-Elevation Myocardial Infarction
- The 2014 ACC/AHA Guideline for Non-ST-Elevation Acute Coronary Syndromes
- The 2014 ACC/AHA Guideline on Perioperative Cardiovascular Evaluation and Management of Patients Undergoing Noncardiac Surgery.
FDA approves drug to treat VOD after HSCT
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has approved the use of defibrotide sodium (Defitelio).
The product can now be used to treat adult and pediatric patients who develop hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome, with renal or pulmonary dysfunction after receiving a hematopoietic stem cell transplant (HSCT).
Defibrotide sodium is the first FDA-approved therapy for patients with this rare, potentially fatal complication.
Defibrotide sodium is a product of Jazz Pharmaceuticals, Inc. The company said shipments of the drug to distribution channels will begin within a week.
The recommended dose and schedule for defibrotide sodium is 6.25 mg/kg every 6 hours, given as a 2-hour intravenous infusion, for at least 21 days, and continued until VOD resolution or up to 60 days of treatment.
In vitro defibrotide sodium has profibrinolytic activity. The use of defibrotide sodium is contraindicated in patients receiving concurrent anticoagulants or fibrinolytic therapies. Hemorrhage and hypersensitivity reactions are the major potential adverse reactions.
The FDA previously granted the defibrotide sodium application priority review status, and the drug received orphan drug designation from the FDA for the treatment of hepatic VOD.
Full prescribing information for defibrotide sodium can be found on the FDA website.
Trial results
The FDA’s approval of defibrotide sodium is supported by data in 528 patients treated on 3 studies: a phase 2 trial, a phase 3 trial, and an expanded access study. Data from the expanded access study were presented at the 2015 BMT Tandem Meetings, and data from the phase 3 trial were published in Blood earlier this year.
The 528 patients all had hepatic VOD with multi-organ dysfunction after HSCT. They received defibrotide sodium at 6.25 mg/kg intravenously every 6 hours until resolution of VOD.
The approval was based on survival at day +100 after HSCT. The day +100 survival rates for Study 1 (phase 3, n=102), Study 2 (phase 2, n=75), and Study 3 (expanded access, n=351) were 38%, 44%, and 45%, respectively.
Based on published reports and analyses of patient-level data, the day +100 survival rates were 21% to 31% for patients with hepatic VOD with renal or pulmonary dysfunction who received supportive care or interventions other than defibrotide sodium.
The safety of defibrotide sodium to support approval is based on data from 176 patients in the clinical development program for the treatment of VOD with renal and/or pulmonary dysfunction following HSCT.
The most common adverse events (incidence ≥10% and independent of causality) were hypotension, diarrhea, vomiting, nausea, and epistaxis. The most common serious adverse events (incidence ≥5% and independent of causality) were hypotension (11%) and pulmonary alveolar hemorrhage (7%).
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has approved the use of defibrotide sodium (Defitelio).
The product can now be used to treat adult and pediatric patients who develop hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome, with renal or pulmonary dysfunction after receiving a hematopoietic stem cell transplant (HSCT).
Defibrotide sodium is the first FDA-approved therapy for patients with this rare, potentially fatal complication.
Defibrotide sodium is a product of Jazz Pharmaceuticals, Inc. The company said shipments of the drug to distribution channels will begin within a week.
The recommended dose and schedule for defibrotide sodium is 6.25 mg/kg every 6 hours, given as a 2-hour intravenous infusion, for at least 21 days, and continued until VOD resolution or up to 60 days of treatment.
In vitro defibrotide sodium has profibrinolytic activity. The use of defibrotide sodium is contraindicated in patients receiving concurrent anticoagulants or fibrinolytic therapies. Hemorrhage and hypersensitivity reactions are the major potential adverse reactions.
The FDA previously granted the defibrotide sodium application priority review status, and the drug received orphan drug designation from the FDA for the treatment of hepatic VOD.
Full prescribing information for defibrotide sodium can be found on the FDA website.
Trial results
The FDA’s approval of defibrotide sodium is supported by data in 528 patients treated on 3 studies: a phase 2 trial, a phase 3 trial, and an expanded access study. Data from the expanded access study were presented at the 2015 BMT Tandem Meetings, and data from the phase 3 trial were published in Blood earlier this year.
The 528 patients all had hepatic VOD with multi-organ dysfunction after HSCT. They received defibrotide sodium at 6.25 mg/kg intravenously every 6 hours until resolution of VOD.
The approval was based on survival at day +100 after HSCT. The day +100 survival rates for Study 1 (phase 3, n=102), Study 2 (phase 2, n=75), and Study 3 (expanded access, n=351) were 38%, 44%, and 45%, respectively.
Based on published reports and analyses of patient-level data, the day +100 survival rates were 21% to 31% for patients with hepatic VOD with renal or pulmonary dysfunction who received supportive care or interventions other than defibrotide sodium.
The safety of defibrotide sodium to support approval is based on data from 176 patients in the clinical development program for the treatment of VOD with renal and/or pulmonary dysfunction following HSCT.
The most common adverse events (incidence ≥10% and independent of causality) were hypotension, diarrhea, vomiting, nausea, and epistaxis. The most common serious adverse events (incidence ≥5% and independent of causality) were hypotension (11%) and pulmonary alveolar hemorrhage (7%).
![]()
Photo by Chad McNeeley
The US Food and Drug Administration (FDA) has approved the use of defibrotide sodium (Defitelio).
The product can now be used to treat adult and pediatric patients who develop hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome, with renal or pulmonary dysfunction after receiving a hematopoietic stem cell transplant (HSCT).
Defibrotide sodium is the first FDA-approved therapy for patients with this rare, potentially fatal complication.
Defibrotide sodium is a product of Jazz Pharmaceuticals, Inc. The company said shipments of the drug to distribution channels will begin within a week.
The recommended dose and schedule for defibrotide sodium is 6.25 mg/kg every 6 hours, given as a 2-hour intravenous infusion, for at least 21 days, and continued until VOD resolution or up to 60 days of treatment.
In vitro defibrotide sodium has profibrinolytic activity. The use of defibrotide sodium is contraindicated in patients receiving concurrent anticoagulants or fibrinolytic therapies. Hemorrhage and hypersensitivity reactions are the major potential adverse reactions.
The FDA previously granted the defibrotide sodium application priority review status, and the drug received orphan drug designation from the FDA for the treatment of hepatic VOD.
Full prescribing information for defibrotide sodium can be found on the FDA website.
Trial results
The FDA’s approval of defibrotide sodium is supported by data in 528 patients treated on 3 studies: a phase 2 trial, a phase 3 trial, and an expanded access study. Data from the expanded access study were presented at the 2015 BMT Tandem Meetings, and data from the phase 3 trial were published in Blood earlier this year.
The 528 patients all had hepatic VOD with multi-organ dysfunction after HSCT. They received defibrotide sodium at 6.25 mg/kg intravenously every 6 hours until resolution of VOD.
The approval was based on survival at day +100 after HSCT. The day +100 survival rates for Study 1 (phase 3, n=102), Study 2 (phase 2, n=75), and Study 3 (expanded access, n=351) were 38%, 44%, and 45%, respectively.
Based on published reports and analyses of patient-level data, the day +100 survival rates were 21% to 31% for patients with hepatic VOD with renal or pulmonary dysfunction who received supportive care or interventions other than defibrotide sodium.
The safety of defibrotide sodium to support approval is based on data from 176 patients in the clinical development program for the treatment of VOD with renal and/or pulmonary dysfunction following HSCT.
The most common adverse events (incidence ≥10% and independent of causality) were hypotension, diarrhea, vomiting, nausea, and epistaxis. The most common serious adverse events (incidence ≥5% and independent of causality) were hypotension (11%) and pulmonary alveolar hemorrhage (7%).