User login
Alveolar proteinosis: A slow drowning in mud
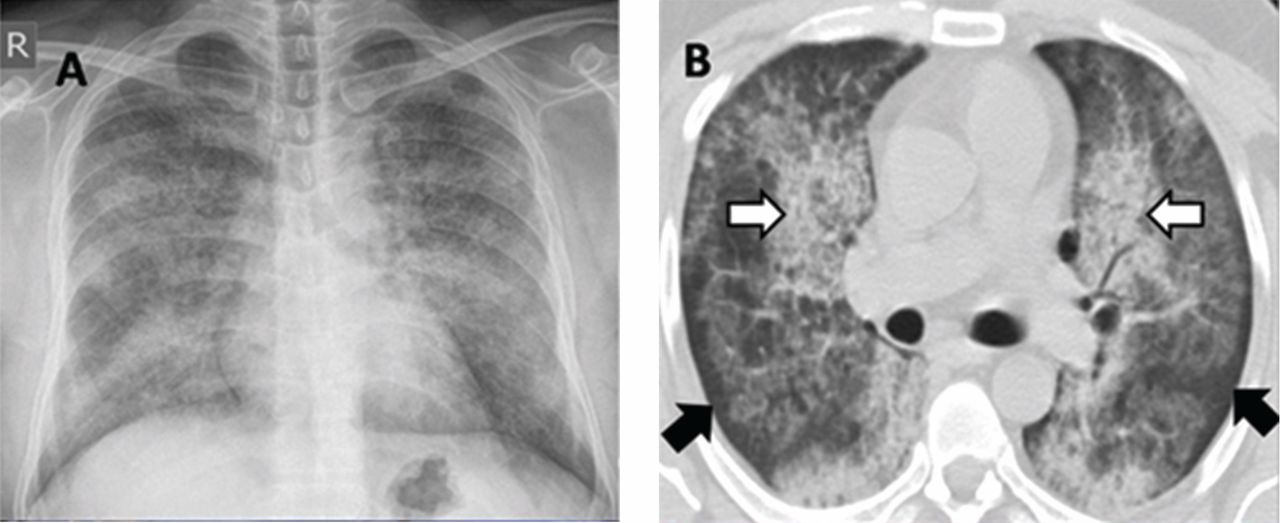
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
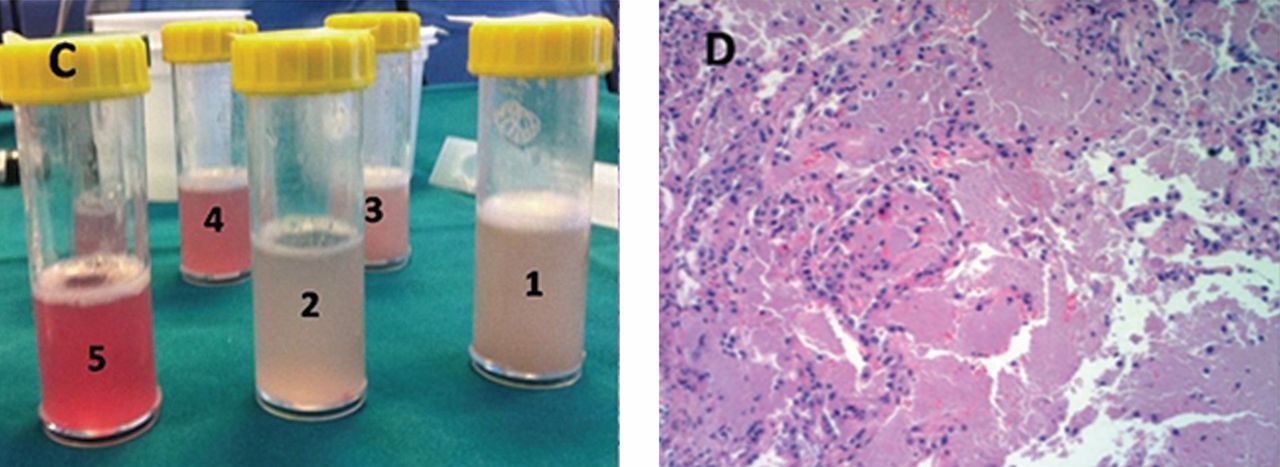
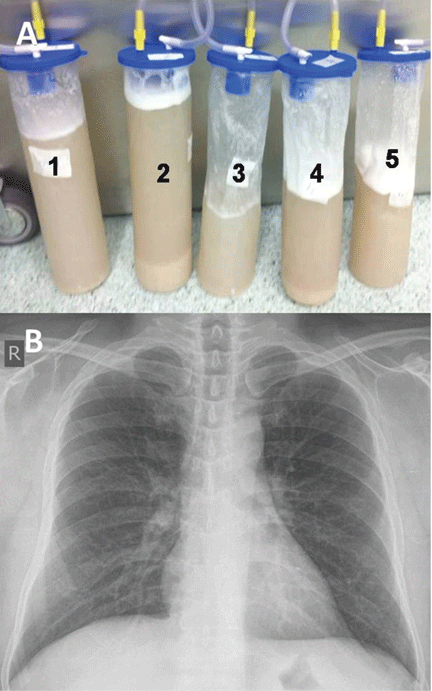
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
A 30-year-old man presented with progressive dyspnea and dry cough, which had developed over the last 6 months. His oxygen saturation was 88% on room air, and he had diffuse bilateral crackles on auscultation. Imaging showed a mixture of diffuse airspace and interstitial abnormalities (Figure 1).
He underwent bronchoscopy. The bronchoalveolar lavage fluid had a turbid appearance that gradually cleared with successive aliquots. Transbronchial biopsy studies confirmed the diagnosis of pulmonary alveolar proteinosis (Figure 2). Sequential whole-lung lavage recovered significant amounts of thick, proteinaceous effluent that slowly cleared. After the procedure, the patient’s symptoms, oxygen saturation, and chest radiographic appearance (Figure 3) improved markedly, with no recurrence at 1 year of follow-up.
ALVEOLAR PROTEINOSIS
Pulmonary alveolar proteinosis is a rare disease characterized by the accumulation of lipoproteinaceous material in the alveolar space secondary to alveolar macrophage dysfunction. The condition can be congenital, secondary, or acquired. Patients typically present with progressive exertional dyspnea, nonproductive cough, variable restrictive ventilatory defects, and diffusion limitation on pulmonary function testing.
Plain chest radiographs usually resemble those seen in pulmonary edema but without features of heart failure, ie, cardiomegaly, Kerley B lines, and effusion.
A “crazy-paving” pattern on computed tomography—a combination of geographic ground-glass appearance and interseptal thickening—suggests alveolar proteinosis, but is not specific for it. Other differential diagnoses for the crazy-paving pattern include Pneumocystis jirovecii infection, invasive mucinous adenocarcinoma, cardiogenic pulmonary edema, alveolar hemorrhage, sarcoidosis, cryptogenic organizing pneumonia, exogenous lipoid pneumonia, drug-induced lung disease, acute radiation pneumonitis, and nonspecific interstitial pneumonia.1
Laboratory testing is not very helpful in the diagnosis, although the serum lactate dehydrogenase level may be mildly elevated. Circulating antibodies to granulocyte macrophage colony-stimulating factor may support the diagnosis, but they are only present in the acquired form. Communication with a research laboratory is usually needed to test for these antibodies.
The bronchoalveolar lavage fluid typically has an opaque, milky, or muddy appearance. The diagnosis is confirmed by demonstration of alveolar filling with material that is periodic acid-Schiff-positive and that is amorphous, eosinophilic, and granular.
Whole-lung lavage2 is the physical removal of surfactant by repeated flooding of the lungs with warmed saline, done under general anesthesia with single-lung ventilation. It remains the standard of care and is indicated in patients with the confirmed diagnosis and one of the following: severe dyspnea, resting hypoxemia (Pao2 < 60 mm Hg at sea level), alveolar-arterial gradient > 40 mm Hg, or a shunt fraction of more than 10%. Successful bronchoscopic lavage has also been reported.3
Other treatments include granulocyte-macrophage colony-stimulating factor, rituximab (Rituxan, an anti-CD20 monoclonal antibody), plasmapheresis, and lung transplantation. Systemic corticosteroids are usually ineffective unless indicated for secondary types of alveolar proteinosis.
Inhalation rather than subcutaneous administration of granulocyte-macrophage colony-stimulating factor seems preferred as it ensures a high concentration in the target organ, avoids systemic complications (injection-site edema, erythema, neutropenia, malaise, and shortness of breath) and achieves lower levels of autoantibodies in bronchoalveolar lavage fluid, which correlates with disease activity.
Data are sparse as to the recurrence of autoimmune pulmonary alveolar proteinosis after whole-lung lavage, yet about 40% of patients require a repeat procedure within 18 months. Recurrence has also been reported after double-lung transplantation.4
Adjuvant therapy with rituximab or, to a lesser extent, with inhaled granulocyte-macrophage colony-stimulating factor has recently been shown to diminish the need for repeated lavage.5 These treatments can also be used when whole-lung lavage cannot be performed or proves to be ineffective.5
Acknowledgment: I would like to thank Dr. Kamelia Velikova for providing the pathology image.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.
- Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. ‘Crazy-paving’ pattern at thin-section CT of the lungs: radiologic-pathologic overview. Radiographics 2003; 23:1509–1519.
- Michaud G, Reddy C, Ernst A. Whole-lung lavage for pulmonary alveolar proteinosis. Chest 2009; 136:1678–1681.
- Cheng SL, Chang HT, Lau HP, Lee LN, Yang PC. Pulmonary alveolar proteinosis: treatment by bronchofiberscopic lobar lavage. Chest 2002; 122:1480–1485.
- Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest 1997; 111:1457–1458.
- Leth S, Bendstrup E, Vestergaard H, Hilberg O. Autoimmune pulmonary alveolar proteinosis: treatment options in year 2013. Respirology 2013; 18:82–91.
SUGGESTED READING
Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis.Eur Respir Rev 2011; 20:98–107.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolarproteinosis. Clin Immunol 2010; 135:223–235.
Ioachimescu OC, Kavuru MS. Pulmonary alveolar proteinosis.Chron Respir Dis 2006; 3:149–159.
Luisetti M, Kadija Z, Mariani F, Rodi G, Campo I, Trapnell BC.Therapy options in pulmonary alveolar proteinosis. TherAdv Respir Dis 2010; 4:239–248.
