User login
Vaccine protects mice from malaria

An RNA replicon-based vaccine can fight malaria infection in mice, according to research published in Nature Communications.
The vaccine targets a protein, Plasmodium macrophage migration inhibitory factor (PMIF), which is produced by malaria parasites and suppresses memory T cells.
The vaccine provided improved control of existing malaria infection as well as protection from reinfection in mice.
This work was funded by National Institutes of Health grants and Novartis Vaccines, Inc.
The research began with a strain of Plasmodium berghei in which PMIF was genetically deleted. Investigators found that mice infected with this strain developed memory T cells and showed stronger anti-parasite immunity.
This led the researchers to test a vaccine targeting PMIF in 2 mouse models of malaria. One model was an early stage liver infection, and the other was a severe, late-stage blood infection.
The vaccine reduced parasitemia in both models and prolonged survival in mice with the late-stage blood infection.
The investigators then cured vaccinated mice of malaria (via treatment with chloroquine) and reinfected them.
Mice reinfected with late-stage malaria had no evidence of parasites in the blood or organs after reinfection.
The mice reinfected with liver-stage malaria had a 70% reduction in liver parasites (compared to control mice) upon reinfection, and they did not develop blood-stage infection.
“If you vaccinate with this specific protein used by the malaria parasite to evade an immune response, you can elicit protection against reinfection,” said study author Richard Bucala, MD, of Yale School of Medicine in New Haven, Connecticut.
“To our knowledge, this has never been shown using a single antigen in fulminant blood-stage infection.”
The researchers also transferred memory T cells from immunized mice to naïve mice that had never been exposed to malaria. The T-cell recipients were completely protected from malaria infection.
The investigators’ next step with this work is to develop a vaccine for humans who have never had malaria, primarily young children.
“The vaccine would be used in children so that they would already have an immune response to this particular malaria product, and when they became infected with malaria, they would have a normal T-cell response, clear the parasite, and be protected from future infection,” Dr Bucala said.
An RNA replicon-based vaccine can fight malaria infection in mice, according to research published in Nature Communications.
The vaccine targets a protein, Plasmodium macrophage migration inhibitory factor (PMIF), which is produced by malaria parasites and suppresses memory T cells.
The vaccine provided improved control of existing malaria infection as well as protection from reinfection in mice.
This work was funded by National Institutes of Health grants and Novartis Vaccines, Inc.
The research began with a strain of Plasmodium berghei in which PMIF was genetically deleted. Investigators found that mice infected with this strain developed memory T cells and showed stronger anti-parasite immunity.
This led the researchers to test a vaccine targeting PMIF in 2 mouse models of malaria. One model was an early stage liver infection, and the other was a severe, late-stage blood infection.
The vaccine reduced parasitemia in both models and prolonged survival in mice with the late-stage blood infection.
The investigators then cured vaccinated mice of malaria (via treatment with chloroquine) and reinfected them.
Mice reinfected with late-stage malaria had no evidence of parasites in the blood or organs after reinfection.
The mice reinfected with liver-stage malaria had a 70% reduction in liver parasites (compared to control mice) upon reinfection, and they did not develop blood-stage infection.
“If you vaccinate with this specific protein used by the malaria parasite to evade an immune response, you can elicit protection against reinfection,” said study author Richard Bucala, MD, of Yale School of Medicine in New Haven, Connecticut.
“To our knowledge, this has never been shown using a single antigen in fulminant blood-stage infection.”
The researchers also transferred memory T cells from immunized mice to naïve mice that had never been exposed to malaria. The T-cell recipients were completely protected from malaria infection.
The investigators’ next step with this work is to develop a vaccine for humans who have never had malaria, primarily young children.
“The vaccine would be used in children so that they would already have an immune response to this particular malaria product, and when they became infected with malaria, they would have a normal T-cell response, clear the parasite, and be protected from future infection,” Dr Bucala said.
An RNA replicon-based vaccine can fight malaria infection in mice, according to research published in Nature Communications.
The vaccine targets a protein, Plasmodium macrophage migration inhibitory factor (PMIF), which is produced by malaria parasites and suppresses memory T cells.
The vaccine provided improved control of existing malaria infection as well as protection from reinfection in mice.
This work was funded by National Institutes of Health grants and Novartis Vaccines, Inc.
The research began with a strain of Plasmodium berghei in which PMIF was genetically deleted. Investigators found that mice infected with this strain developed memory T cells and showed stronger anti-parasite immunity.
This led the researchers to test a vaccine targeting PMIF in 2 mouse models of malaria. One model was an early stage liver infection, and the other was a severe, late-stage blood infection.
The vaccine reduced parasitemia in both models and prolonged survival in mice with the late-stage blood infection.
The investigators then cured vaccinated mice of malaria (via treatment with chloroquine) and reinfected them.
Mice reinfected with late-stage malaria had no evidence of parasites in the blood or organs after reinfection.
The mice reinfected with liver-stage malaria had a 70% reduction in liver parasites (compared to control mice) upon reinfection, and they did not develop blood-stage infection.
“If you vaccinate with this specific protein used by the malaria parasite to evade an immune response, you can elicit protection against reinfection,” said study author Richard Bucala, MD, of Yale School of Medicine in New Haven, Connecticut.
“To our knowledge, this has never been shown using a single antigen in fulminant blood-stage infection.”
The researchers also transferred memory T cells from immunized mice to naïve mice that had never been exposed to malaria. The T-cell recipients were completely protected from malaria infection.
The investigators’ next step with this work is to develop a vaccine for humans who have never had malaria, primarily young children.
“The vaccine would be used in children so that they would already have an immune response to this particular malaria product, and when they became infected with malaria, they would have a normal T-cell response, clear the parasite, and be protected from future infection,” Dr Bucala said.
Turbulence aids platelet production
Turbulence promotes large-scale production of functional platelets from human induced pluripotent stem cells (hiPSCs), according to research published in Cell.
Exposure to turbulent energy in a bioreactor stimulated hiPSC-derived megakaryocytes to produce 100 billion platelets.
Transfusion of these platelets in 2 animal models promoted blood clotting and prevented bleeding just as well as platelets from human donors.
“The discovery of turbulent energy provides a new physical mechanism and ex vivo production strategy for the generation of platelets that should impact clinical-scale cell therapies for regenerative medicine,” said study author Koji Eto, MD, PhD, of Kyoto University in Japan.
Previous attempts to generate platelets from hiPSC-derived megakaryocytes failed to achieve a scale suitable for clinical manufacturing.
While searching for a solution to this problem, Dr Eto and his colleagues noticed that hiPSC-derived megakaryocytes produced more platelets when being rotated in a flask than under static conditions in a petri dish.
This observation suggested that physical stress from horizontal shaking under liquid conditions enhances platelet generation.
Following up on this discovery, the researchers tested a rocking-bag-based bioreactor followed by a new microfluidic system with a flow chamber and multiple pillars, but these devices generated fewer than 20 platelets per hiPSC-derived megakaryocyte.
To examine the ideal physical conditions for generating platelets, Dr Eto and his team conducted live-imaging studies of mouse bone marrow.
These experiments revealed that megakaryocytes release platelets only when they are exposed to turbulent blood flow. In support of this idea, simulations confirmed that the bioreactor and microfluidic system the researchers previously tested lacked sufficient turbulent energy.
“The discovery of the crucial role of turbulence in platelet production significantly extends past research showing that shear stress from blood flow is also a key physical factor in this process,” Dr Eto said.
“Our findings also show that iPS cells are not the end-all be-all for producing platelets. Understanding fluid dynamics in addition to iPS cell technology was necessary for our discovery.”
After testing various devices, the researchers discovered that large-scale production of high-quality platelets was possible using a bioreactor called VerMES. This system consists of 2 oval-shaped, horizontally oriented mixing blades that generate relatively high levels of turbulence by moving up and down in a cylinder.
With the optimal level of turbulent energy and shear stress created by the blade motion, the hiPSC-derived megakaryocytes generated 100 billion platelets—enough to satisfy clinical requirements.
Transfusion experiments in 2 animal models of thrombocytopenia showed that these platelets perform similarly to platelets from human donors. Both types of platelets promoted blood clotting and reduced bleeding times to a comparable extent after ear vein incisions in rabbits and tail artery punctures in mice.
Dr Eto and his team are taking this work further by designing automated protocols, lowering manufacturing costs, and optimizing platelet yields. They are also developing universal platelets lacking human leukocyte antigens in order to reduce the risk of immune-mediated transfusion reactions.
“We expect clinical trials to begin within a year or two,” Dr Eto said. “We believe these findings will be a last scientific step to receiving permission for clinical trials using our platelets.”
Turbulence promotes large-scale production of functional platelets from human induced pluripotent stem cells (hiPSCs), according to research published in Cell.
Exposure to turbulent energy in a bioreactor stimulated hiPSC-derived megakaryocytes to produce 100 billion platelets.
Transfusion of these platelets in 2 animal models promoted blood clotting and prevented bleeding just as well as platelets from human donors.
“The discovery of turbulent energy provides a new physical mechanism and ex vivo production strategy for the generation of platelets that should impact clinical-scale cell therapies for regenerative medicine,” said study author Koji Eto, MD, PhD, of Kyoto University in Japan.
Previous attempts to generate platelets from hiPSC-derived megakaryocytes failed to achieve a scale suitable for clinical manufacturing.
While searching for a solution to this problem, Dr Eto and his colleagues noticed that hiPSC-derived megakaryocytes produced more platelets when being rotated in a flask than under static conditions in a petri dish.
This observation suggested that physical stress from horizontal shaking under liquid conditions enhances platelet generation.
Following up on this discovery, the researchers tested a rocking-bag-based bioreactor followed by a new microfluidic system with a flow chamber and multiple pillars, but these devices generated fewer than 20 platelets per hiPSC-derived megakaryocyte.
To examine the ideal physical conditions for generating platelets, Dr Eto and his team conducted live-imaging studies of mouse bone marrow.
These experiments revealed that megakaryocytes release platelets only when they are exposed to turbulent blood flow. In support of this idea, simulations confirmed that the bioreactor and microfluidic system the researchers previously tested lacked sufficient turbulent energy.
“The discovery of the crucial role of turbulence in platelet production significantly extends past research showing that shear stress from blood flow is also a key physical factor in this process,” Dr Eto said.
“Our findings also show that iPS cells are not the end-all be-all for producing platelets. Understanding fluid dynamics in addition to iPS cell technology was necessary for our discovery.”
After testing various devices, the researchers discovered that large-scale production of high-quality platelets was possible using a bioreactor called VerMES. This system consists of 2 oval-shaped, horizontally oriented mixing blades that generate relatively high levels of turbulence by moving up and down in a cylinder.
With the optimal level of turbulent energy and shear stress created by the blade motion, the hiPSC-derived megakaryocytes generated 100 billion platelets—enough to satisfy clinical requirements.
Transfusion experiments in 2 animal models of thrombocytopenia showed that these platelets perform similarly to platelets from human donors. Both types of platelets promoted blood clotting and reduced bleeding times to a comparable extent after ear vein incisions in rabbits and tail artery punctures in mice.
Dr Eto and his team are taking this work further by designing automated protocols, lowering manufacturing costs, and optimizing platelet yields. They are also developing universal platelets lacking human leukocyte antigens in order to reduce the risk of immune-mediated transfusion reactions.
“We expect clinical trials to begin within a year or two,” Dr Eto said. “We believe these findings will be a last scientific step to receiving permission for clinical trials using our platelets.”
Turbulence promotes large-scale production of functional platelets from human induced pluripotent stem cells (hiPSCs), according to research published in Cell.
Exposure to turbulent energy in a bioreactor stimulated hiPSC-derived megakaryocytes to produce 100 billion platelets.
Transfusion of these platelets in 2 animal models promoted blood clotting and prevented bleeding just as well as platelets from human donors.
“The discovery of turbulent energy provides a new physical mechanism and ex vivo production strategy for the generation of platelets that should impact clinical-scale cell therapies for regenerative medicine,” said study author Koji Eto, MD, PhD, of Kyoto University in Japan.
Previous attempts to generate platelets from hiPSC-derived megakaryocytes failed to achieve a scale suitable for clinical manufacturing.
While searching for a solution to this problem, Dr Eto and his colleagues noticed that hiPSC-derived megakaryocytes produced more platelets when being rotated in a flask than under static conditions in a petri dish.
This observation suggested that physical stress from horizontal shaking under liquid conditions enhances platelet generation.
Following up on this discovery, the researchers tested a rocking-bag-based bioreactor followed by a new microfluidic system with a flow chamber and multiple pillars, but these devices generated fewer than 20 platelets per hiPSC-derived megakaryocyte.
To examine the ideal physical conditions for generating platelets, Dr Eto and his team conducted live-imaging studies of mouse bone marrow.
These experiments revealed that megakaryocytes release platelets only when they are exposed to turbulent blood flow. In support of this idea, simulations confirmed that the bioreactor and microfluidic system the researchers previously tested lacked sufficient turbulent energy.
“The discovery of the crucial role of turbulence in platelet production significantly extends past research showing that shear stress from blood flow is also a key physical factor in this process,” Dr Eto said.
“Our findings also show that iPS cells are not the end-all be-all for producing platelets. Understanding fluid dynamics in addition to iPS cell technology was necessary for our discovery.”
After testing various devices, the researchers discovered that large-scale production of high-quality platelets was possible using a bioreactor called VerMES. This system consists of 2 oval-shaped, horizontally oriented mixing blades that generate relatively high levels of turbulence by moving up and down in a cylinder.
With the optimal level of turbulent energy and shear stress created by the blade motion, the hiPSC-derived megakaryocytes generated 100 billion platelets—enough to satisfy clinical requirements.
Transfusion experiments in 2 animal models of thrombocytopenia showed that these platelets perform similarly to platelets from human donors. Both types of platelets promoted blood clotting and reduced bleeding times to a comparable extent after ear vein incisions in rabbits and tail artery punctures in mice.
Dr Eto and his team are taking this work further by designing automated protocols, lowering manufacturing costs, and optimizing platelet yields. They are also developing universal platelets lacking human leukocyte antigens in order to reduce the risk of immune-mediated transfusion reactions.
“We expect clinical trials to begin within a year or two,” Dr Eto said. “We believe these findings will be a last scientific step to receiving permission for clinical trials using our platelets.”
Study links gut bacteria and TRALI

A new study has revealed a previously unknown link between gastrointestinal flora and transfusion-related acute lung injury (TRALI).
“We observed that the composition of the gastrointestinal flora drives the pathogenic immune response in the lungs during TRALI,” said Rick Kapur, PhD, of Lund University in Lund, Sweden.
Dr Kapur and his colleagues described this discovery in Blood Advances.
The researchers compared 2 groups of mice. One group was kept in a strictly sterile environment, allowing the gastrointestinal flora to be minimally affected by external factors. The other group was raised in a normal, less sterile environment.
“We saw that the mice kept in a more sterile environment were resistant to TRALI development, while the less sterile-raised mice developed severe TRALI,” said study author John W. Semple, PhD, of Lund University.
The composition of the gastrointestinal flora was significantly different between the 2 groups of mice, as determined by genetic sequencing of stool.
When the researchers wiped out the gastrointestinal flora with several types of antibiotics, they found that mice in the less sterile environment did not develop TRALI.
The researchers also transplanted stool from mice that developed TRALI into TRALI-resistant mice. After the stool transplant, the resistant mice were also able to develop TRALI, which confirmed the link between the composition of the gastrointestinal flora and the onset of TRALI.
The researchers still need to clarify which specific gut bacteria are directly involved, but the knowledge that intestinal bacteria may affect the lungs could facilitate diagnostics and the development of potential new drugs.
The ability to easily assess the risk for TRALI via analysis of gastrointestinal flora is equally important, according to the researchers.
“Knowing the composition of the gastrointestinal flora of people who will receive blood transfusions, an analysis which can be easily performed today, would allow you to assess who may be at increased risk for developing TRALI,” Dr Kapur said.
“The TRALI model in mice is very similar to the condition in humans, and the next step will be to validate these findings in humans,” Dr Semple said. “It’s not often that these types of findings in mice can lead directly to clinical studies in humans, but that will be our aim.”
A new study has revealed a previously unknown link between gastrointestinal flora and transfusion-related acute lung injury (TRALI).
“We observed that the composition of the gastrointestinal flora drives the pathogenic immune response in the lungs during TRALI,” said Rick Kapur, PhD, of Lund University in Lund, Sweden.
Dr Kapur and his colleagues described this discovery in Blood Advances.
The researchers compared 2 groups of mice. One group was kept in a strictly sterile environment, allowing the gastrointestinal flora to be minimally affected by external factors. The other group was raised in a normal, less sterile environment.
“We saw that the mice kept in a more sterile environment were resistant to TRALI development, while the less sterile-raised mice developed severe TRALI,” said study author John W. Semple, PhD, of Lund University.
The composition of the gastrointestinal flora was significantly different between the 2 groups of mice, as determined by genetic sequencing of stool.
When the researchers wiped out the gastrointestinal flora with several types of antibiotics, they found that mice in the less sterile environment did not develop TRALI.
The researchers also transplanted stool from mice that developed TRALI into TRALI-resistant mice. After the stool transplant, the resistant mice were also able to develop TRALI, which confirmed the link between the composition of the gastrointestinal flora and the onset of TRALI.
The researchers still need to clarify which specific gut bacteria are directly involved, but the knowledge that intestinal bacteria may affect the lungs could facilitate diagnostics and the development of potential new drugs.
The ability to easily assess the risk for TRALI via analysis of gastrointestinal flora is equally important, according to the researchers.
“Knowing the composition of the gastrointestinal flora of people who will receive blood transfusions, an analysis which can be easily performed today, would allow you to assess who may be at increased risk for developing TRALI,” Dr Kapur said.
“The TRALI model in mice is very similar to the condition in humans, and the next step will be to validate these findings in humans,” Dr Semple said. “It’s not often that these types of findings in mice can lead directly to clinical studies in humans, but that will be our aim.”
A new study has revealed a previously unknown link between gastrointestinal flora and transfusion-related acute lung injury (TRALI).
“We observed that the composition of the gastrointestinal flora drives the pathogenic immune response in the lungs during TRALI,” said Rick Kapur, PhD, of Lund University in Lund, Sweden.
Dr Kapur and his colleagues described this discovery in Blood Advances.
The researchers compared 2 groups of mice. One group was kept in a strictly sterile environment, allowing the gastrointestinal flora to be minimally affected by external factors. The other group was raised in a normal, less sterile environment.
“We saw that the mice kept in a more sterile environment were resistant to TRALI development, while the less sterile-raised mice developed severe TRALI,” said study author John W. Semple, PhD, of Lund University.
The composition of the gastrointestinal flora was significantly different between the 2 groups of mice, as determined by genetic sequencing of stool.
When the researchers wiped out the gastrointestinal flora with several types of antibiotics, they found that mice in the less sterile environment did not develop TRALI.
The researchers also transplanted stool from mice that developed TRALI into TRALI-resistant mice. After the stool transplant, the resistant mice were also able to develop TRALI, which confirmed the link between the composition of the gastrointestinal flora and the onset of TRALI.
The researchers still need to clarify which specific gut bacteria are directly involved, but the knowledge that intestinal bacteria may affect the lungs could facilitate diagnostics and the development of potential new drugs.
The ability to easily assess the risk for TRALI via analysis of gastrointestinal flora is equally important, according to the researchers.
“Knowing the composition of the gastrointestinal flora of people who will receive blood transfusions, an analysis which can be easily performed today, would allow you to assess who may be at increased risk for developing TRALI,” Dr Kapur said.
“The TRALI model in mice is very similar to the condition in humans, and the next step will be to validate these findings in humans,” Dr Semple said. “It’s not often that these types of findings in mice can lead directly to clinical studies in humans, but that will be our aim.”
Protein could be target for MLL-rearranged AML
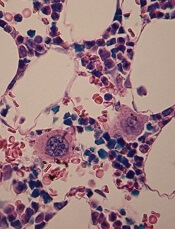
Preclinical research has revealed a potential therapeutic target for MLL-rearranged acute myeloid leukemia (AML).
The target—F-box protein S-phase kinase-associated protein 2 (Skp2)—degrades another protein called p27Kip1 that is important to the formation of healthy blood cells.
This finding was published in the Journal of Experimental Medicine.
“Our work provides a complete mechanistic look into the function of genetic and molecular programs driving this leukemia, and it exploits these processes to identify actionable therapeutic targets,’’ said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
For this work, Dr Grimes and his colleagues performed biochemical analyses of cells from AML patients. This gave the researchers comprehensive information about the targets and functions of the miR-196 molecular signaling pathway.
The team inserted mimics of miR-196 into MLL-AF9 leukemia cells to incorporate them into the cellular machinery. The group then lysed the cells for analyses, which revealed molecular targets of miR-196 in the leukemia cells.
Next, the researchers screened AML cells in mice for miR-196 targets. These experiments showed that certain microRNA targets are more important than others in the maintenance and spread of leukemia stem cells (LSCs).
Computer-assisted analysis of the Molecular Signature Database (a shared multi-institutional resource) allowed the researchers to identify sets of genes that show up in high numbers in MLL-AF9 leukemia.
Additional biochemical testing revealed that miR-196 directly targets and inhibits Cdkn1b/p27Kip1, which controls molecular programming in LSCs that allows them to maintain aggressive MLL-AF9 leukemia.
When miR-196 targets Cdkn1b/p27Kip1, it accelerates MLL-AF9 progression by abnormally linking stem cell activity with the growth of leukemia cells.
With the data suggesting that elevation of p27Kip1 protein levels may be therapeutic to AML patients, the researchers investigated a related molecular pathway that also regulates p27Kip1.
This investigation yielded the treatment target Skp2, which degrades the p27 protein and lowers its expression.
The researchers tested an experimental Skp2 inhibitor, SLZ P1041, on human AML cell lines and found the drug killed AML cells in a dose-dependent manner.
The team also tested SLZ P1041 in combination with other inhibitors—IBET-151, palbociclib, and MI-1. The most consistent synergies were with the combination of SLZ P1041 and MI-1, an inhibitor of the interaction between Menin and MLL.
“We still have extensive additional testing to conduct in laboratory animal models of AML before knowing if [targeting Skp2] will translate to patient care,” Dr Grimes said.
Preclinical research has revealed a potential therapeutic target for MLL-rearranged acute myeloid leukemia (AML).
The target—F-box protein S-phase kinase-associated protein 2 (Skp2)—degrades another protein called p27Kip1 that is important to the formation of healthy blood cells.
This finding was published in the Journal of Experimental Medicine.
“Our work provides a complete mechanistic look into the function of genetic and molecular programs driving this leukemia, and it exploits these processes to identify actionable therapeutic targets,’’ said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
For this work, Dr Grimes and his colleagues performed biochemical analyses of cells from AML patients. This gave the researchers comprehensive information about the targets and functions of the miR-196 molecular signaling pathway.
The team inserted mimics of miR-196 into MLL-AF9 leukemia cells to incorporate them into the cellular machinery. The group then lysed the cells for analyses, which revealed molecular targets of miR-196 in the leukemia cells.
Next, the researchers screened AML cells in mice for miR-196 targets. These experiments showed that certain microRNA targets are more important than others in the maintenance and spread of leukemia stem cells (LSCs).
Computer-assisted analysis of the Molecular Signature Database (a shared multi-institutional resource) allowed the researchers to identify sets of genes that show up in high numbers in MLL-AF9 leukemia.
Additional biochemical testing revealed that miR-196 directly targets and inhibits Cdkn1b/p27Kip1, which controls molecular programming in LSCs that allows them to maintain aggressive MLL-AF9 leukemia.
When miR-196 targets Cdkn1b/p27Kip1, it accelerates MLL-AF9 progression by abnormally linking stem cell activity with the growth of leukemia cells.
With the data suggesting that elevation of p27Kip1 protein levels may be therapeutic to AML patients, the researchers investigated a related molecular pathway that also regulates p27Kip1.
This investigation yielded the treatment target Skp2, which degrades the p27 protein and lowers its expression.
The researchers tested an experimental Skp2 inhibitor, SLZ P1041, on human AML cell lines and found the drug killed AML cells in a dose-dependent manner.
The team also tested SLZ P1041 in combination with other inhibitors—IBET-151, palbociclib, and MI-1. The most consistent synergies were with the combination of SLZ P1041 and MI-1, an inhibitor of the interaction between Menin and MLL.
“We still have extensive additional testing to conduct in laboratory animal models of AML before knowing if [targeting Skp2] will translate to patient care,” Dr Grimes said.
Preclinical research has revealed a potential therapeutic target for MLL-rearranged acute myeloid leukemia (AML).
The target—F-box protein S-phase kinase-associated protein 2 (Skp2)—degrades another protein called p27Kip1 that is important to the formation of healthy blood cells.
This finding was published in the Journal of Experimental Medicine.
“Our work provides a complete mechanistic look into the function of genetic and molecular programs driving this leukemia, and it exploits these processes to identify actionable therapeutic targets,’’ said study author H. Leighton Grimes, PhD, of Cincinnati Children’s Hospital Medical Center in Ohio.
For this work, Dr Grimes and his colleagues performed biochemical analyses of cells from AML patients. This gave the researchers comprehensive information about the targets and functions of the miR-196 molecular signaling pathway.
The team inserted mimics of miR-196 into MLL-AF9 leukemia cells to incorporate them into the cellular machinery. The group then lysed the cells for analyses, which revealed molecular targets of miR-196 in the leukemia cells.
Next, the researchers screened AML cells in mice for miR-196 targets. These experiments showed that certain microRNA targets are more important than others in the maintenance and spread of leukemia stem cells (LSCs).
Computer-assisted analysis of the Molecular Signature Database (a shared multi-institutional resource) allowed the researchers to identify sets of genes that show up in high numbers in MLL-AF9 leukemia.
Additional biochemical testing revealed that miR-196 directly targets and inhibits Cdkn1b/p27Kip1, which controls molecular programming in LSCs that allows them to maintain aggressive MLL-AF9 leukemia.
When miR-196 targets Cdkn1b/p27Kip1, it accelerates MLL-AF9 progression by abnormally linking stem cell activity with the growth of leukemia cells.
With the data suggesting that elevation of p27Kip1 protein levels may be therapeutic to AML patients, the researchers investigated a related molecular pathway that also regulates p27Kip1.
This investigation yielded the treatment target Skp2, which degrades the p27 protein and lowers its expression.
The researchers tested an experimental Skp2 inhibitor, SLZ P1041, on human AML cell lines and found the drug killed AML cells in a dose-dependent manner.
The team also tested SLZ P1041 in combination with other inhibitors—IBET-151, palbociclib, and MI-1. The most consistent synergies were with the combination of SLZ P1041 and MI-1, an inhibitor of the interaction between Menin and MLL.
“We still have extensive additional testing to conduct in laboratory animal models of AML before knowing if [targeting Skp2] will translate to patient care,” Dr Grimes said.
FDA releases guidance docs on gene therapy

The US Food and Drug Administration (FDA) has released several draft guidance documents on gene therapy.
Three are disease-specific guidances—for hemophilia, rare diseases, and retinal disorders—and 3 are guidances on manufacturing gene therapies.
These 6 documents are intended to serve as the building blocks of a modern, comprehensive framework for advancing the field of gene therapy, according to FDA Commissioner Scott Gottlieb, MD.
He said the documents are being issued in draft form so the FDA can solicit public input on these new policies. As with all draft guidances, all comments received will be considered before the FDA finalizes the documents.
Disease-specific guidances
The Human Gene Therapy for Rare Diseases Draft Guidance and the Human Gene Therapy for Retinal Disorders Draft Guidance include considerations for product development, preclinical research, clinical trials, expedited programs, and communication with the FDA.
The Human Gene Therapy for Hemophilia Draft Guidance covers the same topics but also includes considerations for measuring factor VIII and factor IX activity.
The draft guidance for rare diseases encompasses diseases affecting fewer than 200,000 people in the US.
Manufacturing guidances
The 3 remaining draft guidances are actually updates to existing guidances that address manufacturing issues related to gene therapy.
The first updated draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to ensure safety, identity, quality, purity, and strength/potency of investigational gene therapy products. This guidance applies to gene therapies alone and products that contain a gene therapy in combination with a drug or device.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who have received retroviral vector-based gene therapy products.
The third draft guidance, Long Term Follow-Up (LTFU) After Administration of Human Gene Therapy Products, provides recommendations regarding the design of LTFU observational studies for the collection of data on delayed adverse events after gene therapy.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
The US Food and Drug Administration (FDA) has released several draft guidance documents on gene therapy.
Three are disease-specific guidances—for hemophilia, rare diseases, and retinal disorders—and 3 are guidances on manufacturing gene therapies.
These 6 documents are intended to serve as the building blocks of a modern, comprehensive framework for advancing the field of gene therapy, according to FDA Commissioner Scott Gottlieb, MD.
He said the documents are being issued in draft form so the FDA can solicit public input on these new policies. As with all draft guidances, all comments received will be considered before the FDA finalizes the documents.
Disease-specific guidances
The Human Gene Therapy for Rare Diseases Draft Guidance and the Human Gene Therapy for Retinal Disorders Draft Guidance include considerations for product development, preclinical research, clinical trials, expedited programs, and communication with the FDA.
The Human Gene Therapy for Hemophilia Draft Guidance covers the same topics but also includes considerations for measuring factor VIII and factor IX activity.
The draft guidance for rare diseases encompasses diseases affecting fewer than 200,000 people in the US.
Manufacturing guidances
The 3 remaining draft guidances are actually updates to existing guidances that address manufacturing issues related to gene therapy.
The first updated draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to ensure safety, identity, quality, purity, and strength/potency of investigational gene therapy products. This guidance applies to gene therapies alone and products that contain a gene therapy in combination with a drug or device.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who have received retroviral vector-based gene therapy products.
The third draft guidance, Long Term Follow-Up (LTFU) After Administration of Human Gene Therapy Products, provides recommendations regarding the design of LTFU observational studies for the collection of data on delayed adverse events after gene therapy.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
The US Food and Drug Administration (FDA) has released several draft guidance documents on gene therapy.
Three are disease-specific guidances—for hemophilia, rare diseases, and retinal disorders—and 3 are guidances on manufacturing gene therapies.
These 6 documents are intended to serve as the building blocks of a modern, comprehensive framework for advancing the field of gene therapy, according to FDA Commissioner Scott Gottlieb, MD.
He said the documents are being issued in draft form so the FDA can solicit public input on these new policies. As with all draft guidances, all comments received will be considered before the FDA finalizes the documents.
Disease-specific guidances
The Human Gene Therapy for Rare Diseases Draft Guidance and the Human Gene Therapy for Retinal Disorders Draft Guidance include considerations for product development, preclinical research, clinical trials, expedited programs, and communication with the FDA.
The Human Gene Therapy for Hemophilia Draft Guidance covers the same topics but also includes considerations for measuring factor VIII and factor IX activity.
The draft guidance for rare diseases encompasses diseases affecting fewer than 200,000 people in the US.
Manufacturing guidances
The 3 remaining draft guidances are actually updates to existing guidances that address manufacturing issues related to gene therapy.
The first updated draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to ensure safety, identity, quality, purity, and strength/potency of investigational gene therapy products. This guidance applies to gene therapies alone and products that contain a gene therapy in combination with a drug or device.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who have received retroviral vector-based gene therapy products.
The third draft guidance, Long Term Follow-Up (LTFU) After Administration of Human Gene Therapy Products, provides recommendations regarding the design of LTFU observational studies for the collection of data on delayed adverse events after gene therapy.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
Identifying patients with a high risk of AML

Researchers say they have found ways to identify patients with a high risk of developing acute myeloid leukemia (AML).
The researchers used basic clinical and laboratory data to identify patients 6 to 12 months before AML presentation.
Using genetic information, the researchers were able to identify high-risk patients several years before AML presentation.
“This long time window gives us the first opportunity to think about how to prevent AML,” said John Dick, PhD, of Princess Margaret Cancer Centre in Toronto, Ontario, Canada.
However, Dr Dick and his colleagues noted that neither the genetic method nor the clinical data method were entirely accurate in identifying patients who would develop AML.
The researchers described both methods in Nature.
The team’s work began with the goal of distinguishing patients who will develop AML from patients who simply develop age-related clonal hematopoiesis.
“We wanted to know if there was any difference between these 2 groups in the genetics of their ‘normal’ blood samples taken at enrollment,” Dr Dick said. “To find out, we developed a gene-sequencing tool that captured the most common genes that get altered in AML and sequenced all the 500 blood samples.”
The researchers analyzed samples from 95 patients who ultimately developed AML and 414 age- and gender-matched controls. The samples from AML patients were obtained an average of 6.3 years before AML diagnosis.
The researchers found that pre-AML cases had more mutations per sample, higher variant allele frequencies, and a higher frequency of certain mutations than controls. Specifically, pre-AML cases had a significantly greater frequency of DNMT3A, TET2, SRSF2, ASXL1, TP53, U2AF1, JAK2, RUNX1, and IDH2 mutations.
The researchers used these findings to develop a model for predicting AML. They tested the model in a validation cohort of 29 pre-AML cases and 262 controls, as well as in a cohort combining the validation and discovery cohorts. The model predicted AML development with 41.9% sensitivity and 95.7% specificity.
However, the researchers said widespread use of this model would not be practical because AML is so rare. The team said millions of people would need to undergo screening to identify a few pre-AML cases, and there would be “many” false-positives.
Therefore, the researchers developed a model using clinical information from electronic health records—particularly blood counts. This model was able to predict AML development 6 to 12 months before diagnosis with a sensitivity of 25.7% and specificity of 98.2%.
The researchers said these results suggest clinical data can be used to identify patients with a high risk of AML who may benefit from targeted genetic screening. And combining clinical and genetic information in a single model could improve predictive accuracy.
“Our study provides, for the first time, evidence that we can identify people at risk of developing AML many years before they actually develop this life-threatening disease,” said study author George Vassiliou, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“We hope to build on these findings to develop robust screening tests for identifying those at risk and drive research into how to prevent or stall progression towards AML. Our aspiration is that, one day, AML prevention would provide a compelling alternative to treatment.”
Researchers say they have found ways to identify patients with a high risk of developing acute myeloid leukemia (AML).
The researchers used basic clinical and laboratory data to identify patients 6 to 12 months before AML presentation.
Using genetic information, the researchers were able to identify high-risk patients several years before AML presentation.
“This long time window gives us the first opportunity to think about how to prevent AML,” said John Dick, PhD, of Princess Margaret Cancer Centre in Toronto, Ontario, Canada.
However, Dr Dick and his colleagues noted that neither the genetic method nor the clinical data method were entirely accurate in identifying patients who would develop AML.
The researchers described both methods in Nature.
The team’s work began with the goal of distinguishing patients who will develop AML from patients who simply develop age-related clonal hematopoiesis.
“We wanted to know if there was any difference between these 2 groups in the genetics of their ‘normal’ blood samples taken at enrollment,” Dr Dick said. “To find out, we developed a gene-sequencing tool that captured the most common genes that get altered in AML and sequenced all the 500 blood samples.”
The researchers analyzed samples from 95 patients who ultimately developed AML and 414 age- and gender-matched controls. The samples from AML patients were obtained an average of 6.3 years before AML diagnosis.
The researchers found that pre-AML cases had more mutations per sample, higher variant allele frequencies, and a higher frequency of certain mutations than controls. Specifically, pre-AML cases had a significantly greater frequency of DNMT3A, TET2, SRSF2, ASXL1, TP53, U2AF1, JAK2, RUNX1, and IDH2 mutations.
The researchers used these findings to develop a model for predicting AML. They tested the model in a validation cohort of 29 pre-AML cases and 262 controls, as well as in a cohort combining the validation and discovery cohorts. The model predicted AML development with 41.9% sensitivity and 95.7% specificity.
However, the researchers said widespread use of this model would not be practical because AML is so rare. The team said millions of people would need to undergo screening to identify a few pre-AML cases, and there would be “many” false-positives.
Therefore, the researchers developed a model using clinical information from electronic health records—particularly blood counts. This model was able to predict AML development 6 to 12 months before diagnosis with a sensitivity of 25.7% and specificity of 98.2%.
The researchers said these results suggest clinical data can be used to identify patients with a high risk of AML who may benefit from targeted genetic screening. And combining clinical and genetic information in a single model could improve predictive accuracy.
“Our study provides, for the first time, evidence that we can identify people at risk of developing AML many years before they actually develop this life-threatening disease,” said study author George Vassiliou, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“We hope to build on these findings to develop robust screening tests for identifying those at risk and drive research into how to prevent or stall progression towards AML. Our aspiration is that, one day, AML prevention would provide a compelling alternative to treatment.”
Researchers say they have found ways to identify patients with a high risk of developing acute myeloid leukemia (AML).
The researchers used basic clinical and laboratory data to identify patients 6 to 12 months before AML presentation.
Using genetic information, the researchers were able to identify high-risk patients several years before AML presentation.
“This long time window gives us the first opportunity to think about how to prevent AML,” said John Dick, PhD, of Princess Margaret Cancer Centre in Toronto, Ontario, Canada.
However, Dr Dick and his colleagues noted that neither the genetic method nor the clinical data method were entirely accurate in identifying patients who would develop AML.
The researchers described both methods in Nature.
The team’s work began with the goal of distinguishing patients who will develop AML from patients who simply develop age-related clonal hematopoiesis.
“We wanted to know if there was any difference between these 2 groups in the genetics of their ‘normal’ blood samples taken at enrollment,” Dr Dick said. “To find out, we developed a gene-sequencing tool that captured the most common genes that get altered in AML and sequenced all the 500 blood samples.”
The researchers analyzed samples from 95 patients who ultimately developed AML and 414 age- and gender-matched controls. The samples from AML patients were obtained an average of 6.3 years before AML diagnosis.
The researchers found that pre-AML cases had more mutations per sample, higher variant allele frequencies, and a higher frequency of certain mutations than controls. Specifically, pre-AML cases had a significantly greater frequency of DNMT3A, TET2, SRSF2, ASXL1, TP53, U2AF1, JAK2, RUNX1, and IDH2 mutations.
The researchers used these findings to develop a model for predicting AML. They tested the model in a validation cohort of 29 pre-AML cases and 262 controls, as well as in a cohort combining the validation and discovery cohorts. The model predicted AML development with 41.9% sensitivity and 95.7% specificity.
However, the researchers said widespread use of this model would not be practical because AML is so rare. The team said millions of people would need to undergo screening to identify a few pre-AML cases, and there would be “many” false-positives.
Therefore, the researchers developed a model using clinical information from electronic health records—particularly blood counts. This model was able to predict AML development 6 to 12 months before diagnosis with a sensitivity of 25.7% and specificity of 98.2%.
The researchers said these results suggest clinical data can be used to identify patients with a high risk of AML who may benefit from targeted genetic screening. And combining clinical and genetic information in a single model could improve predictive accuracy.
“Our study provides, for the first time, evidence that we can identify people at risk of developing AML many years before they actually develop this life-threatening disease,” said study author George Vassiliou, PhD, of the Wellcome Trust Sanger Institute in Hinxton, UK.
“We hope to build on these findings to develop robust screening tests for identifying those at risk and drive research into how to prevent or stall progression towards AML. Our aspiration is that, one day, AML prevention would provide a compelling alternative to treatment.”
FDA grants EUA for freeze-dried plasma product

The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a freeze-dried plasma product to be used by the military.
The product is Pathogen-Reduced Leukocyte-Depleted Freeze-Dried Plasma manufactured by the Centre de Transfusion Sanguine des Armées, referred to in the EUA as “French FDP.”
Under the EUA, French FDP is authorized for the treatment of hemorrhage or coagulopathy in US military personnel during an emergency involving agents of military combat (eg, firearms, projectiles, and explosive devices) when plasma is not available or when the use of plasma is not practical.
The use of plasma in combat settings is severely limited by logistical and operational challenges, such as the need for refrigeration and, in the case of frozen plasma, a long thawing period.
French FDP is a powdered, freeze-dried product that can be used following reconstitution in settings where refrigeration is not available, thus enabling the rapid availability of plasma for use at the point of injury.
The FDA issued the EUA for French FDP in response to a request from the US Department of Defense (DoD) and after receiving the required determination by DoD and a declaration by the Secretary of the Department of Health and Human Services.
This action is the result of a collaboration between the FDA and the DoD to prioritize development of medical products intended to help save the lives of American military personnel. The FDA outlined its approach to advancing the development of such products in a work plan developed with DoD.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country by expediting the development and availability of safe and effective priority medical products that are essential to the health of our military service members,” said FDA Commissioner Scott Gottlieb, MD.
“Through our collaborative program with the DoD, they’ve made clear the importance of access to freeze-dried plasma in initial efforts to control hemorrhage from battlefield trauma. Granting this authorization will support access to this important product in the event it’s needed. The FDA remains deeply committed to implementing an enduring pathway to ensure that these potentially life-saving medical products are made available in the most expeditious, safe, and effective manner possible.”
The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a freeze-dried plasma product to be used by the military.
The product is Pathogen-Reduced Leukocyte-Depleted Freeze-Dried Plasma manufactured by the Centre de Transfusion Sanguine des Armées, referred to in the EUA as “French FDP.”
Under the EUA, French FDP is authorized for the treatment of hemorrhage or coagulopathy in US military personnel during an emergency involving agents of military combat (eg, firearms, projectiles, and explosive devices) when plasma is not available or when the use of plasma is not practical.
The use of plasma in combat settings is severely limited by logistical and operational challenges, such as the need for refrigeration and, in the case of frozen plasma, a long thawing period.
French FDP is a powdered, freeze-dried product that can be used following reconstitution in settings where refrigeration is not available, thus enabling the rapid availability of plasma for use at the point of injury.
The FDA issued the EUA for French FDP in response to a request from the US Department of Defense (DoD) and after receiving the required determination by DoD and a declaration by the Secretary of the Department of Health and Human Services.
This action is the result of a collaboration between the FDA and the DoD to prioritize development of medical products intended to help save the lives of American military personnel. The FDA outlined its approach to advancing the development of such products in a work plan developed with DoD.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country by expediting the development and availability of safe and effective priority medical products that are essential to the health of our military service members,” said FDA Commissioner Scott Gottlieb, MD.
“Through our collaborative program with the DoD, they’ve made clear the importance of access to freeze-dried plasma in initial efforts to control hemorrhage from battlefield trauma. Granting this authorization will support access to this important product in the event it’s needed. The FDA remains deeply committed to implementing an enduring pathway to ensure that these potentially life-saving medical products are made available in the most expeditious, safe, and effective manner possible.”
The US Food and Drug Administration (FDA) has granted emergency use authorization (EUA) for a freeze-dried plasma product to be used by the military.
The product is Pathogen-Reduced Leukocyte-Depleted Freeze-Dried Plasma manufactured by the Centre de Transfusion Sanguine des Armées, referred to in the EUA as “French FDP.”
Under the EUA, French FDP is authorized for the treatment of hemorrhage or coagulopathy in US military personnel during an emergency involving agents of military combat (eg, firearms, projectiles, and explosive devices) when plasma is not available or when the use of plasma is not practical.
The use of plasma in combat settings is severely limited by logistical and operational challenges, such as the need for refrigeration and, in the case of frozen plasma, a long thawing period.
French FDP is a powdered, freeze-dried product that can be used following reconstitution in settings where refrigeration is not available, thus enabling the rapid availability of plasma for use at the point of injury.
The FDA issued the EUA for French FDP in response to a request from the US Department of Defense (DoD) and after receiving the required determination by DoD and a declaration by the Secretary of the Department of Health and Human Services.
This action is the result of a collaboration between the FDA and the DoD to prioritize development of medical products intended to help save the lives of American military personnel. The FDA outlined its approach to advancing the development of such products in a work plan developed with DoD.
“Earlier this year, we reaffirmed our commitment to the Department of Defense and to the dedicated men and women protecting our country by expediting the development and availability of safe and effective priority medical products that are essential to the health of our military service members,” said FDA Commissioner Scott Gottlieb, MD.
“Through our collaborative program with the DoD, they’ve made clear the importance of access to freeze-dried plasma in initial efforts to control hemorrhage from battlefield trauma. Granting this authorization will support access to this important product in the event it’s needed. The FDA remains deeply committed to implementing an enduring pathway to ensure that these potentially life-saving medical products are made available in the most expeditious, safe, and effective manner possible.”
New approach to AE reporting needed, group says

A group of experts has called for improvements in reporting adverse events (AEs) that occur in patients with hematologic malignancies.
The group highlighted deficiencies in capturing chronic, cumulative, and late AEs; collecting patient-reported outcomes (PROs); reporting AEs associated with hematopoietic stem cell transplant (HSCT); assessing long-term toxicity in survivors; reporting AEs to regulatory agencies; and tracking AEs that occur in routine clinical practice.
The experts discussed these problems and made recommendations for fixing them in The Lancet Haematology.
“The success in outcomes and survival in many hematological malignancies is historically unparalleled and fueled by scientific discovery and implementation,” said author Gita Thanarajasingam, MD, of the Mayo Clinic in Rochester, Minnesota.
“Patients are now living with the challenge of managing not just their hematological malignancy but also managing chronic therapy for their illness, with new types of acute, chronic, cumulative, and late toxicities. Measures to address the broad facets of toxicity assessment must be prioritized and further developed to ultimately enhance accurate, comprehensive, patient-centered toxicity reporting that will meaningfully inform the care of patients with blood cancers.”
Dr Thanarajasingam and a group of clinicians, investigators, regulators, biostatisticians, and patient advocates analyzed the evidence on AEs and proposed recommendations to policy makers, researchers, industry, and regulators.
First, the group noted that chronic, delayed, and cumulative AEs may go unreported in patients with hematologic malignancies. Therefore, longitudinal methods for AE analysis are needed, and phase 1 trials should have longer periods for evaluating dose-limiting toxicity.
The experts also said PROs are often overlooked, but it should be standard to assess PROs in clinical trials of patients with hematologic malignancies.
Another of the group’s concerns is the “cumbersome” reporting of AEs associated with HSCT acting as a barrier to clinical trials. The experts recommended using registry data to develop a consensus on expected AEs after HSCT.
The group also highlighted deficiencies in the “description and management” of cumulative and late toxicities in survivors of hematologic malignancies. Potential solutions to this problem include developing infrastructure to collect data for adult survivors and standardizing the use and content of survivorship care plans.
The experts said “meaningful” AEs are often underreported to regulatory agencies, so better systems are needed for collecting and analyzing AE data, and the electronic submission of AEs should be simplified.
Finally, the group said AEs occurring in routine clinical practice are difficult to capture and analyze on a large scale. This suggests a need to optimize the systematic, objective collection of toxicity data at multiple points in real-world databases, according to the experts.
Additional details and recommendations are available in the full report.
A group of experts has called for improvements in reporting adverse events (AEs) that occur in patients with hematologic malignancies.
The group highlighted deficiencies in capturing chronic, cumulative, and late AEs; collecting patient-reported outcomes (PROs); reporting AEs associated with hematopoietic stem cell transplant (HSCT); assessing long-term toxicity in survivors; reporting AEs to regulatory agencies; and tracking AEs that occur in routine clinical practice.
The experts discussed these problems and made recommendations for fixing them in The Lancet Haematology.
“The success in outcomes and survival in many hematological malignancies is historically unparalleled and fueled by scientific discovery and implementation,” said author Gita Thanarajasingam, MD, of the Mayo Clinic in Rochester, Minnesota.
“Patients are now living with the challenge of managing not just their hematological malignancy but also managing chronic therapy for their illness, with new types of acute, chronic, cumulative, and late toxicities. Measures to address the broad facets of toxicity assessment must be prioritized and further developed to ultimately enhance accurate, comprehensive, patient-centered toxicity reporting that will meaningfully inform the care of patients with blood cancers.”
Dr Thanarajasingam and a group of clinicians, investigators, regulators, biostatisticians, and patient advocates analyzed the evidence on AEs and proposed recommendations to policy makers, researchers, industry, and regulators.
First, the group noted that chronic, delayed, and cumulative AEs may go unreported in patients with hematologic malignancies. Therefore, longitudinal methods for AE analysis are needed, and phase 1 trials should have longer periods for evaluating dose-limiting toxicity.
The experts also said PROs are often overlooked, but it should be standard to assess PROs in clinical trials of patients with hematologic malignancies.
Another of the group’s concerns is the “cumbersome” reporting of AEs associated with HSCT acting as a barrier to clinical trials. The experts recommended using registry data to develop a consensus on expected AEs after HSCT.
The group also highlighted deficiencies in the “description and management” of cumulative and late toxicities in survivors of hematologic malignancies. Potential solutions to this problem include developing infrastructure to collect data for adult survivors and standardizing the use and content of survivorship care plans.
The experts said “meaningful” AEs are often underreported to regulatory agencies, so better systems are needed for collecting and analyzing AE data, and the electronic submission of AEs should be simplified.
Finally, the group said AEs occurring in routine clinical practice are difficult to capture and analyze on a large scale. This suggests a need to optimize the systematic, objective collection of toxicity data at multiple points in real-world databases, according to the experts.
Additional details and recommendations are available in the full report.
A group of experts has called for improvements in reporting adverse events (AEs) that occur in patients with hematologic malignancies.
The group highlighted deficiencies in capturing chronic, cumulative, and late AEs; collecting patient-reported outcomes (PROs); reporting AEs associated with hematopoietic stem cell transplant (HSCT); assessing long-term toxicity in survivors; reporting AEs to regulatory agencies; and tracking AEs that occur in routine clinical practice.
The experts discussed these problems and made recommendations for fixing them in The Lancet Haematology.
“The success in outcomes and survival in many hematological malignancies is historically unparalleled and fueled by scientific discovery and implementation,” said author Gita Thanarajasingam, MD, of the Mayo Clinic in Rochester, Minnesota.
“Patients are now living with the challenge of managing not just their hematological malignancy but also managing chronic therapy for their illness, with new types of acute, chronic, cumulative, and late toxicities. Measures to address the broad facets of toxicity assessment must be prioritized and further developed to ultimately enhance accurate, comprehensive, patient-centered toxicity reporting that will meaningfully inform the care of patients with blood cancers.”
Dr Thanarajasingam and a group of clinicians, investigators, regulators, biostatisticians, and patient advocates analyzed the evidence on AEs and proposed recommendations to policy makers, researchers, industry, and regulators.
First, the group noted that chronic, delayed, and cumulative AEs may go unreported in patients with hematologic malignancies. Therefore, longitudinal methods for AE analysis are needed, and phase 1 trials should have longer periods for evaluating dose-limiting toxicity.
The experts also said PROs are often overlooked, but it should be standard to assess PROs in clinical trials of patients with hematologic malignancies.
Another of the group’s concerns is the “cumbersome” reporting of AEs associated with HSCT acting as a barrier to clinical trials. The experts recommended using registry data to develop a consensus on expected AEs after HSCT.
The group also highlighted deficiencies in the “description and management” of cumulative and late toxicities in survivors of hematologic malignancies. Potential solutions to this problem include developing infrastructure to collect data for adult survivors and standardizing the use and content of survivorship care plans.
The experts said “meaningful” AEs are often underreported to regulatory agencies, so better systems are needed for collecting and analyzing AE data, and the electronic submission of AEs should be simplified.
Finally, the group said AEs occurring in routine clinical practice are difficult to capture and analyze on a large scale. This suggests a need to optimize the systematic, objective collection of toxicity data at multiple points in real-world databases, according to the experts.
Additional details and recommendations are available in the full report.
Health Canada expands approval of obinutuzumab

Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
Health Canada has expanded the approved use of obinutuzumab (Gazyva®).
The anti-CD20 monoclonal antibody is now approved for use in combination with chemotherapy to treat patients with previously untreated follicular lymphoma (FL) that is advanced (stage II bulky, stage III, or stage IV).
In patients who respond to this treatment, obinutuzumab monotherapy can be given as maintenance.
Health Canada previously approved obinutuzumab for the following indications:
- In combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia
- First in combination with bendamustine, then as monotherapy, in FL patients who relapsed after or are refractory to a rituximab-containing regimen.
Phase 3 results
Health Canada’s latest approval of obinutuzumab is based on results from the phase 3 GALLIUM study, which were published in NEJM in October 2017. The following are updated data from the product monograph.
GALLIUM included 1385 patients with previously untreated non-Hodgkin lymphoma, and 1202 of these patients had previously untreated, advanced FL.
Half of the FL patients (n=601) were randomized to receive obinutuzumab plus chemotherapy (followed by obinutuzumab maintenance for up to 2 years), and half were randomized to rituximab plus chemotherapy (followed by rituximab maintenance for up to 2 years).
The different chemotherapies used were CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), CVP (cyclophosphamide, vincristine, and prednisone), and bendamustine.
At a median observation time of 41.1 months, the overall response rate was 91% in the obinutuzumab arm and 88% in the rituximab arm. The complete response rates were 28% and 27%, respectively.
The median progression-free survival was not reached in either arm. The hazard ratio, for obinutuzumab compared to rituximab, was 0.72 (95% CI, 0.56-0.93, P=0.0118).
The estimated 3-year progression-free survival was 78.9% in the rituximab arm and 83.4% in the obinutuzumab arm.
Safety was evaluated based on all 1385 patients in the study, 86% of whom had FL and 14% of whom had marginal zone lymphoma.
Serious adverse events (AEs) occurred in 50% of patients in the obinutuzumab arm and 43% in the rituximab arm. Fatal AEs occurred in 5% and 4%, respectively. Infections and second malignancies were the leading causes of these deaths.
During the monotherapy period, the most common AEs (≥ 5%) in patients treated with obinutuzumab were cough (21%), neutropenia (19%), upper respiratory tract infection (15%), viral upper respiratory tract infection (15%), diarrhea (13%), arthralgia (10%), fatigue (9%), sinusitis (9%), infusion reactions (8%), pneumonia (8%), herpes zoster (8%), lower respiratory tract infection (7%), pyrexia (7%), back pain (6%), headache (6%), urinary tract infection (6%), nausea (6%), bronchitis (5%), and vomiting (5%).
Grade 3-4 AEs (≥1%) in patients treated with obinutuzumab included neutropenia (17%), pneumonia (3%), and febrile neutropenia (2%). There were 2 deaths due to pneumonia in the obinutuzumab arm.
A new use for ibrutinib?

Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”
Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”
Preclinical research suggests ibrutinib could treat G-CSFR-mutant myeloid disorders.
“Mutations in G-CSFR have a harmful effect on the production of neutrophils and are reported in patients with several blood disorders, including severe congenital neutropenia, chronic neutrophilic leukemia, and acute myeloid leukemia,” said Ken Greis, PhD, of the University of Cincinnati in Ohio.
“Unfortunately, despite years of research, the malignant signaling of the mutated G-CSFRs is not well understood.”
With this in mind, Dr Greis and his colleagues created a comprehensive signaling network of normal and mutated G-CSFR. Their goal was to understand how abnormal cellular signaling from the mutant receptors results in disease development.
The researchers described this work in Leukemia.
“We are able to look at . . . phosphorylation that results in phosphate groups being attached to the amino acid tyrosine (Tyr) in proteins,” Dr Greis explained. “These phosphorylation events (pTyr) can act as switches to activate or inactivate proteins and/or specific cellular processes.”
“By evaluating pTyr activity in the normal versus mutant receptor cells, we can produce a network similar to a wiring diagram of cellular regulation. Observed disruptions at any of the nodes in the network for the mutated receptors can then be investigated further to understand and perhaps target the abnormal signaling corresponding to the disease.”
This analysis of pTyr activity revealed that G-CSFR mutants had aberrant activation of BTK, as well as abnormal kinetics of canonical STAT3, STAT5, and MAPK phosphorylation.
“When we first got these results, one of the most exciting things was that BTK was already the target of an FDA-approved drug, ibrutinib . . .,” said study author H. Leighton Grimes, PhD, of the University of Cincinnati.
The researchers tested ibrutinib in cells with mutant and wild-type G-CSFR and found the drug killed the mutant cells but not the wild-type cells. This was the case in myeloid progenitor 32D cell lines and primary human CD34+ umbilical cord blood cells.
“Progenitor cells expressing mutated G-CSFR in animal models and in human blood cells also showed enhanced sensitivity to ibrutinib compared to the normal G-CSFR, thus confirming that the mutated cells could likely be eliminated by treatment with ibrutinib and may represent an effective therapy for these patients,” Dr Grimes said.
Ibrutinib also demonstrated synergy with the JAK1/2 inhibitor ruxolitinib. G-CSFR-mutant CD34+ cells were sensitive to each drug alone, but combining them “dramatically enhanced” the sensitivity, according to the researchers.
“These data demonstrate the strength of global proteomics approaches, like the pTyr profiling used here, in dissecting cancer-forming pathways and points to the possibility that ibrutinib could be an effective therapy for myeloid leukemias with G-CSFR mutations,” Dr Greis said.
“Further studies are needed to determine if these findings will be applicable in patient samples, but the hope is that clinical trials are just around the corner, since we’re investigating a drug that has already been found to be safe by the FDA.”