User login
Novel gene-based therapies for neuromuscular diseases
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5

Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.

Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
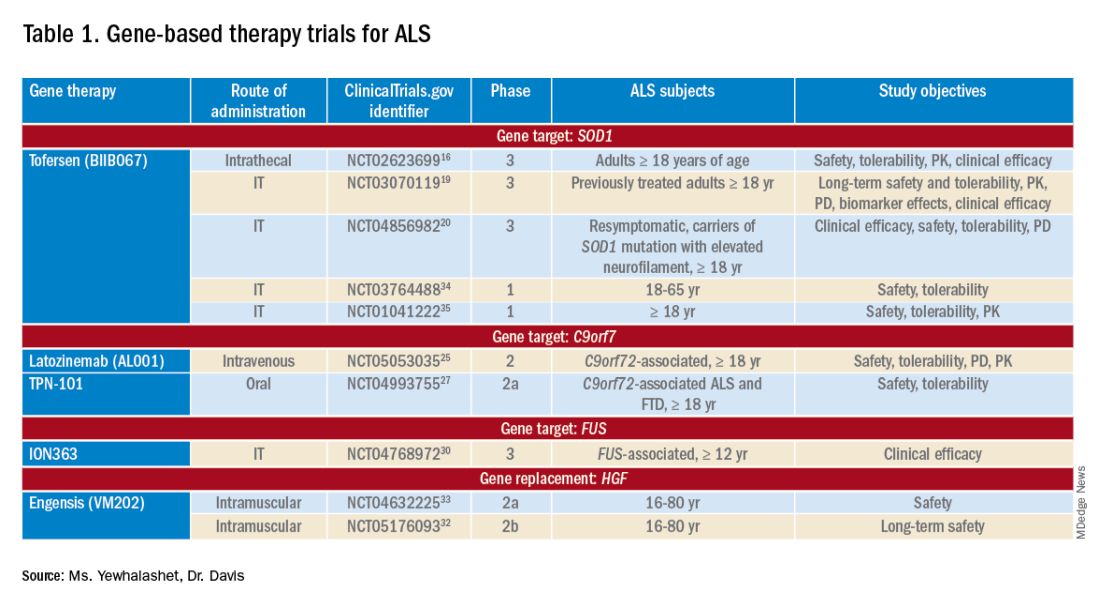
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
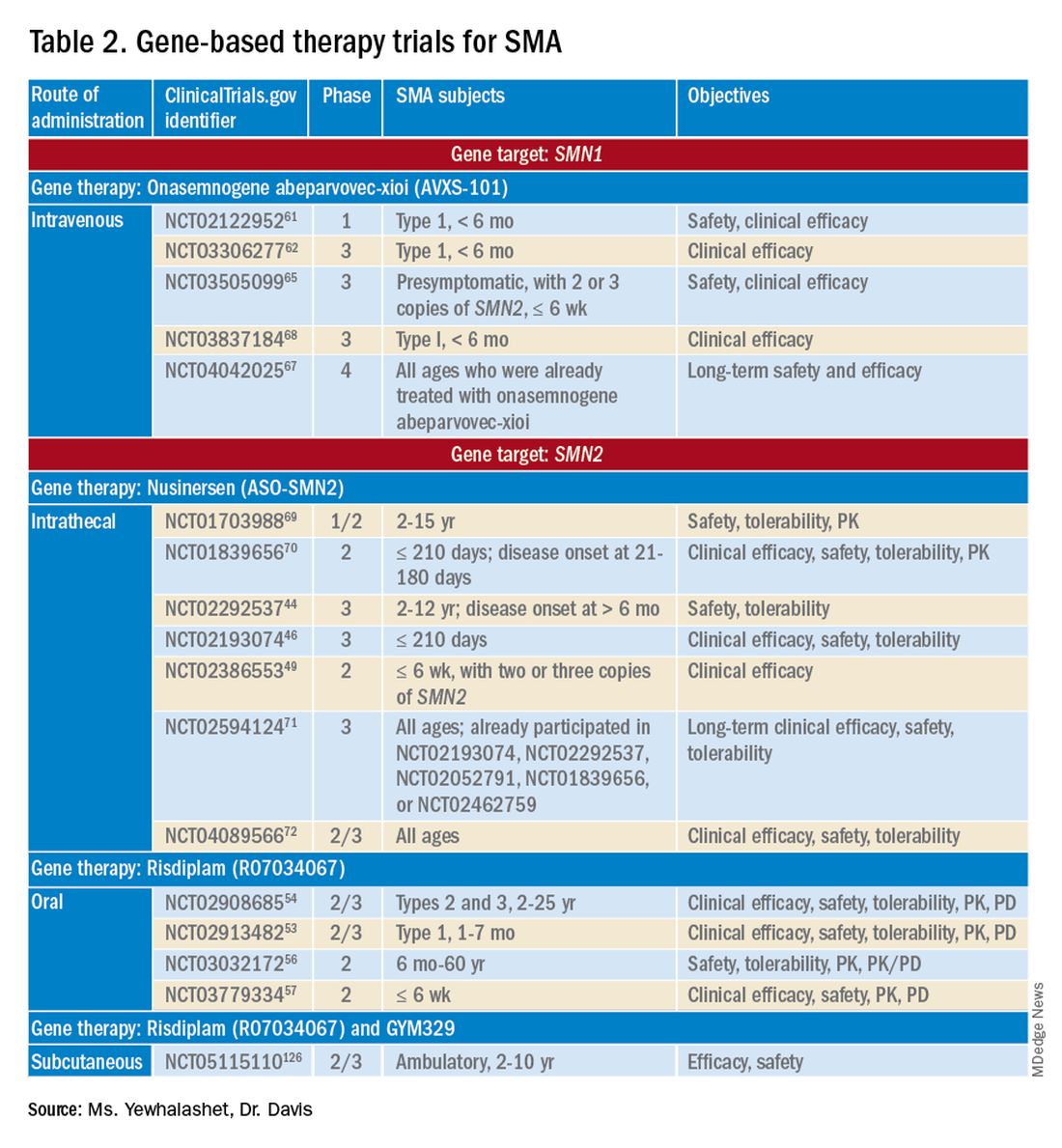
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
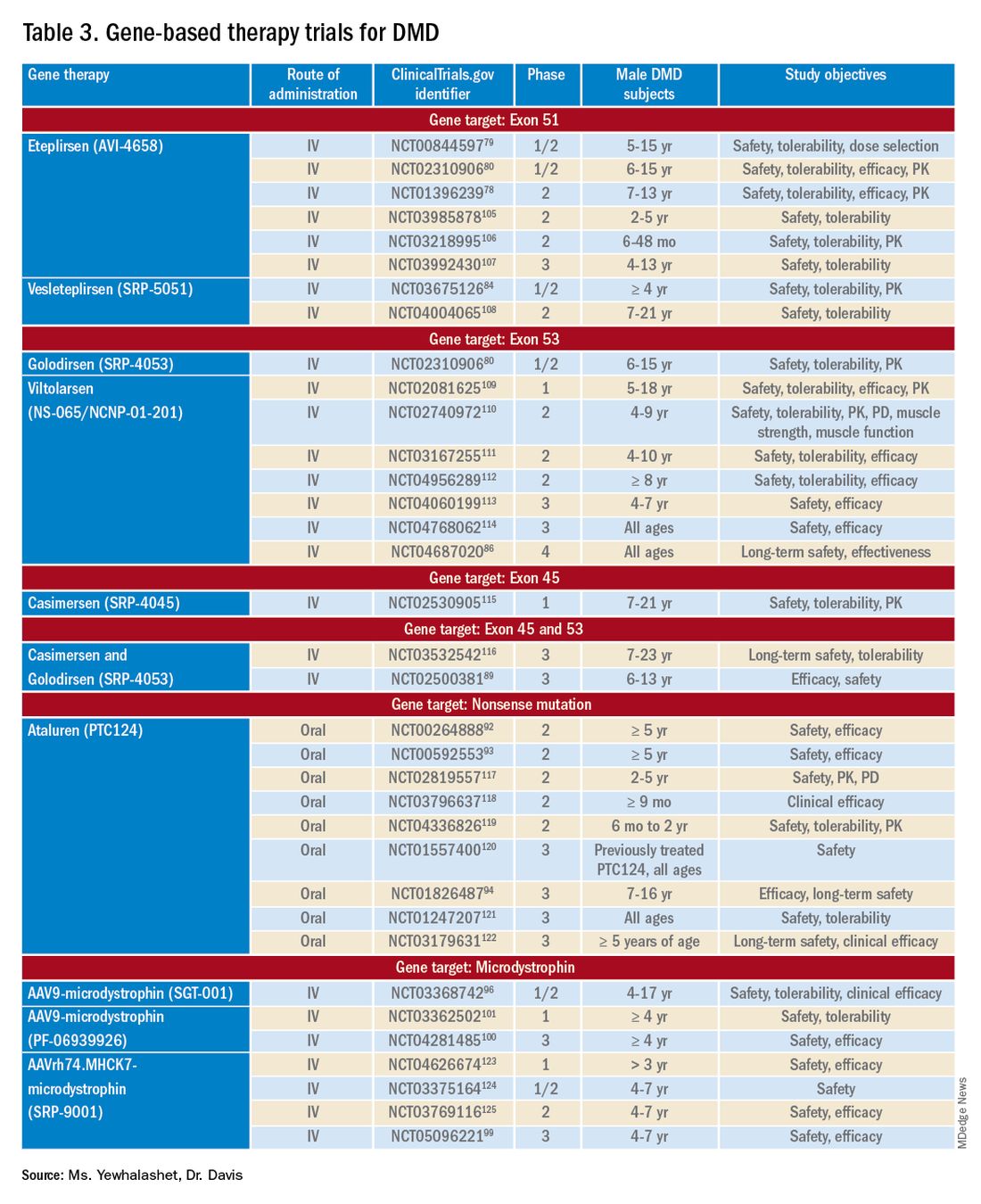
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110