User login
Out of Sight, Not Out of Mind
A 73-year-old man presented to clinic with 6 weeks of headache. He occasionally experienced generalized headaches throughout his life that resolved with naproxen. His new headache was characterized by a progressively worsening sensation of left-eye pressure with radiation to the left temple. Over the previous week, he had intermittent diplopia, left ptosis, and left lacrimation. He denied head trauma, fever, vision loss, photophobia, dysphagia, dysarthria, nausea, vomiting, or jaw claudication.
Primary headaches include tension type, migraine, and trigeminal autonomic cephalalgias (eg, cluster headache). A new headache in an older patient, particularly if protracted and progressive, prioritizes consideration of a secondary headache, which may reflect pathology within the brain parenchyma (eg, intracranial mass), blood vessels (eg, giant cell arteritis), meninges (eg, meningitis), or ventricles (eg, intraventricular cyst). Eye pain may arise from ocular and extraocular disease. Corneal abrasions, infectious keratitis, scleritis, uveitis, or acute angle-closure glaucoma are painful, although the latter is less likely given the prolonged duration of symptoms. Thyroid eye disease or other infiltrative disorders of the orbit can also cause eye discomfort.
Ptosis commonly results from degeneration of the levator aponeurosis. Other causes include third cranial nerve palsy and myasthenia gravis. Interruption of sympathetic innervation of the eyelid by lesions in the brain stem, spinal cord, lung (eg, Pancoast tumor), or cavernous sinus also can result in ptosis.
Whether the patient has monocular or binocular diplopia is uncertain. Monocular diplopia persists with only one eye open and can arise from uncorrected refractive error, corneal irregularities, lenticular opacities, or unilateral macular disease. Binocular diplopia develops from ocular misalignment due to neuromuscular weakness, extraocular muscle entrapment, or an orbital mass displacing the globe. An orbital mass would also explain the unilateral headache and unilateral ptosis.
His medical history included coronary artery disease, seronegative rheumatoid arthritis, osteoporosis, benign prostatic hypertrophy, and ureteral strictures from chronic nephrolithiasis. Following a cholecystectomy for gallstone pancreatitis 13 years earlier, he was hospitalized five more times for pancreatitis. The last episode was 6 years prior to this presentation. At that time, magnetic resonance cholangiopancreatography (MRCP) did not reveal pancreatic divisum, annular pancreas, biliary strictures, or a pancreatic mass. Esophagogastroduodenoscopy peformed during the same hospitalization showed mild gastritis. His recurrent pancreatitis was deemed idiopathic.
His medications were folic acid, cholecalciferol, lisinopril, metoprolol, omeprazole, simvastatin, aspirin, and weekly methotrexate. His sister had breast and ovarian cancer, and his brother had gastric cancer. He had two subcentimeter tubular adenomas removed during a screening colonoscopy 3 years prior. He had a 30 pack-year smoking history and quit 28 years earlier. He did not use alcohol or drugs. He was a retired chemical plant worker.
Choledocholithiasis (as discrete stones or biliary sludge) can trigger pancreatitis despite a cholecystectomy, but the recurrent episodes and negative MRCP should prompt consideration of other causes, such as alcohol. Hypercalcemia, hypertriglyceridemia, and medications are infrequent causes of pancreatic inflammation. IgG4-related disease (IgG4-RD) causes autoimmune pancreatitis and can infiltrate the eyelids, lacrimal glands, extraocular muscles, or orbital connective tissue. Malignancy of the pancreas or ampulla can trigger pancreatitis by causing pancreatic duct obstruction but would not go undetected for 13 years.
The patient was evaluated by an ophthalmologist and a neurologist. His heart rate was 52 beats per minute and blood pressure, 174/70 mm Hg; other vital signs were normal. He had conjunctival chemosis, ptosis, and nonpulsatile proptosis of the left eye with tenderness and increased resistance to retropulsion compared to the right eye (Figure 1). Visual acuity was 20/25 for the right eye and hand motions only in the left eye. The pupils were reactive and symmetric without afferent pupillary defect. There was no optic nerve swelling or pallor. Abduction, adduction, and elevation of the left eye were restricted and associated with diplopia. Movement of the right eye was unrestricted. There was no other facial asymmetry. Facial sensation was normal. Corneal reflexes were intact. Shoulder shrug strength was equal and symmetric. Tongue protrusion was midline. Olfaction and hearing were not assessed. Strength, sensation, and deep tendon reflexes were normal in all extremities. The plantar response was flexor bilaterally.

Unilateral ptosis, chemosis, proptosis, ophthalmoplegia, eye tenderness, and visual loss collectively point to a space-occupying orbital disease. Orbital masses are caused by cancers, infections such as mucormycosis (usually in an immunocompromised host), and inflammatory disorders such as thyroid orbitopathy, sarcoidosis, IgG4-related orbitopathy, granulomatosis with polyangiitis, and orbital pseudotumor (idiopathic inflammation of the orbit). Chemosis reflects edema of the conjunctiva, which can arise from direct conjunctival injury (eg, allergy, infection, or trauma), interruption of the venous drainage of the conjunctiva by vascular disorders (eg, cavernous sinus thrombosis or carotid-cavernous fistula), or space-occupying diseases of the orbit. Monocular visual loss arises from a prechiasmal lesion, and acute monocular visual loss is more commonly caused by posterior ocular pathology (eg, retina or optic nerve) than anterior disease (eg, keratitis). Visual loss in the presence of an orbital process suggests a compressive or infiltrative disease of the optic nerve.
Complete blood count, comprehensive metabolic panel, erythrocyte sedimentation rate, C-reactive protein, and thyroid function tests were normal. Interferon-gamma release assay, HIV antibody, rapid plasma reagin, Lyme antibody, antinuclear antibody, and antineutrophil cytoplasmic antibody (ANCA) tests were negative. A noncontrast computed tomography (CT) scan of the head revealed thickening of the left inferior rectus muscle. Orbital magnetic resonance imaging (MRI) with gadolinium and fluid-attenuated inversion recovery imaging demonstrated a T2 hyperintense, heterogeneous 1.4-cm mass in the left inferior rectus muscle (Figure 2). There was no carotid-cavernous fistula, brain mass, or meningeal enhancement.
 with gadolinium and fluid-attenuated inversion recovery imaging showed a hyperintense, heterogeneous 1.4×1.2×1.2-cm mass in the left inferior rectus muscle")
An isolated mass in one ocular muscle raises the probability of a cancer. The most common malignant orbital tumor is B-cell lymphoma. Metastatic cancer to the eye is rare; breast, prostate, and lung cancer account for the majority of cases. The family history of breast and ovarian cancer raises the possibility of a BRCA mutation, which is also associated with gastric, pancreatic, and prostate malignancies. Granulomatosis with polyangiitis may be ANCA negative in localized sino-orbital disease. Biopsy of the orbital mass is the next step.
The patient underwent transconjunctival orbitotomy with excision of the left inferior rectus mass. Two days later, he presented to the emergency department with acute onset epigastric pain, nausea, and vomiting. A comprehensive review of systems, which had not been performed until this visit, revealed an unintentional 20-lb weight loss over the previous 3 months. He had a progressive ache in the left anterior groin that was dull, tender, nonradiating, and worse with weight bearing. He denied melena or hematochezia.
His temperature was 37 °C; heart rate, 98 beats per minute; and blood pressure, 128/63 mm Hg. He had midepigastric tenderness and point tenderness over the anterior iliac spine. White blood cell count was 12,600/μL; hemo globin, 14.5 g/dL; and platelet count, 158,000/μL. Serum lipase was 7,108 U/L. Serum creatinine, calcium, and triglyceride levels were normal. Alkaline phosphatase was 117 U/L (normal, 34-104 U/L); total bilirubin, 1.1 mg/dL; alanine aminotransferase (ALT), 119 U/L (normal, 7-52 U/L); and aspartate aminotransferase (AST), 236 U/L (normal, 13-39 U/L). Troponin I was undetectable, and an electrocardiogram demonstrated sinus tachycardia. Urinalysis was normal.
Concomitant pancreatitis and hepatitis with an elevated AST-to-ALT ratio should prompt evaluation of recurrent choledocholithiasis and a repeat inquiry about alcohol use. His medications should be reviewed for an association with pancreatitis. Anterior groin discomfort usually reflects osteoarthritis of the hip joint, inguinal hernia, or inguinal lymphadenopathy. Groin pain may be referred from spinal nerve root compression, aortoiliac occlusion, or nephrolithiasis. Weight loss in the presence of an inferior rectus mass suggests one of the aforementioned systemic diseases with orbital manifestations. Pancreatitis and groin discomfort may be important clues, but the chronicity of the recurrent pancreatitis and the high prevalence of hip osteoarthritis make it equally likely that they are unrelated to the eye disease.
CT scan of the abdomen and pelvis with contrast showed peripancreatic edema with fat stranding but no pancreatic or hepatobiliary mass. The common bile duct was normal. A 2.2×1.3-cm mass in the right posterior subphrenic space, a lytic lesion in the left anterior inferior iliac spine, and right nonobstructive nephrolithiasis were identified. CT scan of the chest with contrast showed multiple subpleural nodules and innumerable parenchymal nodules. Subcentimeter hilar, mediastinal, and prevascular lymphadenopathy were present, as well as multiple sclerotic lesions in the right fourth and sixth ribs. Prostate-specific antigen was 0.7 ng/mL (normal, ≤ 4.0 ng/mL). Cancer antigen 19-9 level was 5.5 U/mL (normal, < 37.0 U/mL), and carcinoembryonic antigen (CEA) was 100.1 ng/mL (normal, 0-3 U/mL).
Widespread pulmonary nodules, diffuse lymphadenopathy, and bony lesions raise concern for a metastatic malignancy. There is no evidence of a primary carcinoma. The lack of hepatic involvement reduces the likelihood of a gastrointestinal tumor, although a rectal cancer, which may drain directly into the inferior vena cava and bypass the portal circulation, could present as lung metastases on CT imaging. Lymphoma is plausible given the diffuse lymphadenopathy and orbital mass. Sarcoidosis and histiocytic disorders (eg, Langerhans cell histiocytosis) also cause orbital disease, pulmonary nodules, lymphadenopathy, and bone lesions, although a subphrenic mass would be atypical for both disorders; furthermore, the majority of patients with adult Langerhans cell histiocytosis smoke cigarettes. The elevated CEA makes a metastatic solid tumor more likely than lymphoma but does not specify the location of the primary tumor.
Pathology of the inferior rectus muscle mass showed well-differentiated adenocarcinoma (Figure 3A and 3B). A CT-guided biopsy of the left anterior inferior iliac spine revealed well-differentiated adenocarcinoma (Figure 3C). Adenocarcinoma of unknown primary wasdiagnosed.
Subsequent immunohistochemical (IHC) staining was positive for cytokeratin 7 (CK7) and mucicarmine (Figure 3D and 3E) and negative for cytokeratin 20 (CK20) and thyroid transcription factor 1 (TTF1). This IHC profile suggested pancreatic or upper gastrointestinal tract lineage. Positron emission tomography–CT (PET-CT) scan was aborted because of dyspnea and chest pressure following contrast administration. He declined further imaging or endoscopy. He received palliative radiation and three cycles of paclitaxel and gemcitabine for cancer of unknown primary (CUP). Two months later, he developed bilateral upper-arm weakness due to C7 and T2 cord compression from vertebral and epidural metastases; his symptoms progressed despite salvage chemotherapy. He was transitioned to comfort care and died at home 9 months after diagnosis.
 with gadolinium and fluid-attenuated inversion recovery imaging showed a hyperintense, heterogeneous 1.4×1.2×1.2-cm mass in the left inferior rectus muscle")
DISCUSSION
This patient’s new headache and ocular abnormalities led to the discovery of an inferior rectus muscle mass. Initially unrecognized unintentional weight loss and hip pain recast a localized orbital syndrome as a systemic disease with pancreatic, ocular, pulmonary, lymph node, and skeletal pathology. Biopsies of the orbital rectus muscle and iliac bone demonstrated metastatic adenocarcinoma. Imaging studies did not identify a primary cancer, but IHC analysis suggested carcinoma of upper gastrointestinal or pancreatic origin.
Acute and chronic pancreatitis are both associated with pancreatic cancer.1 Chronic pancreatitis is associated with an increasing cumulative risk of pancreatic cancer; a potential mechanism is chronic inflammation with malignant transformation.2,3 There is also a 20-fold increased risk of pancreatic cancer in the first 2 years following an episode of acute pancreatitis,4 which may develop from malignant pancreatic duct obstruction. Although the post–acute pancreatitis risk of pancreatic cancer attenuates over time, a two-fold increased risk of pancreatic cancer remains after 10 years,4 which suggests that acute pancreatitis (particularly when idiopathic) either contributes to or shares pathogenesis with pancreatic adenocarcinoma. In elderly patients without gallstones or alcohol use, an abdominal CT scan or MRI shortly after resolution of the acute pancreatitis may be considered to assess for an underlying pancreatic tumor.5
CUP is a histologically defined malignancy without a known primary anatomic site despite an extensive evaluation. CUP accounts for up to 10% of all cancer diagnoses.6 CUP is ascribed to a primary cancer that remains too small to be detected or spontaneous regression of the primary cancer.7 Approximately 70% of autopsies of patients with CUP identify the primary tumor, which most commonly originates in the lung, gastrointestinal tract, breast, or pancreas.8
When a metastatic focus of cancer is found but the initial diagnostic evaluation (including CT scan of the chest, abdomen, and pelvis) fails to locate a primary cancer, the next step in searching for the tissue of origin is an IHC analysis of the tumor specimen. IHC analysis is a multistep staining process that can identify major categories of cancer, including carcinoma (adenocarcinoma, squamous cell carcinoma, and neuroendocrine carcinoma) and poorly or undifferentiated neoplasms (including carcinoma, lymphoma, sarcoma, or melanoma). Eighty-five percent of CUP cases are adenocarcinoma, 10% are squamous cell carcinoma, and the remaining 5% are undifferentiated neoplasms.9
There are no consensus guidelines for imaging in patients with CUP who have already undergone a CT scan of the chest, abdomen, and pelvis. Mammography is indicated in women with metastatic adenocarcinoma or axillary lymphadenopathy.7 MRI of the breast is obtained when mammography is nondiagnostic and the suspicion for breast cancer is high. Small clinical studies and meta-analyses support the use of PET-CT scans,7 although one study found that a PET-CT scan was not superior to CT imaging in identifying the primary tumor site in CUP.10 Endoscopy of the upper airway or gastrointestinal tract is rarely diagnostic in the absence of referable symptoms or a suggestive IHC profile (eg, CK7−, CK20+ suggestive of colon cancer).6
Molecular cancer classification has emerged as a useful diagnostic technique in CUP. Cancer cells retain gene expression patterns based on cellular origin, and a tumor’s profile can be compared with a reference database of known cancers, aiding in the identification of the primary tumor type. Molecular cancer classifier assays that use gene expression profiling can accurately determine a primary site11 and have been shown to be concordant with IHC testing.12 Molecular cancer classification is distinct from genetic assays that identify mutations for which there are approved therapies. Serum tumor markers are generally not useful in establishing the primary tumor and should be considered based on the clinical presentation (eg, prostate-specific antigen testing in a man with adenocarcinoma of unknown primary and osteoblastic metastases).
CUP is classified as favorable or unfavorable based on the IHC, pattern of spread, and serum markers in certain cases.6 Approximately 20% of CUP patients can be categorized into favorable subsets, such as adenocarcinoma in a single axillary lymph node in a female patient suggestive of a breast primary cancer, or squamous cell carcinoma in a cervical lymph node suggestive of a head or neck primary cancer.7 The remaining 80% of cases are categorized as unfavorable CUP and often have multiple metastases. Our patient’s pattern of spread and limited response to chemotherapy is characteristic of the unfavorable subset of CUP. The median survival of this group is 9 months, and only 25% of patients survive longer than 1 year.13
Biomarker-driven treatment of specific molecular targets independent of the tissue of origin (tissue-agnostic therapy) has shown promising results in the treatment of skin, lung, thyroid, colorectal, and gastric cancers.14 Pembrolizumab was the first drug approved by the US Food and Drug Administration based on a tumor’s biomarker without regard to its primary location. Data to support this approach for treating CUP are evolving and offer hope for patients with specific molecular targets.
Following the focused neuro-ophthalmologic evaluations, with focused examination and imaging, the hospitalist’s review of systems at the time of the final admission for pancreatitis set in motion an evaluation that led to a diagnosis of metastatic cancer. The risk factor of recurrent pancreatitis and IHC results suggested that pancreatic adenocarcinoma was the most likely primary tumor. As the focus of cancer treatment shifts away from the tissue of origin and toward molecular and genetic profiles, the search for the primary site may decrease in importance. In the future, even when we do not know the cancer’s origin, we may still know precisely what to do. But for now, as in this patient, our treatments continue to be based on a tumor that is out of sight, but not out of mind.
KEY TEACHING POINTS
- Acute and chronic pancreatitis are associated with an increased risk of pancreatic adenocarcinoma.
- CUP is a cancer in which diagnostic testing does not identify a primary tumor site. Immunohistochemistry and molecular analysis, imaging, and endoscopy are utilized selectively to identify a primary tumor type.
- Treatment of CUP currently depends on the suspected tissue of origin and pattern of spread.
- Tissue-agnostic therapy could allow for treatment for CUP patients independent of the tissue of origin.
Acknowledgments
We thank Andrew Mick, OD, for his review of an earlier version of this manuscript and Peter Phillips, MD, for his interpretation of the pathologic images.
1. Sadr-Azodi O, Oskarsson V, Discacciati A, Videhult P, Askling J, Ekbom A. Pancreatic cancer following acute pancreatitis: a population-based matched cohort study. Am J Gastroenterol. 2018;113(111):1711-1719. https://doi.org/10.1038/s41395-018-0255-9
2. Duell EJ, Lucenteforte E, Olson SH, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23(11):2964-2970. https://doi.org/10.1093/annonc/mds140
3. Ekbom A, McLaughlin JK, Nyren O. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;329(20):1502-1503. https://doi.org/10.1056/NEJM199311113292016
4. Kirkegard J, Cronin-Fenton D, Heide-Jorgensen U, Mortensen FV. Acute pancreatitis and pancreatic cancer risk: a nationwide matched-cohort study in Denmark. Gastroenterology. 2018;154(156):1729-1736. https://doi.org/10.1053/j.gastro.2018.02.011
5. Frampas E, Morla O, Regenet N, Eugene T, Dupas B, Meurette G. A solid pancreatic mass: tumour or inflammation? Diagn Interv Imaging. 2013;94(7-8):741-755. https://doi.org/10.1016/j.diii.2013.03.013
6. Varadhachary GR, Raber MN. Cancer of unknown primary site. N Engl J Med. 2014;371(8):757-765. https://doi.org/10.1056/NEJMra1303917
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017. 2018;35(3):199-206. https://doi.org//10.1053/j.semdp.2017.11.013
8. Pentheroudakis G, Golfinopoulos V, Pavlidis N. Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer. 2007;43(14):2026-2036. https://doi.org/10.1016/j.ejca.2007.06.023
9. Pavlidis N, Fizazi K. Carcinoma of unknown primary (CUP). Crit Rev Oncol Hematol. 2009;69(3):271-278. https://doi.org/10.1016/j.critrevonc.2008.09.005
10. Moller AK, Loft A, Berthelsen AK, et al. A prospective comparison of 18F-FDG PET/CT and CT as diagnostic tools to identify the primary tumor site in patients with extracervical carcinoma of unknown primary site. Oncologist. 2012;17(9):1146-1154. https://doi.org/10.1634/theoncologist.2011-0449
11. Economopoulou P, Mountzios G, Pavlidis N, Pentheroudakis G. Cancer of unknown primary origin in the genomic era: elucidating the dark box of cancer. Cancer Treat Rev. 2015;41(7):598-604. https://doi.org/10.1016/j.ctrv.2015.05.010
12. Greco FA. Molecular diagnosis of the tissue of origin in cancer of unknown primary site: useful in patient management. Curr Treat Options Oncol. 2013;14(4):634-642. https://doi.org/10.1007/s11864-013-0257-1
13. Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin—diagnosis and treatment. Nat Rev Clin Oncol. 2011;8(12):701-710. https://doi.org/10.1038/nrclinonc.2011.158
14. Luoh SW, Flaherty KT. When tissue is no longer the issue: tissue-agnostic cancer therapy comes of age. Ann Intern Med. 2018;169(4):233-239. https://doi.org/10.7326/M17-2832
A 73-year-old man presented to clinic with 6 weeks of headache. He occasionally experienced generalized headaches throughout his life that resolved with naproxen. His new headache was characterized by a progressively worsening sensation of left-eye pressure with radiation to the left temple. Over the previous week, he had intermittent diplopia, left ptosis, and left lacrimation. He denied head trauma, fever, vision loss, photophobia, dysphagia, dysarthria, nausea, vomiting, or jaw claudication.
Primary headaches include tension type, migraine, and trigeminal autonomic cephalalgias (eg, cluster headache). A new headache in an older patient, particularly if protracted and progressive, prioritizes consideration of a secondary headache, which may reflect pathology within the brain parenchyma (eg, intracranial mass), blood vessels (eg, giant cell arteritis), meninges (eg, meningitis), or ventricles (eg, intraventricular cyst). Eye pain may arise from ocular and extraocular disease. Corneal abrasions, infectious keratitis, scleritis, uveitis, or acute angle-closure glaucoma are painful, although the latter is less likely given the prolonged duration of symptoms. Thyroid eye disease or other infiltrative disorders of the orbit can also cause eye discomfort.
Ptosis commonly results from degeneration of the levator aponeurosis. Other causes include third cranial nerve palsy and myasthenia gravis. Interruption of sympathetic innervation of the eyelid by lesions in the brain stem, spinal cord, lung (eg, Pancoast tumor), or cavernous sinus also can result in ptosis.
Whether the patient has monocular or binocular diplopia is uncertain. Monocular diplopia persists with only one eye open and can arise from uncorrected refractive error, corneal irregularities, lenticular opacities, or unilateral macular disease. Binocular diplopia develops from ocular misalignment due to neuromuscular weakness, extraocular muscle entrapment, or an orbital mass displacing the globe. An orbital mass would also explain the unilateral headache and unilateral ptosis.
His medical history included coronary artery disease, seronegative rheumatoid arthritis, osteoporosis, benign prostatic hypertrophy, and ureteral strictures from chronic nephrolithiasis. Following a cholecystectomy for gallstone pancreatitis 13 years earlier, he was hospitalized five more times for pancreatitis. The last episode was 6 years prior to this presentation. At that time, magnetic resonance cholangiopancreatography (MRCP) did not reveal pancreatic divisum, annular pancreas, biliary strictures, or a pancreatic mass. Esophagogastroduodenoscopy peformed during the same hospitalization showed mild gastritis. His recurrent pancreatitis was deemed idiopathic.
His medications were folic acid, cholecalciferol, lisinopril, metoprolol, omeprazole, simvastatin, aspirin, and weekly methotrexate. His sister had breast and ovarian cancer, and his brother had gastric cancer. He had two subcentimeter tubular adenomas removed during a screening colonoscopy 3 years prior. He had a 30 pack-year smoking history and quit 28 years earlier. He did not use alcohol or drugs. He was a retired chemical plant worker.
Choledocholithiasis (as discrete stones or biliary sludge) can trigger pancreatitis despite a cholecystectomy, but the recurrent episodes and negative MRCP should prompt consideration of other causes, such as alcohol. Hypercalcemia, hypertriglyceridemia, and medications are infrequent causes of pancreatic inflammation. IgG4-related disease (IgG4-RD) causes autoimmune pancreatitis and can infiltrate the eyelids, lacrimal glands, extraocular muscles, or orbital connective tissue. Malignancy of the pancreas or ampulla can trigger pancreatitis by causing pancreatic duct obstruction but would not go undetected for 13 years.
The patient was evaluated by an ophthalmologist and a neurologist. His heart rate was 52 beats per minute and blood pressure, 174/70 mm Hg; other vital signs were normal. He had conjunctival chemosis, ptosis, and nonpulsatile proptosis of the left eye with tenderness and increased resistance to retropulsion compared to the right eye (Figure 1). Visual acuity was 20/25 for the right eye and hand motions only in the left eye. The pupils were reactive and symmetric without afferent pupillary defect. There was no optic nerve swelling or pallor. Abduction, adduction, and elevation of the left eye were restricted and associated with diplopia. Movement of the right eye was unrestricted. There was no other facial asymmetry. Facial sensation was normal. Corneal reflexes were intact. Shoulder shrug strength was equal and symmetric. Tongue protrusion was midline. Olfaction and hearing were not assessed. Strength, sensation, and deep tendon reflexes were normal in all extremities. The plantar response was flexor bilaterally.
Unilateral ptosis, chemosis, proptosis, ophthalmoplegia, eye tenderness, and visual loss collectively point to a space-occupying orbital disease. Orbital masses are caused by cancers, infections such as mucormycosis (usually in an immunocompromised host), and inflammatory disorders such as thyroid orbitopathy, sarcoidosis, IgG4-related orbitopathy, granulomatosis with polyangiitis, and orbital pseudotumor (idiopathic inflammation of the orbit). Chemosis reflects edema of the conjunctiva, which can arise from direct conjunctival injury (eg, allergy, infection, or trauma), interruption of the venous drainage of the conjunctiva by vascular disorders (eg, cavernous sinus thrombosis or carotid-cavernous fistula), or space-occupying diseases of the orbit. Monocular visual loss arises from a prechiasmal lesion, and acute monocular visual loss is more commonly caused by posterior ocular pathology (eg, retina or optic nerve) than anterior disease (eg, keratitis). Visual loss in the presence of an orbital process suggests a compressive or infiltrative disease of the optic nerve.
Complete blood count, comprehensive metabolic panel, erythrocyte sedimentation rate, C-reactive protein, and thyroid function tests were normal. Interferon-gamma release assay, HIV antibody, rapid plasma reagin, Lyme antibody, antinuclear antibody, and antineutrophil cytoplasmic antibody (ANCA) tests were negative. A noncontrast computed tomography (CT) scan of the head revealed thickening of the left inferior rectus muscle. Orbital magnetic resonance imaging (MRI) with gadolinium and fluid-attenuated inversion recovery imaging demonstrated a T2 hyperintense, heterogeneous 1.4-cm mass in the left inferior rectus muscle (Figure 2). There was no carotid-cavernous fistula, brain mass, or meningeal enhancement.
An isolated mass in one ocular muscle raises the probability of a cancer. The most common malignant orbital tumor is B-cell lymphoma. Metastatic cancer to the eye is rare; breast, prostate, and lung cancer account for the majority of cases. The family history of breast and ovarian cancer raises the possibility of a BRCA mutation, which is also associated with gastric, pancreatic, and prostate malignancies. Granulomatosis with polyangiitis may be ANCA negative in localized sino-orbital disease. Biopsy of the orbital mass is the next step.
The patient underwent transconjunctival orbitotomy with excision of the left inferior rectus mass. Two days later, he presented to the emergency department with acute onset epigastric pain, nausea, and vomiting. A comprehensive review of systems, which had not been performed until this visit, revealed an unintentional 20-lb weight loss over the previous 3 months. He had a progressive ache in the left anterior groin that was dull, tender, nonradiating, and worse with weight bearing. He denied melena or hematochezia.
His temperature was 37 °C; heart rate, 98 beats per minute; and blood pressure, 128/63 mm Hg. He had midepigastric tenderness and point tenderness over the anterior iliac spine. White blood cell count was 12,600/μL; hemo globin, 14.5 g/dL; and platelet count, 158,000/μL. Serum lipase was 7,108 U/L. Serum creatinine, calcium, and triglyceride levels were normal. Alkaline phosphatase was 117 U/L (normal, 34-104 U/L); total bilirubin, 1.1 mg/dL; alanine aminotransferase (ALT), 119 U/L (normal, 7-52 U/L); and aspartate aminotransferase (AST), 236 U/L (normal, 13-39 U/L). Troponin I was undetectable, and an electrocardiogram demonstrated sinus tachycardia. Urinalysis was normal.
Concomitant pancreatitis and hepatitis with an elevated AST-to-ALT ratio should prompt evaluation of recurrent choledocholithiasis and a repeat inquiry about alcohol use. His medications should be reviewed for an association with pancreatitis. Anterior groin discomfort usually reflects osteoarthritis of the hip joint, inguinal hernia, or inguinal lymphadenopathy. Groin pain may be referred from spinal nerve root compression, aortoiliac occlusion, or nephrolithiasis. Weight loss in the presence of an inferior rectus mass suggests one of the aforementioned systemic diseases with orbital manifestations. Pancreatitis and groin discomfort may be important clues, but the chronicity of the recurrent pancreatitis and the high prevalence of hip osteoarthritis make it equally likely that they are unrelated to the eye disease.
CT scan of the abdomen and pelvis with contrast showed peripancreatic edema with fat stranding but no pancreatic or hepatobiliary mass. The common bile duct was normal. A 2.2×1.3-cm mass in the right posterior subphrenic space, a lytic lesion in the left anterior inferior iliac spine, and right nonobstructive nephrolithiasis were identified. CT scan of the chest with contrast showed multiple subpleural nodules and innumerable parenchymal nodules. Subcentimeter hilar, mediastinal, and prevascular lymphadenopathy were present, as well as multiple sclerotic lesions in the right fourth and sixth ribs. Prostate-specific antigen was 0.7 ng/mL (normal, ≤ 4.0 ng/mL). Cancer antigen 19-9 level was 5.5 U/mL (normal, < 37.0 U/mL), and carcinoembryonic antigen (CEA) was 100.1 ng/mL (normal, 0-3 U/mL).
Widespread pulmonary nodules, diffuse lymphadenopathy, and bony lesions raise concern for a metastatic malignancy. There is no evidence of a primary carcinoma. The lack of hepatic involvement reduces the likelihood of a gastrointestinal tumor, although a rectal cancer, which may drain directly into the inferior vena cava and bypass the portal circulation, could present as lung metastases on CT imaging. Lymphoma is plausible given the diffuse lymphadenopathy and orbital mass. Sarcoidosis and histiocytic disorders (eg, Langerhans cell histiocytosis) also cause orbital disease, pulmonary nodules, lymphadenopathy, and bone lesions, although a subphrenic mass would be atypical for both disorders; furthermore, the majority of patients with adult Langerhans cell histiocytosis smoke cigarettes. The elevated CEA makes a metastatic solid tumor more likely than lymphoma but does not specify the location of the primary tumor.
Pathology of the inferior rectus muscle mass showed well-differentiated adenocarcinoma (Figure 3A and 3B). A CT-guided biopsy of the left anterior inferior iliac spine revealed well-differentiated adenocarcinoma (Figure 3C). Adenocarcinoma of unknown primary wasdiagnosed.
Subsequent immunohistochemical (IHC) staining was positive for cytokeratin 7 (CK7) and mucicarmine (Figure 3D and 3E) and negative for cytokeratin 20 (CK20) and thyroid transcription factor 1 (TTF1). This IHC profile suggested pancreatic or upper gastrointestinal tract lineage. Positron emission tomography–CT (PET-CT) scan was aborted because of dyspnea and chest pressure following contrast administration. He declined further imaging or endoscopy. He received palliative radiation and three cycles of paclitaxel and gemcitabine for cancer of unknown primary (CUP). Two months later, he developed bilateral upper-arm weakness due to C7 and T2 cord compression from vertebral and epidural metastases; his symptoms progressed despite salvage chemotherapy. He was transitioned to comfort care and died at home 9 months after diagnosis.
DISCUSSION
This patient’s new headache and ocular abnormalities led to the discovery of an inferior rectus muscle mass. Initially unrecognized unintentional weight loss and hip pain recast a localized orbital syndrome as a systemic disease with pancreatic, ocular, pulmonary, lymph node, and skeletal pathology. Biopsies of the orbital rectus muscle and iliac bone demonstrated metastatic adenocarcinoma. Imaging studies did not identify a primary cancer, but IHC analysis suggested carcinoma of upper gastrointestinal or pancreatic origin.
Acute and chronic pancreatitis are both associated with pancreatic cancer.1 Chronic pancreatitis is associated with an increasing cumulative risk of pancreatic cancer; a potential mechanism is chronic inflammation with malignant transformation.2,3 There is also a 20-fold increased risk of pancreatic cancer in the first 2 years following an episode of acute pancreatitis,4 which may develop from malignant pancreatic duct obstruction. Although the post–acute pancreatitis risk of pancreatic cancer attenuates over time, a two-fold increased risk of pancreatic cancer remains after 10 years,4 which suggests that acute pancreatitis (particularly when idiopathic) either contributes to or shares pathogenesis with pancreatic adenocarcinoma. In elderly patients without gallstones or alcohol use, an abdominal CT scan or MRI shortly after resolution of the acute pancreatitis may be considered to assess for an underlying pancreatic tumor.5
CUP is a histologically defined malignancy without a known primary anatomic site despite an extensive evaluation. CUP accounts for up to 10% of all cancer diagnoses.6 CUP is ascribed to a primary cancer that remains too small to be detected or spontaneous regression of the primary cancer.7 Approximately 70% of autopsies of patients with CUP identify the primary tumor, which most commonly originates in the lung, gastrointestinal tract, breast, or pancreas.8
When a metastatic focus of cancer is found but the initial diagnostic evaluation (including CT scan of the chest, abdomen, and pelvis) fails to locate a primary cancer, the next step in searching for the tissue of origin is an IHC analysis of the tumor specimen. IHC analysis is a multistep staining process that can identify major categories of cancer, including carcinoma (adenocarcinoma, squamous cell carcinoma, and neuroendocrine carcinoma) and poorly or undifferentiated neoplasms (including carcinoma, lymphoma, sarcoma, or melanoma). Eighty-five percent of CUP cases are adenocarcinoma, 10% are squamous cell carcinoma, and the remaining 5% are undifferentiated neoplasms.9
There are no consensus guidelines for imaging in patients with CUP who have already undergone a CT scan of the chest, abdomen, and pelvis. Mammography is indicated in women with metastatic adenocarcinoma or axillary lymphadenopathy.7 MRI of the breast is obtained when mammography is nondiagnostic and the suspicion for breast cancer is high. Small clinical studies and meta-analyses support the use of PET-CT scans,7 although one study found that a PET-CT scan was not superior to CT imaging in identifying the primary tumor site in CUP.10 Endoscopy of the upper airway or gastrointestinal tract is rarely diagnostic in the absence of referable symptoms or a suggestive IHC profile (eg, CK7−, CK20+ suggestive of colon cancer).6
Molecular cancer classification has emerged as a useful diagnostic technique in CUP. Cancer cells retain gene expression patterns based on cellular origin, and a tumor’s profile can be compared with a reference database of known cancers, aiding in the identification of the primary tumor type. Molecular cancer classifier assays that use gene expression profiling can accurately determine a primary site11 and have been shown to be concordant with IHC testing.12 Molecular cancer classification is distinct from genetic assays that identify mutations for which there are approved therapies. Serum tumor markers are generally not useful in establishing the primary tumor and should be considered based on the clinical presentation (eg, prostate-specific antigen testing in a man with adenocarcinoma of unknown primary and osteoblastic metastases).
CUP is classified as favorable or unfavorable based on the IHC, pattern of spread, and serum markers in certain cases.6 Approximately 20% of CUP patients can be categorized into favorable subsets, such as adenocarcinoma in a single axillary lymph node in a female patient suggestive of a breast primary cancer, or squamous cell carcinoma in a cervical lymph node suggestive of a head or neck primary cancer.7 The remaining 80% of cases are categorized as unfavorable CUP and often have multiple metastases. Our patient’s pattern of spread and limited response to chemotherapy is characteristic of the unfavorable subset of CUP. The median survival of this group is 9 months, and only 25% of patients survive longer than 1 year.13
Biomarker-driven treatment of specific molecular targets independent of the tissue of origin (tissue-agnostic therapy) has shown promising results in the treatment of skin, lung, thyroid, colorectal, and gastric cancers.14 Pembrolizumab was the first drug approved by the US Food and Drug Administration based on a tumor’s biomarker without regard to its primary location. Data to support this approach for treating CUP are evolving and offer hope for patients with specific molecular targets.
Following the focused neuro-ophthalmologic evaluations, with focused examination and imaging, the hospitalist’s review of systems at the time of the final admission for pancreatitis set in motion an evaluation that led to a diagnosis of metastatic cancer. The risk factor of recurrent pancreatitis and IHC results suggested that pancreatic adenocarcinoma was the most likely primary tumor. As the focus of cancer treatment shifts away from the tissue of origin and toward molecular and genetic profiles, the search for the primary site may decrease in importance. In the future, even when we do not know the cancer’s origin, we may still know precisely what to do. But for now, as in this patient, our treatments continue to be based on a tumor that is out of sight, but not out of mind.
KEY TEACHING POINTS
- Acute and chronic pancreatitis are associated with an increased risk of pancreatic adenocarcinoma.
- CUP is a cancer in which diagnostic testing does not identify a primary tumor site. Immunohistochemistry and molecular analysis, imaging, and endoscopy are utilized selectively to identify a primary tumor type.
- Treatment of CUP currently depends on the suspected tissue of origin and pattern of spread.
- Tissue-agnostic therapy could allow for treatment for CUP patients independent of the tissue of origin.
Acknowledgments
We thank Andrew Mick, OD, for his review of an earlier version of this manuscript and Peter Phillips, MD, for his interpretation of the pathologic images.
A 73-year-old man presented to clinic with 6 weeks of headache. He occasionally experienced generalized headaches throughout his life that resolved with naproxen. His new headache was characterized by a progressively worsening sensation of left-eye pressure with radiation to the left temple. Over the previous week, he had intermittent diplopia, left ptosis, and left lacrimation. He denied head trauma, fever, vision loss, photophobia, dysphagia, dysarthria, nausea, vomiting, or jaw claudication.
Primary headaches include tension type, migraine, and trigeminal autonomic cephalalgias (eg, cluster headache). A new headache in an older patient, particularly if protracted and progressive, prioritizes consideration of a secondary headache, which may reflect pathology within the brain parenchyma (eg, intracranial mass), blood vessels (eg, giant cell arteritis), meninges (eg, meningitis), or ventricles (eg, intraventricular cyst). Eye pain may arise from ocular and extraocular disease. Corneal abrasions, infectious keratitis, scleritis, uveitis, or acute angle-closure glaucoma are painful, although the latter is less likely given the prolonged duration of symptoms. Thyroid eye disease or other infiltrative disorders of the orbit can also cause eye discomfort.
Ptosis commonly results from degeneration of the levator aponeurosis. Other causes include third cranial nerve palsy and myasthenia gravis. Interruption of sympathetic innervation of the eyelid by lesions in the brain stem, spinal cord, lung (eg, Pancoast tumor), or cavernous sinus also can result in ptosis.
Whether the patient has monocular or binocular diplopia is uncertain. Monocular diplopia persists with only one eye open and can arise from uncorrected refractive error, corneal irregularities, lenticular opacities, or unilateral macular disease. Binocular diplopia develops from ocular misalignment due to neuromuscular weakness, extraocular muscle entrapment, or an orbital mass displacing the globe. An orbital mass would also explain the unilateral headache and unilateral ptosis.
His medical history included coronary artery disease, seronegative rheumatoid arthritis, osteoporosis, benign prostatic hypertrophy, and ureteral strictures from chronic nephrolithiasis. Following a cholecystectomy for gallstone pancreatitis 13 years earlier, he was hospitalized five more times for pancreatitis. The last episode was 6 years prior to this presentation. At that time, magnetic resonance cholangiopancreatography (MRCP) did not reveal pancreatic divisum, annular pancreas, biliary strictures, or a pancreatic mass. Esophagogastroduodenoscopy peformed during the same hospitalization showed mild gastritis. His recurrent pancreatitis was deemed idiopathic.
His medications were folic acid, cholecalciferol, lisinopril, metoprolol, omeprazole, simvastatin, aspirin, and weekly methotrexate. His sister had breast and ovarian cancer, and his brother had gastric cancer. He had two subcentimeter tubular adenomas removed during a screening colonoscopy 3 years prior. He had a 30 pack-year smoking history and quit 28 years earlier. He did not use alcohol or drugs. He was a retired chemical plant worker.
Choledocholithiasis (as discrete stones or biliary sludge) can trigger pancreatitis despite a cholecystectomy, but the recurrent episodes and negative MRCP should prompt consideration of other causes, such as alcohol. Hypercalcemia, hypertriglyceridemia, and medications are infrequent causes of pancreatic inflammation. IgG4-related disease (IgG4-RD) causes autoimmune pancreatitis and can infiltrate the eyelids, lacrimal glands, extraocular muscles, or orbital connective tissue. Malignancy of the pancreas or ampulla can trigger pancreatitis by causing pancreatic duct obstruction but would not go undetected for 13 years.
The patient was evaluated by an ophthalmologist and a neurologist. His heart rate was 52 beats per minute and blood pressure, 174/70 mm Hg; other vital signs were normal. He had conjunctival chemosis, ptosis, and nonpulsatile proptosis of the left eye with tenderness and increased resistance to retropulsion compared to the right eye (Figure 1). Visual acuity was 20/25 for the right eye and hand motions only in the left eye. The pupils were reactive and symmetric without afferent pupillary defect. There was no optic nerve swelling or pallor. Abduction, adduction, and elevation of the left eye were restricted and associated with diplopia. Movement of the right eye was unrestricted. There was no other facial asymmetry. Facial sensation was normal. Corneal reflexes were intact. Shoulder shrug strength was equal and symmetric. Tongue protrusion was midline. Olfaction and hearing were not assessed. Strength, sensation, and deep tendon reflexes were normal in all extremities. The plantar response was flexor bilaterally.
Unilateral ptosis, chemosis, proptosis, ophthalmoplegia, eye tenderness, and visual loss collectively point to a space-occupying orbital disease. Orbital masses are caused by cancers, infections such as mucormycosis (usually in an immunocompromised host), and inflammatory disorders such as thyroid orbitopathy, sarcoidosis, IgG4-related orbitopathy, granulomatosis with polyangiitis, and orbital pseudotumor (idiopathic inflammation of the orbit). Chemosis reflects edema of the conjunctiva, which can arise from direct conjunctival injury (eg, allergy, infection, or trauma), interruption of the venous drainage of the conjunctiva by vascular disorders (eg, cavernous sinus thrombosis or carotid-cavernous fistula), or space-occupying diseases of the orbit. Monocular visual loss arises from a prechiasmal lesion, and acute monocular visual loss is more commonly caused by posterior ocular pathology (eg, retina or optic nerve) than anterior disease (eg, keratitis). Visual loss in the presence of an orbital process suggests a compressive or infiltrative disease of the optic nerve.
Complete blood count, comprehensive metabolic panel, erythrocyte sedimentation rate, C-reactive protein, and thyroid function tests were normal. Interferon-gamma release assay, HIV antibody, rapid plasma reagin, Lyme antibody, antinuclear antibody, and antineutrophil cytoplasmic antibody (ANCA) tests were negative. A noncontrast computed tomography (CT) scan of the head revealed thickening of the left inferior rectus muscle. Orbital magnetic resonance imaging (MRI) with gadolinium and fluid-attenuated inversion recovery imaging demonstrated a T2 hyperintense, heterogeneous 1.4-cm mass in the left inferior rectus muscle (Figure 2). There was no carotid-cavernous fistula, brain mass, or meningeal enhancement.
An isolated mass in one ocular muscle raises the probability of a cancer. The most common malignant orbital tumor is B-cell lymphoma. Metastatic cancer to the eye is rare; breast, prostate, and lung cancer account for the majority of cases. The family history of breast and ovarian cancer raises the possibility of a BRCA mutation, which is also associated with gastric, pancreatic, and prostate malignancies. Granulomatosis with polyangiitis may be ANCA negative in localized sino-orbital disease. Biopsy of the orbital mass is the next step.
The patient underwent transconjunctival orbitotomy with excision of the left inferior rectus mass. Two days later, he presented to the emergency department with acute onset epigastric pain, nausea, and vomiting. A comprehensive review of systems, which had not been performed until this visit, revealed an unintentional 20-lb weight loss over the previous 3 months. He had a progressive ache in the left anterior groin that was dull, tender, nonradiating, and worse with weight bearing. He denied melena or hematochezia.
His temperature was 37 °C; heart rate, 98 beats per minute; and blood pressure, 128/63 mm Hg. He had midepigastric tenderness and point tenderness over the anterior iliac spine. White blood cell count was 12,600/μL; hemo globin, 14.5 g/dL; and platelet count, 158,000/μL. Serum lipase was 7,108 U/L. Serum creatinine, calcium, and triglyceride levels were normal. Alkaline phosphatase was 117 U/L (normal, 34-104 U/L); total bilirubin, 1.1 mg/dL; alanine aminotransferase (ALT), 119 U/L (normal, 7-52 U/L); and aspartate aminotransferase (AST), 236 U/L (normal, 13-39 U/L). Troponin I was undetectable, and an electrocardiogram demonstrated sinus tachycardia. Urinalysis was normal.
Concomitant pancreatitis and hepatitis with an elevated AST-to-ALT ratio should prompt evaluation of recurrent choledocholithiasis and a repeat inquiry about alcohol use. His medications should be reviewed for an association with pancreatitis. Anterior groin discomfort usually reflects osteoarthritis of the hip joint, inguinal hernia, or inguinal lymphadenopathy. Groin pain may be referred from spinal nerve root compression, aortoiliac occlusion, or nephrolithiasis. Weight loss in the presence of an inferior rectus mass suggests one of the aforementioned systemic diseases with orbital manifestations. Pancreatitis and groin discomfort may be important clues, but the chronicity of the recurrent pancreatitis and the high prevalence of hip osteoarthritis make it equally likely that they are unrelated to the eye disease.
CT scan of the abdomen and pelvis with contrast showed peripancreatic edema with fat stranding but no pancreatic or hepatobiliary mass. The common bile duct was normal. A 2.2×1.3-cm mass in the right posterior subphrenic space, a lytic lesion in the left anterior inferior iliac spine, and right nonobstructive nephrolithiasis were identified. CT scan of the chest with contrast showed multiple subpleural nodules and innumerable parenchymal nodules. Subcentimeter hilar, mediastinal, and prevascular lymphadenopathy were present, as well as multiple sclerotic lesions in the right fourth and sixth ribs. Prostate-specific antigen was 0.7 ng/mL (normal, ≤ 4.0 ng/mL). Cancer antigen 19-9 level was 5.5 U/mL (normal, < 37.0 U/mL), and carcinoembryonic antigen (CEA) was 100.1 ng/mL (normal, 0-3 U/mL).
Widespread pulmonary nodules, diffuse lymphadenopathy, and bony lesions raise concern for a metastatic malignancy. There is no evidence of a primary carcinoma. The lack of hepatic involvement reduces the likelihood of a gastrointestinal tumor, although a rectal cancer, which may drain directly into the inferior vena cava and bypass the portal circulation, could present as lung metastases on CT imaging. Lymphoma is plausible given the diffuse lymphadenopathy and orbital mass. Sarcoidosis and histiocytic disorders (eg, Langerhans cell histiocytosis) also cause orbital disease, pulmonary nodules, lymphadenopathy, and bone lesions, although a subphrenic mass would be atypical for both disorders; furthermore, the majority of patients with adult Langerhans cell histiocytosis smoke cigarettes. The elevated CEA makes a metastatic solid tumor more likely than lymphoma but does not specify the location of the primary tumor.
Pathology of the inferior rectus muscle mass showed well-differentiated adenocarcinoma (Figure 3A and 3B). A CT-guided biopsy of the left anterior inferior iliac spine revealed well-differentiated adenocarcinoma (Figure 3C). Adenocarcinoma of unknown primary wasdiagnosed.
Subsequent immunohistochemical (IHC) staining was positive for cytokeratin 7 (CK7) and mucicarmine (Figure 3D and 3E) and negative for cytokeratin 20 (CK20) and thyroid transcription factor 1 (TTF1). This IHC profile suggested pancreatic or upper gastrointestinal tract lineage. Positron emission tomography–CT (PET-CT) scan was aborted because of dyspnea and chest pressure following contrast administration. He declined further imaging or endoscopy. He received palliative radiation and three cycles of paclitaxel and gemcitabine for cancer of unknown primary (CUP). Two months later, he developed bilateral upper-arm weakness due to C7 and T2 cord compression from vertebral and epidural metastases; his symptoms progressed despite salvage chemotherapy. He was transitioned to comfort care and died at home 9 months after diagnosis.
DISCUSSION
This patient’s new headache and ocular abnormalities led to the discovery of an inferior rectus muscle mass. Initially unrecognized unintentional weight loss and hip pain recast a localized orbital syndrome as a systemic disease with pancreatic, ocular, pulmonary, lymph node, and skeletal pathology. Biopsies of the orbital rectus muscle and iliac bone demonstrated metastatic adenocarcinoma. Imaging studies did not identify a primary cancer, but IHC analysis suggested carcinoma of upper gastrointestinal or pancreatic origin.
Acute and chronic pancreatitis are both associated with pancreatic cancer.1 Chronic pancreatitis is associated with an increasing cumulative risk of pancreatic cancer; a potential mechanism is chronic inflammation with malignant transformation.2,3 There is also a 20-fold increased risk of pancreatic cancer in the first 2 years following an episode of acute pancreatitis,4 which may develop from malignant pancreatic duct obstruction. Although the post–acute pancreatitis risk of pancreatic cancer attenuates over time, a two-fold increased risk of pancreatic cancer remains after 10 years,4 which suggests that acute pancreatitis (particularly when idiopathic) either contributes to or shares pathogenesis with pancreatic adenocarcinoma. In elderly patients without gallstones or alcohol use, an abdominal CT scan or MRI shortly after resolution of the acute pancreatitis may be considered to assess for an underlying pancreatic tumor.5
CUP is a histologically defined malignancy without a known primary anatomic site despite an extensive evaluation. CUP accounts for up to 10% of all cancer diagnoses.6 CUP is ascribed to a primary cancer that remains too small to be detected or spontaneous regression of the primary cancer.7 Approximately 70% of autopsies of patients with CUP identify the primary tumor, which most commonly originates in the lung, gastrointestinal tract, breast, or pancreas.8
When a metastatic focus of cancer is found but the initial diagnostic evaluation (including CT scan of the chest, abdomen, and pelvis) fails to locate a primary cancer, the next step in searching for the tissue of origin is an IHC analysis of the tumor specimen. IHC analysis is a multistep staining process that can identify major categories of cancer, including carcinoma (adenocarcinoma, squamous cell carcinoma, and neuroendocrine carcinoma) and poorly or undifferentiated neoplasms (including carcinoma, lymphoma, sarcoma, or melanoma). Eighty-five percent of CUP cases are adenocarcinoma, 10% are squamous cell carcinoma, and the remaining 5% are undifferentiated neoplasms.9
There are no consensus guidelines for imaging in patients with CUP who have already undergone a CT scan of the chest, abdomen, and pelvis. Mammography is indicated in women with metastatic adenocarcinoma or axillary lymphadenopathy.7 MRI of the breast is obtained when mammography is nondiagnostic and the suspicion for breast cancer is high. Small clinical studies and meta-analyses support the use of PET-CT scans,7 although one study found that a PET-CT scan was not superior to CT imaging in identifying the primary tumor site in CUP.10 Endoscopy of the upper airway or gastrointestinal tract is rarely diagnostic in the absence of referable symptoms or a suggestive IHC profile (eg, CK7−, CK20+ suggestive of colon cancer).6
Molecular cancer classification has emerged as a useful diagnostic technique in CUP. Cancer cells retain gene expression patterns based on cellular origin, and a tumor’s profile can be compared with a reference database of known cancers, aiding in the identification of the primary tumor type. Molecular cancer classifier assays that use gene expression profiling can accurately determine a primary site11 and have been shown to be concordant with IHC testing.12 Molecular cancer classification is distinct from genetic assays that identify mutations for which there are approved therapies. Serum tumor markers are generally not useful in establishing the primary tumor and should be considered based on the clinical presentation (eg, prostate-specific antigen testing in a man with adenocarcinoma of unknown primary and osteoblastic metastases).
CUP is classified as favorable or unfavorable based on the IHC, pattern of spread, and serum markers in certain cases.6 Approximately 20% of CUP patients can be categorized into favorable subsets, such as adenocarcinoma in a single axillary lymph node in a female patient suggestive of a breast primary cancer, or squamous cell carcinoma in a cervical lymph node suggestive of a head or neck primary cancer.7 The remaining 80% of cases are categorized as unfavorable CUP and often have multiple metastases. Our patient’s pattern of spread and limited response to chemotherapy is characteristic of the unfavorable subset of CUP. The median survival of this group is 9 months, and only 25% of patients survive longer than 1 year.13
Biomarker-driven treatment of specific molecular targets independent of the tissue of origin (tissue-agnostic therapy) has shown promising results in the treatment of skin, lung, thyroid, colorectal, and gastric cancers.14 Pembrolizumab was the first drug approved by the US Food and Drug Administration based on a tumor’s biomarker without regard to its primary location. Data to support this approach for treating CUP are evolving and offer hope for patients with specific molecular targets.
Following the focused neuro-ophthalmologic evaluations, with focused examination and imaging, the hospitalist’s review of systems at the time of the final admission for pancreatitis set in motion an evaluation that led to a diagnosis of metastatic cancer. The risk factor of recurrent pancreatitis and IHC results suggested that pancreatic adenocarcinoma was the most likely primary tumor. As the focus of cancer treatment shifts away from the tissue of origin and toward molecular and genetic profiles, the search for the primary site may decrease in importance. In the future, even when we do not know the cancer’s origin, we may still know precisely what to do. But for now, as in this patient, our treatments continue to be based on a tumor that is out of sight, but not out of mind.
KEY TEACHING POINTS
- Acute and chronic pancreatitis are associated with an increased risk of pancreatic adenocarcinoma.
- CUP is a cancer in which diagnostic testing does not identify a primary tumor site. Immunohistochemistry and molecular analysis, imaging, and endoscopy are utilized selectively to identify a primary tumor type.
- Treatment of CUP currently depends on the suspected tissue of origin and pattern of spread.
- Tissue-agnostic therapy could allow for treatment for CUP patients independent of the tissue of origin.
Acknowledgments
We thank Andrew Mick, OD, for his review of an earlier version of this manuscript and Peter Phillips, MD, for his interpretation of the pathologic images.
1. Sadr-Azodi O, Oskarsson V, Discacciati A, Videhult P, Askling J, Ekbom A. Pancreatic cancer following acute pancreatitis: a population-based matched cohort study. Am J Gastroenterol. 2018;113(111):1711-1719. https://doi.org/10.1038/s41395-018-0255-9
2. Duell EJ, Lucenteforte E, Olson SH, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23(11):2964-2970. https://doi.org/10.1093/annonc/mds140
3. Ekbom A, McLaughlin JK, Nyren O. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;329(20):1502-1503. https://doi.org/10.1056/NEJM199311113292016
4. Kirkegard J, Cronin-Fenton D, Heide-Jorgensen U, Mortensen FV. Acute pancreatitis and pancreatic cancer risk: a nationwide matched-cohort study in Denmark. Gastroenterology. 2018;154(156):1729-1736. https://doi.org/10.1053/j.gastro.2018.02.011
5. Frampas E, Morla O, Regenet N, Eugene T, Dupas B, Meurette G. A solid pancreatic mass: tumour or inflammation? Diagn Interv Imaging. 2013;94(7-8):741-755. https://doi.org/10.1016/j.diii.2013.03.013
6. Varadhachary GR, Raber MN. Cancer of unknown primary site. N Engl J Med. 2014;371(8):757-765. https://doi.org/10.1056/NEJMra1303917
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017. 2018;35(3):199-206. https://doi.org//10.1053/j.semdp.2017.11.013
8. Pentheroudakis G, Golfinopoulos V, Pavlidis N. Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer. 2007;43(14):2026-2036. https://doi.org/10.1016/j.ejca.2007.06.023
9. Pavlidis N, Fizazi K. Carcinoma of unknown primary (CUP). Crit Rev Oncol Hematol. 2009;69(3):271-278. https://doi.org/10.1016/j.critrevonc.2008.09.005
10. Moller AK, Loft A, Berthelsen AK, et al. A prospective comparison of 18F-FDG PET/CT and CT as diagnostic tools to identify the primary tumor site in patients with extracervical carcinoma of unknown primary site. Oncologist. 2012;17(9):1146-1154. https://doi.org/10.1634/theoncologist.2011-0449
11. Economopoulou P, Mountzios G, Pavlidis N, Pentheroudakis G. Cancer of unknown primary origin in the genomic era: elucidating the dark box of cancer. Cancer Treat Rev. 2015;41(7):598-604. https://doi.org/10.1016/j.ctrv.2015.05.010
12. Greco FA. Molecular diagnosis of the tissue of origin in cancer of unknown primary site: useful in patient management. Curr Treat Options Oncol. 2013;14(4):634-642. https://doi.org/10.1007/s11864-013-0257-1
13. Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin—diagnosis and treatment. Nat Rev Clin Oncol. 2011;8(12):701-710. https://doi.org/10.1038/nrclinonc.2011.158
14. Luoh SW, Flaherty KT. When tissue is no longer the issue: tissue-agnostic cancer therapy comes of age. Ann Intern Med. 2018;169(4):233-239. https://doi.org/10.7326/M17-2832
1. Sadr-Azodi O, Oskarsson V, Discacciati A, Videhult P, Askling J, Ekbom A. Pancreatic cancer following acute pancreatitis: a population-based matched cohort study. Am J Gastroenterol. 2018;113(111):1711-1719. https://doi.org/10.1038/s41395-018-0255-9
2. Duell EJ, Lucenteforte E, Olson SH, et al. Pancreatitis and pancreatic cancer risk: a pooled analysis in the International Pancreatic Cancer Case-Control Consortium (PanC4). Ann Oncol. 2012;23(11):2964-2970. https://doi.org/10.1093/annonc/mds140
3. Ekbom A, McLaughlin JK, Nyren O. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;329(20):1502-1503. https://doi.org/10.1056/NEJM199311113292016
4. Kirkegard J, Cronin-Fenton D, Heide-Jorgensen U, Mortensen FV. Acute pancreatitis and pancreatic cancer risk: a nationwide matched-cohort study in Denmark. Gastroenterology. 2018;154(156):1729-1736. https://doi.org/10.1053/j.gastro.2018.02.011
5. Frampas E, Morla O, Regenet N, Eugene T, Dupas B, Meurette G. A solid pancreatic mass: tumour or inflammation? Diagn Interv Imaging. 2013;94(7-8):741-755. https://doi.org/10.1016/j.diii.2013.03.013
6. Varadhachary GR, Raber MN. Cancer of unknown primary site. N Engl J Med. 2014;371(8):757-765. https://doi.org/10.1056/NEJMra1303917
7. Bochtler T, Löffler H, Krämer A. Diagnosis and management of metastatic neoplasms with unknown primary. Semin Diagn Pathol. 2017. 2018;35(3):199-206. https://doi.org//10.1053/j.semdp.2017.11.013
8. Pentheroudakis G, Golfinopoulos V, Pavlidis N. Switching benchmarks in cancer of unknown primary: from autopsy to microarray. Eur J Cancer. 2007;43(14):2026-2036. https://doi.org/10.1016/j.ejca.2007.06.023
9. Pavlidis N, Fizazi K. Carcinoma of unknown primary (CUP). Crit Rev Oncol Hematol. 2009;69(3):271-278. https://doi.org/10.1016/j.critrevonc.2008.09.005
10. Moller AK, Loft A, Berthelsen AK, et al. A prospective comparison of 18F-FDG PET/CT and CT as diagnostic tools to identify the primary tumor site in patients with extracervical carcinoma of unknown primary site. Oncologist. 2012;17(9):1146-1154. https://doi.org/10.1634/theoncologist.2011-0449
11. Economopoulou P, Mountzios G, Pavlidis N, Pentheroudakis G. Cancer of unknown primary origin in the genomic era: elucidating the dark box of cancer. Cancer Treat Rev. 2015;41(7):598-604. https://doi.org/10.1016/j.ctrv.2015.05.010
12. Greco FA. Molecular diagnosis of the tissue of origin in cancer of unknown primary site: useful in patient management. Curr Treat Options Oncol. 2013;14(4):634-642. https://doi.org/10.1007/s11864-013-0257-1
13. Massard C, Loriot Y, Fizazi K. Carcinomas of an unknown primary origin—diagnosis and treatment. Nat Rev Clin Oncol. 2011;8(12):701-710. https://doi.org/10.1038/nrclinonc.2011.158
14. Luoh SW, Flaherty KT. When tissue is no longer the issue: tissue-agnostic cancer therapy comes of age. Ann Intern Med. 2018;169(4):233-239. https://doi.org/10.7326/M17-2832
© 2021 Society of Hospital Medicine
More Is Less
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.

The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).

Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).

On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.
The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).
Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).
On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
A 64-year-old man presented with a 2-month history of a nonproductive cough, weight loss, and subjective fevers. He had no chest pain, hemoptysis, or shortness of breath. He also described worsening anorexia and a 15-pound weight loss over the previous 3 months. He had no arthralgias, myalgias, abdominal pain, nausea, emesis, or diarrhea.
Two weeks prior to his presentation, he was diagnosed with pneumonia and given a 5-day course of azithromycin. His symptoms did not improve, so he presented to the emergency room.
He had not been seen regularly by a physician in decades and had no known medical conditions. He did not take any medications. He immigrated from China 3 years prior and lived with his wife in California. He had a 30 pack-year smoking history. He drank a shot glass of liquor daily and denied any drug use.
Weight loss might result from inflammatory disorders like cancer or noninflammatory causes such as decreased oral intake (eg, diminished appetite) or malabsorption (eg, celiac disease). However, his fevers suggest inflammation, which usually reflects an underlying infection, cancer, or autoimmune process. While chronic cough typically results from upper airway cough syndrome (allergic or nonallergic rhinitis), gastroesophageal reflux disease, or asthma, it can also point to pathology of the lung, which may be intrinsic (bronchiectasis) or extrinsic (mediastinal mass). The duration of 2 months makes a typical infectious process like pneumococcal pneumonia unlikely. Atypical infections such as tuberculosis, melioidosis, and talaromycosis are possible given his immigration from East Asia, and coccidioidomycosis given his residence in California. He might have undiagnosed medical conditions, such as diabetes, that could be relevant to his current presentation and classify him as immunocompromised. His smoking history prompts consideration of lung cancer.
His temperature was 36.5 oC, heart rate 70 beats per minute, blood pressure 118/66 mm Hg, respiratory rate 16 breaths per minute, oxygen saturation 98% on room air, and body mass index 23 kg/m2. He was in no acute distress. The findings from the cardiac, lung, abdominal, and neurological exams were normal.
Skin examination found a fixed, symmetric, 5-cm, firm nodule at top of sternum (Figure 1A). In addition, he had two 1-cm, mobile, firm, subcutaneous nodules, one on his anterior left chest and another underneath his right axilla. He also had two 2-cm, erythematous, tender nodules on his left anterior forearm and a 1-cm nodule with a central black plug on the dorsal surface of his right hand (Figure 1B). He did not have any edema.
The white blood cell count was 10,500/mm3 (42% neutrophils, 37% lymphocytes, 16.4% monocytes, and 2.9% eosinophils), hemoglobin was 12.2 g/dL with a mean corpuscular volume of 91 fL, and the platelet count was 441,000/mm3. Basic metabolic panel, aminotransferase, bilirubin, and alkaline phosphatase were within reference ranges. Serum albumin was 3.1 g/dL. Serum total protein was elevated at 8.8 g/dL. Serum calcium was 9.0 mg/dL. Urinalysis results were normal.
The slightly low albumin, mildly elevated platelet count, monocytosis, and normocytic anemia suggest inflammation, although monocytosis might represent a hematologic malignancy like chronic myelomonocytic leukemia (CMML). His subjective fevers and weight loss further corroborate underlying inflammation. What is driving the inflammation? There are two localizing findings: cough and nodular skin lesions.
His lack of dyspnea and normal oxygen saturation, respiratory rate, and lung exam make an extrapulmonary cause of cough such as lymphadenopathy or mediastinal infection possible. The number of nodular skin lesions, wide-spread distribution, and appearance (eg, erythematous, tender) point to either a primary cutaneous disease with systemic manifestations (eg, cutaneous lymphoma) or a systemic disease with cutaneous features (eg, sarcoidosis).
Three categories—inflammatory, infectious, and neoplastic—account for most nodular skin lesions. Usually microscopic evaluation is necessary for definitive diagnosis, though epidemiology, associated symptoms, and characteristics of the nodules help prioritize the differential diagnosis. Tender nodules might reflect a panniculitis; erythema nodosum is the most common type, and while this classically develops on the anterior shins, it may also occur on the forearm. His immigration from China prompts consideration of tuberculosis and cutaneous leishmaniasis. Coccidioidomycosis can lead to inflammation and nodular skin lesions. Other infections such as nontuberculous mycobacteria, nocardiosis, and cryptococcosis may cause disseminated infection with pulmonary and skin manifestations. His smoking puts him at risk of lung cancer, which rarely results in metastatic subcutaneous infiltrates.
A chest radiograph demonstrated a prominent density in the right paratracheal region of the mediastinum with adjacent streaky opacities. A computed tomography scan of the chest with intravenous contrast demonstrated centrilobular emphysematous changes and revealed a 2.6 × 4.7-cm necrotic mass in the anterior chest wall with erosion into the manubrium, a 3.8 × 2.1-cm centrally necrotic soft-tissue mass in the right hilum, a 5-mm left upper-lobe noncalcified solid pulmonary nodule, and prominent subcarinal, paratracheal, hilar, and bilateral supraclavicular lymphadenopathy (Figure 2).
Flow cytometry of the peripheral blood did not demonstrate a lymphoproliferative disorder. Blood smear demonstrated normal red blood cell, white blood cell, and platelet morphology. HIV antibody was negative. Hemoglobin A1c was 6.1%. Smear microscopy for acid-fast bacilli (AFB) was negative and sputum AFB samples were sent for culture. Bacterial, fungal, and AFB blood cultures were collected and pending.
Causes of necrotizing pneumonia include liquid (eg, lymphoma) and solid (eg, squamous cell carcinoma) cancers, infections, and noninfectious inflammatory processes such as granulomatosis with polyangiitis (GPA). Given his subacute presentation and extrapulmonary cutaneous manifestations, consideration of mycobacteria, fungi (eg, Coccidioides, Aspergillus, and Cryptococcus), and filamentous bacteria (eg, Nocardia and Actinomyces) is prioritized among the myriad of infections that can cause a lung cavity. His smoking history and centrilobular emphysematous changes are highly suggestive of chronic obstructive pulmonary disease, which puts him at increased risk of bacterial colonization and recurrent pulmonary infections. Tuberculosis is still possible despite three negative AFB-sputa smears given the sensitivity of smear microscopy (with three specimens) is roughly 70% in an immunocompetent host.
The lymphadenopathy likely reflects spread from the necrotic lung mass. The frequency of non-Hodgkin lymphoma increases with age. The results of the peripheral flow cytometry do not exclude the possibility of an aggressive lymphoma with pulmonary and cutaneous manifestations.
The erosive property of the chest wall mass makes an autoimmune process like GPA unlikely. An aggressive and disseminated infection or cancer is most likely. A pathologic process that originated in the lung and then spread to the lymph nodes and skin is more likely than a disorder which started in the skin. It would be unlikely for a primary cutaneous disorder to cause such a well-defined necrotic lung mass. Lung cancer rarely metastasizes to the skin and, instead, preferentially involves the chest. Ultimately, ascertaining what the patient experienced first (ie, respiratory or cutaneous symptoms) will determine where the pathology originated.
Computed tomography scan of the abdomen and pelvis with intravenous contrast demonstrated multiple ill-defined lytic lesions in the pelvis, including a 12-mm lesion of the left sacral ala and multiple subcentimeter lesions in the medial left iliac bone and superior right acetabulum. In addition, there were two 1-cm, rim-enhancing, hypodense nodules in the subcutaneous fat of the right flank at the level of L5 and the left lower quadrant, respectively. There was also a 2.2 × 1.9-cm faintly rim-enhancing hypodensity within the left iliopsoas muscle belly.
These imaging findings further corroborate a widely metastatic process probably originating in the lung and spreading to the lymph nodes, skin, muscles, and bones. The characterization of lesions as lytic as opposed to blastic is less helpful because many diseases can cause both. It does prompt consideration of multiple myeloma; however, multiple myeloma less commonly manifests with extramedullary plasmacytomas and is less likely given his normal renal function and calcium level. Bone lesions lessen the likelihood of GPA, and his necrotic lung mass makes sarcoidosis unlikely. Atypical infections and cancers are the prime suspect of his multisystemic disease.
There are no data yet to suggest a weakened immune system, which would increase his risk for atypical infections. His chronic lung disease, identified on imaging, is a risk factor for nocardiosis. This gram-positive, weakly acid-fast bacterium can involve any organ, although lung, brain, and skin are most commonly involved. Disseminated nocardiosis can result from a pulmonary or cutaneous site of origin. Mycobacteria; Actinomyces; dimorphic fungi like Histoplasma, Coccidioides, and Blastomyces; and molds such as Aspergillus can also cause disseminated disease with pulmonary, cutaneous, and musculoskeletal manifestations.
While metastases to muscle itself are rare, they can occur with primary lung cancers. Primary lung cancer with extrapulmonary features is feasible. Squamous cell lung cancer is the most likely to cavitate, although it rarely spreads to the skin. An aggressive lymphoma like diffuse large B-cell lymphoma or cutaneous T-cell lymphoma (higher occurrence in Asians) might also explain his constellation of findings. If culture data remain negative, then biopsy of the chest wall mass might be the safest and highest-yield target.
On hospital day 2, the patient developed new-onset severe neck pain. Magnetic resonance imaging of the cervical, thoracic, and lumbar spine revealed multilevel, bony, lytic lesions with notable cortical breakthrough of the C2 and C3 vertebrae into the prevertebral space, as well as epidural extension and paraspinal soft-tissue extension of the thoracic and lumbar vertebral lesions (Figure 3).
On hospital day 3, the patient reported increased tenderness in his skin nodules with one on his left forearm spontaneously draining purulent fluid. Repeat complete blood count demonstrated a white blood cell count of 12,600/mm3 (45% neutrophils, 43% lymphocytes, 8.4% monocytes, and 4.3% eosinophils), hemoglobin of 16 g/dL, and platelet count of 355,000/mm3.
The erosion into the manubrium and cortical destruction of the cervical spine attests to the aggressiveness of the underlying disease process. Noncutaneous lymphoma and lung cancer are unlikely to have such prominent skin findings; the visceral pathology, necrotizing lung mass, and bone lesions make cutaneous lymphoma less likely. At this point, a disseminated infectious process is most likely. Leading considerations based on his emigration from China and residence in California are tuberculosis and coccidioidomycosis, respectively. Tuberculous spondylitis most commonly involves the lower thoracic and upper lumbar region, and less commonly the cervical spine. His three negative AFB sputa samples further reduce its posttest probability. Ultimately microbiologic data are needed to distinguish between a disseminated fungal process, like coccidioidomycosis, or tuberculosis.
Given the concern for malignancy, a fine needle aspiration of the left supraclavicular lymph node was pursued. This revealed fungal microorganisms morphologically compatible with Coccidioides spp. with a background of necrotizing granulomas and acute inflammation. Fungal blood cultures grew Coccidioides immitis. AFB blood cultures were discontinued due to overgrowth of mold. The Coccidioides immitis antibody immunodiffusion titer was positive at 1:256.
During the remainder of the hospitalization, the patient was treated with oral fluconazole 800 mg daily. The patient underwent surgical debridement of the manubrium. In addition, given the concern for cervical spine instability, neurosurgery recommended follow-up with interval imaging. Since his discharge from the hospital, the patient continues to take oral fluconazole with resolution of his cutaneous lesions and respiratory symptoms. His titers have incrementally decreased from 1:256 to 1:16 after 8 months of treatment.
COMMENTARY
This elderly gentleman from China presented with subacute symptoms and was found to have numerous cutaneous nodules, lymphadenopathy, and diffuse osseous lesions. This multisystem illness posed a diagnostic challenge, forcing our discussant to search for a disease process that could lead to such varied findings. Ultimately, epidemiologic and clinical clues suggested a diagnosis of disseminated coccidioidomycosis, which was later confirmed on lymph node biopsy.
Coccidioides species are important fungal pathogens in the Western Hemisphere. This organism exhibits dimorphism, existing as mycelia (with arthroconidia) in soil and spherules in tissues. Coccidioides spp are endemic to the Southwestern United States, particularly California’s central valley and parts of Arizona; it additionally remains an important pathogen in Mexico, Central America, and South America.1 Newer epidemiologic studies have raised concerns that the incidence of coccidioidomycosis is increasing and that its geographic range may be more extensive than previously appreciated, with it now being found as far north as Washington state.2
Coccidioidal infection can take several forms. One-half to two-thirds of infections may be asymptomatic.3 Clinically significant infections can include an acute self-limiting respiratory illness, pulmonary nodules and cavities, chronic fibrocavitary pneumonia, and infections with extrapulmonary dissemination. Early respiratory infection is often indistinguishable from typical community-acquired pneumonia (10%-15% of pneumonia in endemic areas) but can be associated with certain suggestive features, such as erythema nodosum, erythema multiforme, prominent arthralgias (ie, “desert rheumatism”), and a peripheral eosinophilia.4,5
Extrapulmonary dissemination is rare and most commonly associated with immunocompromising states.6 However, individuals of African or Filipino ancestry also appear to be at increased risk for disseminated disease, which led to a California court decision that excluded African American inmates from state prisons located in Coccidioides endemic areas.7 The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the central nervous system (CNS).6 CNS disease has a predilection to manifest as a chronic basilar meningitis, most often complicated by hydrocephalus, vasculitic infarction, and spinal arachnoiditis.8
Cutaneous manifestations of coccidioidomycosis can occur as immunologic phenomenon associated with pulmonary disease or represent skin and soft tissue foci of disseminated infection.9 In primary pulmonary infection, skin findings can range from a nonspecific exanthem to erythema nodosum and erythema multiforme, which are thought to represent hypersensitivity responses. In contrast, Coccidioides spp can infect the skin either through direct inoculation (as in primary cutaneous coccidioidomycosis) or via hematogenous dissemination.9,10 A variety of lesions have been described, with painless nodules being the most frequently encountered morphotype in one study.11,12 On histopathologic examination, these lesions often have features of granulomatous dermatitis, eosinophilic infiltration, gummatous necrosis, microabscesses, or perivascular inflammation.13
Another common and highly morbid site of extrapulmonary dissemination is the musculoskeletal system. Bone and joint coccidioidomycosis most frequently affect the axial skeleton, although peripheral skeletal structures and joints can also be involved.6,12 Vertebral coccidioidomycosis is associated with significant morbidity. A study describing the magnetic resonance imaging findings of patients with vertebral coccidioidomycosis found that Coccidioides spp appeared to have a predilection for the thoracic vertebrae (in up to 80% of the study’s cohort).14 Skip lesions with noncontiguously involved vertebrae occurred in roughly half of patients, highlighting the usefulness of imaging the total spine in suspected cases.
The diagnosis of coccidioidomycosis is often established through serologic testing or by isolation of Coccidioides spp. on histopathology or culture. Obtaining sputum or tissue may be difficult, so clinicians often rely on noninvasive diagnostic tests such as coccidioidal antigen and serologies by enzyme immunoassays, immunodiffusion, and complement fixation. Enzyme immunoassays IgM and IgG results are positive early in the disease process and need to be confirmed with immunodiffusion or complement fixation testing. Complement fixation IgG is additionally useful to monitor disease activity over time and can help inform risk of disseminated disease.15 The gold standard of diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation either by direct visualization of a spherule or growth in fungal cultures.16 Polymerase chain reaction testing of sputum samples is an emerging diagnostic technique that has been found to have similar sensitivity rates to fungal culture.17
Treatment decisions in coccidioidomycosis are complex and vary by site of infection, immune status of the host, and extent of disease.16 While uncomplicated primary pulmonary infections can often be managed with observation alone, prolonged medical therapy with azole antifungals is often recommended for complicated pulmonary infections, symptomatic cavitary disease, and virtually all forms of extrapulmonary disease. Intravenous liposomal amphotericin is often used as initial therapy in immunosuppressed individuals, pregnant women, and those with extensive disease. CNS disease represents a particularly challenging treatment scenario and requires lifelong azole therapy.8,16
The patient in this case initially presented with vague inflammatory symptoms, with each aliquot revealing further evidence of a metastatic disease process. Such multisystem presentations are diagnostically challenging and force clinicians to reach for some feature around which to build their differential diagnosis. It is with this in mind that we are often taught to “localize the lesion” in order to focus our search for a unifying diagnosis. Yet, in this case, the sheer number of disease foci ultimately helped the discussant to narrow the range of diagnostic possibilities because only a limited number of conditions could present with such widespread, multisystem manifestations. Therefore, this case serves as a reminder that, sometimes in clinical reasoning, “more is less.”
KEY TEACHING POINTS
- Coccidioidomycosis is a fungal infection that can present with pulmonary or extrapulmonary disease. Risk of extrapulmonary dissemination is greatest among immunocompromised individuals and those of African or Filipino ancestry.3,7
- The most common sites of extrapulmonary dissemination include the skin and soft tissues, bones and joints, and the CNS.6
- While serologic testing can be diagnostically useful, the gold standard for diagnosis of disseminated coccidioidomycosis infection remains histopathologic confirmation with direct visualization of a spherule or growth in fungal cultures.16
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
1. Benedict K, McCotter OZ, Brady S, et al. Surveillance for Coccidioidomycosis - United States, 2011-2017. MMWR Surveill Summ. 2019;68(No. SS-7):1-15. http://dx.doi.org/10.15585/mmwr.ss6807a1
2. McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Suppl 1):S30-s40. https://doi.org/10.1093/mmy/myy095
3. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis. Clin Infect Dis. 2005;41(9):1217-1223. https://doi.org/10.1086/496991
4. Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14(7):1053-1059. https://doi.org/10.3201/eid1407.070832
5. Saubolle MA, McKellar PP, Sussland D. Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. 2007;45(1):26-30. https://doi.org/10.1128/jcm.02230-06
6. Adam RD, Elliott SP, Taljanovic MS. The spectrum and presentation of disseminated coccidioidomycosis. Am J Med. 2009;122(8):770-777. https://doi.org/10.1016/j.amjmed.2008.12.024
7. Wheeler C, Lucas KD, Mohle-Boetani JC. Rates and risk factors for Coccidioidomycosis among prison inmates, California, USA, 2011. Emerg Infect Dis. 2015;21(1):70-75. https://doi.org/10.3201/eid2101.140836
8. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42(1):103-107. https://doi.org/10.1086/497596
9. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections. Ann N Y Acad Sci. 2007;1111:411-421. https://doi.org/10.1196/annals.1406.010
10. Chang A, Tung RC, McGillis TS, Bergfeld WF, Taylor JS. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49(5):944-949. https://doi.org/10.1016/s0190-9622(03)00462-6
11. Quimby SR, Connolly SM, Winkelmann RK, Smilack JD. Clinicopathologic spectrum of specific cutaneous lesions of disseminated coccidioidomycosis. J Am Acad Dermatol. 1992;26(1):79-85. https://doi.org/10.1016/0190-9622(92)70011-4
12. Crum NF, Lederman ER, Stafford CM, Parrish JS, Wallace MR. Coccidioidomycosis: a descriptive survey of a reemerging disease. clinical characteristics and current controversies. Medicine (Baltimore). 2004;83(3):149-175. https://doi.org/10.1097/01.md.0000126762.91040.fd
13. Carpenter JB, Feldman JS, Leyva WH, DiCaudo DJ. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62(5):831-837. https://doi.org/10.1016/j.jaad.2008.07.031
14. Crete RN, Gallmann W, Karis JP, Ross J. Spinal coccidioidomycosis: MR imaging findings in 41 patients. AJNR Am J Neuroradiol. 2018;39(11):2148-2153. https://doi.org/10.3174/ajnr.a5818
15. McHardy IH, Dinh BN, Waldman S, et al. Coccidioidomycosis complement fixation titer trends in the age of antifungals. J Clin Microbiol. 2018;56(12):e01318-18. https://doi.org/10.1128/jcm.01318-18
16. Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63(6):e112-e146. https://doi.org/10.1093/cid/ciw360
17. Vucicevic D, Blair JE, Binnicker MJ, et al. The utility of Coccidioides polymerase chain reaction testing in the clinical setting. Mycopathologia. 2010;170(5):345-351. https://doi.org/10.1007/s11046-010-9327-0
© 2020 Society of Hospital Medicine
Left Out in the Cold
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
A previously healthy 4-year-old boy presented to his pediatrician for nasal congestion, left ear pain, and intermittent fevers, which he’d been experiencing for 2 days. His exam was consistent with acute otitis media. Cefdinir was prescribed given a rash allergy to amoxicillin. His fever, congestion, and otalgia improved the next day.
Three days later he developed abdominal pain, fever, and labored breathing; his mother brought him to the emergency department (ED). His temperature was 38.0 °C, heart rate 141 beats per minute, blood pressure 117/71 mm Hg, respiratory rate 22 breaths per minute; he had oxygen saturation of 96% on ambient air. Despite mild accessory muscle use, he appeared comfortable and interactive. His left tympanic membrane was bulging without erythema. His neck was supple and mucous membranes moist. He had neither cervical lymphadenopathy nor conjunctival pallor. The cardiopulmonary exam was normal except for tachycardia. His abdomen was soft and not distended without organomegaly or tenderness.
Upper respiratory tract symptoms are commonly encountered in pediatrics and most often result from self-limited viral processes. Evaluation of a child with upper respiratory tract symptoms aims to identify serious causes like meningitis, as well as assessing the need for antimicrobial therapy. Supportive management is often appropriate in otitis media. His new, more concerning symptoms portend either a progression of the original process causing his upper respiratory tract symptoms or a separate etiology. It is key to determine which signs and symptoms are associated with the primary process and which are compensatory or secondary. If he were to be more ill appearing, for example, it is possible that his respiratory distress may be related to an underlying systemic illness rather than a primary lung process. Respiratory distress, abdominal pain, and fever could be a result of sepsis from an intrabdominal process such as ruptured appendicitis, intussusception, or malrotation with volvulus. Other causes of sepsis, such as meningitis or severe mastoiditis, both rare complications of otitis media, should be considered, although he does not appear severely ill. Acute myelogenous leukemia or other malignancies and illnesses associated with immunodeficiency can present with sepsis and chloromas in the middle ear that can be misconstrued as otitis media.
A chest radiograph demonstrated left lower lobe patchy opacities concerning for pneumonia. Rapid respiratory syncytial virus and influenza antigen test results were negative. Laboratory testing for general bloodwork was not obtained. He was administered a single dose of intramuscular ceftriaxone, prescribed a 5-day course of azithromycin, and discharged home. The child’s breathing gradually improved, but he continued to have subjective fevers. Two days later, he developed dark red urine. His mother brought him back to the outpatient clinic.
At the time of the ED visit, a diagnosis of community-acquired pneumonia was plausible given fever, mildly increased work of breathing, and an opacification on chest radiography. Most community-acquired pneumonia is caused by viruses; common bacterial causes for his age include Streptococcus pneumoniae and Moraxella catarrhalis. The first-line treatment for uncomplicated community-acquired pneumonia in children is amoxicillin, but this was appropriately avoided given his allergy.
The persistent fevers are surprising. The improvement in breathing corresponds to the treatment (and resolution) of community-acquired pneumonia. However, the development of dark urine does not. Red urine—in the absence of ingested pigments (such as those found in beets)—usually results from hematuria, hemoglobinuria, or myoglobinuria. Gross hematuria can originate from the kidneys to the urethral meatus. Abdominal masses, kidney trauma, or underlying kidney disease may all present with gross hematuria (or microscopic hematuria, seen only on urinalysis). The urine should be examined for the presence of heme, protein, and for evidence of infection; microscopy should be performed to examine for cellular casts and dysmorphic red cells. Tests of renal function, a comprehensive metabolic panel, evaluation of hematologic indexes, and assessments of inflammatory markers should be performed.
The child lived with his parents and had no siblings. He experienced no physical trauma, and there was no family history of kidney disease or hematuria. His father had a persistent cough and fever for 1 month, but recovered around the time the patient began to experience his initial symptoms. This was the patient’s third diagnosis of pneumonia. He had not traveled and was up to date with immunizations. He attended day care.
The fact that this is not the first episode of “pneumonia” raises important possibilities. The most likely one is that the child has had multiple viral infections; however, he could have an underlying primary immunodeficiency (PI) that predisposes him to recurrent infections. More severe PIs often present with recurrent sepsis, bacteremia, and failure to thrive, none of which were present in this case. Less severe PIs (such as selective IgA deficiency) could be possible. Another possibility is that these recurrent episodes of pneumonia are a relapsing and remitting noninfectious process, such as an antineutrophil cytoplasmic antibodies–associated vasculitis or anti–glomerular basement membrane disease. The patient’s father’s recent prolonged respiratory symptoms may be suggestive of pertussis or a “walking pneumonia” potentially caused by Mycoplasma or another atypical bacterium.
His temperature was 36.9 °C, heart rate 107 beats per minute, blood pressure was 106/67 mm Hg, and respiratory rate was 24 breaths per minute with oxygen saturation of 100% on ambient air. He was well appearing. His mucous membranes were moist, and oropharynx was clear. He had scleral icterus. The cardiopulmonary exam was normal. He had no significant lymphadenopathy, hepatosplenomegaly, or rashes.
The finding of jaundice is an important diagnostic pivot point, especially when combined with hematuria. The next step is determining if the jaundice is resulting from unconjugated or conjugated hyperbilirubinemia; the former most often stems from hemolysis or impairment in conjugation, while the latter results from intrahepatic or extrahepatic biliary defects. Tests for hepatobiliary injury including evaluations of alanine and aspartate aminotransferases and alkaline phosphatase, as well as for hepatic function such as tests of coagulation, should be performed.
The patient was referred to the ED and admitted for further evaluation. A complete blood count revealed a white blood cell (WBC) count of 10,700/µL (61% polymorphonuclear neutrophils, 30% lymphocytes, 5% monocytes, 3% eosinophils, 1% basophils), hemoglobin count was 10.3 g/dL (reticulocyte 2% with absolute reticulocyte count 58,400/μL), and platelet count was 265,000/µL. Components of the basic metabolic panel were within reference ranges except for a mildly elevated blood urea nitrogen level of 14 mg/dL with normal creatinine level of 0.3 mg/dL. Total protein was 6.7 g/dL (reference range, 6.4-8.3) and albumin 3.9 g/dL (reference range, 3.4-4.8). Alkaline phosphatase level was 188 U/L (reference range, 44-147), aspartate aminotransferase level 76 U/L (reference range, 0-40), and alanine aminotransferase level 12 U/L (reference range, 7-40). Total bilirubin level was 2.4 mg/dL (reference range, less than 1.5) with direct bilirubin level of 0.4 mg/dL. His C-reactive protein level was 1.5 mg/mL (reference range, 0-0.75). Creatinine kinase (CK) level was 2,550 U/L (reference range, 2-198). International Normalized Ratio (INR) was 1.0. Urinalysis was notable for 2+ proteinuria, large hemoglobin pigment, and 6 red blood cells per high power field (reference range, 0-4).
His blood urea nitrogen is elevated, reflecting either prerenal azotemia or increased absorption of nitrogenous products. Unconjugated hyperbilirubinemia may result from impaired hepatic bilirubin uptake (such as in heart failure or portosystemic shunts), impaired bilirubin conjugation (resulting from genetic conditions or drugs), or excess bilirubin production (such as in hemolysis); his anemia and lack of other evidence of hepatic dysfunction point to hemolysis as the etiology. The reticulocyte production index is approximately 1%, which suggests that an increase in erythrocyte generation is present but inadequate. This, however, does not mean that an erythrocyte production abnormality is present since reticulocytosis can be delayed in many cases of acute hemolytic anemia. It is also possible that the same hemolytic process is affecting mature and immature erythrocytes. A peripheral blood smear should be reviewed for evidence of intravascular hemolysis and testing for autoimmune hemolysis should be performed. Notably, his white blood cell and platelet counts are preserved, which makes a bone marrow–involved malignancy or infiltrative process less likely. The alkaline phosphatase elevation may result from either intrahepatic or extrahepatic biliopathy; bone damage is also possible. The elevation of aspartate aminotransferase, CK, and potassium, along with marked urinary heme pigment, may indicate muscle damage; the most common myositis in children is benign acute childhood myositis resulting from viral infection. However, the moderate level of CK elevation seen in this case is nonspecific and can result from many different etiologies. A metabolic myopathy, such as carnitine palmitoyltransferase II deficiency, can be made worse by metabolic stress and result in rhabdomyolysis; the presentations of inborn errors of metabolism are varied and a planned-out, stepwise approach in evaluation is fundamental.
Lactic acid dehydrogenase (LDH) level was 1,457 U/L (reference range, 140-280), and haptoglobin level was less than 6 mg/dL (reference range, 30-200). Peripheral blood smear demonstrated occasional atypical, reactive-appearing lymphocytes with red cell clumping and agglutination, as well as rare target, burr, and fragmented red cells. Test results for urine myoglobin were negative. Results of urine culture were negative. No blood culture was collected.
The elevated LDH, decreased haptoglobin, and findings on the peripheral blood smear confirm hemolysis. The clumping of erythrocytes can be artifactual in the preparation of peripheral smears, but when considered in the context of hemolysis, may be clinically important. Clumping of erythrocytes on the peripheral smear indicates the binding of a protein to antigens on the erythrocyte membrane; when this occurs below body temperature, this is consistent with the presence of a “cold agglutinin,” usually an IgM antibody directed at erythrocyte surface antigens that causes agglutination and destruction, especially in cooler areas of the body. This is a well-known complication of Mycoplasma pneumoniae infections as well as Epstein-Barr virus (EBV) infections; it may also occur with lymphoid malignancies or autoimmune disease.
Direct Coombs IgG test findings were negative, direct Coombs C3 test was positive, and direct Coombs polyspecific test was positive. M pneumoniae IgG antibody level was 1.4 mg/dL (reference ranges: <0.9, negative; 0.91-1.09, equivocal; >1.1, positive); M pneumoniae IgM level was 529 U/mL (reference range: <770, negative). EBV capsid IgM and IgG levels were undetectable. EBV nuclear antigen IgG level was also undetectable. EBV viral load was fewer than 10 copies/mL. Antinuclear antibodies (ANA) level was negative. General IgE and IgM levels were normal, at 11 and 81 mg/dL, respectively. Repeat complete blood count showed WBC of 7,800/µL, hemoglobin of 8.7 g/dL, and platelet count of 341,000/µL. The patient’s hemoglobin remained stable during hospitalization.
This directed testing is helpful in further classifying the patient’s hemolytic anemia. Autoimmune hemolytic anemias are classified into warm antibody–mediated, cold antibody–mediated, and mixed-type forms; drug-induced and alloimmune hemolytic anemias also occur. In addition, both systemic lupus erythematosus and antiphospholipid antibody syndrome can have hemolytic anemia with variable Coombs testing results; neither fit well in this case. The absence of red blood cell–directed IgG antibodies substantially decreases the likelihood of warm antibody–mediated hemolytic anemia. In cold antibody–mediated hemolytic anemia, antibodies bind to the erythrocyte membrane and then adhere to complement C3, which leads to both intravascular and extravascular hemolysis. Important types of cold antibody–mediated hemolytic anemia in children are primary and secondary cold agglutinin disease, along with paroxysmal cold hemoglobinuria. The Donath-Landsteiner test can be helpful in differentiating these conditions. Antibodies to Mycoplasma may be delayed in response to acute infection, and a child who is reinfected may only produce IgG antibodies. Given the patient’s clinical stability and previous health, the most likely diagnosis is Mycoplasma-induced cold antibody–mediated hemolytic anemia. It may be helpful to check convalescent titers to Mycoplasma in 2 to 4 weeks.
Donath-Landsteiner (D-L) antibody test results were positive. Medication-derived hemolytic anemia testing was conducted, but the presence of positive D-L antibody makes the test results inconclusive. This ultimately led to a diagnosis of paroxysmal cold hemoglobinuria (PCH), presumably triggered by a viral syndrome. Convalescent titers to Mycoplasma were not checked given clinical improvement. Because the patient’s hemoglobin was stable during hospitalization, he was not treated with steroids. His parents were counseled on avoiding cold temperatures for several days. Within 1 month, his hemoglobin had recovered without further evidence of hemolysis.
DISCUSSION
Hemolytic anemia refers to the accelerated destruction of red blood cells and can be further classified as acquired or hereditary.1 Hereditary conditions causing hemolytic anemia include enzymopathies (eg, glucose-6-phosphate dehydrogenase deficiency), hemoglobinopathies (eg, sickle cell disease), and membrane abnormalities (eg, hereditary spherocytosis). Acquired pathologies include microangiopathic hemolytic anemia (MAHA), anemias directly caused by certain infections such as malaria, and immune-mediated (Coombs-positive) hemolytic anemias.
MAHA can sometimes be life-threatening and is therefore important to identify quickly. In the right clinical context, such processes may be rapidly recognized by the presence of schistocytes on blood smear in addition to an elevated serum LDH level. Schistocytes suggest mechanical destruction of erythrocytes in the vasculature, the hallmark of MAHA. Important MAHAs include thrombocytopenic purpura, hemolytic-uremic syndrome, and disseminated intravascular coagulation. Though this patient did have a mildly elevated LDH, MAHA was less likely because there were no schistocytes on the blood smear.
Autoimmune hemolytic anemias (AIHAs) are another important subset of acquired hemolytic anemias. AIHAs occur when there is antibody-mediated destruction of erythrocytes. The direct Coombs test evaluates for antibody- or complement-coated erythrocytes. After administration of anti-IgG and anti-C3 serum, the test evaluates for agglutination of the red cells caused by attached antibodies or complement. Coombs-positive AIHA can also be categorized by the temperature of agglutination. “Warm” hemolysis often involves IgG autoantibodies (ie, warm agglutinins), while “cold” antibodies, usually IgM autoantibodies, bind at colder temperatures (0-4 °C) and activate complements, including C3. In this patient, the Coombs C3 was positive while the Coombs IgG was negative, which is more suggestive of a cold complement–mediated pathway.
Cold AIHA can be further categorized into primary cold agglutinin disease, secondary cold agglutinin disease, and PCH. Primary cold agglutinin disease is an autoimmune disorder that mostly occurs in adults. Secondary cold AIHA can often be triggered by bacterial infection (commonly M pneumoniae) or viruses including EBV, measles, and mumps.2 Medications, including penicillin and cephalosporins, can also be implicated. Secondary cold AIHA is also linked with autoimmune diseases, such as systemic lupus erythematosus and lymphoproliferative disorders. PCH can be identified with the unique presence of a specific autoantibody (ie, D-L autoantibody) that agglutinates at cold temperatures but dissociates on subsequent rewarming.3 Complement remains affixed and activates hemolysis.
The D-L antibody responsible for PCH is an IgG antibody to the P-antigen present on the erythrocyte surface. Since the Coombs test is conducted at normal temperature, it will be positive for the affixed complement but not for IgG. The underlying mechanism for PCH was proposed by Julius Donath, MD, and Karl Landsteiner, MD, in 1904 and is considered to be the first description of autoimmune disease being precipitated by antibodies.4 The D-L antibody test itself is uncommonly performed and somewhat difficult to interpret, particularly in adults, and may lead to false-negative results.5
PCH is an acquired, cold AIHA more common to children6,7 and may account for up to 33% of pediatric AIHA cases.8 Typical presentation is after an upper respiratory tract illness; however, the trigger is often not identified. Implicated triggers include a number of viruses.9 Clinical presentation includes findings of intravascular hemolysis similar to those in our patient. The pathogenic IgG autoantibody is polyclonal and is likely formed because of immune stimulation, which is consistent with the predominance of nonmalignant triggers of this disease process.10 Hemolysis and associated symptoms are often exacerbated with cold exposure; both typically resolve within 2 weeks. In recurrent cases, which are a minority, immunosuppression may be considered.10
PCH remains an often-understated cause of hemolytic anemia particularly in children. Lacking obvious pathognomonic clinical symptoms, it may be overlooked for other forms of AIHA or MAHA. However, with a structured approach to evaluation, as with this patient who had hematuria and jaundice, early diagnosis can prevent an unnecessarily extensive workup and can provide reassurance to patient and parents. By understanding the basic categories of hemolytic anemia, the relevant blood testing available, and interpretation of Coombs test results, clinicians can ensure that PCH is a diagnosis that is not left out in the cold.
KEY TEACHING POINTS
- Examination for schistocytes on a blood smear can help identify life-threatening causes of hemolytic anemia.
- Characterization of cold AIHA includes defining the underlying etiology as primary cold agglutinin disease, secondary cold agglutinin disease, or PCH.
- PCH is a cold AIHA that is an underrecognized cause of hemolytic anemia in children. The diagnosis of PCH is made by testing for the presence of the D-L antibody.
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
1. Dhaliwal G, Cornett PA, Tierney LM Jr. Hemolytic anemia. Am Fam Physician. 2004;69(11):2599-2606.
2. Djaldetti M. Paroxysmal cold hemoglobinuria. CRC Crit Rev Clin Lab Sci. 1978;9(1):49-83. https://doi.org/10.3109/10408367809150915
3. Levine P, Celano MJ, Falkowski F. The specificity of the antibody in paroxysmal cold hemoglobinuria (P.C.H.). Transfusion. 1963;3(4):278-280. https://doi.org/10.1111/j.1537-2995.1963.tb04643.x
4. Donath J, Landsteiner K. Uber Paroxysmale Hamoglobinurie. Munch Med Wochenschr. 1904;51:1590-1593
5. Zeller MP, Arnold DM, Al Habsi K, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. 2017;57(1):137-143. https://doi.org/10.1111/trf.13888
6. Göttsche B, Salama A, Mueller-Eckhardt C. Donath-Landsteiner autoimmune hemolytic anemia in children. a study of 22 cases. Vox Sang. 1990;58(4):281-286. https://doi.org/10.1111/j.1423-0410.1990.tb05000.x
7. Sokol RJ, Booker DJ, Stamps R. Erythropoiesis: paroxysmal cold haemoglobinuria: a clinico-pathological study of patients with a positive Donath-Landsteiner test. Hematology. 1999;4(2):137-164. https://doi.org/10.1080/10245332.1999.11746439
8. Petz LD. Cold antibody autoimmune hemolytic anemias. Blood Rev. 2008;22(1):1-15. https://doi.org/10.1016/j.blre.2007.08.002
9. Leibrandt R, Angelino K, Vizel-Schwartz M, Shapira I. Paroxysmal cold hemoglobinuria in an adult with respiratory syncytial virus. Case Rep Hematol. 2018;2018:1-3. https://doi.org/10.1155/2018/7586719
10. Gertz MA. Management of cold haemolytic syndrome. Br J Haematol. 2007;138(4):422-429. https://doi.org/10.1111/j.1365-2141.2007.06664.x
© 2020 Society of Hospital Medicine
The Right Frame
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.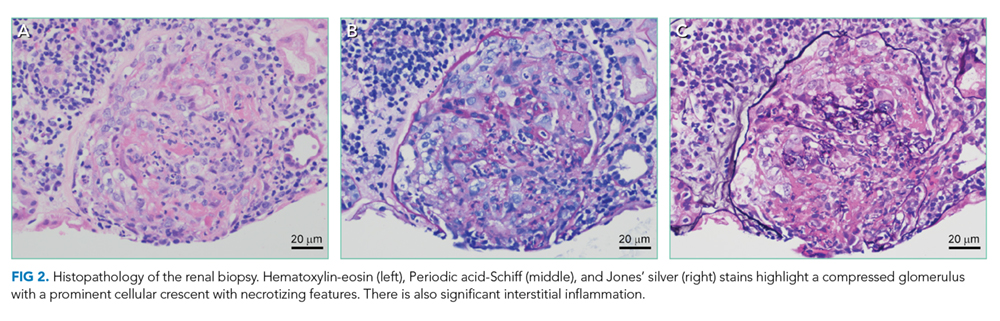
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
A 65-year-old man was transferred to a tertiary academic medical center with one week of progressive shortness of breath, dry cough, and fevers. He reported no weight loss or night sweats but had experienced mild right upper quadrant pain and anorexia for the preceding three weeks. Several years had passed since he had consulted a physician, and he did not take any medications. He immigrated to the United States from Mexico four decades prior. He traveled back frequently to visit his family, most recently one month before his presentation. He worked as a farming supervisor in the Central Valley of California. He smoked tobacco and had a 30 pack-year history. He drank alcohol occasionally and denied any drug use.
Causes of subacute cough and dyspnea include bronchitis, pneumonia, heart failure, and asthma. Pneumonia and heart failure might cause right upper quadrant pain from diaphragmatic irritation and hepatic congestion, respectively. Metastatic cancer or infection may lead to synchronous pulmonary and hepatic involvement. The patient is at increased risk of lung cancer, given his extensive smoking history.
The patient’s place of residence in the Southwestern United States places him at risk of respiratory illness from coccidioidomycosis. His exact involvement with animals and their products should be further explored. For example, consumption of unpasteurized milk might result in pneumonia, hepatitis, or both from M. bovis, Brucella species, or C. burnetii. His travel to Mexico prompts consideration of tuberculosis, histoplasmosis, and paracoccidiomycosis as causes of respiratory and possible hepatic illness.
Two weeks prior, the patient had initially presented to another hospital with one week of intermittent right upper quadrant pain unrelated to eating. An abdominal ultrasound and hepatobiliary iminodiacetic acid (HIDA) scan were normal. Computed tomography (CT) of the chest, abdomen, and pelvis with contrast demonstrated a left upper lobe lung mass measuring 5.5 × 4.4 × 3.7 cm3 and scattered right-sided pulmonary nodules (Figure 1). He underwent CT-guided biopsy of the mass and was discharged with a presumed diagnosis of primary pulmonary malignancy with plans for outpatient follow-up.
Over the next four days, the patient developed progressive dyspnea with cough and subjective fevers. The patient was readmitted with a diagnosis of postobstructive pneumonia and acute kidney injury (creatinine increased from 0.7 mg/dL to 2.9 mg/dL between admissions), and this finding was attributed to contrast-induced nephropathy from his recent CT scan. He was treated with vancomycin and piperacillin/tazobactam for two days but wished to transfer to a tertiary care hospital for a second opinion.
Postobstructive pneumonia, pulmonary embolism, and pleural effusion are common causes of dyspnea in patients with lung cancer. The patient’s travel and occupational history, lung nodules, acute renal insufficiency, and rapidly progressive respiratory symptoms prompt consideration for radiographic mimickers of lung cancer. Tuberculosis might present as a lung mass (pulmonary tuberculoma) during primary infection or reactivation. Noninfectious causes of pulmonary masses and nodules include metastatic cancer (eg, colon cancer), sarcoidosis, IgG4-related disease, and granulomatous polyangiitis (GPA).
Contrast-induced nephropathy is unusual in patients with normal renal function. More probable explanations include hypovolemia or acute tubular necrosis (ATN) from underlying inflammation. The patient’s CT-negative right upper quadrant pain may be a distinct process or represent another facet of a disseminated illness such as hepatic infiltration from lymphoma.
Upon arrival, the patient’s temperature was 38°C, heart rate (HR) 107 beats per minute, blood pressure (BP) 159/89 mm Hg, respiratory rate 25 breaths per minute, and oxygen saturation 92% on 2 L of oxygen per minute. He showed no signs of distress. Mild scleral icterus was noted. The cardiac exam was normal. Auscultation revealed scattered wheezes and crackles in the left upper lobe. Mild right upper quadrant tenderness without hepatosplenomegaly was noted on the abdominal exam. The patient’s lower extremities exhibited bilateral trace edema. No rash was observed, and his neurologic exam was normal.
The white blood cell (WBC) count was 28,300 per cubic millimeter (87% neutrophils, 3.6% lymphocytes, and 0.03% eosinophils), hemoglobin 11.1 g per deciliter, and platelet count 789,000 per cubic millimeter. Sodium was 127 mmol per liter, potassium 4.6 mmol per liter, chloride 101 mmol per liter, bicarbonate 13 mmol per liter, blood urea nitrogen 60 mg per deciliter, and creatinine 3.4 mg per deciliter. Aspartate aminotransferase and alanine aminotransferase levels were normal. Alkaline phosphatase was 283 units per liter (normal range, 31-95), and total bilirubin was 4.5 mg per deciliter (normal range, 0.2-1.3) with a direct bilirubin of 2.7 mg per deciliter. Urinalysis demonstrated urine protein of 30 mg/dL, specific gravity of 1.013, negative nitrites, 10-21 white cells per high-powered field (normal, < 5), and 21-50 red cells per high-powered field (normal, < 3). Urine microscopy revealed muddy brown casts but no cellular casts or dysmorphic red cells. A chest radiograph (CXR) showed patchy consolidations in the bilateral upper lobes and left lower lobe along with Kerley B lines, a small left pleural effusion, and thickened right horizontal fissure; the left upper lobe mass was re-demonstrated. Vancomycin, piperacillin-tazobactam, and azithromycin were administered.
At this point, the most likely source of sepsis is multifocal pneumonia. The patient is at risk for S. aureus and P. aeruginosa given his recent hospitalization. A severe form of leptospirosis (Weil’s disease) is associated with pulmonary disease, hyperbilirubinemia, and renal failure. Repeat abdominal imaging is necessary to evaluate for cholangitis given the patient’s right upper quadrant pain, fever, and jaundice. It would also help categorize his cholestatic pattern of liver injury as intrahepatic or extrahepatic (eg, stricture). An infiltrative disease such as sarcoidosis may cause both intrahepatic cholestasis and parenchymal lung disease, although the pleural pathology is uncommon.
His normal cardiac exam does not exclude cardiogenic pulmonary edema, a common cause of interstitial edema and pleural effusion. In this setting of systemic inflammation (neutrophilia, thrombocytosis, and hypoalbuminemia), the thickened right horizontal fissure and interlobular septa might represent an infiltrative process, such as lymphangitic carcinomatosis, lymphoma, or sarcoidosis.
Muddy brown casts are characteristic of ATN. The patient’s risk factors for ATN include sepsis and previously administered iodinated contrast. Fluid retention from oliguric renal failure is likely contributing to his hyponatremia and lower extremity edema. Pathology isolated to the tubules, however, would not cause hematuria and pyuria and suggests glomerular or interstitial disease. The lack of cellular casts on a single urinary specimen does not eliminate the likelihood of either disease. Hematuria and diffuse parenchymal lung disease prompt consideration of pulmonary-renal syndromes, such as anti-glomerular basement membrane disease, GPA, and systemic lupus erythematosus, which can all be triggered by infection.
On the night of transfer, the patient experienced acute respiratory distress. Heart rate was 130 beats per minute, BP 170/95 mm Hg, respiratory rate 40 breaths per minute, and oxygen saturation 88% on six liters of supplemental oxygen by nasal cannula. His arterial blood gas demonstrated a pH of 7.23, PaCO2 of 32 mm Hg, and PaO2 of 65 mm Hg. He was emergently intubated for progressive hypoxemic respiratory failure. A small amount of blood was noted in the endotracheal tube. A noncontrast CT of the chest demonstrated multifocal airspace opacities and bilateral pleural effusions. The previously noted left upper lobe mass was unchanged.
Rapid respiratory decline and diffuse alveolar disease commonly result from aspiration, flash pulmonary edema, and acute respiratory distress syndrome (ARDS). Necrotizing pneumonia (eg, S. aureus) and trauma during intubation are possible causes of blood in his endotracheal tube. However, in the setting of multifocal airspace opacity, renal insufficiency, hematuria, and rapid respiratory decline, the blood might represent diffuse alveolar hemorrhage (DAH). Bronchoscopy with bronchioalveolar lavage to evaluate for pulmonary edema, infection, and hemorrhage would be indicated.
The patient subsequently developed oliguria, requiring continuous renal replacement therapy. An echocardiogram demonstrated impaired left ventricular relaxation and a reduced ejection fraction of 45% without segmental wall motion abnormalities or valvular disease, and a right ventricular systolic pressure of 36 mm Hg. Over the next 12 hours, his respiratory status improved, and he was extubated to 15 L per minute of supplemental oxygen by high-flow nasal cannula (HFNC).
The pathology report of the lung biopsy from the other hospital disclosed chronic inflammation and fibrosis with ill-defined areas of necrosis and myxoid degeneration surrounded by nuclear palisading suggestive of granulomatous inflammation. Staining for acid-fast bacilli (AFB) and fungal organisms was negative.
The rapid pulmonary recovery is inconsistent with multifocal pneumonia or ARDS. Flash pulmonary edema might result in sudden hypoxemic respiratory failure that resolves with positive pressure ventilation and ultrafiltration. However, this condition would not explain the biopsy results. Granulomatous lung pathology often results from mycobacterial or fungal disease. Tuberculosis and fungal pneumonia are not excluded with negative staining alone. However, neither would cause self-limited respiratory failure. Histologic evidence of necrosis lessens the likelihood of sarcoidosis, which rarely causes fulminant pulmonary disease. Lymphoma can result in granulomatous inflammation but would not cause transient pulmonary disease. GPA, a cause of necrotizing granulomatous lung disease, might result in a lung mass and worsened hypoxemia through DAH.
The patient continued to require 15 L of oxygen per minute by HFNC. He had persistent bilateral perihilar alveolar and interstitial opacities on CXR. Repeat WBC count was 29,200 per cubic millimeter, hemoglobin 7.8 g per deciliter, and platelets 656,000 per cubic millimeter. The C-reactive protein was 300 mg per L (normal range, <6.3) and erythrocyte sedimentation rate 100 mm per hour (normal range, <10). Legionella urinary antigen, serum immunodiffusion for Coccidiodes imitus, human immunodeficiency virus antibody, respiratory viral panel, and beta-D glucan were negative. Rare acid-fast bacilli were visualized in one out of three concentrated AFB sputum smears. He was started on empiric antituberculous therapy with rifampin, isoniazid, pyrazinamide, and ethambutol.
The sputum sample is suggestive of pulmonary tuberculosis. The salient features of this case include systemic inflammation, pulmonary nodules and mass, necrotizing granulomatous lung pathology, renal insufficiency, and hematuria. Disseminated tuberculosis might explain all these findings. However, a positive AFB smear may signal the presence of a nontuberculous mycobacteria, which is less likely to cause this clinical syndrome.
M. tuberculosis complex polymerase chain reaction (MTB PCR) assay returned negative for M. tuberculosis. Antiproteinase 3 antibody was 1,930 units (normal range, <20). Antimyeloperoxidase and antiglomerular basement membrane antibodies were negative.
Tuberculosis and GPA share several overlapping features, such as necrotizing lung pathology and less commonly antineutrophil cytoplasmic autoantibody (ANCA)-associated antibodies. However, the lung mass, acute renal and respiratory failure, hematuria, and the degree of anti-proteinase 3 level elevation are highly suggestive of GPA. The negative MTB PCR raises the possibility that a nontuberculous mycobacterium was detected on the sputum smear. Nevertheless, continued treatment until finalization of culture results is appropriate given that tuberculosis is endemic in Mexico.
The patient’s presenting features of right upper quadrant tenderness, jaundice, and cholestatic hepatitis remain poorly explained by either of these diagnoses. Neither tuberculosis nor GPA commonly presents with accompanying hepatic involvement, though both have been occasionally described as causing hepatitis. As the greatest concern in this patient remains his progressive renal failure and accompanying pulmonary hemorrhage, a renal biopsy to assess for glomerulonephritis associated with GPA is warranted before further investigation into the cause of his cholestatic hepatitis.
A core renal biopsy demonstrated pauci-immune focal crescentic and necrotizing glomerulonephritis with mixed tubulointerstitial inflammation (Figure 2). In conjunction with the pulmonary syndrome and positive antiproteinase 3 serology, a diagnosis of granulomatosis with polyangiitis was made. The patient was treated with pulse dose steroids, rituximab, and plasma exchange. Two weeks later, the sputum mycobacterial culture returned positive for Mycobacterium llatzerense and anti-tuberculous treatment was discontinued.
Over the following weeks, the patient improved and was transitioned off dialysis prior to hospital discharge. By six months later, he had resolution of his hemoptysis, shortness of breath, liver biochemical test abnormalities, and significant improvement in his renal function. Repeat sputum mycobacterial cultures were negative.
DISCUSSION
A 65-year-old man from Mexico with a significant smoking history presented with an apical lung mass and cough, prioritizing tuberculosis and pulmonary malignancy. As the case unfolded, renal failure, multifocal lung opacities, conflicting tuberculosis test results, positive anti-proteinase 3 antibody, and ultimately a renal biopsy led to the diagnosis of granulomatosis with polyangiitis (GPA).
The correct interpretation of occasionally conflicting mycobacterial testing is crucial. Mycobacterial cultures remain the gold standard for diagnosing tuberculosis. However, results take weeks to return. Rapid tests include acid-fast bacilli (AFB) smear microscopy and nucleic acid-amplification tests (NAAT) of sputum or bronchoalveolar samples.1 When three sputum smears are performed, the sensitivity of AFB smear microscopy for tuberculosis in immunocompetent hosts is 70%.1 The AFB smear does not distinguish between different mycobacterial organisms. Thus, a positive result must be interpreted with the relative prevalence of tuberculosis and nontuberculous mycobacteria (NTM) in mind. The addition of NAAT-based assays has allowed for enhanced sensitivity and specificity in the diagnosis of tuberculosis, such that a negative NAAT in a patient with a positive AFB smear strongly argues for the presence of a NTM.2-4
NTM are widely prevalent environmental microbes, with over 140 species described, and careful consideration is required to determine if an isolate is pathogenic.5 Given their ubiquitous nature, a high rate of asymptomatic respiratory and cutaneous colonization occurs. Correspondingly, the diagnosis of NTM disease requires multiple positive cultures or pathologic features on tissue biopsy, compatible clinical findings, and diligent exclusion of other causes.5 A retrospective study of all NTM isolates in Oregon from 2005-2006 revealed that only 47% of patients met the guideline criteria for having symptomatic NTM disease.6 In our case, the patient’s sputum grew M. llatzerense, an aerobic, nonfermenting mycobacterium found in water sources that has only infrequently been implicated as a human pathogen.7,8 Subsequent AFB sputum cultures were negative, and serial imaging showed resolution of the pulmonary findings without additional antimycobacterial therapy, suggesting that this organism was not responsible for the disease process.
Along with microscopic polyangiitis (MPA) and eosinophilic granulomatosis with polyangiitis (EGPA), GPA is an antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis that predominantly affects small to medium sized vessels. Although it can occur at any age, GPA most commonly afflicts older adults, with men and women being diagnosed at roughly equal rates.9 GPA is a multisystem disease with a wide array of clinical manifestations. The most frequently involved sites of disease are the respiratory tract and kidneys, although virtually any organ can be affected. Sino-nasal disease, such as destructive sinusitis, or ear involvement are nearly universal. Lower respiratory manifestations occur in 60% of patients, but are highly diverse and reflect the inherent difficulty in diagnosing this condition.9-11 Additionally, GPA is a frequent cause of the pulmonary-renal syndromes, with glomerulonephritis occurring in 80% of patients.9
The diagnosis of GPA in this case was delayed, in part, due to features suggestive of malignancy and pulmonary tuberculosis. While sino-nasal disease was not noted during this hospitalization, the patient had many different respiratory manifestations, including a dominant pulmonary mass, diffuse nodules, and hypoxemic respiratory failure due to suspected diffuse alveolar hemorrhage (DAH), all of which have been reported in GPA.12 Dysmorphic red cells and red blood cell casts are not sensitive for renal involvement in GPA; their absence does not exclude the possibility of an ANCA-associated vasculitis.13 Hematuria and rapid progression to oliguric renal failure are characteristic of a vasculitic process and should sway clinicians away from a working diagnosis of ATN.
The diagnosis of GPA involves the synthesis of clinical data, radiographic findings, serologic testing, and histopathology. ANCA testing is an essential step in the diagnosis of GPA but has limitations. Patients with GPA more commonly have ANCAs targeting the enzyme proteinase-3 (PR3-ANCA), with MPA being more closely associated with myeloperoxidase (MPO-ANCA), although cross-reactivity and antibody-negative disease can occur.14 Although 90% of patients with GPA with multiorgan involvement will have a positive ANCA, a negative test is more common in localized upper airway disease, where only 50% have a positive ANCA.15 A number of drugs, medications, infections, and nonvasculitic autoimmune diseases have been associated with positive ANCA serologies in the absence of systemic vasculitis.14,16,17 As such, pathologic demonstration of vasculitis is necessary for establishing the diagnosis. Typical sites for biopsy include the kidneys and lungs.9
This case illustrates how clinicians often find themselves at a diagnostic crossroads—being forced to choose which clinical elements to prioritize. At various points, our patient’s presentation could have been framed as “a man from a Tb-endemic country with hemoptysis and an apical opacity,” “an elderly man with extensive smoking history and lung mass,” or “a patient with elevated inflammatory markers and pulmonary-renal syndrome”. In such situations, it is incumbent on the clinician to evaluate how well a given problem representation encompasses or highlights the salient features of a case. As with painting or photography, an essential aspect of appreciating the whole picture involves carefully selecting the right frame.
KEY TEACHING POINTS
- The diagnosis of tuberculosis relies on smear microscopy, nucleic-acid amplification testing (NAAT), and cultures. A positive AFB smear with negative NAAT suggests the presence of a nontuberculous mycobacteria (NTM).
- NTM are common environmental organisms and often exist as nonpathogenic colonizers.6 The diagnosis of NTM disease requires exclusion of other causes and careful clinical, microbiologic, and radiographic correlation.
- Granulomatosis with polyangiitis is a multisystem disease often involving the respiratory track and kidney. Pulmonary disease can present with airway involvement, parenchymal nodules, opacities, pleural findings, and diffuse alveolar hemorrhage.12
Disclosures
Drs. Minter, Geha, Boslett, Chung, and Ramani have no disclosures. Dr. Manesh is supported by the Jeremiah A. Barondess Fellowship in the Clinical Transaction of the New York Academy of Medicine, in collaboration with the Accreditation Council for Graduate Medical Education (ACGME).
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
1. Lewinsohn DM, Leonard MK, LoBue PA, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention clinical practice guidelines: diagnosis of tuberculosis in adults and children. Clin Infect Dis. 2017;64(2):e1-e33. PubMed
2. Steingart KR, Sohn H, Schiller I, et al. Xpert(R) MTB/RIF assay for pulmonary tuberculosis and rifampicin resistance in adults. Cochrane Database Syst Rev. 2013;(1):Cd009593. PubMed
3. Luetkemeyer AF, Firnhaber C, Kendall MA, et al. Evaluation of Xpert MTB/RIF versus afb smear and culture to identify pulmonary tuberculosis in patients with suspected tuberculosis from low and higher prevalence settings. Clin Infect Dis. 2016;62(9):1081-1088. PubMed
4. Boehme CC, Nabeta P, Hillemann D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363(11):1005-1015. PubMed
5. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367-416. PubMed
6. Winthrop KL, McNelley E, Kendall B, et al. Pulmonary nontuberculous mycobacterial disease prevalence and clinical features: an emerging public health disease. Am J Respir Crit Care Med. 2010;182(7):977-982. PubMed
7. Teixeira L, Avery RK, Iseman M, et al. Mycobacterium llatzerense lung infection in a liver transplant recipient: case report and review of the literature. Am J Transplant. 2013;13(8):2198-2200. PubMed
8. Cárdenas AM, Gomila M, Lalucat J, Edelstein PH. Abdominal abscess caused by Mycobacterium llatzerense. J Clin Microbiol. 2014;52(4):1287-1289. PubMed
9. Jennette JC, Falk RJ. Small-vessel vasculitis. N Engl J Med. 1997;337(21):1512-1523. PubMed
10. Mahr A, Katsahian S, Varet H, et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: a cluster analysis. Ann Rheum Dis. 2013;72(6):1003-1010. PubMed
11. Holle JU, Gross WL, Latza U, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis Rheum. 2011;63(1):257-266. PubMed
12. Cordier JF, Valeyre D, Guillevin L, Loire R, Brechot JM. Pulmonary Wegener’s granulomatosis. A clinical and imaging study of 77 cases. Chest. 1990;97(4):906-912. PubMed
13. Hamadah AM, Gharaibeh K, Mara KC, et al. Urinalysis for the diagnosis of glomerulonephritis: role of dysmorphic red blood cells. Nephrol Dial Transplant. 2018;33(8):1397-1403. PubMed
14. Jennette JC, Falk RJ. Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol. 2014;10(8):463-473. PubMed
15. Borner U, Landis BN, Banz Y, et al. Diagnostic value of biopsies in identifying cytoplasmic antineutrophil cytoplasmic antibody-negative localized Wegener’s granulomatosis presenting primarily with sinonasal disease. Am J Rhinol Allergy. 2012;26(6):475-480. PubMed
16. Mahr A, Batteux F, Tubiana S, et al. Brief report: prevalence of antineutrophil cytoplasmic antibodies in infective endocarditis. Arthritis Rheumatol. 2014;66(6):1672-1677. PubMed
17. Sherkat R, Mostafavizadeh K, Zeydabadi L, Shoaei P, Rostami S. Antineutrophil cytoplasmic antibodies in patients with pulmonary tuberculosis. Iran J Immunol. 2011;8(1):52-57. PubMed
© 2019 Society of Hospital Medicine