User login
Fatigue, arthalgia, amenorrhea—Dx?
THE CASE
A 46-year-old Caucasian female with a history of epilepsy came into our family medicine center complaining of weakness, fatigue, and arthralgia that made it difficult for her to walk. She’d had these symptoms for 6 months and reported having amenorrhea and hot flashes for the past 2 years.
The patient’s blood pressure was 133/72 mm Hg, heart rate was 82 beats per min, and respiratory rate was 20 breaths per min. Her skin was dry without hyperpigmentation, and her sclerae were anicteric. A musculoskeletal examination revealed tenderness of the metacarpophalangeal and metatarsophalangeal joints without edema, deformity, or evidence of synovitis.
She had no history of skin bronzing, jaundice, transfusions, hepatitis, abdominal pain, or diabetes and denied using tobacco, alcohol, or illicit drugs. Her medications included lamotrigine (250 mg BID) and over-the-counter iron supplementation. She had no family history of rheumatoid arthritis, lupus, cirrhosis, hemochromatosis, or other liver disease. Her mother died from colorectal cancer and her father’s cause of death was unknown; her sisters did not have any medical issues. The patient’s lab tests were normal, except for the following: aspartate aminotransferase, 89 U/L (normal, 13-45 U/L); alanine aminotransferase, 80 U/L (normal, 5-57 U/L); and alkaline phosphatase, 132 U/L (normal, 39-117 U/L). Her coagulation panel revealed a prothrombin time of 13.1 seconds, and an international normalized ratio of 1.3. Serology was negative for hepatitis A, B, and C. Additional testing revealed the following: ferritin, 4014.1 ng/dL (normal, 7-282 ng/dL); iron, 210 mg/dL (normal, 40-170 mg/dL); total iron binding capacity, 258 mg/dL (normal, 260-445 mg/dL); and transferrin saturation, 81% (normal, 20%-55%).
Abdominal ultrasonography revealed gallstones, an enlarged spleen, a dilated portal vein, and a fatty liver consistent with cirrhosis. X-rays showed soft-tissue swelling and demineralization in her hands consistent with osteopenia and degenerative arthritis in both feet.
THE DIAGNOSIS
Based on our patient’s complaints of fatigue, weakness, arthralgia, and amenorrhea, as well as her abnormal iron levels, we suspected hereditary hemochromatosis (HH). We ordered HFE genotyping, and the results indicated that the patient was homozygous for the C282Y mutation, confirming our diagnosis.
DISCUSSION
HH is an autosomal recessive disorder of iron homeostasis characterized by increased gastrointestinal iron absorption and tissue deposition of iron. It is caused by mutations in the HFE gene (C282Y or H63D) located on chromosome 6 (locus 6p21) and commonly seen in Northern European Caucasians.1 Approximately 85% of patients with HH are homozygous for C282Y; the H63D mutation can cause HH when in the presence of a single C282Y mutation.1 Men manifest HH symptoms usually between the ages of 40 and 60 years,2 although women may be affected at a later age than men because physiologic blood loss from menstruation and parturition limit the rate at which excess iron is accumulated.2
|
Signs and symptoms of HH include depression, fatigue, restless legs syndrome, weakness, and weight loss.3 In advanced HH, patients may develop progressive skin pigmentation or bronzing, and hypogonadism. Advanced HH can affect the patient’s organs, including the pancreas (diabetes), liver (hepatomegaly, abnormal liver function tests), pituitary gland (amenorrhea, decreased libido, erectile dysfunction), and heart (arrhythmias, congestive heart failure), as well as the musculoskeletal system (joint pain).3,4 The spleen can also be affected after cirrhosis develops. Cirrhosis, hepatocellular carcinoma, and cardiomyopathy can reduce life expectancy.4
Testing for HH
Because symptoms of HH are common and nonspecific, a high degree of clinical suspicion is required for early diagnosis. The differential diagnosis includes conditions related to chronic liver disease or iron overload (TABLE).5 If the diagnosis goes undetected until complications arise, the risk of morbidity and mortality are greatly increased.5
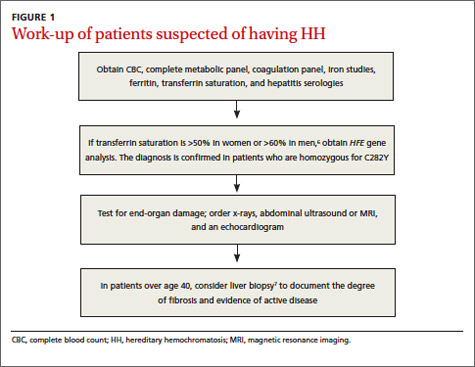
If HH is suspected, serum ferritin concentration and fasting serum transferrin saturation (the ratio of serum iron level to total iron-binding capacity × 100) are recommended as initial tests.5 The normal range of transferrin saturation for males is 15% to 50% and the normal range for females is 12% to 45%. If the transferrin saturation exceeds 50% in women or 60% in men, further evaluation is warranted (FIGURE 1).6,7 The sensitivity and specificity of elevated transferrin saturation for HH are 92% and 93%, respectively.5 These transferrin saturation cutoffs don’t apply to patients with a history of frequent blood transfusion (ie, patients with sickle cell disease or thalassemia).
Additional testing for patients in whom you suspect HH includes:
• a complete blood count, metabolic panel, and coagulation panel
• hepatitis serologies
• imaging (abdominal ultrasound, skeletal radiographs, echocardiogram, abdominal magnetic resonance imaging [MRI])
• a liver biopsy with iron staining and quantitative iron measurements.
The gold standard. Performing a liver biopsy to measure hepatic iron concentration by staining is considered the gold standard test for HH.8 But since genetic testing has become more readily available, liver biopsies aren’t widely used to confirm the diagnosis.8 The diagnosis of HH usually is confirmed by molecular testing for the C282Y and H63D mutations. Liver biopsy may be recommended to document the degree of fibrosis in all homozygotes over age 40 with elevated serum transaminase levels, clinical evidence of liver disease, or a serum ferritin level >1000 mcg/L.7
Phlebotomy helps lower iron levels
Treatment should not be delayed until symptoms develop.3 The mainstay of therapy is phlebotomy.9 If phlebotomy is started before the onset of organ damage, patients can anticipate a normal lifespan.9 Without treatment death may occur from cirrhosis, hepatocellular carcinoma, or cardiomyopathy.
Removal of 1 unit of red blood cells (450-500 mL) results in the loss of approximately 200 mg of iron. Serum ferritin level testing is the most reliable and least expensive method to monitor therapy.9 Iron depletion is complete when the serum ferritin level is 10 to 20 g/L, when the hemoglobin concentration is <11 g/dL, or the hematocrit is <33% for >3 weeks. HH patients need to undergo lifelong phlebotomy to maintain a serum ferritin level <50 g/L. Encourage patients to take in an adequate amount of dietary protein, vitamin B12, and folate to support the accelerated level of erythropoiesis that occurs during therapy.9
Chelation therapy is reserved for patients with advanced disease (eg, those with organ damage) or those who do not respond to phlebotomy.10 Deferoxamine given intravenously (IV) or subcutaneously has been the standard chelation agent. It’s usually administered by continuous subcutaneous infusion using a battery-operated pump at a dose of 40 mg/kg/d for 8 to 12 hours nightly for 5 to 7 nights weekly. A dose of approximately 2 g per 24 hours usually achieves maximal urinary iron excretion.
The use of deferoxamine therapy is limited by cost as well as the need for parenteral therapy, discomfort, inconvenience, and neurotoxicity.5 The US Food and Drug Administration recently approved an oral ironchelating agent, deferasirox, for the treatment of secondary iron overload due to ineffective erythropoiesis. Studies are ongoing to evaluate its potential use in HH.5,9
Our patient’s outcome
Our patient declined liver biopsy and her sisters declined HFE genotyping. Our patient did, however, complete 7 phlebotomies over 4 months. Two months later, she reported shortness of breath during exertion, leg swelling, and palpitations. A chest x-ray revealed a right-sided pleural effusion and an electrocardiogram showed atrial fibrillation with rapid ventricular response. Our patient was admitted for telemetry monitoring and started on diltiazem IV. Echocardiogram showed a restrictive cardiomyopathy, with an ejection fraction of 15% (normal range >55%).
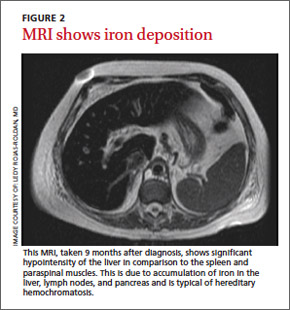
Six weeks later, her ejection fraction decreased to 10%. An MRI of her abdomen showed iron deposition in her liver, pancreas, and lymph nodes (FIGURE 2). She was started on deferoxamine IV and transferred to the coronary care unit for 3 weeks. She was discharged with a diagnosis of class IV heart failure and admitted 2 weeks later for exacerbation of heart failure symptoms. She did not want to pursue a heart transplant. Her condition deteriorated and she expired after a fatal cardiac arrhythmia.
THE TAKEAWAY
Patients with abnormal iron studies and those with evidence of liver disease should be evaluated for HH5 (strength of recommendation [SOR]: A). Fasting serum transferrin saturation and serum ferritin concentration are recommended as initial tests for HH11 (SOR C). Liver biopsy is the gold standard for diagnosis of HH, but the diagnosis usually is confirmed by genetic testing8 (SOR C). Phlebotomy is the mainstay of therapy9 (SOR B). Chelation therapy is reserved for patients with advanced disease or for those who do not respond to phlebotomy10 (SOR C).
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Matthews AL, Grimes SJ, Wiesner GL, et al. Clinical consult: iron overload--hereditary hemochromatosis. Prim Care. 2004;31:767-770,xii-xiii.
2. Gochee PA, Powell LW. What’s new in hemochromatosis. Curr Opin Hematol. 2001;8:98-104.
3. Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256-1262.
4. Adams PC. Hemochromatosis. Clin Liver Dis. 2004;8:735-753,vii.
5. Bacon BR, Adams PC, Kowdley KV, et al; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
6. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65:853-860.
7. Hash RB. Hereditary hemochromatosis. J Am Board Fam Pract. 2001;14:266-273.
8. Qaseem A, Aronson M, Fitterman N, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for hereditary hemochromatosis: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2005;143:517-521.
9. Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program. 2006:36-41.
10. US Preventive Services Task Force. Screening for hemochromatosis: recommendation statement. Ann Intern Med. 2006;145:204-208.
11. Borwein S, Ghent CN, Valberg LS. Diagnostic efficacy of screening tests for hereditary hemochromatosis. Can Med Assoc J. 1984;131:895-901.
THE CASE
A 46-year-old Caucasian female with a history of epilepsy came into our family medicine center complaining of weakness, fatigue, and arthralgia that made it difficult for her to walk. She’d had these symptoms for 6 months and reported having amenorrhea and hot flashes for the past 2 years.
The patient’s blood pressure was 133/72 mm Hg, heart rate was 82 beats per min, and respiratory rate was 20 breaths per min. Her skin was dry without hyperpigmentation, and her sclerae were anicteric. A musculoskeletal examination revealed tenderness of the metacarpophalangeal and metatarsophalangeal joints without edema, deformity, or evidence of synovitis.
She had no history of skin bronzing, jaundice, transfusions, hepatitis, abdominal pain, or diabetes and denied using tobacco, alcohol, or illicit drugs. Her medications included lamotrigine (250 mg BID) and over-the-counter iron supplementation. She had no family history of rheumatoid arthritis, lupus, cirrhosis, hemochromatosis, or other liver disease. Her mother died from colorectal cancer and her father’s cause of death was unknown; her sisters did not have any medical issues. The patient’s lab tests were normal, except for the following: aspartate aminotransferase, 89 U/L (normal, 13-45 U/L); alanine aminotransferase, 80 U/L (normal, 5-57 U/L); and alkaline phosphatase, 132 U/L (normal, 39-117 U/L). Her coagulation panel revealed a prothrombin time of 13.1 seconds, and an international normalized ratio of 1.3. Serology was negative for hepatitis A, B, and C. Additional testing revealed the following: ferritin, 4014.1 ng/dL (normal, 7-282 ng/dL); iron, 210 mg/dL (normal, 40-170 mg/dL); total iron binding capacity, 258 mg/dL (normal, 260-445 mg/dL); and transferrin saturation, 81% (normal, 20%-55%).
Abdominal ultrasonography revealed gallstones, an enlarged spleen, a dilated portal vein, and a fatty liver consistent with cirrhosis. X-rays showed soft-tissue swelling and demineralization in her hands consistent with osteopenia and degenerative arthritis in both feet.
THE DIAGNOSIS
Based on our patient’s complaints of fatigue, weakness, arthralgia, and amenorrhea, as well as her abnormal iron levels, we suspected hereditary hemochromatosis (HH). We ordered HFE genotyping, and the results indicated that the patient was homozygous for the C282Y mutation, confirming our diagnosis.
DISCUSSION
HH is an autosomal recessive disorder of iron homeostasis characterized by increased gastrointestinal iron absorption and tissue deposition of iron. It is caused by mutations in the HFE gene (C282Y or H63D) located on chromosome 6 (locus 6p21) and commonly seen in Northern European Caucasians.1 Approximately 85% of patients with HH are homozygous for C282Y; the H63D mutation can cause HH when in the presence of a single C282Y mutation.1 Men manifest HH symptoms usually between the ages of 40 and 60 years,2 although women may be affected at a later age than men because physiologic blood loss from menstruation and parturition limit the rate at which excess iron is accumulated.2
|
Signs and symptoms of HH include depression, fatigue, restless legs syndrome, weakness, and weight loss.3 In advanced HH, patients may develop progressive skin pigmentation or bronzing, and hypogonadism. Advanced HH can affect the patient’s organs, including the pancreas (diabetes), liver (hepatomegaly, abnormal liver function tests), pituitary gland (amenorrhea, decreased libido, erectile dysfunction), and heart (arrhythmias, congestive heart failure), as well as the musculoskeletal system (joint pain).3,4 The spleen can also be affected after cirrhosis develops. Cirrhosis, hepatocellular carcinoma, and cardiomyopathy can reduce life expectancy.4
Testing for HH
Because symptoms of HH are common and nonspecific, a high degree of clinical suspicion is required for early diagnosis. The differential diagnosis includes conditions related to chronic liver disease or iron overload (TABLE).5 If the diagnosis goes undetected until complications arise, the risk of morbidity and mortality are greatly increased.5
If HH is suspected, serum ferritin concentration and fasting serum transferrin saturation (the ratio of serum iron level to total iron-binding capacity × 100) are recommended as initial tests.5 The normal range of transferrin saturation for males is 15% to 50% and the normal range for females is 12% to 45%. If the transferrin saturation exceeds 50% in women or 60% in men, further evaluation is warranted (FIGURE 1).6,7 The sensitivity and specificity of elevated transferrin saturation for HH are 92% and 93%, respectively.5 These transferrin saturation cutoffs don’t apply to patients with a history of frequent blood transfusion (ie, patients with sickle cell disease or thalassemia).
Additional testing for patients in whom you suspect HH includes:
• a complete blood count, metabolic panel, and coagulation panel
• hepatitis serologies
• imaging (abdominal ultrasound, skeletal radiographs, echocardiogram, abdominal magnetic resonance imaging [MRI])
• a liver biopsy with iron staining and quantitative iron measurements.
The gold standard. Performing a liver biopsy to measure hepatic iron concentration by staining is considered the gold standard test for HH.8 But since genetic testing has become more readily available, liver biopsies aren’t widely used to confirm the diagnosis.8 The diagnosis of HH usually is confirmed by molecular testing for the C282Y and H63D mutations. Liver biopsy may be recommended to document the degree of fibrosis in all homozygotes over age 40 with elevated serum transaminase levels, clinical evidence of liver disease, or a serum ferritin level >1000 mcg/L.7
Phlebotomy helps lower iron levels
Treatment should not be delayed until symptoms develop.3 The mainstay of therapy is phlebotomy.9 If phlebotomy is started before the onset of organ damage, patients can anticipate a normal lifespan.9 Without treatment death may occur from cirrhosis, hepatocellular carcinoma, or cardiomyopathy.
Removal of 1 unit of red blood cells (450-500 mL) results in the loss of approximately 200 mg of iron. Serum ferritin level testing is the most reliable and least expensive method to monitor therapy.9 Iron depletion is complete when the serum ferritin level is 10 to 20 g/L, when the hemoglobin concentration is <11 g/dL, or the hematocrit is <33% for >3 weeks. HH patients need to undergo lifelong phlebotomy to maintain a serum ferritin level <50 g/L. Encourage patients to take in an adequate amount of dietary protein, vitamin B12, and folate to support the accelerated level of erythropoiesis that occurs during therapy.9
Chelation therapy is reserved for patients with advanced disease (eg, those with organ damage) or those who do not respond to phlebotomy.10 Deferoxamine given intravenously (IV) or subcutaneously has been the standard chelation agent. It’s usually administered by continuous subcutaneous infusion using a battery-operated pump at a dose of 40 mg/kg/d for 8 to 12 hours nightly for 5 to 7 nights weekly. A dose of approximately 2 g per 24 hours usually achieves maximal urinary iron excretion.
The use of deferoxamine therapy is limited by cost as well as the need for parenteral therapy, discomfort, inconvenience, and neurotoxicity.5 The US Food and Drug Administration recently approved an oral ironchelating agent, deferasirox, for the treatment of secondary iron overload due to ineffective erythropoiesis. Studies are ongoing to evaluate its potential use in HH.5,9
Our patient’s outcome
Our patient declined liver biopsy and her sisters declined HFE genotyping. Our patient did, however, complete 7 phlebotomies over 4 months. Two months later, she reported shortness of breath during exertion, leg swelling, and palpitations. A chest x-ray revealed a right-sided pleural effusion and an electrocardiogram showed atrial fibrillation with rapid ventricular response. Our patient was admitted for telemetry monitoring and started on diltiazem IV. Echocardiogram showed a restrictive cardiomyopathy, with an ejection fraction of 15% (normal range >55%).
Six weeks later, her ejection fraction decreased to 10%. An MRI of her abdomen showed iron deposition in her liver, pancreas, and lymph nodes (FIGURE 2). She was started on deferoxamine IV and transferred to the coronary care unit for 3 weeks. She was discharged with a diagnosis of class IV heart failure and admitted 2 weeks later for exacerbation of heart failure symptoms. She did not want to pursue a heart transplant. Her condition deteriorated and she expired after a fatal cardiac arrhythmia.
THE TAKEAWAY
Patients with abnormal iron studies and those with evidence of liver disease should be evaluated for HH5 (strength of recommendation [SOR]: A). Fasting serum transferrin saturation and serum ferritin concentration are recommended as initial tests for HH11 (SOR C). Liver biopsy is the gold standard for diagnosis of HH, but the diagnosis usually is confirmed by genetic testing8 (SOR C). Phlebotomy is the mainstay of therapy9 (SOR B). Chelation therapy is reserved for patients with advanced disease or for those who do not respond to phlebotomy10 (SOR C).
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
THE CASE
A 46-year-old Caucasian female with a history of epilepsy came into our family medicine center complaining of weakness, fatigue, and arthralgia that made it difficult for her to walk. She’d had these symptoms for 6 months and reported having amenorrhea and hot flashes for the past 2 years.
The patient’s blood pressure was 133/72 mm Hg, heart rate was 82 beats per min, and respiratory rate was 20 breaths per min. Her skin was dry without hyperpigmentation, and her sclerae were anicteric. A musculoskeletal examination revealed tenderness of the metacarpophalangeal and metatarsophalangeal joints without edema, deformity, or evidence of synovitis.
She had no history of skin bronzing, jaundice, transfusions, hepatitis, abdominal pain, or diabetes and denied using tobacco, alcohol, or illicit drugs. Her medications included lamotrigine (250 mg BID) and over-the-counter iron supplementation. She had no family history of rheumatoid arthritis, lupus, cirrhosis, hemochromatosis, or other liver disease. Her mother died from colorectal cancer and her father’s cause of death was unknown; her sisters did not have any medical issues. The patient’s lab tests were normal, except for the following: aspartate aminotransferase, 89 U/L (normal, 13-45 U/L); alanine aminotransferase, 80 U/L (normal, 5-57 U/L); and alkaline phosphatase, 132 U/L (normal, 39-117 U/L). Her coagulation panel revealed a prothrombin time of 13.1 seconds, and an international normalized ratio of 1.3. Serology was negative for hepatitis A, B, and C. Additional testing revealed the following: ferritin, 4014.1 ng/dL (normal, 7-282 ng/dL); iron, 210 mg/dL (normal, 40-170 mg/dL); total iron binding capacity, 258 mg/dL (normal, 260-445 mg/dL); and transferrin saturation, 81% (normal, 20%-55%).
Abdominal ultrasonography revealed gallstones, an enlarged spleen, a dilated portal vein, and a fatty liver consistent with cirrhosis. X-rays showed soft-tissue swelling and demineralization in her hands consistent with osteopenia and degenerative arthritis in both feet.
THE DIAGNOSIS
Based on our patient’s complaints of fatigue, weakness, arthralgia, and amenorrhea, as well as her abnormal iron levels, we suspected hereditary hemochromatosis (HH). We ordered HFE genotyping, and the results indicated that the patient was homozygous for the C282Y mutation, confirming our diagnosis.
DISCUSSION
HH is an autosomal recessive disorder of iron homeostasis characterized by increased gastrointestinal iron absorption and tissue deposition of iron. It is caused by mutations in the HFE gene (C282Y or H63D) located on chromosome 6 (locus 6p21) and commonly seen in Northern European Caucasians.1 Approximately 85% of patients with HH are homozygous for C282Y; the H63D mutation can cause HH when in the presence of a single C282Y mutation.1 Men manifest HH symptoms usually between the ages of 40 and 60 years,2 although women may be affected at a later age than men because physiologic blood loss from menstruation and parturition limit the rate at which excess iron is accumulated.2
|
Signs and symptoms of HH include depression, fatigue, restless legs syndrome, weakness, and weight loss.3 In advanced HH, patients may develop progressive skin pigmentation or bronzing, and hypogonadism. Advanced HH can affect the patient’s organs, including the pancreas (diabetes), liver (hepatomegaly, abnormal liver function tests), pituitary gland (amenorrhea, decreased libido, erectile dysfunction), and heart (arrhythmias, congestive heart failure), as well as the musculoskeletal system (joint pain).3,4 The spleen can also be affected after cirrhosis develops. Cirrhosis, hepatocellular carcinoma, and cardiomyopathy can reduce life expectancy.4
Testing for HH
Because symptoms of HH are common and nonspecific, a high degree of clinical suspicion is required for early diagnosis. The differential diagnosis includes conditions related to chronic liver disease or iron overload (TABLE).5 If the diagnosis goes undetected until complications arise, the risk of morbidity and mortality are greatly increased.5
If HH is suspected, serum ferritin concentration and fasting serum transferrin saturation (the ratio of serum iron level to total iron-binding capacity × 100) are recommended as initial tests.5 The normal range of transferrin saturation for males is 15% to 50% and the normal range for females is 12% to 45%. If the transferrin saturation exceeds 50% in women or 60% in men, further evaluation is warranted (FIGURE 1).6,7 The sensitivity and specificity of elevated transferrin saturation for HH are 92% and 93%, respectively.5 These transferrin saturation cutoffs don’t apply to patients with a history of frequent blood transfusion (ie, patients with sickle cell disease or thalassemia).
Additional testing for patients in whom you suspect HH includes:
• a complete blood count, metabolic panel, and coagulation panel
• hepatitis serologies
• imaging (abdominal ultrasound, skeletal radiographs, echocardiogram, abdominal magnetic resonance imaging [MRI])
• a liver biopsy with iron staining and quantitative iron measurements.
The gold standard. Performing a liver biopsy to measure hepatic iron concentration by staining is considered the gold standard test for HH.8 But since genetic testing has become more readily available, liver biopsies aren’t widely used to confirm the diagnosis.8 The diagnosis of HH usually is confirmed by molecular testing for the C282Y and H63D mutations. Liver biopsy may be recommended to document the degree of fibrosis in all homozygotes over age 40 with elevated serum transaminase levels, clinical evidence of liver disease, or a serum ferritin level >1000 mcg/L.7
Phlebotomy helps lower iron levels
Treatment should not be delayed until symptoms develop.3 The mainstay of therapy is phlebotomy.9 If phlebotomy is started before the onset of organ damage, patients can anticipate a normal lifespan.9 Without treatment death may occur from cirrhosis, hepatocellular carcinoma, or cardiomyopathy.
Removal of 1 unit of red blood cells (450-500 mL) results in the loss of approximately 200 mg of iron. Serum ferritin level testing is the most reliable and least expensive method to monitor therapy.9 Iron depletion is complete when the serum ferritin level is 10 to 20 g/L, when the hemoglobin concentration is <11 g/dL, or the hematocrit is <33% for >3 weeks. HH patients need to undergo lifelong phlebotomy to maintain a serum ferritin level <50 g/L. Encourage patients to take in an adequate amount of dietary protein, vitamin B12, and folate to support the accelerated level of erythropoiesis that occurs during therapy.9
Chelation therapy is reserved for patients with advanced disease (eg, those with organ damage) or those who do not respond to phlebotomy.10 Deferoxamine given intravenously (IV) or subcutaneously has been the standard chelation agent. It’s usually administered by continuous subcutaneous infusion using a battery-operated pump at a dose of 40 mg/kg/d for 8 to 12 hours nightly for 5 to 7 nights weekly. A dose of approximately 2 g per 24 hours usually achieves maximal urinary iron excretion.
The use of deferoxamine therapy is limited by cost as well as the need for parenteral therapy, discomfort, inconvenience, and neurotoxicity.5 The US Food and Drug Administration recently approved an oral ironchelating agent, deferasirox, for the treatment of secondary iron overload due to ineffective erythropoiesis. Studies are ongoing to evaluate its potential use in HH.5,9
Our patient’s outcome
Our patient declined liver biopsy and her sisters declined HFE genotyping. Our patient did, however, complete 7 phlebotomies over 4 months. Two months later, she reported shortness of breath during exertion, leg swelling, and palpitations. A chest x-ray revealed a right-sided pleural effusion and an electrocardiogram showed atrial fibrillation with rapid ventricular response. Our patient was admitted for telemetry monitoring and started on diltiazem IV. Echocardiogram showed a restrictive cardiomyopathy, with an ejection fraction of 15% (normal range >55%).
Six weeks later, her ejection fraction decreased to 10%. An MRI of her abdomen showed iron deposition in her liver, pancreas, and lymph nodes (FIGURE 2). She was started on deferoxamine IV and transferred to the coronary care unit for 3 weeks. She was discharged with a diagnosis of class IV heart failure and admitted 2 weeks later for exacerbation of heart failure symptoms. She did not want to pursue a heart transplant. Her condition deteriorated and she expired after a fatal cardiac arrhythmia.
THE TAKEAWAY
Patients with abnormal iron studies and those with evidence of liver disease should be evaluated for HH5 (strength of recommendation [SOR]: A). Fasting serum transferrin saturation and serum ferritin concentration are recommended as initial tests for HH11 (SOR C). Liver biopsy is the gold standard for diagnosis of HH, but the diagnosis usually is confirmed by genetic testing8 (SOR C). Phlebotomy is the mainstay of therapy9 (SOR B). Chelation therapy is reserved for patients with advanced disease or for those who do not respond to phlebotomy10 (SOR C).
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
1. Matthews AL, Grimes SJ, Wiesner GL, et al. Clinical consult: iron overload--hereditary hemochromatosis. Prim Care. 2004;31:767-770,xii-xiii.
2. Gochee PA, Powell LW. What’s new in hemochromatosis. Curr Opin Hematol. 2001;8:98-104.
3. Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256-1262.
4. Adams PC. Hemochromatosis. Clin Liver Dis. 2004;8:735-753,vii.
5. Bacon BR, Adams PC, Kowdley KV, et al; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
6. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65:853-860.
7. Hash RB. Hereditary hemochromatosis. J Am Board Fam Pract. 2001;14:266-273.
8. Qaseem A, Aronson M, Fitterman N, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for hereditary hemochromatosis: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2005;143:517-521.
9. Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program. 2006:36-41.
10. US Preventive Services Task Force. Screening for hemochromatosis: recommendation statement. Ann Intern Med. 2006;145:204-208.
11. Borwein S, Ghent CN, Valberg LS. Diagnostic efficacy of screening tests for hereditary hemochromatosis. Can Med Assoc J. 1984;131:895-901.
1. Matthews AL, Grimes SJ, Wiesner GL, et al. Clinical consult: iron overload--hereditary hemochromatosis. Prim Care. 2004;31:767-770,xii-xiii.
2. Gochee PA, Powell LW. What’s new in hemochromatosis. Curr Opin Hematol. 2001;8:98-104.
3. Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256-1262.
4. Adams PC. Hemochromatosis. Clin Liver Dis. 2004;8:735-753,vii.
5. Bacon BR, Adams PC, Kowdley KV, et al; American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328-343.
6. Brandhagen DJ, Fairbanks VF, Baldus W. Recognition and management of hereditary hemochromatosis. Am Fam Physician. 2002;65:853-860.
7. Hash RB. Hereditary hemochromatosis. J Am Board Fam Pract. 2001;14:266-273.
8. Qaseem A, Aronson M, Fitterman N, et al; Clinical Efficacy Assessment Subcommittee of the American College of Physicians. Screening for hereditary hemochromatosis: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2005;143:517-521.
9. Brissot P, de Bels F. Current approaches to the management of hemochromatosis. Hematology Am Soc Hematol Educ Program. 2006:36-41.
10. US Preventive Services Task Force. Screening for hemochromatosis: recommendation statement. Ann Intern Med. 2006;145:204-208.
11. Borwein S, Ghent CN, Valberg LS. Diagnostic efficacy of screening tests for hereditary hemochromatosis. Can Med Assoc J. 1984;131:895-901.
