User login
Intravascular large B-cell lymphoma: an elusive diagnosis with challenging management
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.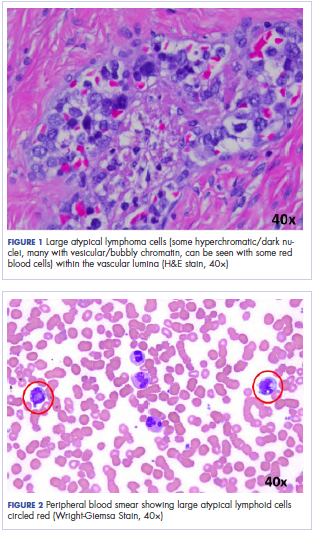
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.